(EU) 2021/808Prováděcí nařízení Komise (EU) 2021/808 ze dne 22. března 2021 o provádění analytických metod pro rezidua farmakologicky účinných látek používaných u zvířat určených k produkci potravin a o interpretaci výsledků, jakož i o metodách, které se mají používat pro odběr vzorků, a o zrušení rozhodnutí 2002/657/ES a 98/179/ES (Text s významem pro EHP)
| Publikováno: | Úř. věst. L 180, 21.5.2021, s. 84-109 | Druh předpisu: | Prováděcí nařízení |
| Přijato: | 22. března 2021 | Autor předpisu: | Evropská komise |
| Platnost od: | 10. června 2021 | Nabývá účinnosti: | 10. června 2021 |
| Platnost předpisu: | Ano | Pozbývá platnosti: | |
Text předpisu s celou hlavičkou je dostupný pouze pro registrované uživatele.
|
21.5.2021 |
CS |
Úřední věstník Evropské unie |
L 180/84 |
PROVÁDĚCÍ NAŘÍZENÍ KOMISE (EU) 2021/808
ze dne 22. března 2021
o provádění analytických metod pro rezidua farmakologicky účinných látek používaných u zvířat určených k produkci potravin a o interpretaci výsledků, jakož i o metodách, které se mají používat pro odběr vzorků, a o zrušení rozhodnutí 2002/657/ES a 98/179/ES
(Text s významem pro EHP)
EVROPSKÁ KOMISE,
s ohledem na Smlouvu o fungování Evropské unie,
s ohledem na nařízení Evropského parlamentu a Rady (EU) 2017/625 ze dne 15. března 2017 o úředních kontrolách a jiných úředních činnostech prováděných s cílem zajistit uplatňování potravinového a krmivového práva a pravidel týkajících se zdraví zvířat a dobrých životních podmínek zvířat, zdraví rostlin a přípravků na ochranu rostlin, o změně nařízení Evropského parlamentu a Rady (ES) č. 999/2001, (ES) č. 396/2005, (ES) č. 1069/2009, (ES) č. 1107/2009, (EU) č. 1151/2012, (EU) č. 652/2014, (EU) 2016/429 a (EU) 2016/2031, nařízení Rady (ES) č. 1/2005 a (ES) č. 1099/2009 a směrnic Rady 98/58/ES, 1999/74/ES, 2007/43/ES, 2008/119/ES a 2008/120/ES a o zrušení nařízení Evropského parlamentu a Rady (ES) č. 854/2004 a (ES) č. 882/2004, směrnic Rady 89/608/EHS, 89/662/EHS, 90/425/EHS, 91/496/EHS, 96/23/ES, 96/93/ES a 97/78/ES a rozhodnutí Rady 92/438/EHS (nařízení o úředních kontrolách) (1), a zejména na čl. 34 odst. 6 uvedeného nařízení,
vzhledem k těmto důvodům:
|
(1) |
Nařízení (EU) 2017/625 stanoví pravidla pro provádění úředních kontrol a jiných úředních činností příslušnými orgány členských států ve všech fázích produkce, zpracování a distribuce za účelem ověření souladu s právními předpisy Unie mimo jiné v oblasti bezpečnosti potravin. Poskytuje konkrétní pravidla pro úřední kontroly v souvislosti s látkami, jejichž používání může vést k přítomnosti reziduí v potravinách a krmivu, a stanoví obecné požadavky pro metody, které se mají používat pro odběr vzorků, laboratorní analýzy a zkoušky během úředních kontrol a jiných úředních činností. |
|
(2) |
Rozhodnutí Komise 2002/657/ES (2) stanoví požadavky na provádění analytických metod a interpretaci výsledků analýz určitých látek a jejich reziduí v živých zvířatech a živočišných produktech a rozhodnutí Komise 98/179/ES (3) stanoví prováděcí pravidla k úřednímu odběru vzorků pro zjišťování některých látek a jejich reziduí v živých zvířatech a živočišných produktech. Obě rozhodnutí byla přijata na základě směrnice Rady 96/23/ES (4), která byla zrušena nařízením (EU) 2017/625. Ve světle nového vědeckého vývoje by se měla tato pravidla aktualizovat a měla by se začlenit do rámce pro úřední kontroly definovaného nařízením (EU) 2017/625. |
|
(3) |
V souladu s čl. 1 odst. 2 rozhodnutí 2002/657/ES se uvedené rozhodnutí nevztahuje na látky, pro něž jsou v jiných předpisech Unie stanovena zvláštní pravidla. Těmito látkami jsou mykotoxiny v potravinách, dioxiny a polychlorované bifenyly (PCB) s dioxinovým efektem v potravinách a olovo, kadmium, rtuť a benzo[a]pyren v potravinách. Mykotoxiny v potravinách musí splňovat požadavky stanovené nařízením Komise (ES) č. 401/2006 (5), kterým se stanoví metody odběru vzorků a metody analýzy pro úřední kontrolu množství mykotoxinů v potravinách. V případě dioxinů a PCB s dioxinovým efektem se uplatňuje nařízení Komise (EU) 2017/644 (6), kterým se stanoví metody odběru vzorků a analýzy pro kontrolu obsahu dioxinů, PCB s dioxinovým efektem a PCB bez dioxinového efektu v některých potravinách. Ustanovení pro odběr vzorků a analýzu pro úřední kontroly obsahu olova, kadmia, rtuti a benzo[a]pyrenu v potravinách jsou stanovena v nařízení Komise (ES) č. 333/2007 (7). |
|
(4) |
Z důvodů jasnosti a právní jistoty je vhodné sloučit ustanovení týkající se odběru vzorků a analýzy farmakologicky účinných látek do jednoho právního aktu, jak je tomu v případě mykotoxinů, dioxinů, PCB s dioxinovým efektem, olova, kadmia, rtuti a benzo[a]pyrenu v potravinách. |
|
(5) |
Rozhodnutí 98/179/ES a 2002/657/ES by proto měla být zrušena a nahrazena tímto nařízením. |
|
(6) |
V souladu s nařízením Evropského parlamentu a Rady (ES) č. 1831/2003 (8) lze kokcidiostatika a histomonostatika použít jako doplňkové látky v krmivech, a proto se na analýzy jejich obsahu v krmivech vztahuje nařízení Komise (ES) č. 152/2009 (9), kterým se stanoví metody odběru vzorků a laboratorního zkoušení pro úřední kontrolu krmiv. Toto nařízení by se však mělo uplatňovat v případech, kdy se krmivo analyzuje jako součást následných opatření při vyšetřování zdroje vzorků, které nejsou vyhovující v případech podezření na nesoulad nebo zjištěného nesouladu s pravidly Unie pro používání nebo rezidua farmakologicky účinných látek, které jsou povoleny ve veterinárních léčivých přípravcích nebo jako doplňkové látky, nebo s pravidly Unie pro používání nebo rezidua zakázaných nebo nepovolených farmakologicky účinných látek. |
|
(7) |
Aby se zajistila kontinuita provádění úředních kontrol a jiných úředních činností týkajících se reziduí farmakologicky účinných látek a aby se zamezilo nutnosti opětovně validovat všechny metody naráz, mohou se metody, které byly validovány před datem vstupu tohoto nařízení v platnost, nadále používat po omezenou dobu pod podmínkou splnění požadavků uvedených v bodech 2 a 3 přílohy I rozhodnutí 2002/657/ES. Je proto vhodné poskytnout členským státům dostatek času na uplatnění požadavků stanovených tímto nařízením na všechny analytické metody. |
|
(8) |
Opatření stanovená tímto nařízením jsou v souladu se stanoviskem Stálého výboru pro rostliny, zvířata, potraviny a krmiva, |
PŘIJALA TOTO NAŘÍZENÍ:
Článek 1
Předmět a oblast působnosti
Toto nařízení stanoví pravidla týkající se analytických metod používaných pro odběr vzorků a laboratorní analýzy v souvislosti s rezidui farmakologicky účinných látek v živých zvířatech určených k produkci potravin, jejich tělesných částech a tekutinách, exkrementech, tkáních, produktech živočišného původu, vedlejších produktech živočišného původu, krmivu a vodě. Rovněž stanoví pravidla pro interpretaci analytických výsledků těchto laboratorních analýz.
Toto nařízení se vztahuje na úřední kontroly, jejichž cílem je ověřit soulad s požadavky na přítomnost reziduí farmakologicky účinných látek.
Článek 2
Definice
Pro účely tohoto nařízení se použijí definice uvedené v článku 2 nařízení Komise v přenesené pravomoci (EU) 2019/2090 (10), v nařízení Komise (EU) 2019/1871 (11), v článku 2 nařízení Evropského parlamentu a Rady (ES) č. 470/2009 (12) a v nařízení Rady (EHS) č. 315/93 (13).
Použijí se rovněž tyto definice:
|
1) |
„absolutní výtěžností“ se rozumí výtěžek analytu ze závěrečné fáze analytického procesu vydělený množstvím analytu v původním vzorku a vyjadřuje se v procentech; |
|
2) |
„přesností“ se rozumí těsnost shody mezi výsledkem zkoušky a přijatou skutečnou referenční hodnotou stanovenou odhadem pravdivosti a preciznosti (14); |
|
3) |
„chybou alfa (α)“ se rozumí pravděpodobnost, že zkoušený vzorek je vyhovující, ačkoli byla naměřena nevyhovující hodnota; |
|
4) |
„analytem“ se rozumí složka systému, jež se má analyzovat; |
|
5) |
„povolenou látkou“ se rozumí farmakologicky účinná látka povolená k použití u zvířat určených k produkci potravin v souladu se směrnicí Evropského parlamentu a Rady 2001/82/ES (15); |
|
6) |
„chybou beta (β)“ se rozumí pravděpodobnost, že zkoušený vzorek je ve skutečnosti nevyhovující, ačkoli byla naměřena vyhovující hodnota; |
|
7) |
„systematickou chybou (vychýlením)“ se rozumí rozdíl mezi odhadovanou hodnotou výsledku zkoušky a přijatou referenční hodnotou; |
|
8) |
„kalibračním standardem“ se rozumí sledovatelná reference pro měření, která reprezentuje určité množství sledované látky navázané na referenční stav; |
|
9) |
„certifikovaným referenčním materiálem“ (CRM) se rozumí referenční materiál doprovázený dokumentem vydaným pověřeným subjektem a poskytující jednu nebo více specifikovaných hodnot vlastností s přidruženými nejistotami a návaznostmi s použitím platných postupů (16); |
|
10) |
„simultánní chromatografií“ se rozumí technika, ve které se neznámá látka nanese na chromatografický nosič spolu s jednou nebo více známými sloučeninami v očekávání, že relativní chování neznámé látky a známých látek napomůže identifikaci neznámé látky; |
|
11) |
„kolaborativní studií“ se rozumí analyzování vzorku (vzorků) za použití stejné metody ke stanovení pracovních charakteristik metody v různých laboratořích, kde tato studie umožňuje vypočítat náhodnou chybu měření a laboratorní systematickou chybu pro používanou metodu; |
|
12) |
„konfirmační metodou“ se rozumí metoda, která poskytuje úplné nebo doplňující informace umožňující jednoznačnou identifikaci látky a podle potřeby její kvantifikaci jedním z těchto způsobů:
|
|
13) |
„koeficientem rozšíření (k)“ se rozumí číslo, které vyjadřuje požadovanou úroveň spolehlivosti a které je spojeno s rozšířenou nejistotou měření; |
|
14) |
„limitem rozhodnutí pro konfirmaci (CCα)“ se rozumí limitní hodnota, při jejímž dosažení nebo překročení lze s pravděpodobností chyby α usuzovat, že uvedený vzorek je nevyhovující, a hodnota 1 – α znamená statistickou jistotu v procentech, že byla překročena přípustná hodnota; |
|
15) |
„detekční schopností (CCβ)“ se rozumí nejmenší množství analytu, které může být s pravděpodobností chyby β ve vzorku detekováno nebo kvantifikováno:
|
|
16) |
„uměle obohaceným vzorkem“ se rozumí vzorek obohacený o známé množství analytu, který má být detekován nebo kvantifikován; |
|
17) |
„mezilaboratorní studií“ se rozumí zorganizování, provedení a vyhodnocení zkoušek na tomtéž vzorku (vzorcích) dvěma či více laboratořemi za předem stanovených podmínek s cílem vyhodnotit provádění zkoušky buď formou kolaborativní studie, nebo zkoušky odborné způsobilosti; |
|
18) |
„vnitřním standardem (IS)“ se rozumí látka, která není obsažena ve vzorku a která má fyzikálně-chemické vlastnosti co možná nejvíce podobné vlastnostem analytu, který má být identifikován nebo kvantifikován; |
|
19) |
„sledovanou úrovní“ se rozumí koncentrace látky nebo analytu ve vzorku, která je významná pro rozhodování o tom, zda vzorek splňuje požadavky právních předpisů, pokud jde o:
|
|
20) |
„nejnižší kalibrovanou úrovní“ (LCL) se rozumí nejnižší koncentrace, na které byl systém měření kalibrován; |
|
21) |
„matricí“ se rozumí materiál, ze kterého je vzorek odebrán; |
|
22) |
„matricovým efektem“ se rozumí rozdíl v analytické odezvě mezi standardem rozpuštěným v rozpouštědle a standardem s odpovídající matricí buď bez korekce za použití vnitřního standardu, nebo s korekcí za použití vnitřního standardu; |
|
23) |
„standardem s odpovídající matricí“ se rozumí slepá matrice (tj. matrice neobsahující analyt), ke které se po zpracování vzorku přidá analyt v určitém rozsahu koncentrací; |
|
24) |
„standardem s obohacenou matricí“ se rozumí slepá matrice (tj. matrice neobsahující analyt), ke které se před extrakcí rozpouštědlem a zpracováním vzorku přidá analyt v určitém rozsahu koncentrací; |
|
25) |
„měřenou veličinou“ se rozumí konkrétní množství, které je předmětem měření; |
|
26) |
„nejistotou měření“ se rozumí nezáporný parametr spojený s výsledkem měření, který charakterizuje rozptyl hodnot, který lze rozumně přisoudit měřené veličině na základě použitých informací; |
|
27) |
„pracovními kritérii“ se rozumí požadavky na pracovní charakteristiky, podle nichž lze rozhodnout, zda je analytická metoda vhodná k zamýšlenému použití a zda poskytuje spolehlivé výsledky; |
|
28) |
„precizností“ se rozumí těsnost shody mezi nezávislými výsledky zkoušek získanými za stanovených podmínek a vyjadřuje se jako směrodatná odchylka nebo variační koeficient výsledků zkoušky; |
|
29) |
„kvalitativní metodou“ se rozumí analytická metoda, kterou se detekuje nebo identifikuje látka nebo skupina látek na základě jejích chemických, biologických nebo fyzikálních vlastností; |
|
30) |
„kvantitativní metodou“ se rozumí analytická metoda, kterou se stanoví množství nebo hmotnostní podíl látky takovým způsobem, že je možné jej vyjádřit jako číselnou hodnotu příslušných jednotek; |
|
31) |
„výtěžností“ se rozumí množství analytu korigované na výtěžnost vydělené množstvím analytu, kterým byl uměle obohacen vzorek matrice, a vyjadřuje se v procentech; |
|
32) |
„korekcí na výtěžnost“ se rozumí použití vnitřních standardů, použití kalibrační křivky matrice a rovněž použití korekčního faktoru výtěžnosti a rovněž kombinace těchto přístupů; |
|
33) |
„referenčním materiálem“ se rozumí materiál dostatečně homogenní a stabilní s ohledem na jednu nebo více specifikovaných vlastností, u něhož bylo prokázáno, že je vhodný pro své zamýšlené použití v procesu měření nebo při zkoumání jmenovitých vlastností (19); |
|
34) |
„relativním matricovým efektem“ se rozumí rozdíl v analytické odezvě mezi standardem rozpuštěným v rozpouštědle a standardem s odpovídající matricí s korekcí za použití vnitřního standardu; |
|
35) |
„opakovatelností“ se rozumí preciznost za podmínek, za nichž jsou nezávislé výsledky zkoušky získány toutéž metodou, na téže zkoušené položce, v téže laboratoři tímtéž pracovníkem při použití téhož zařízení v krátkých časových intervalech; |
|
36) |
„reprodukovatelností“ se rozumí preciznost za podmínek, za nichž jsou výsledky zkoušky získány toutéž metodou, na téže zkoušené položce v různých laboratořích různými pracovníky při použití různého zařízení (20); |
|
37) |
„robustností“ se rozumí citlivost analytické metody na změny experimentálních podmínek, za nichž lze metodu použít tak, jak je popsána, nebo se specifikovanými malými úpravami; |
|
38) |
„screeningovou metodou“ se rozumí metoda sloužící ke screeningu látky nebo třídy látek na sledované úrovni; |
|
39) |
„cílovou koncentrací screeningu (STC)“ se rozumí koncentrace nižší než nebo rovnající se CCβ, při níž screeningové měření vede ke kategorizaci vzorku jako potenciálně nevyhovující „pozitivní screening“ a vyvolává nutnost provést konfirmační zkoušku; |
|
40) |
„selektivitou“ se rozumí schopnost metody rozlišit mezi měřeným analytem a jinými látkami; |
|
41) |
„vnitrolaboratorní studií“ nebo „vnitrolaboratorní validací“ se rozumí analytická studie prováděná po určitou odůvodněnou dobu v rámci jediné laboratoře s použitím jedné metody analýzy na tomtéž zkušebním materiálu nebo na různých zkušebních materiálech za různých podmínek; |
|
42) |
„standardním přídavkem“ se rozumí postup, při němž se jedna část vzorku analyzuje jako taková a k dalším zkušebním podílům se před analýzou přidá známé množství standardu daného analytu; |
|
43) |
„standardem analytu“ se rozumí analyt se známým a certifikovaným obsahem a čistotou určený k použití jako referenční látka při analýze; |
|
44) |
„látkou“ se rozumí hmota neměnného složení charakterizovaná entitami, které ji tvoří, a určitými fyzikálními vlastnostmi; |
|
45) |
„zkušebním podílem“ se rozumí množství materiálu odebrané ze vzorku a určené ke zkoušení nebo pozorování; |
|
46) |
„pravdivostí“ se rozumí těsnost shody mezi průměrnou hodnotou získanou z velkého počtu výsledků zkoušek s přijatou referenční hodnotou; |
|
47) |
„jednotkami“ se rozumí jednotky popsané v normě ISO 80000 (21) a ve směrnici Rady 80/181/EHS (22); |
|
48) |
„validací“ se rozumí proces, při němž se zkoumáním a následným poskytnutím objektivního důkazu prokáže, že jsou splněny specifikované požadavky konkrétního zamýšleného použití (23), prostřednictvím vnitrolaboratorní studie nebo kolaborativní studie; |
|
49) |
„vnitrolaboratorní reprodukovatelností“ nebo „mezilehlou precizností/vnitrolaboratorní reprodukovatelností“ se rozumí preciznost měření za souboru vnitrolaboratorních podmínek v konkrétní laboratoři. |
Článek 3
Metody analýzy
Členské státy zajistí, aby byly vzorky odebírány v souladu s článkem 34 nařízení (EU) 2017/625 a analyzovány za použití metod, které splňují tyto požadavky:
|
1) |
jsou zdokumentovány v pokynech pro zkoušku, přednostně podle příloh normy ISO 78-2:1999 Chemie – Návrhy norem – Část 2: Metody chemického rozboru (24); |
|
2) |
splňují pracovní kritéria a další požadavky pro analytické metody stanovené v kapitole 1 přílohy I tohoto nařízení; |
|
3) |
byly validovány v souladu s požadavky stanovenými v kapitole 2 a 4 přílohy I tohoto nařízení; |
|
4) |
umožňují prosazování referenčních hodnot pro opatření stanovených v nařízení (EU) 2019/1871, identifikaci přítomnosti zakázaných a nepovolených látek a prosazování maximálních limitů, které byly stanoveny na základě nařízení (EHS) č. 315/93 a nařízení (ES) č. 124/2009, a maximálních limitů reziduí (MLR), které byly stanoveny na základě nařízení (ES) č. 1831/2003 a (ES) č. 470/2009. |
Článek 4
Kontrola kvality
Členské státy zajistí kvalitu výsledků prováděných analýz podle nařízení (EU) 2017/625, zejména monitorováním zkoušek nebo výsledků kalibrace v souladu s normou ISO/IEC 17025:2017 Všeobecné požadavky na kompetenci zkušebních a kalibračních laboratoří a s požadavky pro kontrolu kvality během rutinní analýzy, jak jsou stanoveny v kapitole 3 přílohy I tohoto nařízení.
Článek 5
Interpretace výsledků
1) Výsledek analýzy se považuje za nevyhovující, jestliže se rovná limitu rozhodnutí pro konfirmaci (CCα) nebo je vyšší.
2) U povolených látek, pro které byla stanovena hodnota MLR nebo maximální limit, je limitem rozhodnutí pro konfirmaci (CCα) koncentrace, při jejímž dosažení nebo převýšení lze rozhodnout se statistickou jistotou číselné hodnoty 1 – α, že byla překročena přípustná hodnota.
3) U nepovolených nebo zakázaných látek nebo u povolených látek, pro které nebyla stanovena hodnota MLR ani maximální limit u konkrétního druhu nebo produktu, je limitem rozhodnutí pro konfirmaci (CCα) nejnižší úroveň koncentrace, při níž lze rozhodnout se statistickou jistotou číselné hodnoty 1 – α, že je přítomen konkrétní analyt.
4) U nepovolených nebo zakázaných farmakologicky účinných látek má být chyba α 1 % nebo nižší. V případě všech ostatních látek musí být chyba α nejvýše 5 %.
Článek 6
Metody odběru vzorků
Členské státy zajistí, aby se vzorky odebíraly, manipulovalo se s nimi a byly označovány v souladu s podrobnými metodami pro odběr vzorků stanovenými v příloze II tohoto nařízení.
Článek 7
Zrušení a přechodná opatření
Rozhodnutí 2002/657/ES a 98/179/ES se zrušují od data vstupu tohoto nařízení v platnost.
Nicméně do 10. června 2026 se v případě metod, které byly validovány před datem vstupu tohoto nařízení v platnost, nadále uplatňují požadavky stanovené v bodech 2 a 3 přílohy I rozhodnutí 2002/657/ES.
Článek 8
Vstup v platnost
Toto nařízení vstupuje v platnost dvacátým dnem po vyhlášení v Úředním věstníku Evropské unie.
Toto nařízení je závazné v celém rozsahu a přímo použitelné ve všech členských státech.
V Bruselu dne 22. března 2021.
Za Komisi
předsedkyně
Ursula VON DER LEYEN
(1) Úř. věst. L 95, 7.4.2017, s. 1.
(2) Rozhodnutí Komise 2002/657/ES ze dne 14. srpna 2002, kterým se provádí směrnice Rady 96/23/ES, pokud jde o provádění analytických metod a interpretaci výsledků (Úř. věst. L 221, 17.8.2002, s. 8).
(3) Rozhodnutí Komise 98/179/ES ze dne 23. února 1998, kterým se stanoví prováděcí pravidla k úřednímu odběru vzorků pro zjišťování některých látek a jejich reziduí v živých zvířatech a živočišných produktech (Úř. věst. L 65, 5.3.1998, s. 31).
(4) Směrnice Rady 96/23/ES ze dne 29. dubna 1996 o kontrolních opatřeních u některých látek a jejich reziduí v živých zvířatech a živočišných produktech a o zrušení směrnic 85/358/EHS a 86/469/EHS a rozhodnutí 89/187/EHS a 91/664/EHS (Úř. věst. L 125, 23.5.1996, s. 10).
(5) Nařízení Komise (ES) č. 401/2006 ze dne 23. února 2006, kterým se stanoví metody odběru vzorků a metody analýzy pro úřední kontrolu množství mykotoxinů v potravinách (Úř. věst. L 70, 9.3.2006, s. 12).
(6) Nařízení Komise (EU) 2017/644 ze dne 5. dubna 2017, kterým se stanoví metody odběru vzorků a analýzy pro kontrolu obsahu dioxinů, PCB s dioxinovým efektem a PCB bez dioxinového efektu v některých potravinách a kterým se zrušuje nařízení (EU) č. 589/2014 (Úř. věst. L 92, 6.4.2017, s. 9).
(7) Nařízení Komise (ES) č. 333/2007 ze dne 28. března 2007, kterým se stanoví metody odběru vzorků a metody analýzy pro kontrolu obsahu stopových prvků a kontaminujících látek z výroby v potravinách (Úř. věst. L 88, 29.3.2007, s. 29).
(8) Nařízení Evropského parlamentu a Rady (ES) č. 1831/2003 ze dne 22. září 2003 o doplňkových látkách používaných ve výživě zvířat (Úř. věst. L 268, 18.10.2003, s. 29).
(9) Nařízení Komise (ES) č. 152/2009 ze dne 27. ledna 2009, kterým se stanoví metody odběru vzorků a laboratorního zkoušení pro úřední kontrolu krmiv (Úř. věst. L 54, 26.2.2009, s. 1).
(10) Nařízení Komise v přenesené pravomoci (EU) 2019/2090 ze dne 19. června 2019, kterým se doplňuje nařízení Evropského parlamentu a Rady (EU) 2017/625, pokud jde o případy podezření na nesoulad nebo zjištěného nesouladu s pravidly Unie pro používání nebo rezidua farmakologicky účinných látek, které jsou povoleny ve veterinárních léčivých přípravcích nebo jako doplňkové látky, nebo s pravidly Unie pro používání nebo rezidua zakázaných nebo nepovolených farmakologicky účinných látek (Úř. věst. L 317, 9.12.2019, s. 28).
(11) Nařízení Komise (EU) 2019/1871 ze dne 7. listopadu 2019 o referenčních hodnotách pro opatření pro nepovolené farmakologicky účinné látky přítomné v potravinách živočišného původu a o zrušení rozhodnutí 2005/34/ES (Úř. věst. L 289, 8.11.2019, s. 41).
(12) Nařízení Evropského parlamentu a Rady (ES) č. 470/2009 ze dne 6. května 2009, kterým se stanoví postupy Společenství pro stanovení limitů reziduí farmakologicky účinných látek v potravinách živočišného původu, kterým se zrušuje nařízení Rady (EHS) č. 2377/90 a kterým se mění směrnice Evropského parlamentu a Rady 2001/82/ES a nařízení Evropského parlamentu a Rady (ES) č. 726/2004 (Úř. věst. L 152, 16.6.2009, s. 11).
(13) Nařízení Rady (EHS) č. 315/93 ze dne 8. února 1993, kterým se stanoví postupy Společenství pro kontrolu kontaminujících látek v potravinách (Úř. věst. L 37, 13.2.1993, s. 1).
(14) ISO 3534-1: 2006 Statistika – Slovník a značky – Část 1: Obecné statistické termíny a termíny používané v pravděpodobnosti (kapitola 1).
(15) Směrnice Evropského parlamentu a Rady 2001/82/ES ze dne 6. listopadu 2001 o kodexu Společenství týkajícím se veterinárních léčivých přípravků (Úř. věst. L 311, 28.11.2001, s. 1).
(16) JCGM 200:2008, Mezinárodní slovník metrologie – Základní a obecné koncepty a související pojmy (VIM), třetí vydání 2008: https://www.iso.org/sites/JCGM/VIM-JCGM200.htm (Kapitola 5: Standardy měření (Etalony)).
(17) Nařízení Komise (ES) č. 124/2009 ze dne 10. února 2009, kterým se stanoví maximální limity pro přítomnost kokcidiostatik nebo histomonostatik v potravinách, jež je důsledkem nevyhnutelné křížové kontaminace necílových krmiv těmito látkami (Úř. věst. L 40, 11.2.2009, s. 7).
(18) Nařízení Komise (EU) č. 37/2010 ze dne 22. prosince 2009 o farmakologicky účinných látkách a jejich klasifikaci podle maximálních limitů reziduí v potravinách živočišného původu (Úř. věst. L 15, 20.1.2010, s. 1).
(19) Komise pro Codex Alimentarius, organizace OSN pro výživu a zemědělství/Světová zdravotnická organizace, Pokyny pro analytickou terminologii (CAC/GL 72-2009).
(20) ISO 5725-1:1994 Přesnost (správnost a shodnost) metod a výsledků měření – Část 1: Obecné zásady a definice (kapitola 3).
(21) ISO 80000-1:2009 Veličiny a jednotky – Část 1: Obecně (Úvod).
(22) Směrnice Rady 80/181/EHS ze dne 20. prosince 1979 o sbližování právních předpisů členských států týkajících se jednotek měření a o zrušení směrnice 71/354/EHS (Úř. věst. L 39, 15.2.1980, s. 40).
(23) ISO/IEC 17025:2017 Všeobecné požadavky na kompetenci zkušebních a kalibračních laboratoří (kapitola 3).
(24) ISO 78-2:1999 Chemie – Návrhy norem – Část 2: Metody chemického rozboru (přílohy).
PŘÍLOHA I
KAPITOLA 1
PRACOVNÍ KRITÉRIA A JINÉ POŽADAVKY NA ANALYTICKÉ METODY
1.1. Požadavky na screeningové metody
1.1.1. Kategorie vhodných screeningových metod
Jako vhodné screeningové metody se mají použít kvalitativní, semikvantitativní nebo kvantitativní metody.
1.1.2. Požadavky na biologické, biochemické nebo fyzikálně chemické screeningové metody
U zakázaných nebo nepovolených látek má být hodnota CCβ na co nejnižší rozumně dosažitelné úrovni a v každém případě nižší než referenční hodnota pro opatření u látek, pro něž jsou referenční hodnoty pro opatření stanoveny podle nařízení (EU) 2019/1871.
U povolených farmakologicky účinných látek má být hodnota CCβ nižší než MLR nebo maximální limit.
Pro účely screeningu se mají použít pouze ty analytické metody, u nichž lze zpětně v dokumentech prokázat, že jsou validovány a že je u nich pravděpodobnost falešně vyhovujícího výsledku menší než nebo rovna 5 % (chyba β). Při podezření na nevyhovující výsledek se má tento výsledek potvrdit konfirmační metodou.
Kvantitativní screeningové metody používané ke screeningu i konfirmaci mají splňovat stejné požadavky na přesnost, rozpětí a preciznost, jak je popsáno v bodech 1.2.2.1 a 1.2.2.2.
1.2. Požadavky na konfirmační metody
1.2.1. Obecné požadavky na konfirmační metody
U zakázaných nebo nepovolených látek má být hodnota CCα na co nejnižší rozumně dosažitelné úrovni. U zakázaných nebo nepovolených látek, pro něž je stanovena referenční hodnota pro opatření podle nařízení (EU) 2019/1871, má být hodnota CCα rovna referenční hodnotě pro opatření nebo nižší.
U povolených látek je hodnota CCα vyšší než hodnota MLR nebo maximální limit, ale má se jí co možná nejvíce blížit.
Pro účely konfirmace se mají použít pouze ty analytické metody, u nichž lze zpětně v dokumentech prokázat, že jsou validovány a že je u nich pravděpodobnost falešně nevyhovujícího výsledku (chyba α) menší než nebo rovna 1 % u zakázaných či nepovolených látek nebo je menší než nebo rovna 5 % u povolených látek.
Konfirmační metody mají poskytovat informace o strukturním chemickém složení analytu. Proto konfirmační metody založené pouze na chromatografické analýze bez použití hmotnostně spektrometrické detekce nejsou samy o sobě vhodné k použití jako konfirmační metody pro zakázané nebo nepovolené farmakologicky účinné látky. Není-li hmotnostní spektrometrie vhodná pro povolené látky, je možné použít jiné metody, jako je HPLC-DAD a HPLC-FLD, nebo jejich kombinaci.
Pokud to konfirmační metoda vyžaduje, přidává se ke zkušebnímu podílu vhodný vnitřní standard na počátku extrakčního postupu. Podle dostupnosti by měl být použit buď analyt značený stabilními izotopy, který je vhodný zejména pro hmotnostně spektrometrickou detekci, nebo analogické sloučeniny, které mají velmi podobnou strukturu jako tento analyt. Nelze-li použít vhodný vnitřní standard, potvrdí se identifikace analytu simultánní chromatografií (1). V tomto případě se získá jeden pík, přičemž zvětšení výšky (nebo plochy) píku odpovídá množství přidaného analytu. Není-li toto proveditelné, použijí se standardy s odpovídající matricí nebo standardy s obohacenou matricí.
1.2.2. Obecná pracovní kritéria pro konfirmační metody
1.2.2.1. Pravdivost podle výtěžnosti
U opakovaných analýz certifikovaného referenčního materiálu musí odchylka experimentálně stanovené střední hodnoty hmotnostního zlomku korigovaného na výtěžnost od certifikované hodnoty splňovat rozpětí minimální pravdivosti uvedená v tabulce 1.
Tabulka 1
Minimální pravdivost kvantitativních metod
|
Hmotnostní zlomek |
Rozpětí |
|
≤ 1 μg/kg |
–50 % až +20 % |
|
> 1 μg/kg až 10 μg/kg |
–30 % až +20 % |
|
≥ 10 μg/kg |
–20 % až +20 % |
Nejsou-li k dispozici žádné certifikované referenční materiály, je přípustné, aby se pravdivost měření stanovila jinými způsoby, např. za použití materiálů s přiřazenými hodnotami z mezilaboratorních studií nebo přidáním známých množství analytu (analytů) ke slepé matrici.
1.2.2.2. Preciznost
Variační koeficient (CV) pro opakovanou analýzu referenčního nebo obohaceného materiálu za podmínek vnitrolaboratorní reprodukovatelnosti nesmí překročit hodnotu vypočtenou z Horwitzovy rovnice. Jedná se o tuto rovnici:
CV = 2(1 – 0,5 log C),
kde C je hmotnostní zlomek vyjádřený jako mocnina deseti (např. 1 mg/g = 10-3). V případě hmotnostních zlomků pod 120 μg/kg vede použití Horwitzovy rovnice k nepřijatelně vysokým hodnotám. Proto nesmí povolený maximální variační koeficient přesáhnout hodnoty uvedené v tabulce 2.
Tabulka 2
Přípustný variační koeficient
|
Hmotnostní zlomek |
CV reprodukovatelnosti (%) |
|
> 1 000 μg/kg |
16 (odvozeno z Horwitzovy rovnice) |
|
> 120 μg/kg – 1 000 μg/kg |
22 (odvozeno z Horwitzovy rovnice) |
|
10–120 μg/kg |
25 (*) |
|
< 10 μg/kg |
30 (*) |
U analýz prováděných v podmínkách opakovatelnosti má být variační koeficient za podmínek opakovatelnosti roven dvěma třetinám hodnot uvedených v tabulce 2 nebo nižší než tyto hodnoty.
1.2.3. Požadavky na chromatografickou separaci
U kapalinové (LC) nebo plynové chromatografie (GC) má být minimálním přijatelným retenčním časem pro vyšetřovaný analyt (vyšetřované analyty) dvojnásobek retenčního času, který odpovídá mrtvému objemu kolony. Retenční čas analytu v extraktu má odpovídat retenčnímu času kalibračního standardu, standardu s odpovídající matricí nebo standardu s obohacenou matricí s tolerancí ± 0,1 minuty. U rychlé chromatografie, kde je retenční čas kratší než dvě minuty, je přípustná odchylka menší než 5 % retenčního času. Použije-li se vnitřní standard, musí poměr chromatografického retenčního času analytu ku retenčnímu času vnitřního standardu, to znamená relativní retenční čas analytu, odpovídat retenčnímu času kalibračního standardu, standardu s odpovídající matricí nebo standardu s obohacenou matricí s maximální odchylkou 0,5 % v případě plynové chromatografie a 1 % v případě kapalinové chromatografie u metod validovaných od data vstupu tohoto nařízení v platnost.
1.2.4. Specifická pracovní kritéria pro hmotnostní spektrometrii
1.2.4.1. Hmotnostně spektrometrická detekce
Hmotnostně spektrometrická detekce se provede za použití některé z těchto možností:
|
1. |
snímání celého hmotnostního spektra (full scan, FS); |
|
2. |
sledování vybraných iontů (selected ion monitoring, SIM); |
|
3. |
techniky sekvenční hmotnostní spektrometrie (MSn), např. sledování vybrané reakce (selected reaction monitoring, SRM); |
|
4. |
kombinace technik hmotnostní spektrometrie (MS) nebo sekvenční hmotnostní spektrometrie (MSn) s vhodnými ionizačními technikami. |
Je vhodná jak hmotnostní spektrometrie s nízkým rozlišením (LRMS, při rozlišení na hmotnostní jednotku), tak hmotnostní spektrometrie s vysokým rozlišením (HRMS), včetně např. přístrojů typu „double focusing sectors“, „Time of Flight (TOF)“ a „Orbitrap“.
Při konfirmaci identity analytu pomocí hmotnostní spektrometrie s vysokým rozlišením (HRMS) musí být hmotnostní odchylka všech diagnostických iontů pod hodnotou 5 ppm (nebo v případě m/z < 200 pod hodnotou 1 mDa). Na základě toho by se mělo vybrat účinné rozlišení vhodné pro daný účel, které bude obvykle větší než 10 000 pro celé hmotnostní rozpětí při zachování 10 % chromatografického údolí nebo 20 000 při plné šířce v polovině výšky (full width at half maximum, FWHM).
Když se provádí hmotnostně spektrometrické stanovení snímáním celého spektra (LRMS i HMRS), jsou vhodné pouze diagnostické ionty s relativní intenzitou větší než 10 % v referenčním spektru kalibračního standardu, standardu s odpovídající matricí nebo standardu s obohacenou matricí. Diagnostické ionty mají zahrnovat molekulární iont (je-li přítomen při ≥ 10 % intenzitě základního píku) a charakteristické fragmentové nebo produktové ionty.
Výběr prekurzorového iontu: provádí-li se hmotnostně spektrometrické stanovení fragmentací po výběru prekurzorového iontu, provádí se výběr prekurzorového iontu při rozlišení na hmotnostní jednotku nebo lepším. Vybraným prekurzorovým iontem má být molekulární iont, charakteristické adukty molekulárního iontu, charakteristické produktové ionty nebo jeden z jejich izotopových iontů. V případě, že vybraný prekurzor má vybraný rozsah hmotností (mass selection window) větší než jeden dalton (např. v případě datově nezávislého sběru dat, „data independent acquisition“) považuje se tato technika za konfirmační analýzu celého spektra (full-scan).
Fragmentové a produktové ionty: Vybrané fragmentové nebo produktové ionty mají být diagnostické fragmenty pro měřený analyt/produkt. Neselektivní přechody (např. tropyliový kation nebo ztráta vody) se mají vyloučit, kdykoli je to možné. Množství diagnostických iontů se stanoví z plochy píku nebo výšky integrovaných chromatogramů extrahovaných iontů. Tento postup lze rovněž použít, pokud se k identifikaci používají měření celého spektra. Poměr signál/šum (S/N) musí být pro všechny diagnostické ionty roven třem ku jedné (3:1) nebo větší.
Relativní intenzity: relativní intenzity diagnostických iontů (iontový poměr) se vyjadřují jako procento intenzity nejvíce zastoupeného iontu nebo přechodu. Iontový poměr se musí stanovit porovnáním spekter nebo integrováním signálů hmotnostních stop extrahovaných iontů. Iontový poměr analytu určený k potvrzení má odpovídat poměrům standardů s odpovídající matricí nebo standardů s obohacenou matricí nebo standardních roztoků při srovnatelných koncentracích měřených za stejných podmínek v rámci ± 40 % relativní odchylky.
U všech hmotnostně spektrometrických analýz se stanoví nejméně jeden iontový poměr. Tyto ionty jsou pokud možno získány v rámci jediného skenu, avšak ionty mohou rovněž pocházet z různých skenů téhož nástřiku (tj. celý sken a fragmentační sken).
1.2.4.2. Identifikace
K výběru vhodných režimů snímání a hodnotících kritérií se použije systém identifikačních bodů. Při konfirmaci identity látek v matrici, pro kterou je stanoven MLR (povolené použití), se vyžadují minimálně 4 identifikační body. U nepovolených nebo zakázaných látek se vyžaduje 5 identifikačních bodů. Jeden bod může pocházet z chromatografické separace. Tabulka 3 uvádí počet identifikačních bodů, kterého lze u každé z technik dosáhnout. Aby se splnily podmínky pro identifikační body požadované pro konfirmaci, je možné přidat identifikační body získané jinými technikami.
|
1. |
Všechny hmotnostně spektrometrické analýzy se zkombinují se separační technikou, která vykazuje dostatečnou separační sílu a selektivitu pro konkrétní použití. Mezi vhodné separační techniky patří mimo jiné kapalinová a plynová chromatografie, kapilární elektroforéza (CE) a superkritická fluidní chromatografie (SFC). V případě analytu, který představuje jakoukoli izobarickou nebo izomerní sloučeninu, je k potvrzení jeho identity naprosto nutná přijatelnost retenčního času (tj. ± 0,5 % v GC a ± 1 % v LC a SFC). |
|
2. |
Pro účely získání minimálního počtu identifikačních bodů mohou být kombinovány maximálně tři samostatné techniky. |
|
3. |
Různé způsoby ionizace (např. elektronová ionizace a chemická ionizace) se považují za různé techniky. Tabulka 3 Identifikační body u jednotlivých technik
Tabulka 4 Příklady počtu identifikačních bodů u konkrétních technik a kombinací technik (n = celé číslo)
|
1.2.5. Specifická pracovní kritéria pro stanovení analytu za použití kapalinové chromatografie s jinými technikami detekce, než je hmotnostní spektrometrie
Pouze u povolených látek je možné použít jako alternativu k metodám založeným na hmotnostní spektrometrii následující techniky, pod podmínkou, že jsou splněna příslušná kritéria pro tyto techniky:
|
1. |
spektrofotometrie s plným rozsahem spektra s detekcí diodovým polem (DAD) v případě použití s HPLC; |
|
2. |
spektrofotometrie s fluorescenční detekcí (FLD) v případě použití s HPLC. |
Kapalinová chromatografie s UV/VIS detekcí (jedna vlnová délka) není sama o sobě vhodná jako konfirmační metoda.
1.2.5.1. Pracovní kritéria pro spektrofotometrii s plným rozsahem spektra s detekcí diodovým polem
Je nutné splnit pracovní kritéria pro chromatografickou separaci uvedená v kapitole 1.2.3.
Absorpční maxima v UV spektru analytu musí mít v rámci tolerance maxim dané rozlišovací schopností detekčního systému tytéž vlnové délky jako maxima kalibračního standardu v matrici. V případě detekce diodovým polem je tato tolerance maxim obvykle ± 2 nm. Spektrum analytu v oblasti nad 220 nm se nesmí v případě oblastí těch částí spekter, které mají relativní absorbanci rovnající se 10 % nebo větší, vizuálně lišit od spektra kalibračního standardu. Toto kritérium je splněno, jsou-li přítomna stejná maxima a dále není-li rozdíl mezi oběma spektry v žádném bodě větší než 10 % absorbance kalibračního standardu. Je-li vyhledávání a porovnávání prováděno pomocí digitální knihovny, musí být při srovnávání údajů ze spekter úředních vzorků s údaji ze spektra kalibračního roztoku překročen faktor kritické shody. Tento faktor musí být pro každý analyt stanoven při procesu validace, a to na základě spekter, pro něž jsou splněna výše uvedená kritéria. Musí být prozkoumána proměnlivost spekter způsobená matricí vzorku a pracovními charakteristikami detektoru.
1.2.5.2. Pracovní kritéria pro spektrofotometrii s fluorescenční detekcí
Je nutné splnit pracovní kritéria pro chromatografickou separaci uvedená v kapitole 1.2.3.
Výběr excitačních a emisních vlnových délek v kombinaci s chromatografickými podmínkami musí být proveden tak, aby se minimalizovaly efekty rušivých sloučenin v extraktech slepého vzorku. Rozdíl mezi excitační a emisní vlnovou délkou by měl být minimálně 50 nanometrů.
Nejbližší maximum píku v chromatogramu musí být od stanovovaného píku analytu vzdáleno alespoň o jednu šířku píku analytu v 10 % výšky.
Tato metoda je použitelná pro molekuly, které vykazují přirozenou fluorescenci, a pro molekuly, které vykazují fluorescenci po transformaci nebo derivatizaci.
KAPITOLA 2
VALIDACE
2.1. Pracovní charakteristiky, které je nutno stanovit u analytických metod
Validací metody se má prokázat, že uvedená analytická metoda splňuje kritéria pro příslušné pracovní charakteristiky. Různé účely kontroly vyžadují různé kategorie metod. Tabulka 5 uvádí, které pracovní charakteristiky se ověřují u každého typu metody. Další vysvětlení každého parametru je podáno v této kapitole.
Tabulka 5
Klasifikace analytických metod podle pracovních charakteristik, které mají být stanoveny
|
Metoda |
Konfirmační |
Screeningová |
|||
|
Kvalitativní |
Kvantitativní |
Kvalitativní |
Semikvantitativní |
Kvantitativní |
|
|
Látky |
A |
A, B |
A, B |
A, B |
A, B |
|
Identifikace v souladu s kapitolou 1.2 |
x |
x |
|
|
|
|
CCα |
x |
x |
|
|
|
|
CCβ |
– |
|
x |
x |
x |
|
Pravdivost |
x |
|
|
x |
|
|
Preciznost |
x |
|
(x) |
x |
|
|
Relativní matricový efekt/absolutní výtěžnost (*) |
x |
|
|
x |
|
|
Selektivita/specifičnost |
x |
x |
x |
x |
|
|
Stabilita (#) |
x |
x |
x |
x |
|
|
Robustnost |
x |
x |
x |
x |
|
|
x: Prostřednictvím validace je nutné prokázat, že jsou splněny požadavky na pracovní charakteristiky. (x) Požadavky na preciznost dle kapitoly 1.2.2.2 není u semikvantitativních screeningových metod nutné splnit. Preciznost by se však měla stanovit za účelem prokázání vhodnosti metody pro zabránění falešně vyhovujícím analytickým výsledkům. A: zakázané nebo nepovolené látky B: povolené látky |
|||||
2.2. Pravdivost, opakovatelnost a vnitrolaboratorní reprodukovatelnost
V této kapitole jsou uvedeny příklady validačních postupů pro analytické metody a odkazy na tyto postupy. K prokázání toho, že metoda splňuje pracovní kritéria, lze použít i jiné přístupy, pokud lze jimi získat stejnou úroveň a kvalitu informací.
2.2.1. Konvenční validace
Výpočet parametrů konvenčními metodami vyžaduje, aby bylo provedeno několik samostatných experimentů. U všech větších změn musí být stanovena každá pracovní charakteristika (viz oddíl 2.4). V případě metod pro více analytů může být stanoveno několik analytů současně, pokud byly vyloučeny možné rušivé vlivy. Řadu pracovních charakteristik lze stanovit podobným způsobem. Proto se v zájmu minimalizace rozsahu prací doporučuje v co největší možné míře experimenty kombinovat (např. opakovatelnost a vnitrolaboratorní reprodukovatelnost se specifičností, analýzu slepých vzorků pro stanovení limitu rozhodnutí pro konfirmaci a testování specifičnosti).
2.2.1.1. Pravdivost na základě certifikovaného referenčního materiálu
Je vhodnější stanovit pravdivost analytické metody prostřednictvím certifikovaného referenčního materiálu (CRM). Tento postup je popsán v normě ISO 5725-4:1994 (2).
Níže je uveden příklad:
|
1. |
podle zkušebního postupu dané metody se analyzuje šest replikátů CRM; |
|
2. |
stanoví se koncentrace analytu přítomného v každém vzorku replikátů; |
|
3. |
pro těchto šest replikátů se vypočte průměr, směrodatná odchylka a variační koeficient (%); |
|
4. |
pravdivost vyjádřená v procentech se vypočte jako poměr zjištěné průměrné koncentrace a certifikované hodnoty (vyjádřené jako koncentrace) vynásobený 100. |
Pravdivost (%) = (detekovaná průměrná koncentrace korigovaná na výtěžnost) × 100/certifikovaná hodnota.
2.2.1.2. Pravdivost na základě obohacených vzorků
Není-li k dispozici žádný certifikovaný referenční materiál, stanoví se pravdivost metody pomocí experimentů používajících obohacenou slepou matrici, minimálně v souladu s tímto schématem:
|
1. |
V případě metod validovaných od data vstupu tohoto nařízení v platnost se zvolí slepý materiál a obohatí se v koncentraci:
|
|
2. |
Pro každou koncentraci se analýza provádí s šesti replikáty. |
|
3. |
Vzorky se analyzují. |
|
4. |
Vypočte se koncentrace stanovená v každém vzorku. |
|
5. |
Vypočte se pravdivost pro každý vzorek za použití níže uvedené rovnice a následně se vypočte průměrná pravdivost a variační koeficient pro šest výsledků na každé úrovni koncentrace. |
Pravdivost (%) = (detekovaná průměrná koncentrace korigovaná na výtěžnost) × 100/úroveň obohacení.
V případě metod pro povolené látky validovaných před datem použitelnosti tohoto nařízení je dostačující stanovení pravdivosti metody za použití šesti obohacených alikvotů v koncentraci 0,5, 1,0 a 1,5násobku MLR nebo maximálního limitu.
2.2.1.3. Opakovatelnost
|
1. |
V případě metod validovaných od data vstupu tohoto nařízení v platnost se připraví soubor vzorků identických slepých matric stejného druhu. Ty se obohatí analytem za účelem získání koncentrací odpovídajících:
|
|
2. |
Pro každou koncentraci se analýza provádí s nejméně šesti replikáty. |
|
3. |
Vzorky se analyzují. |
|
4. |
Vypočte se koncentrace stanovená v každém vzorku. |
|
5. |
Vypočte se průměrná koncentrace, směrodatná odchylka a variační koeficient (%) obohacených vzorků. |
|
6. |
Tento postup se opakuje alespoň při dvou příležitostech. |
|
7. |
Vypočtou se celkové průměrné koncentrace, směrodatné odchylky (vypočítáním průměru druhých mocnin směrodatné odchylky z jednotlivých případů a jeho odmocněním) a variační koeficienty u obohacených vzorků.
V případě metod pro povolené látky validovaných před datem vstupu tohoto nařízení v platnost je dostačující stanovení opakovatelnosti za použití obohacených matric v koncentracích 0,5, 1,0 a 1,5násobku MLR nebo maximálního limitu. Alternativně lze provést výpočet opakovatelnosti dle normy ISO 5725-2:2019 (7). |
2.2.1.4. Vnitrolaboratorní reprodukovatelnost
|
1. |
V případě validací prováděných po datu vstupu tohoto nařízení v platnost se připraví soubor vzorků specifikovaného zkušebního materiálu (identické nebo odlišné matrice) obohaceného o analyt (analyty) za účelem získání koncentrací odpovídajících:
|
|
2. |
Provede se analýza na každé úrovni koncentrace nejméně s šesti replikáty slepého materiálu. |
|
3. |
Vzorky se analyzují. |
|
4. |
Vypočte se koncentrace stanovená v každém vzorku. |
|
5. |
Tyto kroky se opakují alespoň při dvou dalších příležitostech s různými šaržemi slepého materiálu, různými pracovníky a za co možná nejvíce různých podmínek prostředí, např. s různými šaržemi reakčních činidel, rozpouštědel, při různých laboratorních teplotách, s různými přístroji nebo s variací jiných parametrů. |
|
6. |
U obohacených vzorků se stanoví průměrná koncentrace, směrodatná odchylka a variační koeficient (%).
V případě metod pro povolené látky validovaných před datem vstupu tohoto nařízení v platnost je dostačující vnitrolaboratorní reprodukovatelnost za použití obohacených matric v koncentracích 0,5, 1,0 a 1,5násobku MLR nebo maximálního limitu. Alternativně lze výpočet vnitrolaboratorní reprodukovatelnosti/mezilehlé preciznosti provést rovněž podle normy ISO 5725-2:2019, ISO 11843-1:1997 (8), Codex CAC/GL 59-2006 (9). |
2.2.2. Validace podle alternativních modelů
Výpočet parametrů v souladu s alternativními modely vyžaduje provedení plánu experimentů. Plán experimentů má/musí být navržen v závislosti na počtu různých druhů a různých faktorů, které se zkoumají. Proto je prvním krokem celého postupu validace posouzení souborů vzorků, které se budou v laboratoři v budoucnu analyzovat, za účelem určení nejdůležitějších druhů a faktorů, které mohou ovlivnit výsledky měření. Přístup na základě faktorů umožňuje stanovit nejistotu měření u výsledků zkoušky získaných za různých zkušebních podmínek v dané laboratoři, jako jsou různí pracovníci provádějící analýzu, různé přístroje, různé šarže reakčních činidel, různé matrice, různé doby trvání stanovení a různé teploty stanovení. Následně je nutné podle účelu zvolit rozmezí koncentrace, a sice podle MLR nebo maximálního limitu v případě povolených látek nebo podle referenční hodnoty pro opatření či LCL u zakázaných nebo nepovolených látek.
Přístup na základě faktorů má za cíl stanovit spolehlivé údaje o preciznosti a údaje z měření pomocí simultánně kontrolované variace vybraných faktorů. To umožňuje hodnocení kombinovaného účinku faktoriálních efektů a náhodných efektů. Plán experimentů rovněž umožňuje zkoumat robustnost (10) analytické metody a stanovení směrodatné odchylky vnitrolaboratorní reprodukovatelnosti u všech matric.
Příklad alternativního přístupu za použití ortogonálního plánu experimentů je uveden dále.
Je možné zkoumat až sedm faktorů (faktory šumu). Studie je navržena takovým způsobem, aby se preciznost, pravdivost (na základě obohacených vzorků), citlivost, nejistota měření a kritické koncentrace stanovovaly souběžně díky zavedení plánu experimentů.
Tabulka 6
Příklad ortogonálního plánu experimentů se sedmi faktory (I–VII) pozměňovanými na dvou úrovních (A/B) ve validační studii s osmi měřeními (kombinace úrovní faktorů)
|
Faktor |
I |
II |
III |
IV |
V |
VI |
VII |
|
Měření 01 |
A |
A |
A |
A |
A |
A |
A |
|
Měření 02 |
A |
A |
B |
A |
B |
B |
B |
|
Měření 03 |
A |
B |
A |
B |
A |
B |
B |
|
Měření 04 |
A |
B |
B |
B |
B |
A |
A |
|
Měření 05 |
B |
A |
A |
B |
B |
A |
B |
|
Měření 06 |
B |
A |
B |
B |
A |
B |
A |
|
Měření 07 |
B |
B |
A |
A |
B |
B |
A |
|
Měření 08 |
B |
B |
B |
A |
A |
A |
B |
Výpočet charakteristik metody se provede způsobem popsaným autory Jülicher a kol. (11).
2.2.3. Další validační přístupy
K prokázání toho, že metoda splňuje pracovní kritéria pro pracovní charakteristiky, lze použít i jiné přístupy, pokud jimi lze získat stejnou úroveň a kvalitu informací. Validace může být provedena formou mezilaboratorní studie podle Codex Alimentarius, ISO nebo IUPAC (12) nebo alternativními metodami, jako jsou studie v jediné laboratoři nebo vnitrolaboratorní validace (13). Použijí-li se alternativní validační postupy, musí být ve zprávě o validaci uveden výchozí model a strategie s příslušnými podmínkami, předpoklady a vzorci, nebo musí být alespoň uvedeny odkazy na dostupné literární zdroje.
2.3. Selektivita/specifičnost
Sílu rozlišení mezi analytem a blízce příbuznými látkami je nutné stanovit v co největší možné míře. Stanoví se rušivé vlivy homologů, izomerů, produktů rozkladu, endogenních látek, analogů, metabolických produktů sledovaného rezidua, sloučenin matrice nebo jakékoli jiné interferující látky, a je-li to zapotřebí, upraví se metoda tak, aby se zabránilo zjištěným rušivým vlivům. Ke stanovení specifičnosti metody se použije níže uvedený postup:
|
1. |
Zvolí se okruh chemicky příbuzných sloučenin nebo jiných látek, které se mohou pravděpodobně vyskytovat spolu se sledovanou sloučeninou a které mohou být přítomny ve vzorku, a ověří se, zda mohou narušovat analýzu cílového analytu (cílových analytů). |
|
2. |
Analyzuje se vhodný počet reprezentativních slepých vzorků, např. různé dávky nebo šarže různých zvířecích druhů (n ≥ 20), a v oblasti, v níž podle očekávání dochází k eluci cílového analytu, se zkoumají jakékoli rušivé vlivy (signály, píky, stopy iontů). |
|
3. |
Reprezentativní slepé vzorky se obohatí v příslušné koncentraci látkami, které by možná mohly narušovat identifikaci a/nebo kvantifikaci analytu, a zkoumá se, zda přidaná látka:
|
2.4. Robustnost
Přetrvávající funkčnost analytické metody se má testovat za různých experimentálních podmínek, které zahrnují např. různé podmínky odběru vzorků a menší změny, ke kterým může dojít při rutinním testování. Změny provedené v experimentálních podmínkách pro testování robustnosti metody by měly být menší. Vyhodnotí se důležitost těchto změn. U všech menších změn, u nichž se ukázalo, že mají významný vliv na provedení stanovení, se stanoví každá pracovní charakteristika.
2.5. Stabilita
Stanoví se stabilita kalibračního standardu, standardu s odpovídající matricí a/nebo standardů s obohacenou matricí a analytu nebo složek matrice ve vzorku během uchovávání či analýzy, neboť nestabilita může ovlivnit výsledky testu.
Stabilita analytu za různých podmínek uchovávání je obvykle dobře popsána. Požadované informace je možné získat z experimentů prováděných za účelem monitoringu podmínek uchovávání standardů a vzorků, které se provádějí jako součást běžného postupu akreditace laboratoře a systému kontroly kvality. Jsou-li k dispozici údaje o stabilitě analytů v matrici (např. na základě informací z laboratoří EURL, publikovaných údajů atd.), není nutné, aby tyto údaje stanovovala každá laboratoř. Odkazování se na dostupné údaje o stabilitě analytů v roztoku a v matrici však je přípustné, pouze pokud jsou použity totožné podmínky.
V případě, že požadované údaje o stabilitě nejsou k dispozici, měly by se použít níže uvedené přístupy.
2.5.1. Stanovení stability analytu v roztoku
|
1. |
Připraví se čerstvé zásobní roztoky analytu (analytů) a zředí se podle pokynů pro zkoušku tak, aby se připravil dostatečný počet alikvotů (např. 40) každé vybrané koncentrace. Připraví se vzorky:
|
|
2. |
Podle pokynů pro zkoušku se změří obsah v čerstvě připraveném roztoku. |
|
3. |
Příslušné objemy se rozdělí do vhodných nádob, označí se a uchovávají se ve světelných a teplotních podmínkách uvedených ve schématu zahrnutém v tabulce 7. Doba uchovávání se zvolí s ohledem na použitou analytickou praxi, ideálně dokud nejsou pozorovatelné první známky rozkladu během identifikace a/nebo kvantifikace. Pokud během studie stability není pozorován žádný rozklad, měla by se doba uchovávání ve studii stability rovnat maximální době uchovávání roztoku. |
|
4. |
Vypočte se koncentrace analytu (analytů) v každém alikvotu ve srovnání s koncentrací analytu v čerstvě připraveném roztoku podle níže uvedeného vzorce:
Zbývající analyt (%) = Ci × 100/Cčerstvý Ci = koncentrace v časovém bodě i Cčerstvý = koncentrace čerstvě připraveného roztoku Průměrná hodnota z pěti replikátů roztoku, které byly uchovávány, se nesmí lišit o více než 15 % od průměrné hodnoty z pěti čerstvě připravených replikátů roztoku. Průměrná hodnota z pěti čerstvě připravených roztoků se použije jako základ pro výpočet procentuálního rozdílu. Tabulka 7 Schéma pro stanovení stability analytu v roztoku
|
2.5.2. Stanovení stability analytu (analytů) v matrici
|
1. |
Pokud je to možné, použijí se přirozeně kontaminované vzorky. Není-li k dispozici přirozeně kontaminovaná matrice, použije se slepá matrice obohacená analytem. |
|
2. |
Je-li k dispozici přirozeně kontaminovaná matrice, stanoví se koncentrace v čerstvě získané matrici. Další alikvoty homogenizované přirozeně kontaminované matrice se uchovávají při teplotě minus 20 °C nebo podle potřeby nižší a koncentrace analytu se stanoví, dokud je vzorek uchováván v laboratoři. |
|
3. |
Není-li k dispozici přirozeně kontaminovaná matrice, zhomogenizuje se slepá matrice. Matrice se rozdělí na pět alikvotů. Každý z alikvotů se uměle obohatí o analyt připravený přednostně jako vodný roztok o malém objemu. Jeden alikvot se ihned analyzuje. Zbývající alikvoty se uchovávají při teplotě nejméně minus 20 °C nebo podle potřeby nižší a analyzují se po krátkodobém, střednědobém a dlouhodobém uchovávání s ohledem na použité analytické metody. |
|
4. |
Zaznamená se maximální přijatelná doba uchovávání a optimální podmínky uchovávání.
Průměrná hodnota pěti replikátů roztoku, které byly uchovávány, se od průměrné hodnoty pěti čerstvě připravených replikátů roztoku nesmí lišit o více než vnitrolaboratorní reprodukovatelnost metody. Průměrná hodnota z pěti čerstvě připravených roztoků se použije jako základ pro výpočet procentuálního rozdílu. |
2.6. Limit rozhodnutí (CCα pro konfirmaci)
Pro konfirmační metody se musí stanovit CCα. CCα se stanoví za podmínek splňujících požadavky na identifikaci nebo na identifikaci a kvantitativní stanovení, jak jsou definovány v oddíle „Pracovní kritéria a jiné požadavky na analytické metody“ kapitoly 1.
Za účelem kontroly souladu vzorků je v hodnotě CCα (limit rozhodnutí pro konfirmaci) již zohledněna kombinovaná standardní nejistota měření.
|
1. |
Pro nepovolené nebo zakázané farmakologicky účinné látky se CCα vypočte takto:
Metodu 2 lze použít pro výpočet CCα pouze do 1. ledna 2026 v případě metod validovaných před vstupem tohoto nařízení v platnost. U metod validovaných po vstupu tohoto nařízení v platnost se použije pouze metoda 1 nebo metoda 3. |
|
2. |
Pro povolené látky se CCα vypočte takto:
|
2.7. Detekční schopnost pro screening (CCβ)
Pro screeningové metody se stanoví CCβ. CCβ se stanoví tak, jak je definováno pod názvem „Pracovní kritéria a jiné požadavky na analytické metody“ v kapitole 1 této přílohy, a v souladu s požadavky uvedenými v tabulce 5. Na screeningové metody však není nutné aplikovat všechny požadavky pro identifikaci (srov. 1.2.3, 1.2.4, 1.2.5).
|
1. |
Pro nepovolené nebo zakázané farmakologicky účinné látky se má zajistit maximální chyba β 5 %. CCβ se vypočte takto:
|
|
2. |
Pro povolené látky se musí zajistit maximální chyba β 5 %. CCβ se vypočte takto:
U farmakologicky účinných látek, pro které je MLR stanovena pro součet různých látek, se použije CCβ látky s nejvyšší koncentrací ve vzorku jako CCβ pro posouzení souhrnu látek v měřeném vzorku. |
2.8. Kalibrační křivky
Použijí-li se kalibrační křivky pro kvantifikaci:
|
1) |
musí být sestrojeny alespoň z pěti pokud možno ekvidistantních hodnot (včetně nulové hodnoty); |
|
2) |
popíše se pracovní rozsah křivky; |
|
3) |
popíše se matematická rovnice křivky a shoda proložení dat (koeficient stanovení R2) s křivkou; |
|
4) |
popíšou se přípustná rozpětí parametrů křivky. |
U kalibračních křivek založených na standardním roztoku, standardech s odpovídající matricí nebo standardech s obohacenou matricí se uvedou přípustná rozpětí pro parametry kalibrační křivky, která se mohou u jednotlivých řad lišit.
2.9. Absolutní výtěžnost
Absolutní výtěžnost metody se musí stanovit, jestliže se nepoužije žádný vnitřní standard ani kalibrace s použitím obohacené matrice.
Jsou-li splněny požadavky pro pravdivost uvedené v tabulce 1, je možné použít fixní korekční faktor. Jinak se použije faktor výtěžnosti získaný pro uvedenou konkrétní dávku. Případně se namísto korekčního faktoru výtěžnosti použije postup přidání standardu (16) nebo vnitřní standard.
Absolutní výtěžnost se vypočte pro nejméně šest reprezentativních dávek matrice.
Jeden alikvot slepé matrice se obohatí analytem před extrakcí a druhý alikvot slepé matrice se obohatí po přípravě vzorku na příslušnou úroveň koncentrace a stanoví se koncentrace analytu.
Výtěžnost se vypočte takto:
výtěžnost (analyt) = (standard s obohacenou matricí)/(standard s odpovídající matricí) × 100
2.10. Relativní matricové efekty
Relativní matricový efekt se stanoví ve všech případech. Je to možné provést jako součást validace nebo v samostatných experimentech. Výpočet relativního matricového efektu se provede pro nejméně dvacet různých slepých dávek (matrice/druhu) v souladu s rozsahem metody (např. různé druhy, které mají být zahrnuty).
Slepá matrice se po extrakci obohatí analytem v koncentraci odpovídající referenční hodnotě pro opatření, MLR nebo maximální limit a analyzuje se společně s čistým roztokem analytu.
Relativní matricový efekt neboli matricový faktor (MF) se vypočte jako:
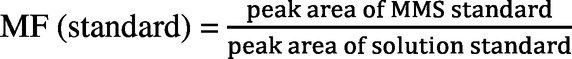
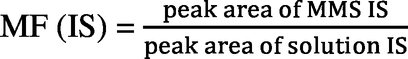
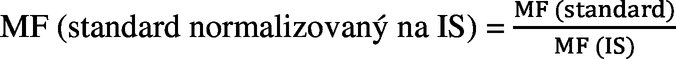
IS: vnitřní standard
MMS: standard s odpovídající matricí
Variační koeficient pro MF nesmí být větší než 20 %.
KAPITOLA 3
KONTROLA KVALITY BĚHEM RUTINNÍ ANALÝZY – PRŮBĚŽNÉ OVĚŘOVÁNÍ ZPŮSOBILOSTI METODY
Je nutné splnit požadavky pro zajištění kvality analytických výsledků uvedené v kapitole 7.7 normy ISO/IEC 17025:2017 (17).
Upřednostňovanou možností pro poskytnutí důkazů o funkční způsobilosti metody během rutinní analýzy je analýza certifikovaného referenčního materiálu (CRM). Vzhledem k tomu, že CRM, které obsahují příslušné analyty v požadovaných koncentracích, jsou zřídkakdy dostupné, lze jako alternativu použít rovněž referenční materiály dodávané a charakterizované laboratořemi EURL nebo laboratořemi, které jsou akreditovány dle normy ISO/IEC 17043:2010 (18). Jako další alternativu je možné použít vnitrolaboratorní referenční materiály, které jsou pravidelně kontrolovány.
Průběžné ověřování funkční způsobilosti metody během rutinní analýzy by se mělo provádět ve screeningovém kroku a konfirmačním kroku.
|
1. |
Pro screeningový krok: pro každou sérii (dávku) prováděných analýz se souběžně analyzuje soubor těchto vzorků ke kontrole kvality:
|
|
2. |
Pro konfirmační krok: pro každou sérii (dávku) prováděných analýz se souběžně analyzuje soubor těchto vzorků ke kontrole kvality:
|
Pro vzorky ke kontrole kvality se doporučuje toto pořadí: kontrolní vzorek pro vhodnost systému přístroje, vyhovující kontrolní vzorek, vzorek (vzorky) k potvrzení, znovu vyhovující kontrolní vzorek a obohacený vzorek pro kontrolu kvality (nevyhovující kontrolní vzorky).
U kvantitativních metod se musí pro každou dávku úředních vzorků analyzovat a měřit kalibrační křivka před výše uvedenými vzorky nebo po nich.
Je-li to proveditelné, vyhodnotí se pravdivost (na základě uměle obohacených vzorků) všech cílových analytů v nevyhovujících kontrolních vzorcích prostřednictvím grafů kontroly kvality v souladu s kapitolou 7.7 normy ISO/IEC 17025:2017. Pokud to vyžaduje neúměrně velký počet stanovení pravdivosti, je možné snížit počet analytů na počet reprezentativních analytů.
KAPITOLA 4
ROZŠÍŘENÍ ROZSAHU VALIDACE DŘÍVE VALIDOVANÉ METODY
Někdy je nezbytné rozšířit rozsah validace metody, která již byla komplexně validována. V těchto případech je třeba provést rozšíření rozsahu účinným a analyticky správným způsobem. Toho lze dosáhnout provedením validace na menším počtu vzorků (např. poloviční počet vzorků), než je tomu při kompletní validaci.
Typ a počet modifikací, které je třeba validovat v jednom redukovaném validačním plánu, musí být vždy založen na odborných znalostech a předchozích zkušenostech. Například změna techniky detekce by každopádně vyžadovala kompletní validaci.
Obecně platí, že k zajištění přetrvávající validity metody je nutné neustále sledovat její funkční způsobilost a porovnávat ji s validačními parametry získanými na začátku. V ideálním případě je tato průběžná kontrola funkční způsobilosti metody navržena takovým způsobem, že lze v průběhu času shromáždit chybějící údaje pro kompletní validaci (např. pomocí několika datových bodů ze vzorků ke kontrole kvality v každé analytické sérii).
4.1. Rozšíření metod s ohledem na rozmezí koncentrací
Kvůli změnám MLR, maximálních limitů a referenčních hodnot pro opatření může být nezbytné upravit rozmezí koncentrace, pro které je metoda validována. V takovém případě je přípustné použít redukovaný validační plán.
Kalibrační křivky pro modifikovaný rozsah by se měly připravit dle validačního postupu. Měly by se analyzovat různé dávky uměle obohacené na různých úrovních koncentrace (srov. 2.2.1, 2.2.2). Pravdivost, opakovatelnost a vnitrolaboratorní reprodukovatelnost/mezilehlá preciznost by se měly nacházet v přípustném rozmezí ve srovnání s hodnotami původně validované metody. Je-li to relevantní, měl by se provést nový výpočet CCβ (screeningové metody) a CCα (konfirmační metody).
4.2. Rozšíření metod s ohledem na další látky
Obecně je možné rozšířit metodu na další sloučeniny pouze v případě analytů, které mají podobnou strukturu a charakteristiky jako ty, které již jsou v analytické metodě zahrnuty. V tomto případě je přípustné použít redukovaný validační plán. Podobně není povolena žádná odchylka od popisu metody.
Kalibrační křivky pro další látky by se měly připravit dle validačního postupu. Měly by se analyzovat různé dávky materiálů matrice uměle obohacené na různých úrovních koncentrace (srov. 2.2.1, 2.2.2). Pravdivost, opakovatelnost a vnitrolaboratorní reprodukovatelnost/mezilehlá preciznost by se měly nacházet v rozmezí srovnatelném s rozmezím ostatních analytů původně validované metody a měly by být v souladu s požadavky uvedenými v bodě 1.2.2. Musí se provést výpočet CCβ (screeningové metody) a CCα (konfirmační metody) pro nové analyty.
4.3. Rozšíření metod s ohledem na matrice/druhy
O zahrnutí nových matric nebo druhů do již validované analytické metody se vždy rozhoduje případ od případu na základě dosavadních znalostí a zkušeností s uvedenou metodou a předběžných experimentů posuzujících potenciální matricové efekty a interference. Obecně to bude možné pouze u matric, které vykazují podobné vlastnosti, a u nekritických analytů (stabilita, detekovatelnost).
Kalibrační křivky (standard nebo matrice) by se měly připravit dle validačního postupu. Měly by se analyzovat různé dávky materiálu matrice uměle obohacené na různých úrovních koncentrace (srov. 2.2.1, 2.2.2). Pravdivost, opakovatelnost a vnitrolaboratorní reprodukovatelnost/mezilehlá preciznost by se měly nacházet v přípustném rozmezí původně validované metody a měly by být v souladu s požadavky uvedenými v bodě 1.2.2. V závislosti na validačním postupu může být nezbytné provést nový výpočet CCβ (screeningové metody) nebo CCα (konfirmační metody).
Nejsou-li výsledky v přípustném rozmezí v porovnání s hodnotami původní matrice, bude nezbytné provést další kompletní validaci, aby se stanovily pracovní parametry specifické pro matrici/druh.
V případě, že se MLR pro konkrétní látku u určitých matric liší, bude velmi pravděpodobně obtížné upravit rozsah metody na další matrici/druh a koncentraci, neboť v tomto případě je nutné zvážit dvě modifikace. V těchto případech se doporučuje kompletní validace.
(1) Simultánní chromatografií se rozumí postup, při němž je extrakt před chromatografií rozdělen na dvě části. U první části se provede chromatografická analýza. Druhá část se smísí se standardem analytu, který má být měřen. U této části se rovněž provede chromatografická analýza. Přidané množství standardu analytu se musí blížit odhadovanému množství analytu v extraktu. Simultánní chromatografie je vhodná pro zlepšení identifikace analytu při použití chromatografických metod, zejména nelze-li použít vhodný vnitřní standard.
(*) Uvedený variační koeficient CV (%) je vodítkem a měl by být co možná rozumně nejnižší.
(a) Jestliže je prekurzorový iont stejný iont (nebo adukt nebo izotop) jako HRMS iont sledovaný v celém skenu, nezískává se žádný další identifikační bod pro výběr prekurzorového iontu.
(#) Jsou-li údaje o analytech v matrici k dispozici ve vědecké literatuře nebo z jiné laboratoře, nemusí dotčená laboratoř tyto údaje znovu stanovovat. Odkazování se na dostupné údaje o stabilitě analytů v roztoku však je přípustné, pouze pokud jsou použity totožné podmínky.
(*) Pro MS metody je důležité prokázat prostřednictvím validace, že jsou splněny požadavky na pracovní charakteristiky. Relativní matricový efekt uvedené metody se musí stanovit, pokud tento efekt nebyl posuzován během validačního postupu. Absolutní výtěžnost metody se musí stanovit, jestliže se nepoužije žádný vnitřní standard ani kalibrace s použitím obohacené matrice.
(2) ISO 5725-4:2020 Přesnost (pravdivost a preciznost) metod a výsledků měření – Část 4: Základní metody pro stanovení pravdivosti normalizované metody měření (klauzule 3).
(3) Pokud u nepovolené farmakologicky účinné látky není validace koncentrace rovnající se 0,5násobku referenční hodnoty pro opatření rozumně dosažitelná, je možné koncentraci rovnající se 0,5násobku referenční hodnoty pro opatření nahradit nejnižší koncentrací v rozmezí 0,5násobku až 1,0násobku referenční hodnoty pro opatření, která je rozumně dosažitelná.
(4) Pokud u konkrétní farmakologicky účinné látky není validace koncentrace rovnající se 0,1násobku MLR rozumně dosažitelná, je možné koncentraci rovnající se 0,1násobku MLR nahradit nejnižší koncentrací v rozmezí 0,1násobku až 0,5násobku MLR, která je rozumně dosažitelná.
(5) Pokud u nepovolené farmakologicky účinné látky není validace koncentrace rovnající se 0,5násobku referenční hodnoty pro opatření rozumně dosažitelná, je možné koncentraci rovnající se 0,5násobku referenční hodnoty pro opatření nahradit nejnižší koncentrací v rozmezí 0,5násobku až 1,0násobku referenční hodnoty pro opatření, která je rozumně dosažitelná.
(6) Pokud u konkrétní farmakologicky účinné látky není validace koncentrace rovnající se 0,1násobku MLR rozumně dosažitelná, je možné koncentraci rovnající se 0,1násobku MLR nahradit nejnižší koncentrací v rozmezí 0,1násobku až 0,5násobku MLR, která je rozumně dosažitelná.
(7) ISO 5725-2:2019 Přesnost (pravdivost a preciznost) metod a výsledků měření – Část 2: Základní metoda pro stanovení opakovatelnosti a reprodukovatelnosti normalizované metody měření (klauzule 3).
(8) ISO 11843-1:1997 Detekční schopnost – Část 1: Termíny a definice.
(9) Komise pro Codex Alimentarius, organizace OSN pro výživu a zemědělství, Světová zdravotnická organizace, Pokyny pro odhad nejistoty výsledků (CAC/GL 59-2006).
(10) Změny experimentálních podmínek, které jsou zde zmiňovány, mohou zahrnovat materiály vzorků, analyty, podmínky uchovávání, environmentální podmínky a/nebo podmínky přípravy vzorků. U všech experimentálních podmínek, které by mohly v praxi kolísat (např. stabilita reakčních činidel, složení vzorku, pH, teplota) se uvedou změny, které by mohly mít vliv na analytický výsledek.
(11) Jülicher, B., Gowik, P. a Uhlig, S. (1998) Assessment of detection methods in trace analysis by means of a statistically based in-house validation concept. Analyst, 120, 173.
(12) IUPAC (1995), Protocol for the design, conduct and interpretation of method-performance studies, Pure & Applied Chem, 67, 331.
(13) Gowik, P., Jülicher, B. and Uhlig, S. (1998) Multi-residue method for non-steroidal anti-inflammatory drugs in plasma using high performance liquid chromatography-photodiode-array detection. Method description and comprehensive in-house validation. J. Chromatogr., 716, 221.
(14) ISO 11843-1:1997 Detekční schopnost – Část 1: Termíny a definice.
(15) Prováděcí nařízení Komise (EU) 2018/470 ze dne 21. března 2018 o podrobných pravidlech pro maximální limit reziduí, jenž má být vzat v úvahu pro kontrolní účely u potravin získaných ze zvířat ošetřených v EU podle článku 11 směrnice 2001/82/ES (Úř. věst. L 79, 22.3.2018, s. 16).
(16) Přidané množství standardu analytu může být např. být dvojnásobkem až pětinásobkem odhadnutého množství analytu ve vzorku. Účelem toho postupu je stanovit obsah analytu ve vzorku, přičemž se zohlední výtěžnost analytického postupu.
(17) Norma ISO/IEC 17025: Obecné požadavky na kompetenci zkušebních a kalibračních laboratoří z roku 2017 (kapitola 7.7).
(18) ISO/IEC 17043:2010 Posuzování shody — Všeobecné požadavky na zkoušení způsobilosti.
PŘÍLOHA II
POSTUPY VZORKOVÁNÍ A OŠETŘENÍ ÚŘEDNÍCH VZORKŮ
1. Množství vzorku
Minimální množství vzorku je definováno v národním kontrolním programu pro rezidua. Minimální množství vzorku musí umožňovat schváleným laboratořím provést analytické postupy nezbytné pro dokončení screeningových a konfirmačních analýz. Konkrétně v případě drůbeže, akvakultury, králíků, farmové zvěře, plazů a hmyzu sestává vzorek z jednoho nebo více zvířat v závislosti na požadavcích analytických metod. V případě vajec je velikost vzorku alespoň dvanáct vajec v závislosti na použitých analytických metodách. Je-li nutné v jednom vzorku analyzovat několik kategorií látek různými analytickými metodami, zvýší se v souladu s tím i velikost vzorku.
2. Rozdělení do podvzorků
Pokud je to technicky možné nebo to je požadováno vnitrostátními právními předpisy, rozdělí se každý vzorek do nejméně dvou rovnocenných podvzorků tak, aby každý umožňoval úplný analytický postup. Rozdělení do podvzorků se může provést v místě odběru vzorku nebo v laboratoři.
3. Sledovatelnost
Každý vzorek se odebere takovým způsobem, aby bylo možné jej vždy zpětně vysledovat až k původnímu hospodářství a šarži zvířat nebo případně k jednotlivému zvířeti. Zejména v případě mléka lze vzorky odebírat dle rozhodnutí příslušného členského státu na kterémkoli z těchto míst:
|
1. |
v hospodářství ze sběrné cisterny; |
|
2. |
na úrovni mlékárenského průmyslu před vyprázdněním přepravní nádoby. |
4. Nádoby na vzorky
Vzorky se odebírají do vhodných nádob, aby byla zachována neporušenost vzorku a zajištěna jeho sledovatelnost. Nádoby mají především vyloučit možnost záměny vzorků, křížovou kontaminaci a znehodnocení. Nádoby musí být úředně zapečetěny.
5. Zpráva o odběru vzorku
Po každém odběru vzorku musí být vyhotovena zpráva.
Inspektor uvede ve zprávě o odběru alespoň tyto údaje:
|
1. |
adresu příslušných orgánů; |
|
2. |
jméno inspektora nebo identifikační kód; |
|
3. |
úřední kódové číslo vzorku; |
|
4. |
datum odběru; |
|
5. |
jméno a adresu vlastníka nebo osoby pověřené péčí o zvířata nebo o živočišné produkty; |
|
6. |
název a adresu hospodářství původu zvířat (při odběru v hospodářství); |
|
7. |
registrační číslo zařízení – číslo jatek; |
|
8. |
označení zvířete nebo produktu; |
|
9. |
druh zvířete; |
|
10. |
matrici vzorku; |
|
11. |
je-li to relevantní, medikaci během posledních čtyř týdnů před odběrem vzorku (při odběru v hospodářství); |
|
12. |
látku nebo skupiny látek pro vyšetření; |
|
13. |
zvláštní poznámky. |
V závislosti na postupu odběru vzorků je nutné poskytnout papírové nebo elektronické exempláře zprávy. Zpráva o odběru vzorku a její kopie se mají vypracovat způsobem, který zajišťuje jejich autentičnost a právní validitu, což může vyžadovat, aby tyto dokumenty podepsal inspektor. V případě odběru vzorků v hospodářství lze požádat farmáře nebo jeho zástupce o podepsání originálu zprávy o odběru vzorku.
Originál zprávy o odběru uchovává příslušný orgán, který musí zabezpečit, aby k tomuto originálu zprávy neměly přístup nepovolané osoby.
Podle potřeby může být o provedeném odběru informován farmář nebo vlastník zařízení.
6. Zpráva o odběru vzorku pro laboratoř
Zpráva o odběru vzorku pro laboratoř zřízenou příslušnými orgány musí být v souladu s požadavky uvedenými v kapitole 7 normy ISO/IEC 17025:2017 (1) a musí obsahovat přinejmenším tyto informace:
|
1. |
adresu příslušných orgánů nebo určených subjektů; |
|
2. |
jméno inspektora nebo identifikační kód; |
|
3. |
úřední kódové číslo vzorku; |
|
4. |
datum odběru; |
|
5. |
druh zvířete; |
|
6. |
matrici vzorku; |
|
7. |
látky nebo skupiny látek pro vyšetření; |
|
8. |
zvláštní poznámky. |
Zpráva o odběru vzorku pro laboratoř musí být přiložena ke vzorku, když se odesílá do laboratoře.
7. Přeprava a uchovávání
Programy kontroly reziduí musí specifikovat vhodné podmínky uchovávání a přepravy pro každou kombinaci analytu/matrice, aby byla zaručena stabilita a celistvost vzorku. Doba přepravy musí být co možná nejkratší a teplota během přepravy musí být dostačující k zajištění stability analytu.
Zvláštní pozornost se věnuje přepravním krabicím, teplotě a době, za kterou musí být odebraný vzorek doručen příslušné laboratoři.
V případě jakéhokoli nedodržení požadavků programu kontroly reziduí laboratoř neprodleně informuje příslušný orgán.
(1) ISO/IEC 17025:2017 Všeobecné požadavky na kompetenci zkušebních a kalibračních laboratoří (kapitola 7.7).