(EU) 2019/1604Prováděcí nařízení Komise (EU) 2019/1604 ze dne 27. září 2019, kterým se mění nařízení (EHS) č. 2568/91 o charakteristikách olivového oleje a olivového oleje z pokrutin a o příslušných metodách analýzy
| Publikováno: | Úř. věst. L 250, 30.9.2019, s. 14-48 | Druh předpisu: | Prováděcí nařízení |
| Přijato: | 27. září 2019 | Autor předpisu: | Evropská komise |
| Platnost od: | 20. října 2019 | Nabývá účinnosti: | 20. října 2019 |
| Platnost předpisu: | Zrušen předpisem (EU) 2022/2104 | Pozbývá platnosti: | 24. listopadu 2022 |
Text předpisu s celou hlavičkou je dostupný pouze pro registrované uživatele.
PROVÁDĚCÍ NAŘÍZENÍ KOMISE (EU) 2019/1604
ze dne 27. září 2019,
kterým se mění nařízení (EHS) č. 2568/91 o charakteristikách olivového oleje a olivového oleje z pokrutin a o příslušných metodách analýzy
EVROPSKÁ KOMISE,
s ohledem na Smlouvu o fungování Evropské unie,
s ohledem na nařízení Evropského parlamentu a Rady (EU) č. 1308/2013 ze dne 17. prosince 2013, kterým se stanoví společná organizace trhů se zemědělskými produkty a zrušují nařízení Rady (EHS) č. 922/72, (EHS) č. 234/79, (ES) č. 1037/2001 a (ES) č. 1234/2007 (1), a zejména na čl. 91 první pododstavec písm. d) uvedeného nařízení,
vzhledem k těmto důvodům:
|
(1) |
Nařízení Komise (EHS) č. 2568/91 (2) definuje fyzikálně-chemické a organoleptické charakteristiky olivového oleje a olivového oleje z pokrutin a stanoví metody hodnocení těchto charakteristik. |
|
(2) |
Tyto metody a limitní hodnoty charakteristik olejů jsou pravidelně aktualizovány na základě stanoviska odborníků v oboru chemie a v souladu s činností probíhající v rámci Mezinárodní rady pro olivy (IOC). |
|
(3) |
Aby bylo na úrovni Unie zajištěno provádění aktuálních mezinárodních norem stanovených IOC, měly by být aktualizovány některé metody analýzy stanovené v nařízení (EHS) č. 2568/91. |
|
(4) |
Obchodní norma IOC byla změněna, pokud jde o stanovení mezní hodnoty volné kyselosti, peroxidového čísla, organoleptického hodnocení (medián vad a medián znaku ovocné chuti a vůně) a rozdílu mezi ECN42 (HPLC) a ECN42 (teoretický výpočet), tak, aby byla v souladu s hodnotami přesnosti analytické metody. |
|
(5) |
V souladu s čl. 2a odst. 5 nařízení (EHS) č. 2568/91 mají členské státy ověřit, zda je vzorek olivového oleje v souladu s deklarovanou kategorií, kontrolou jednotlivých charakteristik stanovených v příloze I uvedeného nařízení, a to buď v jakémkoli pořadí, nebo v pořadí uvedeném v rozhodovacím schématu v příloze Ib uvedeného nařízení. |
|
(6) |
Vzhledem k nejnovějšímu vývoji je vhodné aktualizovat tabulky uvedené v příloze Ib nařízení (EHS) č. 2568/91 a případně v jejím dodatku. Kromě toho se zdá, že pro popis obsahu přílohy Ib je pojem „vývojový diagram“ vhodnější než pojem „rozhodovací schéma“. |
|
(7) |
Bod 9.4 přílohy XII nařízení (EHS) č. 2568/91 definuje medián vad jako medián vady vnímané s největší intenzitou. V souvislosti s oponentními hodnoceními a vzhledem k tomu, že shodu oleje musí posoudit různé zkušební komise, je třeba upřesnit, že rozhodnutí týkající se souladu charakteristik oleje s deklarovanou kategorií se vztahuje výhradně na hodnotu mediánu hlavní vady, bez ohledu na její povahu. |
|
(8) |
Nařízení (EHS) č. 2568/91 by proto mělo být odpovídajícím způsobem změněno. |
|
(9) |
Opatření stanovená tímto nařízením jsou v souladu se stanoviskem Výboru pro společnou organizaci zemědělských trhů, |
PŘIJALA TOTO NAŘÍZENÍ:
Článek 1
Nařízení (EHS) č. 2568/91 se mění takto:
|
1) |
článek 2 se mění takto:
|
|
2) |
v čl. 2a odst. 5 se písmeno b) nahrazuje tímto:
|
|
3) |
tabulka „PŘÍLOHA Obsah“ se nahrazuje tabulkou v příloze I tohoto nařízení; |
|
4) |
příloha I se nahrazuje zněním přílohy II tohoto nařízení; |
|
5) |
bod 2.1 přílohy Ia se nahrazuje tímto:
|
|
6) |
příloha Ib se nahrazuje zněním v příloze III tohoto nařízení; |
|
7) |
příloha V se zrušuje; |
|
8) |
bod 4.2 přílohy VII se nahrazuje tímto:
|
|
9) |
příloha XII se mění v souladu s přílohou IV tohoto nařízení; |
|
10) |
příloha XVII se mění v souladu s přílohou V tohoto nařízení; |
|
11) |
příloha XVIII se mění v souladu s přílohou VI tohoto nařízení; |
|
12) |
příloha XIX se nahrazuje zněním uvedeným v příloze VII tohoto nařízení; |
|
13) |
bod 4.2 přílohy XX se nahrazuje tímto:
|
Článek 2
Toto nařízení vstupuje v platnost dvacátým dnem po vyhlášení v Úředním věstníku Evropské unie.
Toto nařízení je závazné v celém rozsahu a přímo použitelné ve všech členských státech.
V Bruselu dne 27. září 2019.
Za Komisi
předseda
Jean-Claude JUNCKER
(1) Úř. věst. L 347, 20.12.2013, s. 671.
(2) Nařízení Komise (EHS) č. 2568/91 ze dne 11. července 1991 o charakteristikách olivového oleje z pokrutin a o příslušných metodách analýzy (Úř. věst. L 248, 5.9.1991, s. 1).
PŘÍLOHA I
„PŘÍLOHY
OBSAH
|
Příloha I |
Charakteristiky olivového oleje |
|
Příloha Ia |
Odběr vzorků ze šarží olivového oleje a olivového oleje z pokrutin ve spotřebitelských obalech |
|
Příloha Ib |
Vývojový diagram pro ověření souladu vzorku olivového oleje s deklarovanou kategorií |
|
Příloha II |
Stanovení volných mastných kyselin, metoda za studena |
|
Příloha III |
Stanovení peroxidového čísla |
|
Příloha IV |
Stanovení obsahu vosku pomocí kapilární plynové chromatografie |
|
Příloha VII |
Stanovení procentního podílu 2-glyceryl monopalmitátu |
|
Příloha IX |
Spektrofotometrická analýza v ultrafialové oblasti spektra |
|
Příloha X |
Stanovení methylesterů mastných kyselin plynovou chromatografií |
|
Příloha XI |
Stanovení těkavých halogenovaných rozpouštědel v olivovém oleji |
|
Příloha XII |
Metoda mezinárodní rady pro olivový olej pro organoleptické hodnocení panenského olivového oleje |
|
Příloha XV |
Stanovení obsahu oleje v olivových pokrutinách |
|
Příloha XVI |
Stanovení jodového čísla |
|
Příloha XVII |
Metoda pro stanovení stigmastadienolů v rostlinných olejích |
|
Příloha XVIII |
Stanovení rozdílu mezi skutečným a teoretickým obsahem triacylglycerolů s ekvivalentním počtem uhlíkových atomů 42 (ECN 42) |
|
Příloha XIX |
Stanovení složení a obsahu sterolů a alkoholových sloučenin kapilární plynovou chromatografií |
|
Příloha XX |
Metoda stanovení obsahu vosků, methylesterů mastných kyselin a ethylesterů mastných kyselin kapilární plynovou chromatografií |
|
Příloha XXI |
Výsledky kontrol shody provedených u olivového oleje uvedené v čl. 8 odst. 2 |
PŘÍLOHA II
„PŘÍLOHA I
CHARAKTERISTIKY OLIVOVÉHO OLEJE
Jakostní charakteristiky
|
Ethylestery mastných kyselin (mg/kg) |
≤ 35 |
— |
— |
— |
— |
— |
— |
— |
|||||||||||||||||
|
Organoleptické hodnocení |
Medián znaku ovocná chuť a vůně (Mf) |
Mf > 0,0 |
Mf > 0,0 |
— |
— |
— |
— |
— |
— |
||||||||||||||||
|
Medián vad (Md) (*) |
Md = 0,0 |
Md ≤ 3,5 |
Md > 3,5 (1) |
|
|
|
|
|
|||||||||||||||||
|
Delta-K |
≤ 0,01 |
≤ 0,01 |
– |
≤ 0,16 |
≤ 0,15 |
— |
≤ 0,20 |
≤ 0,18 |
|||||||||||||||||
|
K268 nebo K270 |
≤ 0,22 |
≤ 0,25 |
— |
≤ 1,25 |
≤ 1,15 |
— |
≤ 2,00 |
≤ 1,70 |
|||||||||||||||||
|
K232 |
≤ 2,50 |
≤ 2,60 |
— |
— |
— |
— |
— |
— |
|||||||||||||||||
|
Peroxidové číslo (mEq O2/kg) |
≤ 20,0 |
≤ 20,0 |
— |
≤ 5,0 |
≤ 15,0 |
— |
≤ 5,0 |
≤ 15,0 |
|||||||||||||||||
|
Kyselost (%) (*) |
≤ 0,80 |
≤ 2,0 |
> 2,0 |
≤ 0,30 |
≤ 1,00 |
— |
≤ 0,30 |
≤ 1,00 |
|||||||||||||||||
|
Kategorie |
|
|
|
|
|
|
|
|
|||||||||||||||||
Charakteristiky týkající se čistoty
|
2-glyceryl monopalmitát (%) |
≤ 0,9, pokud celkové množství palmitové kyseliny v % ≤ 14,00 % |
≤ 1,0, pokud celkové množství palmitové kyseliny v % > 14,00 % |
≤ 0,9, pokud celkové množství palmitové kyseliny v % ≤ 14,00 % |
≤ 1,0, pokud celkové množství palmitové kyseliny v % > 14,00 % |
≤ 0,9, pokud celkové množství palmitové kyseliny v % ≤ 14,00 % |
≤ 1,1, pokud celkové množství palmitové kyseliny v % > 14,00 % |
≤ 0,9, pokud celkové množství palmitové kyseliny v % ≤ 14,00 % |
≤ 1,1, pokud celkové množství palmitové kyseliny v % > 14,00 % |
≤ 0,9, pokud celkové množství palmitové kyseliny v % ≤ 14,00 % |
≤ 1,0, pokud celkové množství palmitové kyseliny v % > 14,00 % |
≤ 1,4 |
≤ 1,4 |
≤ 1,2 |
|||||||||||||||||
|
Rozdíl: ECN42 (HPLC) a ECN42 (teoretický výpočet) |
≤ |0,20| |
≤ |0,20| |
≤ |0,30| |
≤ |0,30| |
≤ |0,30| |
≤ |0,60| |
≤ |0,50| |
≤ |0,50| |
||||||||||||||||||||||
|
Stigmastadieny (mg/kg) (3) |
≤ 0,05 |
≤ 0,05 |
≤ 0,50 |
— |
— |
— |
— |
— |
||||||||||||||||||||||
|
Úhrn transizomerů kyseliny linolové + linolenové (%) |
≤ 0,05 |
≤ 0,05 |
≤ 0,10 |
≤ 0,30 |
≤ 0,30 |
≤ 0,10 |
≤ 0,35 |
≤ 0,35 |
||||||||||||||||||||||
|
Úhrn transizomerů kyseliny olejové (%) |
≤ 0,05 |
≤ 0,05 |
≤ 0,10 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,40 |
≤ 0,40 |
||||||||||||||||||||||
|
Složení mastných kyselin (2) |
á (%) |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
|||||||||||||||||||||
|
Behenová (%) |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,30 |
≤ 0,30 |
≤ 0,30 |
||||||||||||||||||||||
|
Eikosanová (%) |
≤ 0,50 |
≤ 0,50 |
≤ 0,50 |
≤ 0,50 |
≤ 0,50 |
≤ 0,50 |
≤ 0,50 |
≤ 0,50 |
||||||||||||||||||||||
|
Arachidová (%) |
≤ 0,60 |
≤ 0,60 |
≤ 0,60 |
≤ 0,60 |
≤ 0,60 |
≤ 0,60 |
≤ 0,60 |
≤ 0,60 |
||||||||||||||||||||||
|
Linolenová (%) |
≤ 1,00 |
≤ 1,00 |
≤ 1,00 |
≤ 1,00 |
≤ 1,00 |
≤ 1,00 |
≤ 1,00 |
≤ 1,00 |
||||||||||||||||||||||
|
Myristová (%) |
≤ 0,03 |
≤ 0,03 |
≤ 0,03 |
≤ 0,03 |
≤ 0,03 |
≤ 0,03 |
≤ 0,03 |
≤ 0,03 |
||||||||||||||||||||||
|
Kategorie |
|
|
|
|
|
|
|
|
||||||||||||||||||||||
|
Vosky (mg/kg) (**) |
C42 + C44 + C46 ≤ 150 |
C42 + C44 + C46 ≤ 150 |
C40 + C42 + C44 + C46 ≤ 300 (6) |
C40 + C42 + C44 + C46 ≤ 350 |
C40 + C42 + C44 + C46 ≤ 350 |
C40 + C42 + C44 + C46 > 350 (7) |
C40 + C42 + C44 + C46 > 350 |
C40 + C42 + C44 + C46 > 350 |
|||||||||||||||||
|
Erythrodiol a uvaol (%) (**) |
≤ 4,5 |
≤ 4,5 |
≤ 4,5 (6) |
≤ 4,5 |
≤ 4,5 |
> 4,5 (7) |
4,5 |
> 4,5 |
|||||||||||||||||
|
Steroly celkem (mg/kg) |
≥ 1 000 |
≥ 1 000 |
≥ 1 000 |
≥ 1 000 |
≥ 1 000 |
≥ 2 500 |
≥ 1 800 |
≥ 1 600 |
|||||||||||||||||
|
Složení sterolů |
Delta-7-stigmastenol (4) (%) |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
||||||||||||||||
|
Zjevný β–sitosterol (5) (%) |
≥ 93,0 |
≥ 93,0 |
≥ 93,0 |
≥ 93,0 |
≥ 93,0 |
≥ 93,0 |
≥ 93,0 |
≥ 93,0 |
|||||||||||||||||
|
Stigmasterol (%) |
< kamp. |
< kamp. |
— |
< kamp. |
< kamp. |
— |
< kamp. |
< kamp. |
|||||||||||||||||
|
Kampesterol (4) (%) |
≤ 4,0 |
≤ 4,0 |
≤ 4,0 |
≤ 4,0 |
≤ 4,0 |
≤ 4,0 |
≤ 4,0 |
≤ 4,0 |
|||||||||||||||||
|
Brassikasterol (%) |
≤ 0,1 |
≤ 0,1 |
≤ 0,1 |
≤ 0,1 |
≤ 0,1 |
≤ 0,2 |
≤ 0,2 |
≤ 0,2 |
|||||||||||||||||
|
Cholesterol (%) |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
≤ 0,5 |
|||||||||||||||||
|
Kategorie |
|
|
|
|
|
|
|
|
|||||||||||||||||
Poznámky:
|
a) |
Výsledky zkoušek musí být uvedeny na stejný počet desetinných míst, jaký je předepsán pro každou charakteristiku. Poslední desetinné místo se přitom musí zaokrouhlit nahoru, pokud je číslice na dalším desetinném místě vyšší než 4. |
|
b) |
Pokud jakákoli charakteristika neodpovídá uvedeným hodnotám, může být olivový olej zařazen do jiné kategorie nebo označen jako nesplňující požadavky pro účely tohoto nařízení. |
|
c) |
V případě lampantového olivového oleje se mohou obě jakostní charakteristiky označené hvězdičkou (*) současně lišit od mezních hodnot stanovených pro tuto kategorii. |
|
d) |
Charakteristiky olejů označené dvěma hvězdičkami (**) znamenají, že u surového oleje z pokrutin se od uvedených hodnot mohou lišit obě mezní hodnoty současně. V případě olivového oleje z pokrutin a rafinovaného olivového oleje z pokrutin se od uvedených hodnot může lišit jedna mezní hodnota. |
Dodatek
Rozhodovací schémata
Kampesterol – rozhodovací schéma pro panenské a extra panenské olivové oleje:
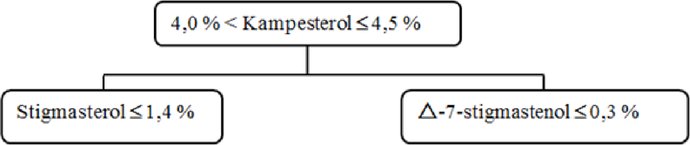
Ostatní parametry musí být v souladu s mezními hodnotami stanovenými v tomto nařízení.
Delta-7-stigmastenol – rozhodovací schéma pro:
|
— |
extra panenské a panenské olivové oleje
|
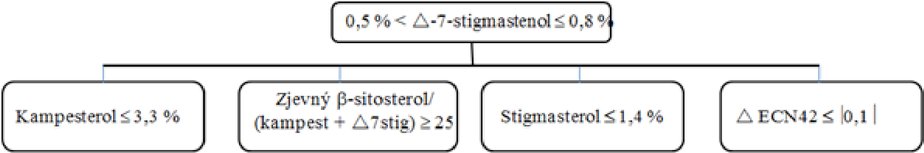
Ostatní parametry musí být v souladu s mezními hodnotami stanovenými v tomto nařízení.
|
— |
olivové oleje z pokrutin (surové a rafinované)
|

Ostatní parametry musí být v souladu s mezními hodnotami stanovenými v tomto nařízení.
(1) Medián vad může být nejvýše roven 3,5, je-li medián znaku ovocná chuť a vůně je roven 0,0.
(2) Obsah ostatních mastných kyselin (%): palmitová: 7,50–20,00; palmitolejová: 0,30–3,50; heptadekanová: ≤ 0,40; heptadecenová ≤ 0,60; stearová: 0,50–5,00; olejová: 55,00–83,00; linolová: 2,50–21,00.
(3) Celkové množství izomerů, které by mohly (nebo nemohly) být separovány kapilární kolonou.
(4) Viz dodatek k této příloze.
(5) Zjevný β-sitosterol: Delta-5,23-stigmastadienol + chlerosterol + beta-sitosterol + sitostanol + delta-5-avenasterol + delta-5,24-stigmastadienol.
(6) Oleje s obsahem vosků od 300 mg/kg do 350 mg/kg jsou považovány za lampantové olivové oleje, pokud je celkový obsah alifatických alkoholů nejvýše roven 350 mg/kg nebo pokud obsah erythrodiolu a uvaolu je nejvýše roven 3,5 %.
(7) Oleje s obsahem vosků od 300 mg/kg do 350 mg/kg jsou považovány za surový olej z pokrutin, pokud je celkový obsah alifatických alkoholů vyšší než 350 mg/kg a pokud je obsah erythrodiolu a uvaolu větší než 3,5 %.
PŘÍLOHA III
„PŘÍLOHA Ib
VÝVOJOVÝ DIAGRAM PRO OVĚŘENÍ SOULADU VZORKU OLIVOVÉHO OLEJE S DEKLAROVANOU KATEGORIÍ
Všeobecná tabulka
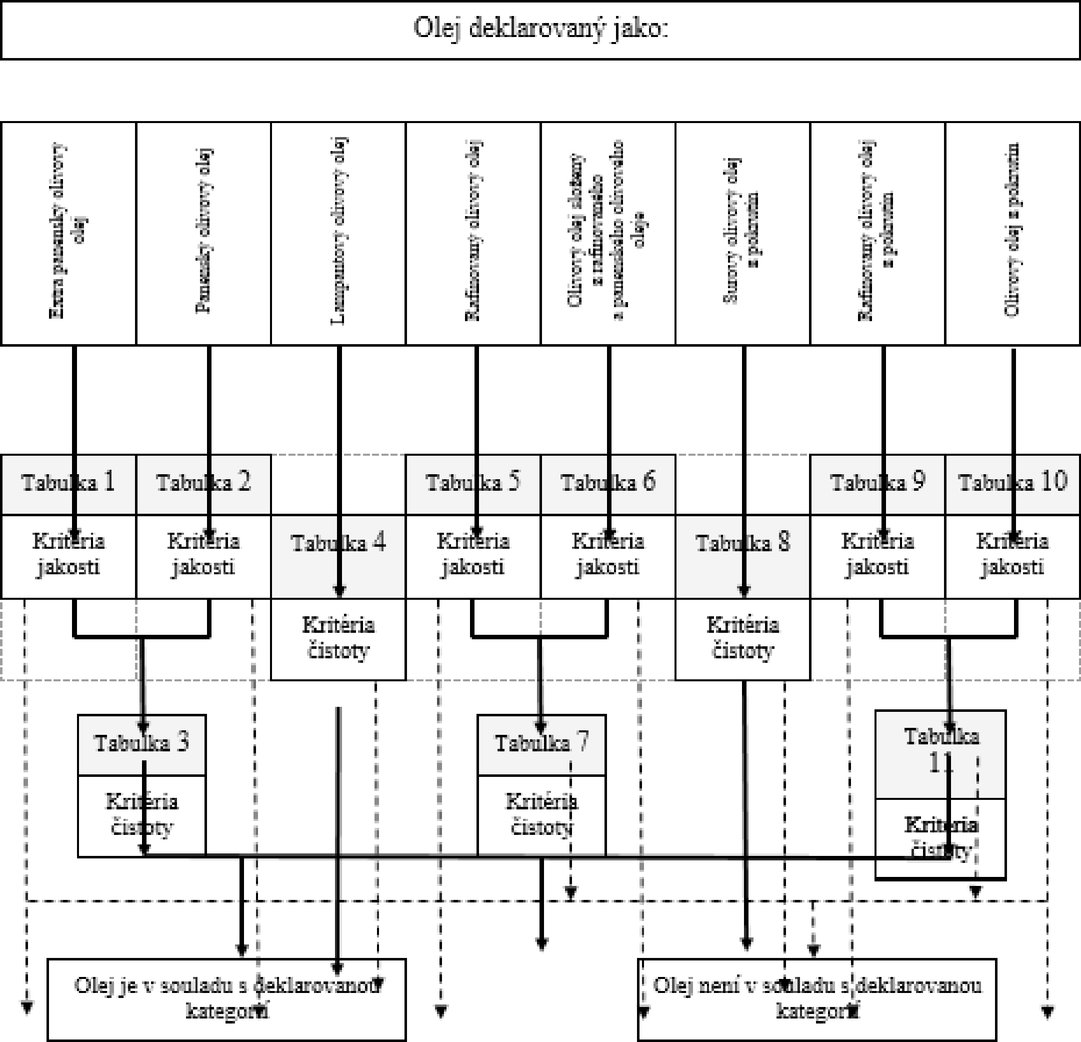
Tabulka 1 – Extra panenský olivový olej – kritéria jakosti
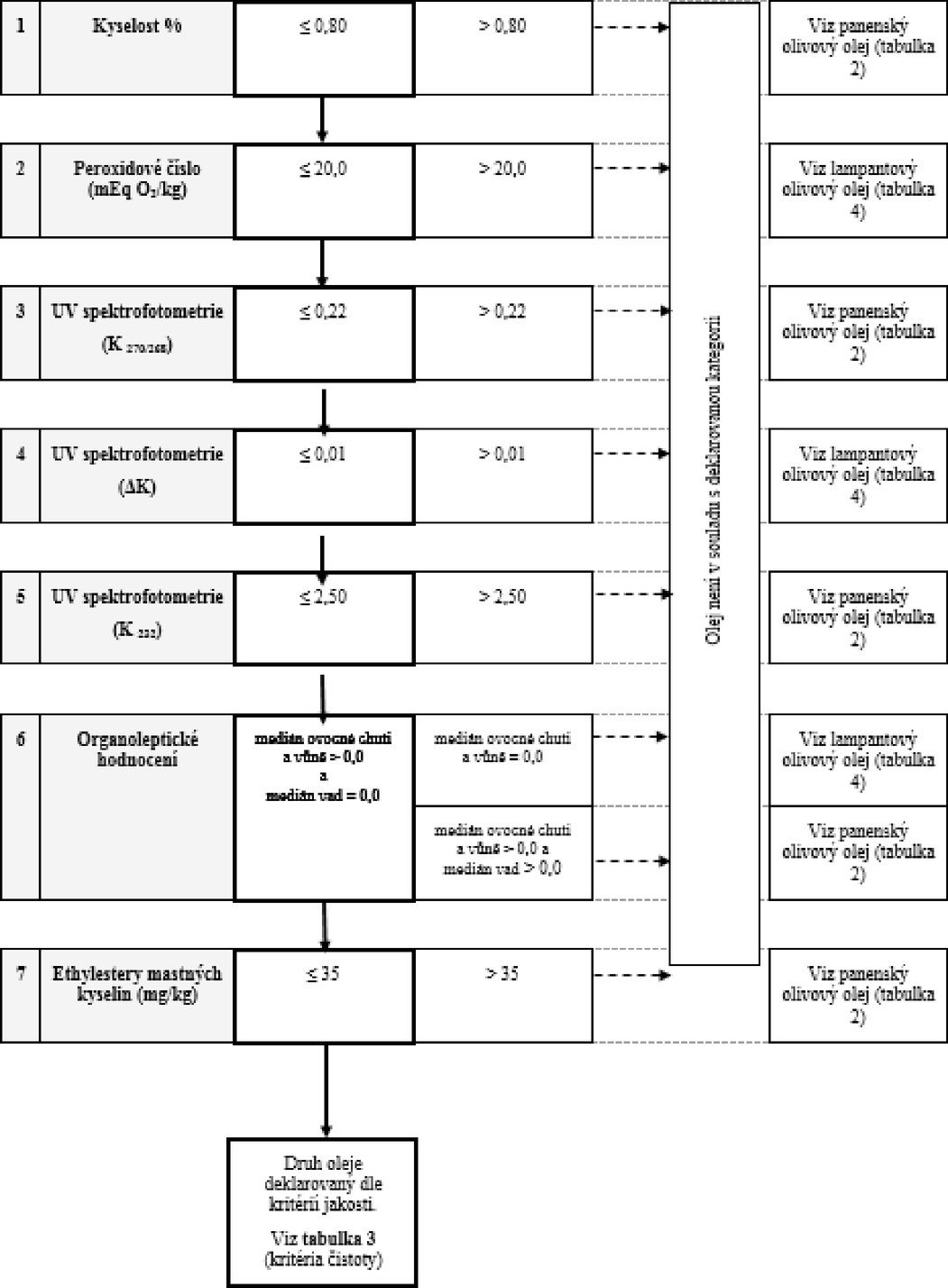
Tabulka 2 – Extra panenský olivový olej – kritéria jakosti
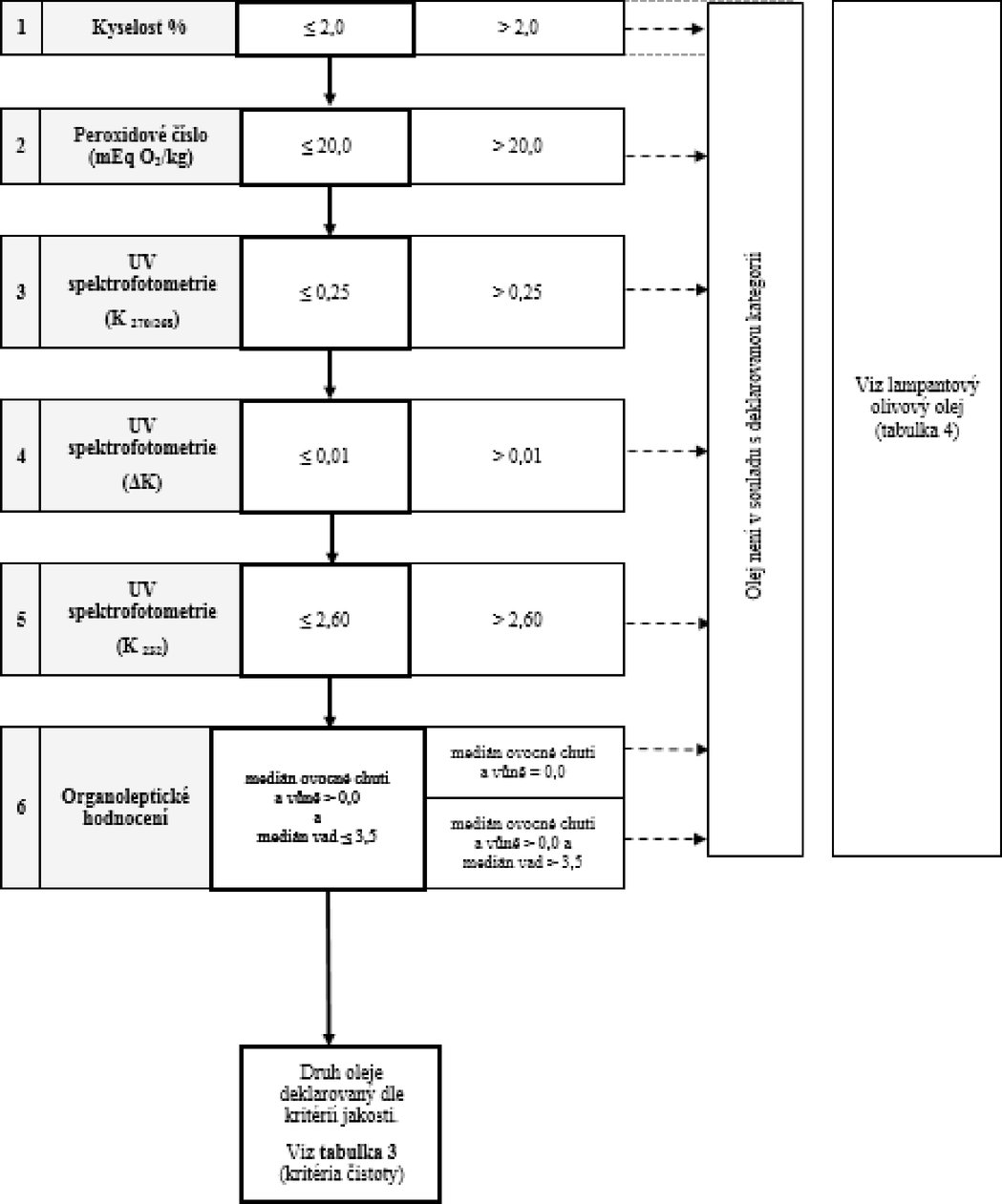
Tabulka 3 – Extra panenský olivový olej a panenský olivový olej – kritéria čistoty
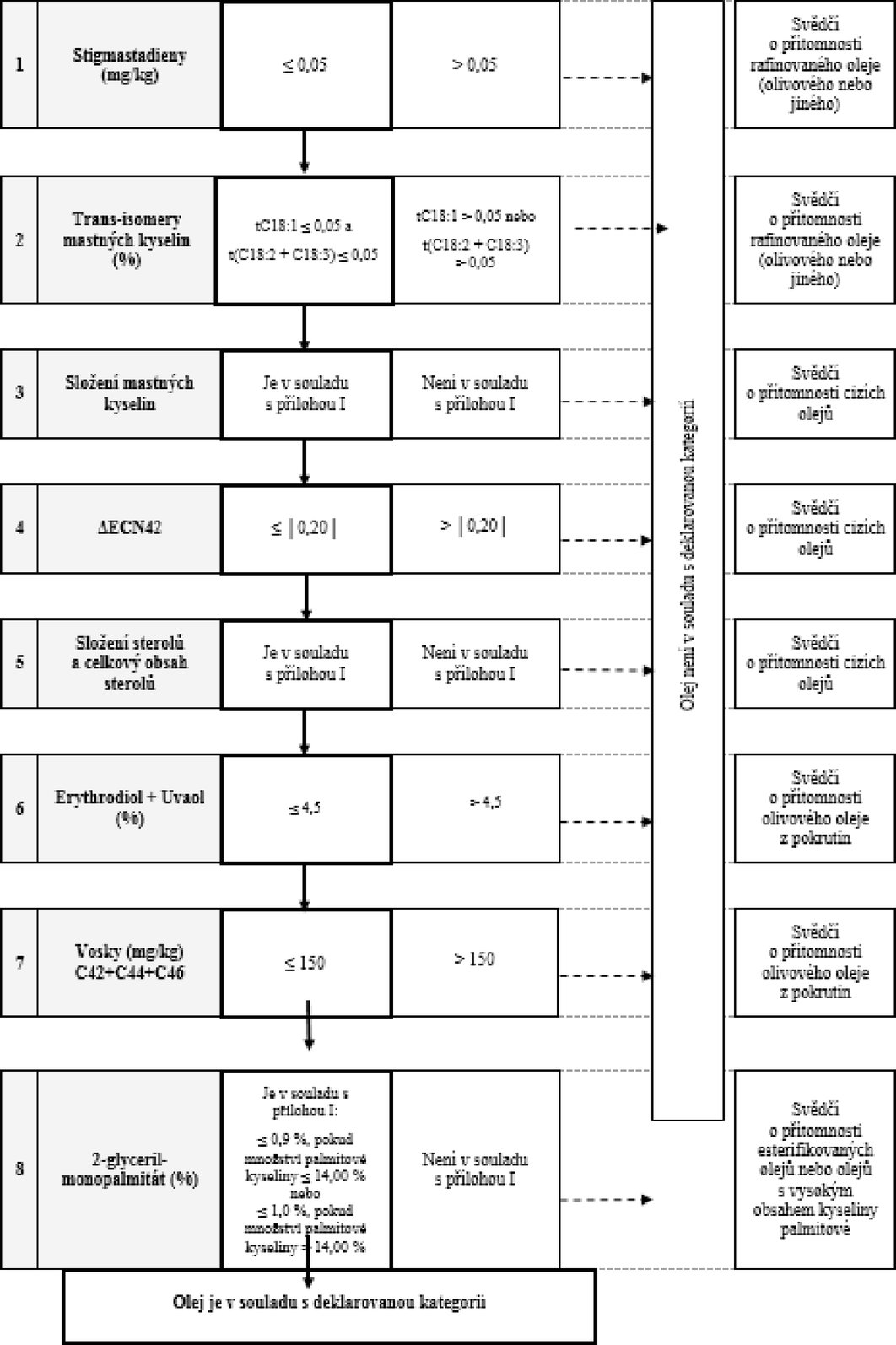
Tabulka 4 – Lampantový olivový olej – kritéria čistoty
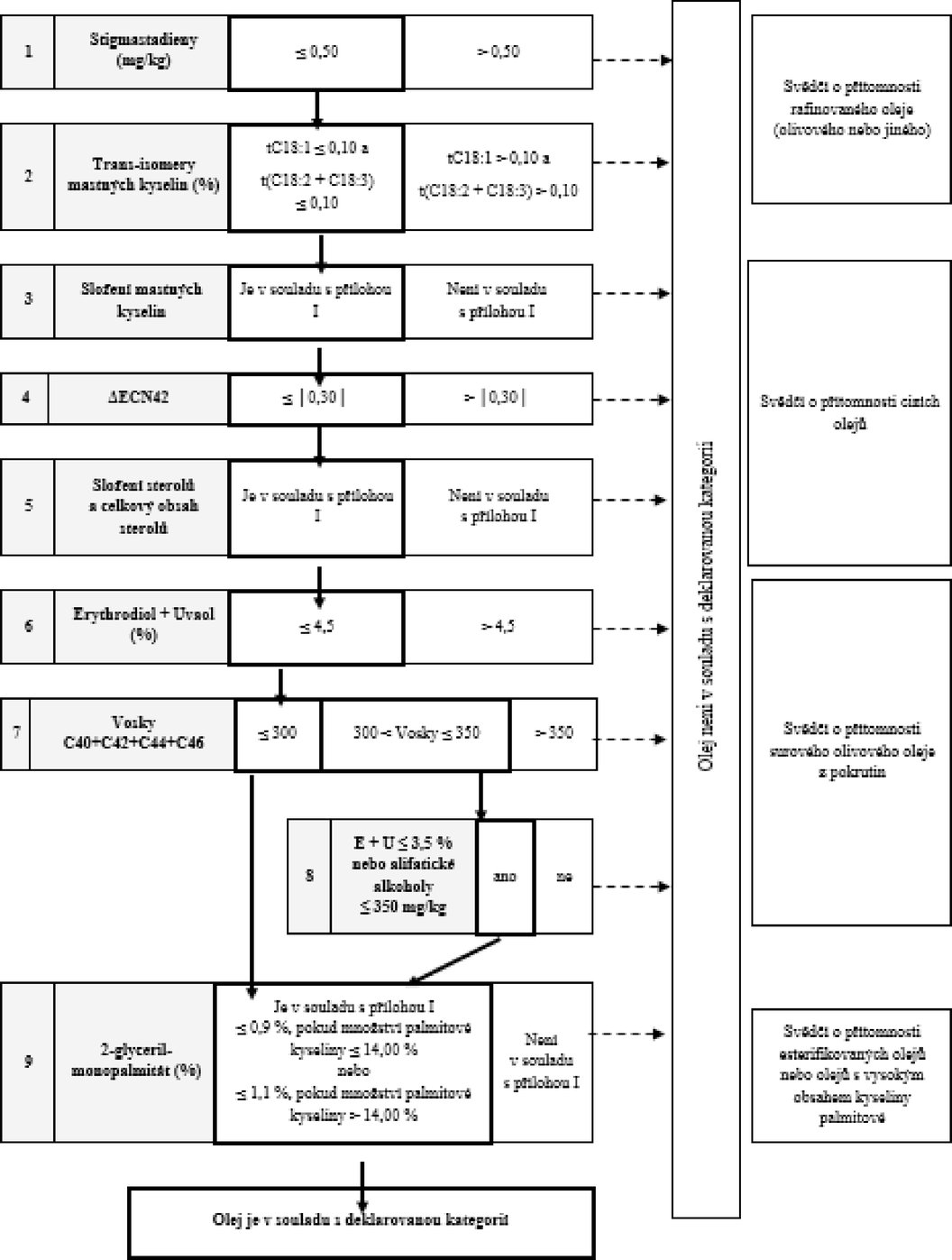
Tabulka 5 – Rafinovaný olivový olej – kritéria jakosti
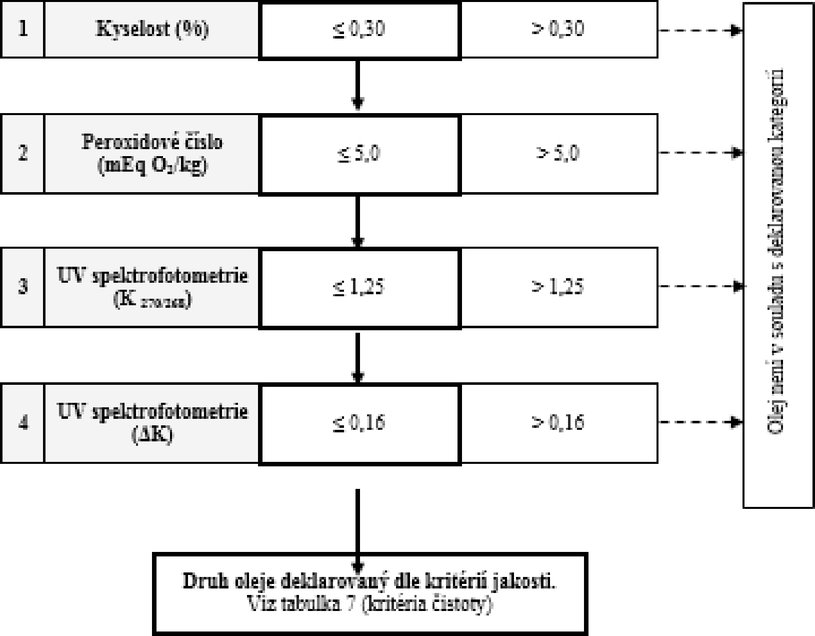
Tabulka 6 – Olivový olej (složený z rafinovaného a panenského olivového oleje) – kritéria jakosti
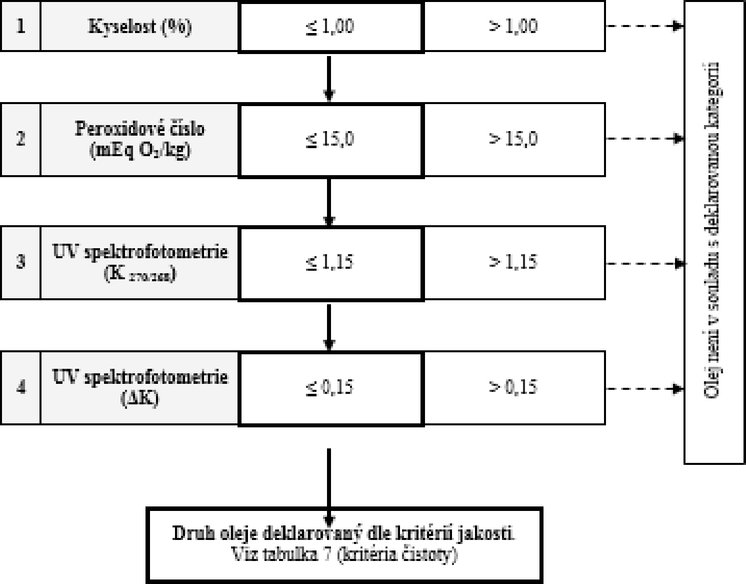
Tabulka 7 – Rafinovaný olivový olej a olivový olej složený z rafinovaného a panenského olivového oleje – kritéria čistoty
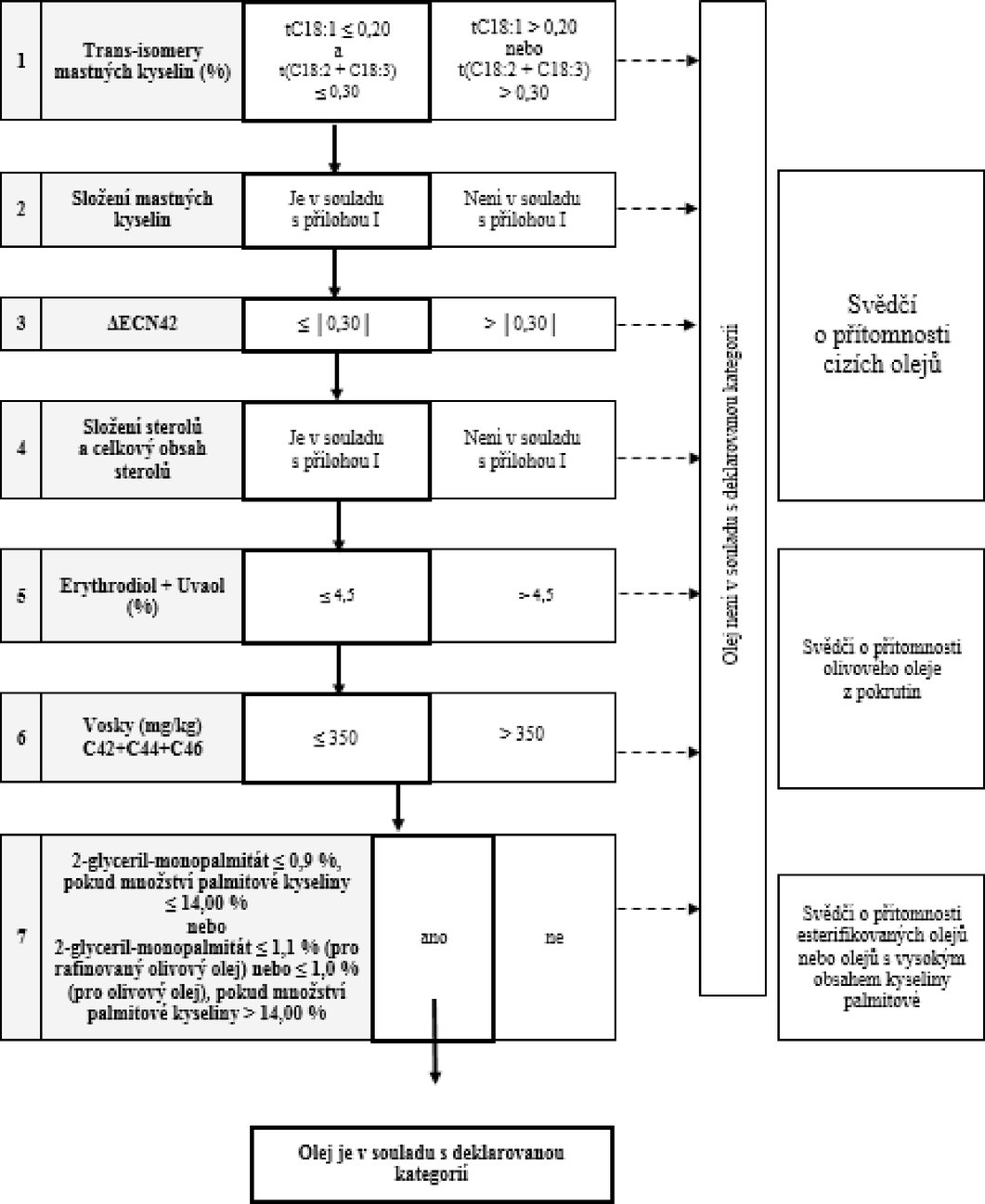
Tabulka 8 – Surový olivový olej z pokrutin – kritéria čistoty
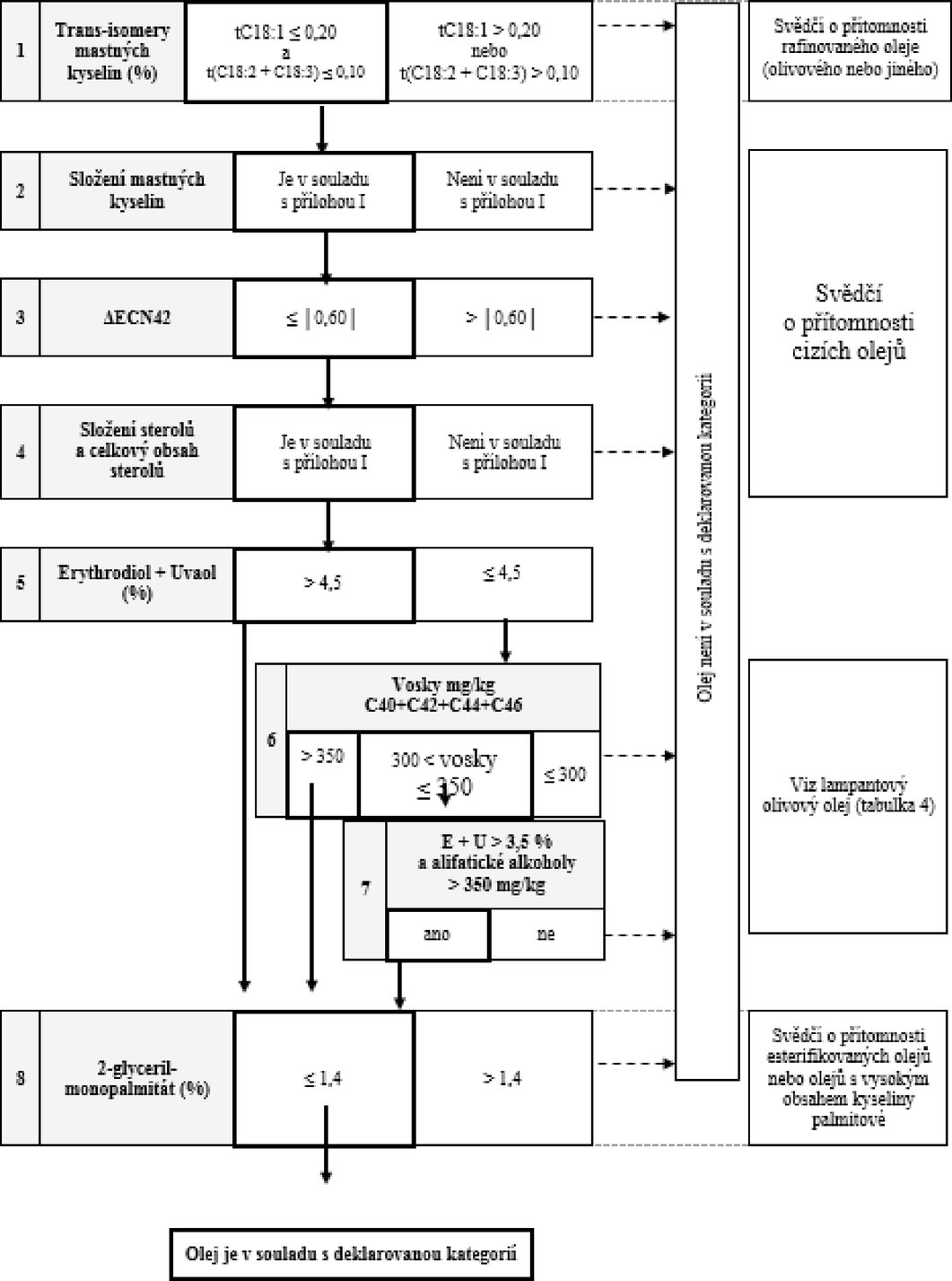
Tabulka 9 – Rafinovaný olivový olej z pokrutin – kritéria jakosti
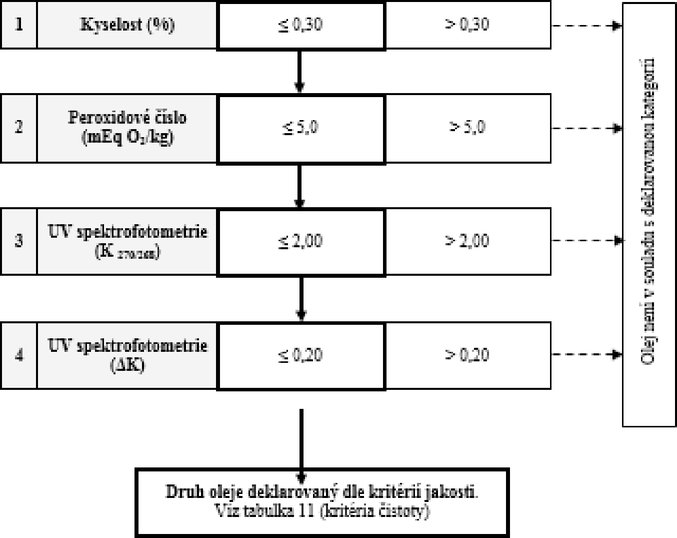
Tabulka 10 – Olivový olej z pokrutin – kritéria jakosti
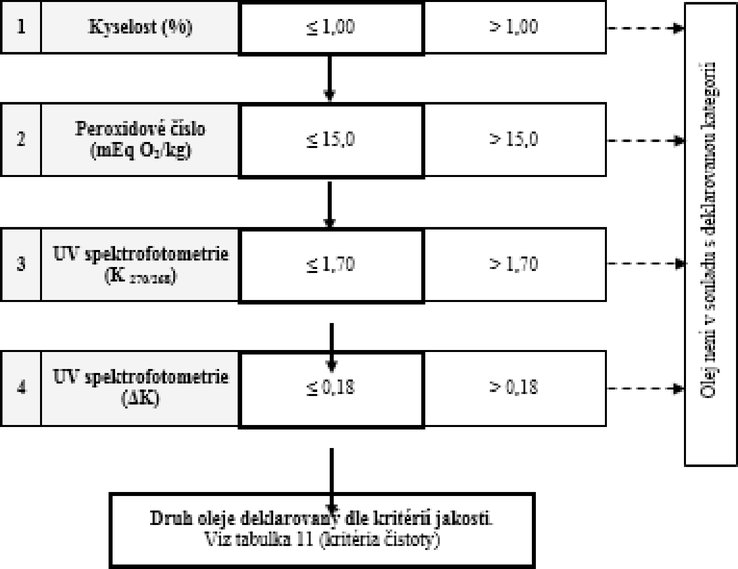
Tabulka 11 – Rafinovaný olivový olej z pokrutin a olivový olej z pokrutin – kritéria čistoty
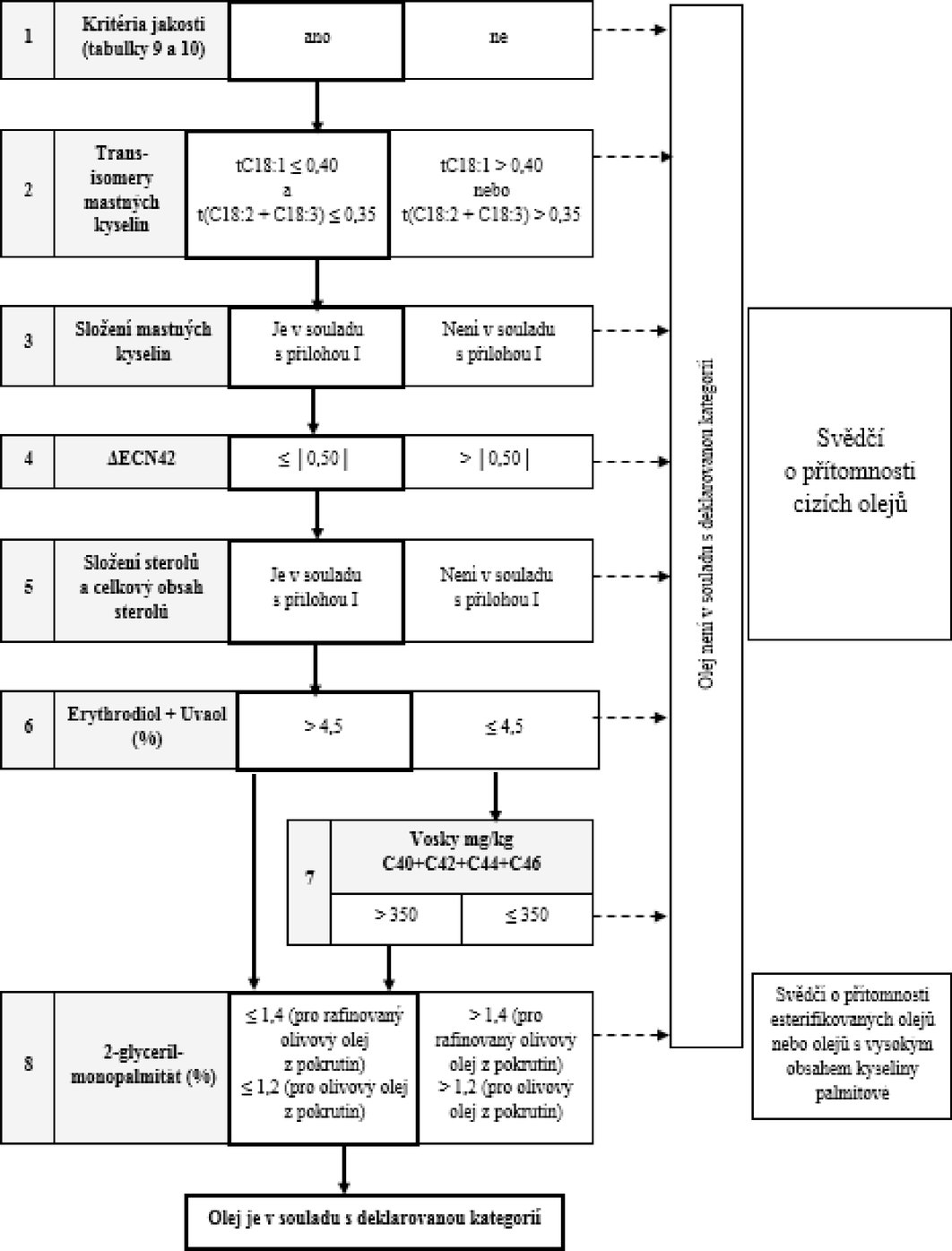
PŘÍLOHA IV
Příloha XII se mění takto:
|
1) |
bod 3.3 se nahrazuje tímto: „3.3 Nepovinná terminologie pro účely označování Vedoucí zkušební komise může na požádání potvrdit, že hodnocené oleje splňují definice a rozpětí odpovídající výhradně následujícím termínům v závislosti na intenzitě a vnímání znaků. Pozitivní znaky (ovocná chuť a vůně, hořký a štiplavý): podle intenzity vnímání:
Seznam termínů podle intenzity vnímání:
|
|
2) |
bod 9.4 se nahrazuje tímto: „9.4 Klasifikace oleje Olej se zařadí do následujících kategorií podle mediánu vad a mediánu znaku ovocné chuti a vůně. Medián vad se stanovuje jako medián vady vnímané s největší intenzitou. Medián vad a medián znaku ovocné chuti a vůně se vyjadřuje s přesností na jedno desetinné místo. Klasifikace oleje se provádí porovnáváním hodnoty mediánu vad a mediánu znaku ovocné chuti a vůně s níže uvedenými referenčními intervaly. Při stanovování hranice těchto intervalů byla zohledněna chyba metody, proto se tyto hranice považují za absolutní. Počítačové programy umožňují zobrazení klasifikace v tabulce statistických údajů nebo na grafu.
V případě hodnocení, která se provádějí za účelem sledování shody, se provádí jedna zkouška. V případě oponentních hodnocení se musí analýza provést dvakrát při různých zkušebních sezeních. Výsledky duplicitní analýzy musí být statisticky homogenní. (Viz bod 9.5). Pokud tomu tak není, musí se vzorek znovu dvakrát analyzovat. Konečná hodnota mediánu klasifikačních znaků se vypočte na základě průměru obou mediánů.“ |
PŘÍLOHA V
Příloha XVII se mění takto:
|
1) |
bod 5.1 se nahrazuje tímto:
|
|
2) |
v bodě 6.3.3 se doplňuje nový text, který zní: „Poznámka 10: Pokud se stigmastadienoly vyskytují v koncentraci vyšší než 4 mg/kg a je-li požadována kvantifikace, musí být použita metoda Mezinárodní rady pro olivy pro stanovení sterenů v rafinovaných olejích.“ |
PŘÍLOHA VI
Příloha XVIII se mění takto:
|
1) |
bod 4.2.1 se nahrazuje tímto:
|
|
2) |
vkládá se nový bod 4.2.12, který zní:
|
PŘÍLOHA VII
„PŘÍLOHA XIX
STANOVENÍ SLOŽENÍ A OBSAHU STEROLŮ A ALKOHOLOVÝCH SLOUČENIN KAPILÁRNÍ PLYNOVOU CHROMATOGRAFIÍ
1. OBLAST PŮSOBNOSTI
Tato metoda popisuje postup pro stanovení obsahu jednotlivých alkoholových sloučenin a celkový obsah alkoholových sloučenin v olivových olejích a olivových olejích z pokrutin a jejich směsích.
Alkoholové sloučeniny v olivových olejích a olivových olejích z pokrutin sestávají z alifatických alkoholů, sterolů a triterpenických dialkoholů.
2. PODSTATA METODY
Oleje, k nimž je přidán α-cholestanol a 1-ikosanol jako vnitřní standard, jsou zmýdelněny pomocí ethanolového roztoku hydroxidu draselného; nezmýdelnitelné látky se následně extrahují pomocí diethyletheru.
Jednotlivé sloučeniny alkoholu se od nezmýdelnitelných látek oddělí tenkovrstvou chromatografií na bazické silikagelové desce (referenční metoda) nebo vysoce účinnou kapalinovou chromatografií pomocí kolony naplněné silikagelem. Frakce oddělená ze silikagelu se převede na trimethylsilylethery a analyzuje se pomocí kapilární plynové chromatografie.
ČÁST 1
PŘÍPRAVA NEZMÝDELNITELNÝCH LÁTEK
1. OBLAST PŮSOBNOSTI
Tato část popisuje přípravu a extrakci nezmýdelnitelných látek. Zahrnuje přípravu a extrakci nezmýdelnitelných látek z olivového oleje a olivového oleje z pokrutin.
2. PODSTATA METODY
Vzorek se zmýdelní varem pod zpětným chladičem s ethanolovým roztokem hydroxidu draselného. Nezmýdelnitelné látky se extrahují pomocí diethyletheru.
3. PŘÍSTROJE A POMŮCKY
Obvyklé laboratorní vybavení a zejména:
|
3.1. |
Baňka s kulatým dnem na 250 ml vybavená zpětným chladičem se zábrusem. |
|
3.2. |
Dělící nálevka na 500 ml. |
|
3.3. |
Baňka na 250 ml. |
|
3.4. |
Mikrostříkačky na 100 μl a 500 μl. |
|
3.5. |
Válcová filtrační nálevka s pórovitou fritou G3 (pórovitost 15 až 40 μm) o průměru přibližně 2 cm a výšce 5 cm, vhodná pro vakuovou filtraci a s vnějším zábrusem. |
|
3.6. |
Erlenmeyerova baňka na 50 ml s vnitřním zábrusem pro použití s filtrační nálevkou (3.5). |
|
3.7. |
Zkumavka na 10 ml s kónickým dnem a skleněnou zátkou. |
|
3.8. |
Exsikátor s chloridem vápenatým. |
4. ČINIDLA
|
4.1. |
Hydroxid draselný, minimální titr 85 %. |
|
4.2. |
Hydroxid draselný ve formě ethanolového roztoku o koncentraci přibližně 2 M. 130 g hydroxidu draselného (4.1) se za současného chlazení rozpustí ve 200 ml destilované vody a doplní se do jednoho litru ethanolem (4.7). Tento roztok je nutno uchovávat v dobře uzavřených skleněných láhvích z tmavého skla a skladovat po dobu maximálně dvou dnů. |
|
4.3. |
Diethylether čistoty p.a. |
|
4.4. |
Bezvodý síran sodný čistoty p.a. |
|
4.5. |
Aceton čistoty pro chromatografii. |
|
4.6. |
Diethylether čistoty pro chromatografii. |
|
4.7. |
Ethanol čistoty p.a. |
|
4.8. |
Ethylacetát čistoty p.a. |
|
4.9. |
Vnitřní standard, α-cholestanol o čistotě vyšší než 99 % (čistota musí být kontrolována pomocí analýzy plynovou chromatografií). |
|
4.10. |
Vnitřní standardní roztok α-cholestanolu, roztok (0,2 m/V) v ethylacetátu (4.8). |
|
4.11. |
Roztok fenolftaleinu 10 g/l v ethanolu (bod 4.7). |
|
4.12. |
0,1 % roztok 1-ikosanolu v ethylacetátu (m/V) (vnitřní standard). |
5. POSTUP
Pomocí mikrostříkačky na 500 μl (3.4) se do baňky na 250 ml (3.1) dá objem vnitřního standardního roztoku α-cholestanolu (4.10) a objem 1-ikosanolu (4.12) obsahující množství cholestanolu a ikosanolu rovnající se přibližně 10 % obsahu sterolu a alkoholu ve vzorku. Například při analýze olivového oleje se na 5 g vzorku olivového oleje přidá 500 μl roztoku α-cholestanolu (4.10) a 250 μl roztoku 1-ikosanolu (4.12). Při analýze olivového oleje z pokrutin se přidá 1 500 μl roztoku α-cholestanolu (4.10) a 1500 μl roztoku 1-ikosanolu (4.12). Vzorek se nechá odpařit do sucha v mírném proudu dusíku a v teplé vodní lázni. Po vychladnutí baňky se do stejné baňky naváží 5,00 ± 0,01 g vysušeného přefiltrovaného vzorku.
|
Poznámka 1: |
Živočišné nebo rostlinné oleje obsahující větší množství cholesterolu mohou vykazovat pík s retenčním časem identickým cholestanolu. Pokud k tomu dojde, musí být sterolová frakce analyzována jednou s vnitřním standardem a jednou bez něj. |
Přidá se 50 ml ethanolového roztoku hydroxidu draselného o koncentraci 2 M (4.2) a malé množství pemzy, připojí se zpětný chladič a zahřeje se do mírného varu, dokud nedojde ke zmýdelnění (roztok se vyčeří). V zahřívání se pokračuje dalších 20 minut a poté se přes chladič přidá 50 ml destilované vody, chladič se pak odpojí a baňka se vychladí na teplotu přibližně 30 °C.
Obsah baňky se kvantitativně převede do dělicí nálevky na 500 ml (3.2) a propláchne se několika dávkami destilované vody (50 ml). Přidá se přibližně 80 ml diethyletheru (4.6), vše se důkladně protřepe po dobu přibližně 60 sekund a pravidelně se uvolňuje tlak obracením dělicí nálevky a otevíráním kohoutku. Nechá se odstát až do úplného oddělení obou fází (viz poznámka 2). Poté se co nejvíce mýdlového roztoku odčerpá do druhé dělicí nálevky. Stejným způsobem se provedou další dvě extrakce vodně-alkoholové fáze, vždy s použitím 60 až 70 ml diethyletheru (4.6).
|
Poznámka 2: |
Všechny emulze je možné odstranit přidáním malých množství ethanolu (4.7). |
Všechny tři etherové extrakty se smíchají v jedné dělicí nálevce obsahující 50 ml vody. Dále se promývá vodou (50 ml), dokud se promývací voda při přidání kapky roztoku fenolftaleinu nepřestane zbarvovat růžově (4.11). Odstraní se vodná fáze, etherová fáze se přefiltruje na bezvodém síranu sodném (4.4) do předem zvážené baňky na 250 ml, přičemž nálevka a filtr se promyjí malými množstvími diethyletheru (4.6).
Rozpouštědlo se nechá odpařit destilací ve vakuu v rotačním odpařovači při 30 °C. Přidá se 5 ml acetonu (4.5) a těkavé rozpouštědlo se zcela odstraní mírným proudem dusíku. Zbytek se suší v sušárně při 103 ± 2 °C po dobu 15 min. Po vychlazení v exsikátorech se zváží s přesností na 0,1 mg.
ČÁST 2
SEPARACE FRAKCÍ ALKOHOLOVÝCH SLOUČENIN
1. OBLAST PŮSOBNOSTI
Nezmýdelnitelné látky, jejichž příprava je popsána v části 1, se nechají frakcionovat v různých alkoholových sloučeninách, alifatických alkoholech, sterolech a triterpenických dialkoholech (erythrodiolu a uvaolu).
2. PODSTATA METODY
Nezmýdelnitelné látky lze frakcionovat pomocí základní tenkovrstvé chromatografie (referenční metoda), zviditelnit a odpovídající pásma seškrábat a extrahovat. Alternativní metodou separace je vysoce účinná kapalinová chromatografie za použití kolony se silikagelem a UV detektoru, jejíž pomocí lze získat jednotlivé frakce. Alifatické a triterpenické alkoholy, sterol a triterpenické dialkoholy se společně izolují.
3. PŘÍSTROJE A POMŮCKY
Obvyklé laboratorní vybavení a zejména:
|
3.1. |
Kompletní zařízení pro tenkovrstvou chromatografii se skleněnými deskami o rozměrech 20 × 20 cm. |
|
3.2. |
Ultrafialová lampa s vlnovou délkou 366 nebo 254 nm. |
|
3.3. |
Mikrostříkačky na 100 μl a 500 μl. |
|
3.4. |
Válcová filtrační nálevka s pórovitou fritou G3 (pórovitost 15 až 40 μm) o průměru přibližně 2 cm a výšce 5 cm, vhodná pro vakuovou filtraci a s vnějším zábrusem. |
|
3.5. |
Erlenmeyerova baňka na 50 ml s vnitřním zábrusem pro použití s filtrační nálevkou (3.4). |
|
3.6. |
Zkumavka na 10 ml s kónickým dnem a skleněnou zátkou. |
|
3.7. |
Exsikátor s chloridem vápenatým. |
|
3.8. |
Systém vysoce účinné kapalinové chromatografie složený z:
|
|
3.9. |
Kolony pro vysoce účinnou kapalinovou chromatografii (25 cm x 4 mm vnitřní průměr) se silikagelem 60 (velikost částic 5 μm). |
|
3.10. |
Injekčního filtru, 0,45 μm. |
|
3.11. |
Erlenmeyerovy baňky na 25 ml. |
4. ČINIDLA
|
4.1. |
Hydroxid draselný, minimální titr 85 %. |
|
4.2. |
Hydroxid draselný ve formě ethanolového roztoku o koncentraci přibližně 2 M. 130 g hydroxidu draselného (4.1) se za současného chlazení rozpustí ve 200 ml destilované vody a doplní se do jednoho litru ethanolem (4.9). Tento roztok je nutno uchovávat v dobře uzavřených skleněných láhvích z tmavého skla a skladovat po dobu maximálně dvou dnů. |
|
4.3. |
Diethylether čistoty p.a. |
|
4.4. |
Hydroxid draselný ve formě ethanolového roztoku o koncentraci přibližně 0,2 M. 13 g hydroxidu draselného (4.1) se rozpustí ve 20 ml destilované vody a doplní se do jednoho litru ethanolem (4.9). |
|
4.5. |
Silikagelem potažené skleněné desky (20 x 20) bez fluorescenčního indikátoru o tloušťce 0,25 mm (obchodně dostupné, připraveny k použití). |
|
4.6. |
Aceton čistoty pro chromatografii. |
|
4.7. |
n-hexan čistoty pro chromatografii. |
|
4.8. |
Diethylether čistoty pro chromatografii. |
|
4.9. |
Ethanol čistoty p.a. |
|
4.10. |
Ethylacetát čistoty p.a. |
|
4.11. |
Referenční roztok pro tenkovrstvou chromatografii: cholesterol, fytosteroly, alkoholy a erythrodiol – 5 % roztok v ethylacetátu (4.10). |
|
4.12. |
Roztok 2,7-dichlor-fluoresceinu, 0,2 % v ethanolovém roztoku. Mírně se alkalizuje přidáním několika kapek 2M alkoholového roztoku hydroxidu draselného (4.2). |
|
4.13. |
n-hexan (4.7)/diethylether (4.8) směs 65:35 (V/V). |
|
4.14. |
Mobilní fáze vysoce účinné kapalinové chromatografie: n-hexan (4.7)/diethylether (4.8) směs 1:1 (V/V). |
5. REFERENČNÍ METODA SEPARACE ALKOHOLOVÝCH SLOUČENIN POMOCÍ ZÁKLADNÍ TENKOVRSTVÉ CHROMATOGRAFICKÉ DESKY
Příprava bazických desek pro tenkovrstvou chromatografii. Silikagelové desky (4.5) se na 10 sekund ponoří nebo namočí zhruba 4 cm hluboko do ethanolového roztoku hydroxidu draselného o koncentraci 0,2 M (4.4), poté se nechají dvě hodiny sušit v digestoři a nakonec na jednu hodinu umístí do sušárny vyhřáté na 100 °C.
Desky se vyjmou ze sušárny a až do okamžiku použití uloží do exsikátoru s chloridem vápenatým (3.7) (takto upravené desky musí být použity během patnácti dnů).
Vyvíjecí komora se naplní směsí hexanu a diethyletheru (4.13) (viz poznámka 3) do výšky přibližně 1 cm. Vyvíjecí komora se uzavře vhodným krytem a nejméně půl hodiny ponechá v klidu a chladnu, aby se mohla vytvořit rovnováha mezi parami a kapalinou. Na vnitřní povrch vyvíjecí komory je možné připojit proužky filtračního papíru namočené v eluční směsi za účelem zkrácení doby vyvolávání přibližně o jednu třetinu a docílení stejnoměrnější, pravidelné eluce složek.
|
Poznámka 3: |
Vyvíjecí roztok musí být pro každou analýzu vyměněn, aby bylo dosaženo dokonale reprodukovatelných podmínek vyvíjení. Lze použít alternativní rozpouštědlo 50:50 (V/V) n-hexanu a diethyletheru. |
Připraví se přibližně 5 % roztok nezmýdelnitelné látky, jejíž příprava je popsána v části 1, v ethylacetátu (4.10) a 0,3 ml tohoto roztoku se pomocí mikrostříkačky na 100 μl (3.3) nastříkne jako úzký a rovnoměrný pruh k dolnímu okraji (2 cm) chromatografické desky (4.5). Ve stejné úrovni se nanese 2 až 3 μl referenčního roztoku materiálu (4.11) za účelem stanovení sterolového pásma a pásma triterpenických dialkoholů a alkoholů po dokončení vyvíjení.
Deska se umístí do vyvíjecí komory (3.1). Teplota okolí se udržuje mezi 15 a 20 °C (viz poznámka 4). Vyvíjecí komora se okamžitě uzavře a vzorek se nechá eluovat, dokud čelo rozpouštědla nedosáhne přibližně 1 cm od horního okraje desky. Deska se poté vyjme z vyvíjecí komory a rozpouštědlo se nechá odpařit pod proudem horkého vzduchu nebo se deska na chvíli nechá v digestoři.
|
Poznámka 4: |
Vyšší teplota může zhoršit oddělování. |
Deska se lehce a rovnoměrně postříká roztokem 2,7-dichlor-fluoresceinu (4.12) a nechá se uschnout. Při pozorování pod UV lampou (3.2) je možné stanovit sterolové pásmo a pásmo triterpenických dialkoholů a alkoholů porovnáním skvrn vytvořených referenčním roztokem (bod 4.11). Obrysy pásem podél okrajů fluorescence se vyznačí černým značkovačem (viz obrázek tenkovrstvé desky č. 1).
Silikagel se z označené oblasti seškrábe pomocí kovové stěrky. Odebraná hmota se rozmělní na malé části a převede do filtrační nálevky (3.4). Přidá se 10 ml horkého ethylacetátu (4.10). Obsah se důkladně promíchá kovovou stěrkou a přefiltruje (v případě potřeby ve vakuu). Filtrát se shromáždí v Erlenmeyerově baňce (3.5) připojené na filtrační nálevku.
Zbytek v baňce se třikrát propláchne diethyletherem (4.3) (přibližně 10 ml při každém propláchnutí). Filtrát se přitom sbírá do stejné baňky připojené k nálevce. Filtrát se odpaří na objem přibližně 4 až 5 ml a zbytkový roztok se převede do předem zvážené zkumavky na 10 ml (3.6). Zkumavka se vysuší pod mírným proudem dusíku. Zbytek se znovu rozpustí několika kapkami acetonu (4.6) a opět vysuší. Zbytek ve zkumavce je tvořen sterolem a triterpenickýcmi dialkoholy nebo frakcemi alkoholů a triterpenických dialkoholů.
6. SEPARACE ALKOHOLOVÉ FRAKCE POMOCÍ VYSOCE ÚČINNÉ KAPALINOVÉ CHROMATOGRAFIE
Nezmýdelnitelné látky získané podle postupu v části 1 se rozpustí ve 3 ml mobilní fáze (4.14), roztok se přefiltruje přes injekční filtr (3.10) a uchová se.
200 μl přefiltrovaného roztoku nezmýdelnitelných látek se vstříkne do zařízení vysoce účinné kapalinové chromatogrofie (3.8).
Nechá se proběhnout separace vysoce účinnou kapalinovou chromatografií při toku 0,8 ml/min. Prvních pět minut se nezohlední; do 25 ml Erlenmeyerových baněk (3.11) se zachytí alifatické a triterpenické alkoholy odseparované mezi 5. a 10. minutou a steroly a erytrodiol a uvaol odseparované mezi 11. a 25. minutou (viz poznámka 5).
Separaci lze sledovat UV detektorem při vlnové délce 210 nm nebo detektorem indexu lomu (viz obrázek 6).
Frakce se odpaří do sucha a připraví se na chromatografickou analýzu.
|
Poznámka 5: |
Je potřeba pečlivě sledovat tlak čerpadla vysoce účinné kapalinové chromatografie, protože diethylether může způsobit zvýšení tlaku – pro udržení tlaku pod kontrolou je zapotřebí upravit průtok. |
ČÁST 3
ANALÝZA FRAKCÍ ALKOHOLOVÝCH SLOUČENIN PLYNOVOU CHROMATOGRAFIÍ
1. OBLAST PŮSOBNOSTI
Tato část popisuje použití plynové chromatografie v kapilární koloně pro stanovení kvalitativního a kvantitativního složení alkoholových sloučenin izolovaných podle metody uvedené v části 2.
2. PODSTATA METODY
Frakce získané z nezmýdelnitelných látek pomocí tenkovrstvé chromatografie nebo vysokoúčinné kapalinové chromatografie se derivatizují na trimethylsilylethery a analyzují pomocí kapilární plynové chromatografie opatřené nástřikovým zařízením s děličem a plameno-ionizačním detektorem.
3. PŘÍSTROJE A POMŮCKY
Obvyklé laboratorní vybavení a zejména:
|
3.1. |
Zkumavka na 10 ml s kónickým dnem a skleněnou zátkou. |
|
3.2. |
Plynový chromatograf vhodný pro použití s kapilární kolonou, opatřený nástřikovým zařízením s děličem složený z:
|
|
3.3. |
Kapilární kolona z taveného křemene dlouhá 20 až 30 m, s vnitřním průměrem 0,25 až 0,32 mm, potažená 5 % difenyl – 95 % dimethylpolysiloxanem (stacionární fáze SE-52 nebo SE-54 nebo ekvivalentní), o jednotné tloušťce 0,10 až 0,30 μm. |
|
3.4. |
Mikrostříkačka na 10 μl pro plynovou chromatografii opatřená tvrzenou jehlou pro nástřik pomocí děliče. |
4. ČINIDLA
|
4.1. |
Bezvodý pyridin pro chromatografii. |
|
4.2. |
Hexamethyl disilazan čistoty p.a. |
|
4.3. |
Trimethyl-chlorsilan čistoty p.a. |
|
4.4. |
Zkušební roztoky sterol(trimethylsilyl)etherů. Připraví se ze sterolů a z erythrodiolu získaných z olejů, které je obsahují, těsně před použitím. |
|
4.5. |
Standardní roztoky trimethylsilyletherů alifatických alkoholů C20 až C28. Mohou se připravit ze směsí čistých alkoholů v době požadovaného použití. |
|
4.6. |
Nosný plyn: vodík nebo helium, čisté pro plynovou chromatografii. |
|
4.7. |
Pomocné plyny: vodík, helium, dusík a vzduch, čisté pro plynovou chromatografii. |
|
4.8. |
Činidlo pro silylaci tvořené směsí pyridinu, hexamethyl-disilazanu a trimethyl-chlorsilanu v poměru 9:3:1 (V/V/V). |
|
4.9. |
n-hexan čistoty pro chromatografii. |
5. PŘÍPRAVA TRIMETHYLSILYLETHERŮ
Do zkumavky (3.1) obsahující frakci alkoholových sloučenin se přidá činidlo pro silylaci (4.8) (viz poznámka 6), v množství 50 μl na každý mg alkoholové sloučeniny. Přitom je nutné zabránit jakékoli absorpci vlhkosti (viz poznámka 7).
|
Poznámka 6: |
Roztoky připravené k použití jsou obchodně dostupné. Dostupná jsou rovněž další silylační činidla, jako například N,O-bis-(trimethylsilyl)trifluoracetamid + 1 % trimethyl-chlor-silan pro smíchání se stejným objemem bezvodého pyridinu. Pyridin lze nahradit stejným množstvím acetonitrilu. |
|
Poznámka 7: |
Vznik slabé opalescence je normální a nezpůsobuje žádnou anomálii. Tvorba bílých vloček nebo existence růžového zabarvení jsou známkami přítomné vlhkosti nebo degradace činidla. V tomto případě se zkouška musí opakovat (pouze v případě, že byl použit hexamethyl-disilazan/trimethyl-chlorsilan). |
Zkumavka (3.1) se uzavře a pečlivě protřepe bez převracení, dokud se sloučeniny nerozpustí. Poté se nechá v klidu nejméně 15 minut při laboratorní teplotě a následně několik minut odstřeďuje. Čirý roztok je připraven pro plynovou chromatografickou analýzu.
6. ANALÝZA PLYNOVOU CHROMATOGRAFIÍ
6.1. Přípravné operace a kondicionace kapilární kolony
Kapilární kolona se připojí (3.3) k plynovému chromatografu tak, že vstup kolony se připojí k nástřikovému zařízení s děličem a výstup kolony se připojí k detektoru.
Provede se kompletní kontrola plynového chromatografu (těsnost plynových okruhů, účinnost detektoru, účinnost oddělovacího a zapisovacího systému atd.).
Kolonu, která má být použita poprvé, je nutno nejprve kondicionovat. Kolonou se nechá protékat malé množství plynu, poté se zapne plynový chromatograf a nechá se postupně zahřívat, dokud není dosaženo teploty nejméně o 20 °C vyšší, než je provozní teplota (viz poznámka 8). Tato teplota se udržuje nejméně dvě hodiny a poté se systém převede do pracovního režimu (regulace průtoku plynu, oddělování, zapálení plamene, připojení výpočetního systému, nastavení teploty kolony, detektoru, injektoru atd.) a zaznamená se signál s citlivostí, která činí nejméně dvojnásobek citlivosti uvažované pro provedení analýzy. Základní linie záznamu musí být rovná, bez jakýchkoli píků nebo driftů. Záporné drifty jsou známkou nedokonalé těsnosti spojů kolony, zatímco kladné svědčí o nedostatečné kondicionaci kolony.
|
Poznámka 8: |
Teplota kondicionace musí být nejméně o 20 °C nižší než maximální teplota použitelná pro uvažovanou stacionární fázi. |
6.2. Provozní podmínky
Je zapotřebí optimalizovat teplotní program a průtok nosného plynu tak, aby se získaly chromatogramy podobné chromatogramům na obrázcích 3 až 6.
Byly testovány a shledány jako užitečné následující parametry:
6.2.1. Alifatické alkoholy
|
Program pece |
180 °C (8 min.) → 260 °C (při 5 °C/min.) → 260 °C (15 min.) |
|
Teplota injektoru |
280 °C |
|
Teplota detektoru: |
290 °C |
|
Lineární rychlost nosného plynu: |
helium (20 až 30 cm/s), vodík (30 až 50 cm/s) |
|
Oddělovací poměr: |
1:50 až 1:100 |
|
Vstříknutý objem: |
0,5 až 1 μl trimethylsilyletherového roztoku |
6.2.2. Sterol a triterpenické dialkoholy
|
Program pece |
260 ± 5 °C isotermický |
|
Teplota injektoru |
280 – 300 °C |
|
Teplota detektoru |
280 – 300 °C |
|
Lineární rychlost nosného plynu |
helium (20 až 30 cm/s), vodík (30 až 50 cm/s) |
|
Oddělovací poměr: |
1:50 až 1:100 |
|
Vstříknutý objem: |
0,5 až 1 μl trimethylsilyletherového roztoku |
Tyto podmínky se mohou měnit podle charakteristik kolony a plynového chromatografu s cílem zajistit, aby chromatogramy splňovaly tyto požadavky:
|
— |
retenční čas alkoholu C26 musí být 18 ± 5 minut, |
|
— |
pík alkoholu C22 musí být u olivového oleje 80 ± 20 % plného rozsahu a u oleje z pokrutin 40 ± 20 % plného rozsahu, |
|
— |
retenční čas β-sitosterolu by měl být 20 ± 5 minut, |
|
— |
pík kampesterolu musí být u olivového oleje (střední obsah 3 %) 20 ± 5 % plného rozsahu, |
|
— |
všechny přítomné steroly musí být odděleny. Kromě separace musí být píky též zcela rozlišeny, tj. dráha záznamu píku se musí vrátit na základní linii dříve, než se zvedne do dalšího píku. Neúplné rozlišení je však tolerováno za předpokladu, že pík s relativním retenčním časem 1,02 (sitostanol) je možné kvantifikovat pomocí kolmé úsečky. |
6.3. Analytický postup
Do mikrostříkačky na 10 μl (3.4) se natáhne 1 μl hexanu, poté 0,5 μl vzduchu a nakonec 0,5 až 1 μl zkušebního roztoku. Píst mikrostříkačky se povytáhne tak, aby se vyprázdnila jehla. Jehla se zavede přes membránu injektoru a přibližně po jedné nebo dvou sekundách se rychle nastříkne roztok. Přibližně po pěti sekundách se jehla pomalu vytáhne. Lze užít rovněž automatický injektor.
Zaznamenávání je prováděno, dokud přítomná trimethylsilyletherová směs příslušných alkoholových sloučenin není zcela eluována. Základní linie musí i nadále splňovat požadavky odpovídajících provozních podmínek (6.2.1 nebo 6.2.2).
6.4. Identifikace píků
Identifikace jednotlivých píků se provádí podle retenčních časů a porovnáním se směsí alifatických a triterpenických alkoholů nebo sterolu a triterpenických dialkoholů TMSE, analyzovanou za stejných podmínek. Chromatogram frakce alifatických a triterpenických dialkoholů je znázorněn na obrázku 3 a odpovídající chromatogramy sterolů a triterpenických dialkoholů na obrázku 2.
Alifatické alkoholy se eluují v tomto pořadí: C20-ol (V.S.), C22-ol, C23-ol, C24-ol, C25-ol, C26-ol, C27-ol a C28-ol.
Steroly a triterpenické dialkoholy se eluují v tomto pořadí: cholesterol, brassikasterol, ergosterol, 24-methylen-cholesterol, kampesterol, kampestanol, stigmasterol, Δ7-kampesterol, Δ5,23-stigmastadienol, chlerosterol, β-sistosterol, sitostanol, Δ5-avenasterol, Δ5,24-stigmastadienol, Δ7-stigmastenol, Δ7-avenasterol, erythrodiol a uvaol.
6.5. Kvantitativní vyhodnocení
Pomocí systému sběru dat se vypočítají plochy píků 1-ikosanolu a alifatických alkoholů C22, C24, C26 a C28. Faktor odezvy 1-eikosanolu by se měl považovat za rovný 1.
Pomocí výpočetního systému se vypočítá plocha píků α-cholestanolu, sterolů a triterpenických dialkoholů. Píky jakýchkoli sloučenin, které nejsou mezi sloučeninami uvedenými v tabulce 1 (výpočet pro ergosterol se provádět nesmí) se ignorují. Faktor odezvy α-cholestanolu by se měl považovat za rovný 1.
Obsah jednotlivých alkoholových sloučenin vyjádřený v mg/kg tuku se vypočítá podle vzorce:
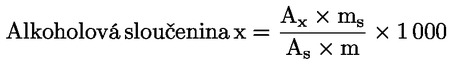
kde:
|
Ax |
= |
plocha píku alkoholové sloučeniny x vypočtená dle výpočetního systému. |
|
As |
= |
plocha píku 1-ikosanolu/α-cholestanolu vypočtená dle výpočetního systému. |
|
ms |
= |
množství přidaného 1-ikosanolu/α-cholestanolu v mg. |
|
m |
= |
hmotnost vzorku použitého pro stanovení v g. |
7. VYJÁDŘENÍ VÝSLEDKŮ
Uvádí se obsah jednotlivých alifatických a triterpenických alkoholů v mg/kg tuku a jejich souhrn jako „celkový obsah alifatických alkoholů“. Celkový obsah je součet C22, C24, C26 a C28.
Složení jednotlivých alkoholových sloučenin by se mělo vyjadřovat s přesností na jedno desetinné místo.
Celková koncentrace sterolů by se měla vyjádřit v celých číslech.
Procentní podíl jednotlivých sterolů z poměru plochy příslušného píku k celkové ploše píků sterolů se vypočítá podle vzorce:
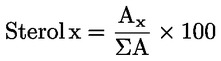
kde:
|
Ax |
= |
plocha píku sterolu x, |
|
ΣA |
= |
celková plocha píků sterolů. |
Zjevný β -sitosterol: Δ5,23-stigmastadienol + chlerosterol + β-sitosterol + sitostanol + Δ5-avenasterol + Δ5,24-stigmastadienol.
Vypočítá se procentní podíl erythrodiolu a uvaolu:
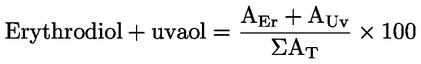
kde:
|
AEr |
= |
plocha erythrodiolu vypočtená dle výpočetního systému, |
|
AUv |
= |
plocha uvaolu vypočtená dle výpočetního systému. |
|
Σ AT |
= |
součet ploch píků sterolu + erythrodiolu + uvaolu vypočtených dle výpočetního systému. |
Kromě výpočtu relativního podílu jednotlivých sterolů a triterpenických dialkoholů a celkové koncentrace sterolů se musí vypočítat také koncentrace erythrodiolu a uvaolu a jejich součet v mg/kg tuku, a to podle následujících vzorců:
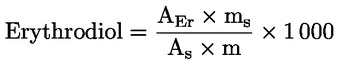
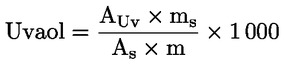
kde:
|
Aer |
= |
plocha píku erythrodiolu vypočtená dle výpočetního systému, |
|
Auv |
= |
plocha uvaolu vypočtená dle výpočetního systému, |
|
As |
= |
plocha píku α-cholestanolu vypočtená dle výpočetního systému, |
|
ms |
= |
množství přidaného α-cholestanolu v mg. |
|
m |
= |
hmotnost vzorku použitého pro stanovení v g. |
Dodatek
|
|
|

Obrázek 1 – tenkovrstvá chromatografie nezmýdelnitelné frakce z olivového oleje z pokrutin dvakrát eluované s hexanem:diethyletherem (65:35), vyvinuté s SO4H2 (50 %) a zahřáté. Pásma, která je třeba seškrábat, se nachází v obdélníku; 1 jsou pásma alifatických alkoholů a 2 sterolů a triterpenických dialkoholů.
Tabulka I – Relativní retenční časy sterolů
|
Pík |
Identifikace |
Relativní retenční čas |
||
|
Kolona SE 54 |
Kolona SE 52 |
|||
|
1 |
Cholesterol |
Δ-5-cholesten-3β-ol |
0,67 |
0,63 |
|
2 |
Cholestanol |
5α-cholestan-3β-ol |
0,68 |
0,64 |
|
3 |
Brassikasterol |
[24S]-24-methyl-Δ-5,22-cholestadien-3β-ol |
0,73 |
0,71 |
|
* |
Ergosterol |
[24S]-24-methyl-Δ-5,7,22 cholestatrien-3β-ol |
0,78 |
0,76 |
|
4 |
24-methylen-cholesterol |
24-methylen-Δ-5,24-cholestadien-3β-o1 |
0,82 |
0,80 |
|
5 |
Kampesterol |
(24R)-24-methyl-Δ-5-cholesten-3β-ol |
0,83 |
0,81 |
|
6 |
Kampestanol |
(24R)-24-methyl-cholestan-3β-ol |
0,85 |
0,82 |
|
7 |
Stigmasterol |
(24S)-24-ethyl-Δ-5,22-cholestadien-3β-ol |
0,88 |
0,87 |
|
8 |
Δ-7-kampesterol |
(24R)-24-methyl-Δ-7-cholesten-3β-ol |
0,93 |
0,92 |
|
9 |
Δ-5,23-stigmastadienol |
(24R,S)-24-ethyl-Δ-5,23-cholestadien-3β-ol |
0,95 |
0,95 |
|
10 |
Chlerosterol |
(24S)-24-ethyl-Δ-5,25-cholestadien-3β-ol |
0,96 |
0,96 |
|
11 |
β-sitosterol |
(24R)-24-ethyl-Δ-5-cholesten-3β-ol |
1,00 |
1,00 |
|
12 |
Sitostanol |
24-ethyl-cholestan-3β-ol |
1,02 |
1,02 |
|
13 |
Δ-5-avenasterol |
(24Z)-24-ethyliden-Δ-cholesten-3β-ol |
1,03 |
1,03 |
|
14 |
Δ-5,24-stigmastadienol |
(24R,S)-24-ethyl-Δ-5,24-cholestadien-3β-ol |
1,08 |
1,08 |
|
15 |
Δ-7-stigmastenol |
(24R,S)-24-ethyl-Δ-7-cholesten-3β-ol |
1,12 |
1,12 |
|
16 |
Δ-7-avenasterol |
(24Z)-24-ethyliden-Δ-7-cholesten-3β-ol |
1,16 |
1,16 |
|
17 |
Erythrodiol |
5α-olean-12-en-3β,28-diol |
1,41 |
1,41 |
|
18 |
Uvaol |
Δ12-ursen-3β,28-diol |
1,52 |
1,52 |
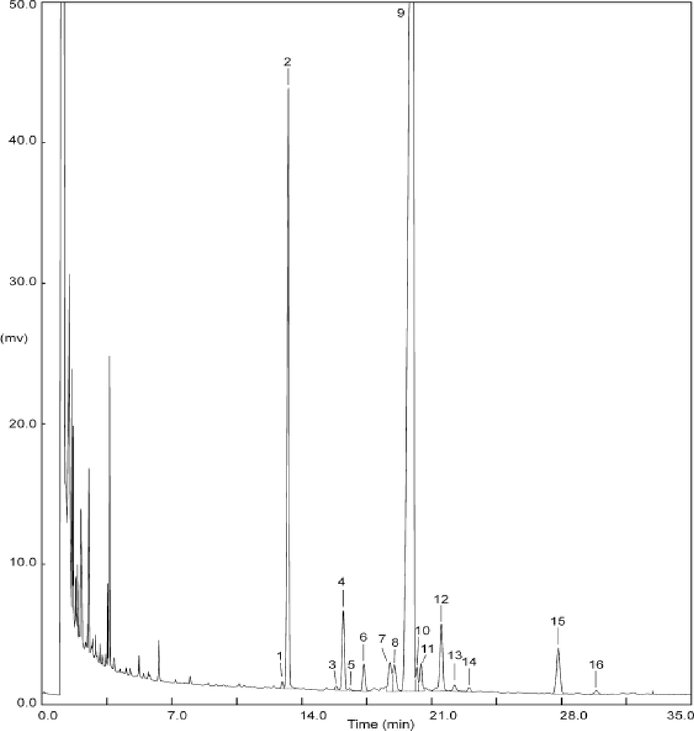
Obrázek 2 – GC-FID chromatografický profil sterolu a triterpenických dialkoholů z rafinovaného olivového oleje. (1) Cholesterol, (2) α-cholestanol (V.S.), (3) 24-methylencholesterol, (4) kampesterol, (5) kampestanol, (6) stigmasterol, (7) Δ5,23-stigmastadienol, (8) chlerosterol, (9) β-sitosterol, (10) sitostanol, (11) Δ5-avenasterol, (12) Δ5,24-stigmastadienol, (13) Δ7-stigmastenol, (14) Δ7-avenasterol, (15) erythrodiol, (16) uvaol.
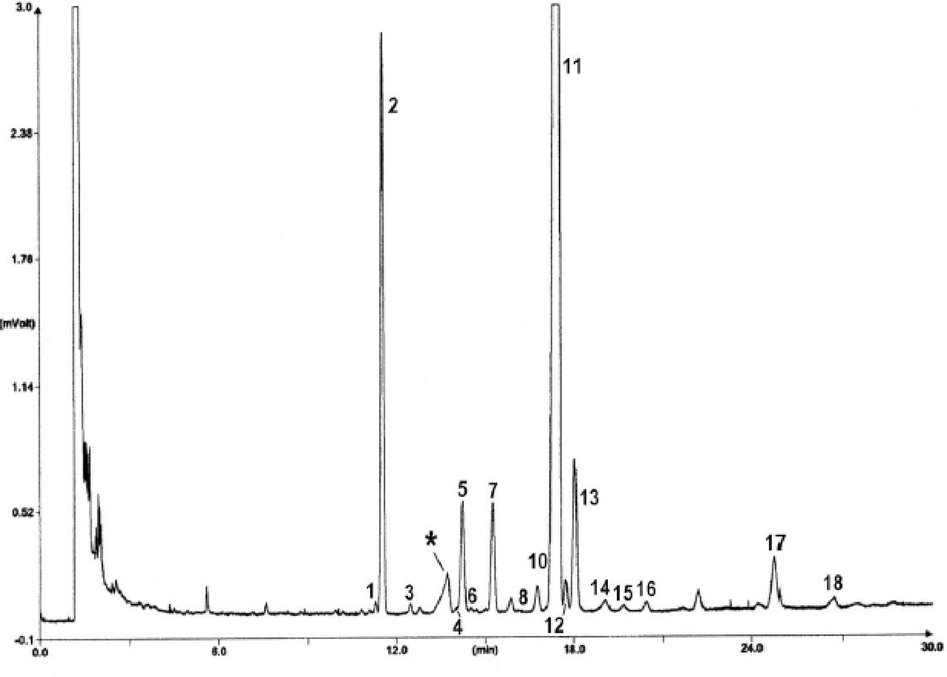
Obrázek 3 – GC-FID chromatografický profil sterolu a triterpenických dialkoholů z lampantového olivového oleje. (1) Cholesterol, (2) α-cholestanol, (3) brassikasterol, (4) 24-methylencholesterol, (5) kampesterol, (6) kampestanol, (7) stigmasterol, (8) Δ7-kampesterol, (9) Δ5,23-stigmastadienol, (10) chlerosterol, (11) β-sitosterol, (12) sitostanol, (13) Δ5-avenasterol, (14) Δ5,24-stigmastadienol, (15) Δ7-stigmastenol, (16) Δ7-avenasterol, (17) erythrodiol, (18) uvaol.
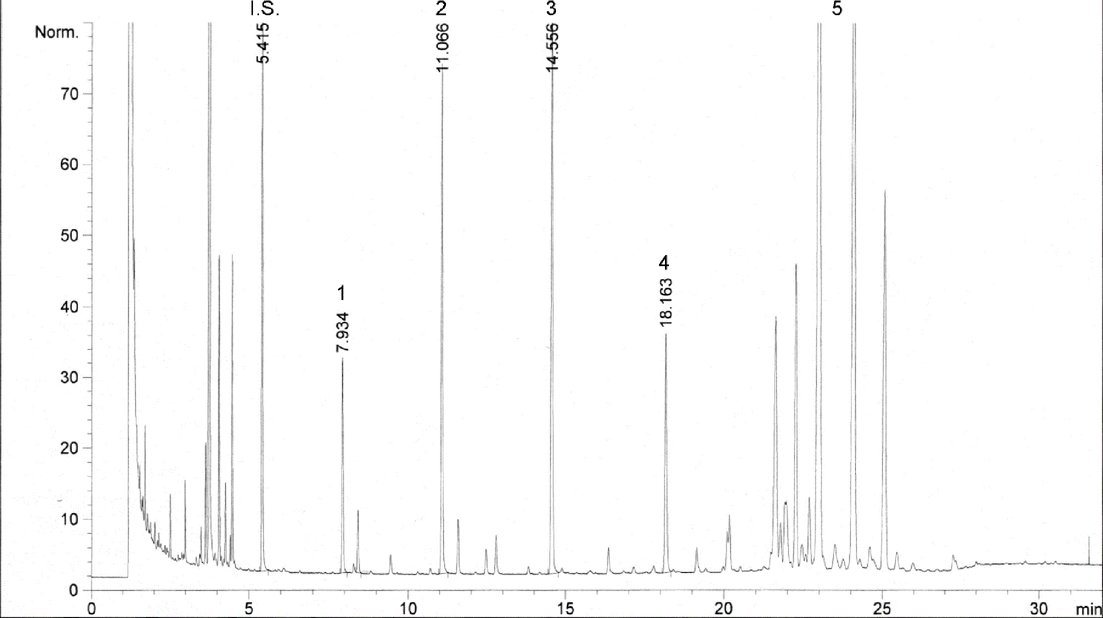
Obrázek 4 – GC-FID chromatografický profil alifatických alkoholů a triterpenických alkoholů z olivového oleje. (V.S.) C20-ol, (1) C22-ol, (2) C24-ol, (3) C26-ol, (4) C28-ol, (5) triterpenické alkoholy.
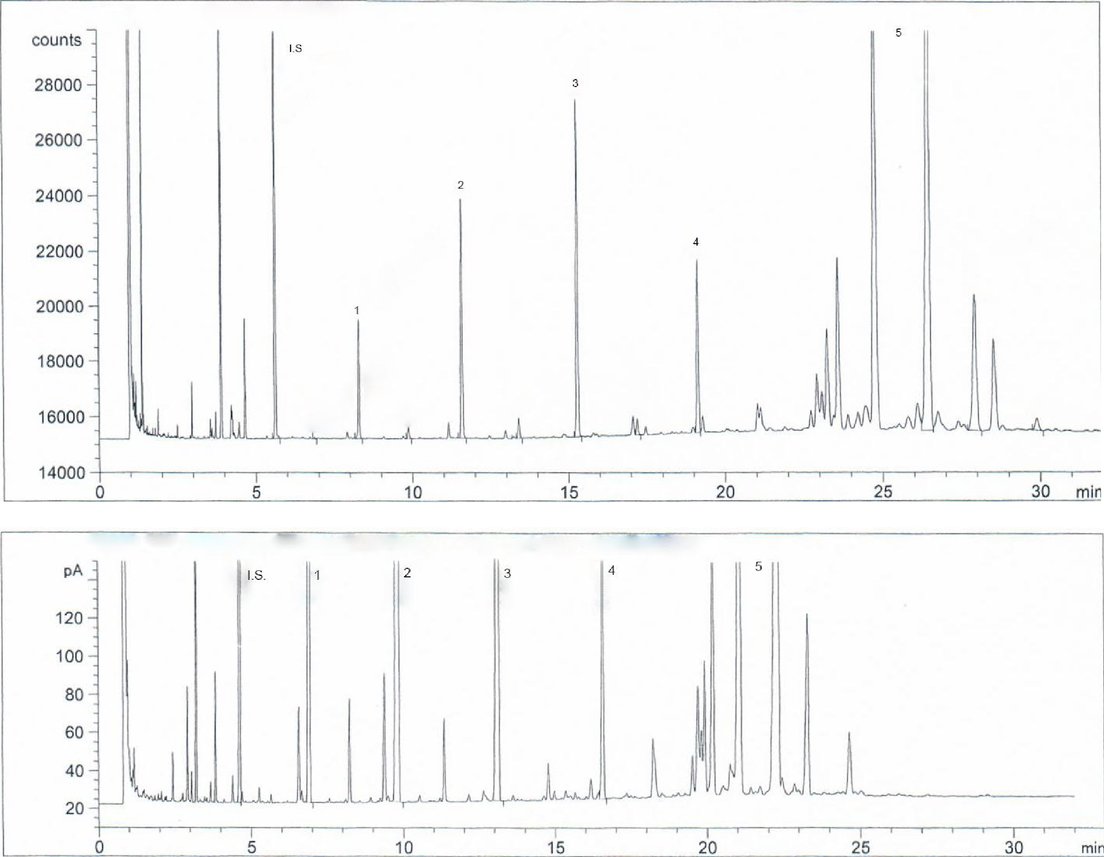
Obrázek 5 – GC-FID chromatografický profil alifatických alkoholů a triterpenických alkoholů z rafinovaného olivového oleje a olivového oleje po druhém odstřeďování. (V.S.) C20-ol, (1) C22-ol, (2) C24-ol, (3) C26-ol, (4) C28-ol, (5) triterpenické alkoholy.
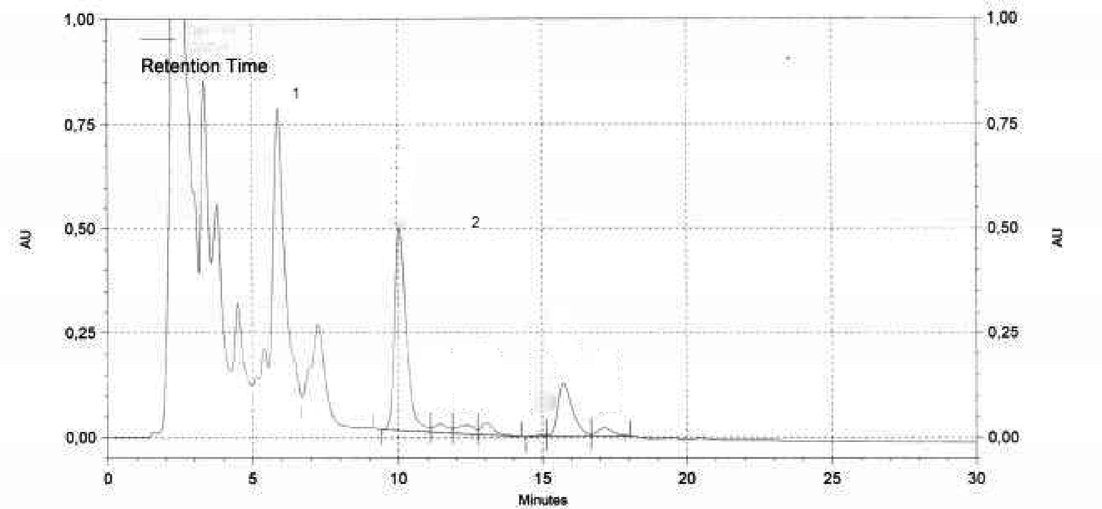
Obrázek 6 – Chromatogram nezmýdelnitelných látek z olivového oleje separovaných vysoce účinnou kapalinovou chromatografií za použití UV detektoru. (1) Alifatické a triterpenické alkoholy, (2) steroly a triterpenické dialkoholy.