(EU) 2018/150Prováděcí nařízení Komise (EU) 2018/150 ze dne 30. ledna 2018, kterým se mění prováděcí nařízení (EU) 2016/1240, pokud jde o metody analýzy a hodnocení jakosti mléka a mléčných výrobků způsobilých pro veřejnou intervenci a podporu soukromého skladování
| Publikováno: | Úř. věst. L 26, 31.1.2018, s. 14-47 | Druh předpisu: | Prováděcí nařízení |
| Přijato: | 30. ledna 2018 | Autor předpisu: | Evropská komise |
| Platnost od: | 7. února 2018 | Nabývá účinnosti: | 7. února 2018 |
| Platnost předpisu: | Ano | Pozbývá platnosti: | |
Text předpisu s celou hlavičkou je dostupný pouze pro registrované uživatele.
PROVÁDĚCÍ NAŘÍZENÍ KOMISE (EU) 2018/150
ze dne 30. ledna 2018,
kterým se mění prováděcí nařízení (EU) 2016/1240, pokud jde o metody analýzy a hodnocení jakosti mléka a mléčných výrobků způsobilých pro veřejnou intervenci a podporu soukromého skladování
EVROPSKÁ KOMISE,
s ohledem na Smlouvu o fungování Evropské unie,
s ohledem na nařízení Evropského parlamentu a Rady (EU) č. 1306/2013 ze dne 17. prosince 2013 o financování, řízení a sledování společné zemědělské politiky a o zrušení nařízení Rady (EHS) č. 352/78, (ES) č. 165/94, (ES) č. 2799/98, (ES) č. 814/2000, (ES) č. 1290/2005 a (ES) č. 485/2008 (1), a zejména na čl. 62 odst. 2 písm. i) uvedeného nařízení,
vzhledem k těmto důvodům:
|
(1) |
Nařízení Komise v přenesené pravomoci (EU) 2016/1238 (2) a prováděcí nařízení Komise (EU) 2016/1240 (3) stanoví pravidla pro veřejnou intervenci a podporu soukromého skladování. Nařízení Komise (ES) č. 273/2008 (4) stanoví metody, které mají být použity při posuzování, zda mléko a mléčné výrobky splňují požadavky na způsobilost stanovené v uvedených nařízeních pro veřejnou intervenci a podporu soukromého skladování. |
|
(2) |
S ohledem na technický vývoj metod analýzy a hodnocení jakosti mléka a mléčných výrobků by měly být provedeny podstatné změny, aby se zjednodušily a aktualizovaly odkazy na normy ISO. V zájmu jasnosti a účinnosti a s ohledem na rozsah a technickou povahu změn ustanovení nařízení (ES) č. 273/2008 by příslušná ustanovení uvedeného nařízení měla být začleněna do prováděcího nařízení (EU) 2016/1240. |
|
(3) |
V zájmu zajištění jednotného dodržování nových norem a metod ve všech členských státech by laboratořím měla být poskytnuta dostatečná lhůta na přezkum postupů a použití aktualizovaných metod. |
|
(4) |
Prováděcí nařízení (EU) 2016/1240 by proto mělo být odpovídajícím způsobem změněno. |
|
(5) |
Nařízení (ES) č. 273/2008 je třeba v zájmu právní jistoty zrušit. |
|
(6) |
Opatření stanovená tímto nařízením jsou v souladu se stanoviskem Výboru pro společnou organizaci zemědělských trhů, |
PŘIJALA TOTO NAŘÍZENÍ:
Článek 1
Prováděcí nařízení (EU) 2016/1240 se mění takto:
|
1) |
Článek 4 se mění takto:
|
|
2) |
Vkládá se nový článek 60a, který zní: „Článek 60a Zvláštní ustanovení o kontrolách týkajících se veřejné intervence a podpory soukromého skladování mléka a mléčných výrobků 1. Způsobilost másla, sušeného odstředěného mléka a sýrů k získání podpory soukromého skladování se určí v souladu s metodami stanovenými v přílohách VI, VII a VIII. Uvedené metody se určí s odkazem na poslední verzi příslušných evropských, případně mezinárodních norem platných nejméně 6 měsíců před prvním dnem období veřejné intervence stanoveného v článku 12 nařízení (EU) č. 1308/2013. 2. Výsledky kontrol, které jsou prováděny s použitím metod stanovených tímto nařízením, se hodnotí v souladu s přílohou IX.“ |
|
3) |
Přílohy se mění v souladu s přílohou tohoto nařízení. |
Článek 2
Nařízení (ES) č. 273/2008 se zrušuje.
Článek 3
Toto nařízení vstupuje v platnost sedmým dnem po vyhlášení v Úředním věstníku Evropské unie.
Toto nařízení je závazné v celém rozsahu a přímo použitelné ve všech členských státech.
V Bruselu dne 30. ledna 2018.
Za Komisi
předseda
Jean-Claude JUNCKER
(1) Úř. věst. L 347, 20.12.2013, s. 549.
(2) Nařízení Komise v přenesené pravomoci (EU) 2016/1238 ze dne 18. května 2016, kterým se doplňuje nařízení Evropského parlamentu a Rady (EU) č. 1308/2013, pokud jde o veřejnou intervenci a podporu soukromého skladování (Úř. věst. L 206, 30.7.2016, s. 15).
(3) Prováděcí nařízení Komise (EU) 2016/1240 ze dne 18. května 2016, kterým se stanoví pravidla pro uplatňování nařízení Evropského parlamentu a Rady (EU) č. 1308/2013, pokud jde o veřejnou intervenci a podporu soukromého skladování (Úř. věst. L 206, 30.7.2016, s. 71).
(4) Nařízení Komise (ES) č. 273/2008 ze dne 5. března 2008, kterým se stanoví prováděcí pravidla k nařízení Rady (ES) č. 1255/1999, pokud jde o metody analýzy a hodnocení jakosti mléka a mléčných výrobků (Úř. věst. L 88, 29.3.2008, s. 1).
PŘÍLOHA
Přílohy prováděcího nařízení (EU) 2016/1240 se mění takto:
|
1) |
Příloha IV se mění takto:
|
||||||||||||||||||||||||||||||||||||||||||||||||||
|
2) |
V příloze V se vkládá nová část Ia, která zní: „ČÁST IA Metody analýzy sušeného odstředěného mléka určeného k veřejné intervenci
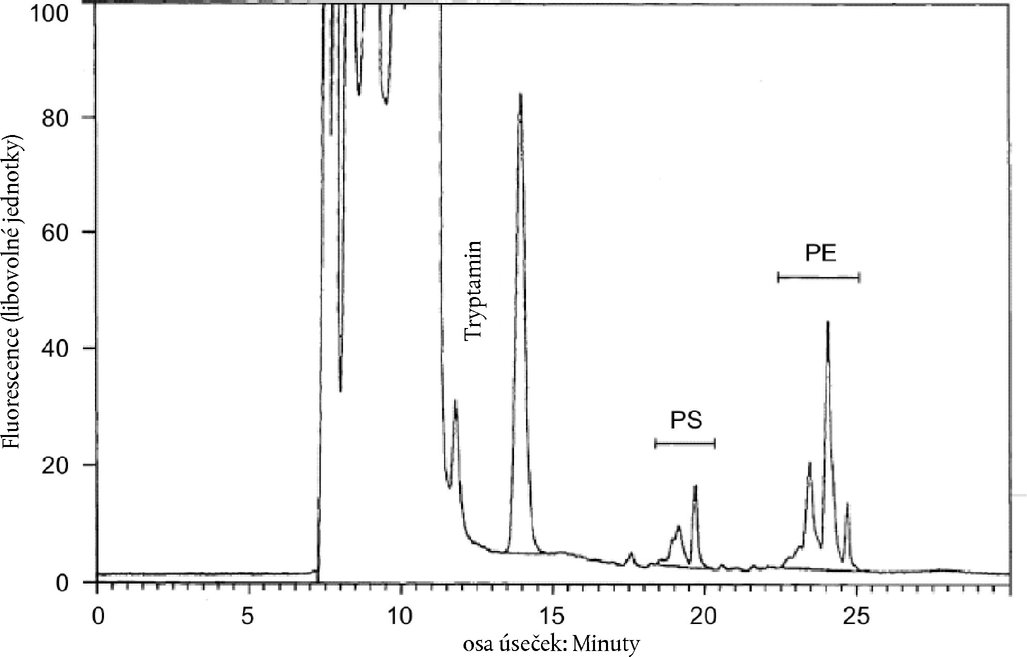
Dodatek I KVANTITATIVNÍ STANOVENÍ FOSFATIDYLSERINU A FOSFATIDYLETHANOLAMINU V SUŠENÉM ODSTŘEDĚNÉM MLÉCE Metoda: HPLC s obrácenými fázemi 1. PŘEDMĚT A OBLAST POUŽITÍ Tato metoda popisuje postup kvantitativního stanovení fosfatidylserinu (PS) a fosfatidylethanolaminu (PE) v sušeném odstředěném mléce (SOM) a je vhodná pro zjišťování sušiny podmáslí v SOM. 2. DEFINICE Obsah PS + PE: hmotnostní zlomek látky stanovené popisovaným postupem. Výsledek se vyjadřuje v miligramech dipalmitoyl-fosfatidylethanolaminu (PEDP) na 100 g prášku. 3. PODSTATA METODY Extrakce aminofosfolipidů methanolem z rekonstituovaného sušeného mléka. Stanovení obsahu PS a PE jako derivátů o-ftaldialdehydu (OPA) metodou HPLC s obrácenými fázemi (RP) a fluorescenční detekcí. Kvantitativní stanovení obsahu PS a PE ve zkušebním vzorku porovnáním se srovnávacím vzorkem, který obsahuje známé množství PEDP. 4. CHEMIKÁLIE Používají se pouze chemikálie čistoty p.a. Pokud není uvedeno jinak, používá se pouze destilovaná voda nebo voda nejméně rovnocenné čistoty. 4.1. Referenční materiál: PEDP o čistotě nejméně 99 % Poznámka: Referenční materiál musí být skladován při teplotě – 18 °C. 4.2. Chemikálie pro přípravu srovnávacích a zkušebních vzorků 4.2.1. Methanol čistoty pro HPLC 4.2.2. Chloroform čistoty pro HPLC 4.2.3. Monohydrochlorid tryptaminu 4.3. Chemikálie pro přípravu o-ftaldialdehydových derivátů 4.3.1. Hydroxid sodný, 12 M vodný roztok 4.3.2. Kyselina boritá, 0,4 M vodný roztok s hodnotou pH upravenou hydroxidem sodným (4.3.1) na 10,0 4.3.3. 2-merkaptoethanol 4.3.4. o-ftaldialdehyd (OPA) 4.4. Eluční rozpouštědla pro HPLC 4.4.1. Eluční rozpouštědla musí být připravena pomocí chemikálií čistoty pro HPLC. 4.4.2. Voda čistoty pro HPLC 4.4.3. Methanol fluorometricky zjištěné čistoty 4.4.4. Tetrahydrofuran 4.4.5. Natrium dihydrogenfosforečnan 4.4.6. Octan sodný 4.4.7. Kyselina octová. 5. PŘÍSTROJE A POMŮCKY 5.1. Analytické váhy s přesností měření na 0,1 mg a s možností odečtu po 1 mg 5.2. Kádinky o objemu 25 a 100 ml 5.3. Pipety schopné dávkovat od 1 do 10 ml 5.4. Magnetická míchačka 5.5. Dělené pipety schopné dávkovat 0,2, 0,5 a 5 ml 5.6. Odměrné baňky o objemu 10, 50 a 100 ml 5.7. Injekční stříkačky o objemu 20 a 100 μl 5.8. Ultrazvuková lázeň 5.9. Odstředivka fungující při 27 000 × g 5.10. Skleněné nádobky o objemu asi 5 ml 5.11. Odměrný válec o objemu 25 ml 5.12. pH metr s přesností na 0,1 jednotky pH 5.13. Zařízení na HPLC 5.13.1. Gradientový čerpací systém schopný fungovat při průtoku 1,0 ml/min při 200 barech 5.13.2. Automatický dávkovač vzorků s možností derivatizace 5.13.3. Termostat kolony schopný kolonu udržovat na teplotě 30 °C ± 1 °C 5.13.4. Fluorescenční detektor schopný fungovat při excitační vlnové délce 330 nm a emisní vlnové délce 440 nm 5.13.5. Integrátor nebo programové vybavení pro zpracování dat schopné měřit plochy píků 5.13.6. Kolona LiChrospher® – 100 (250 × 4,6 mm) nebo rovnocenná kolona naplněná oktadecylsilanem (C 18), velikost částic 5 μm 6. ODBĚR VZORKŮ Vzorky se odebírají v souladu s normou ISO 707. 7. PRACOVNÍ POSTUP 7.1. Příprava roztoku vnitřního standardu 7.1.1. Navažte 30,0 ± 0,1 mg tryptaminhydrochloridu (4.2.3) do odměrné baňky na 100 ml (5.6) a doplňte po značku methanolem (4.2.1). 7.1.2. Pipetou (5.3) přeneste 1 ml tohoto roztoku do odměrné baňky na 10 ml (5.6) a doplňte po značku methanolem (4.2.1) s cílem získat koncentraci tryptaminu 0,15 mM. 7.2. Příprava roztoku zkušebního vzorku 7.2.1. Navažte 1,000 ± 0,001 g vzorku SOM do kádinky na 25 ml (5.2). Pipetou (5.3) přidejte 10 ml destilované vody o teplotě 40 °C ± 1 °C a po dobu 30 minut míchejte magnetickou míchačkou (5.4), aby se rozpustily všechny kousky. 7.2.2. Pipetou (5.5) přeneste 0,2 ml rekonstituovaného mléka do odměrné baňky na 10 ml (5.6), pomocí injekční stříkačky (5.7) přidejte 100 μl 0,15 mM roztoku tryptaminu (7.1) a doplňte po značku methanolem (4.2.1). Převracením opatrně promíchejte a vložte na 15 minut do ultrazvukové lázně (5.8). 7.2.3. Odstřeďujte (5.9) při 27 000 × g po dobu 10 minut a supernatant odeberte do skleněné nádobky (5.10). Poznámka: Do provedení analýzy HP musí být roztok zkušebního vzorku skladován při teplotě 4 °C. 7.3. Příprava roztoku vnějšího standardu 7.3.1. Navažte 55,4 mg PEDP (4.1) do odměrné baňky na 50 ml (5.6) a odměrným válcem (5.11) přidejte asi 25 ml chloroformu (4.2.2). Zazátkovanou baňku zahřejte na 50 °C ± 1 °C a opatrně promíchávejte, dokud se PEDP nerozpustí. Baňku ochlaďte na 20 °C, doplňte po značku methanolem (4.2.1) a promíchejte převracením. 7.3.2. Pipetou (5.3) přeneste 1 ml tohoto roztoku do odměrné baňky na 100 ml (5.6) a doplňte po značku methanolem (4.2.1). Pipetou (5.3) přeneste 1 ml tohoto roztoku do odměrné baňky na 10 ml (5.6), přidejte 100 μl (5.7) 0,15mM roztoku tryptaminu (7.1) a doplňte po značku methanolem (4.2.1). Promíchejte převracením. Poznámka: Do provedení analýzy HP musí být roztok referenčního vzorku skladován při teplotě 4 °C. 7.4. Příprava derivatizačního činidla Do odměrné baňky na 10 ml (5.6) navažte 25,0 ± 0,1 mg OPA (4.3.4), přidejte 0,5 ml (5.5) methanolu (4.2.1) a pečlivě promíchejte, aby se OPA rozpustil. Doplňte po značku roztokem kyseliny borité (4.3.2) a injekční stříkačkou (5.7) přidejte 20 μl 2-merkaptoethanolu (4.3.3). Poznámka: Derivatizační činidlo musí být skladováno v hnědé nádobě při teplotě 4 °C; činidlo zůstává stálé po dobu jednoho týdne. 7.5. Stanovení pomocí HPLC 7.5.1. Eluční rozpouštědla (4.4) Rozpouštědlo A: Roztok 0,3 mM natria dihydrogenfosforečnanu a roztok 3 mM octanu sodného (s hodnotou pH upravenou na 6,5 ± 0,1 kyselinou octovou): methanol: tetrahydrofuran = 558: 440:2 (obj.). Rozpouštědlo B: Methanol 7.5.2. Doporučený eluční gradient:
Poznámka: Může se stát, že eluční gradient bude muset být mírně upraven, aby bylo dosaženo rozlišení znázorněné na obrázku 1. Teplota kolony: 30 °C. 7.5.3. Objem nástřiku: 50 μl derivatizačního činidla a 50 μl roztoku vzorku 7.5.4. Ekvilibrace kolony Při každodenním provádění pokusů kolonu promývejte 100 % rozpouštědlem B po dobu 15 minut, potom připravte poměr A:B = 40:60 a provádějte ekvilibraci po dobu 15 minut při průtoku 1 ml/min. Proveďte slepý pokus nástřikem methanolu (4.2.1). Poznámka: Před uskladněním kolony na delší dobu kolonu promývejte po dobu 30 minut směsí methanolu s chloroformem v poměru 80:20 (obj.). 7.5.5. Stanovení obsahu PS + PE ve zkušebním vzorku 7.5.6. Proveďte sérii chromatografických analýz při zachování stejných časových odstupů mezi jednotlivými pokusy, abyste získali konstantní retenční časy. Pro výpočet odezvového faktoru nastříkněte roztok vnějšího standardu (7.3) na každých 5–10 roztoků zkušebních vzorků. Poznámka: Kolonu je nutné po každých 20–25 pokusech vyčistit nejméně třicetiminutovým promýváním 100 % rozpouštědlem B (7.5.1). 7.6. Integrační režim 7.6.1. Pík PEDP PEDP se vymývá v podobě jediného píku. Plochu píku stanovte integrací mezi sedly. 7.6.2. Pík tryptaminu Tryptamin se vymývá v podobě jediného píku (obrázek 1). Plochu píku stanovte integrací mezi sedly. 7.6.3. Skupiny píků PS a PE Za takto popsaných podmínek (obrázek 1) se PS vymývá v podobě dvou hlavních, částečně nerozlišených píků, před nimiž se nachází jeden menší pík. PE se vymývá v podobě tří hlavních, částečně nerozlišených píků. Celou plochu každé skupiny píků stanovte nastavením základní linie tak, jak je znázorněno na obrázku 1. 8. VÝPOČET A VYJADŘOVÁNÍ VÝSLEDKŮ Obsah PS + PE ve zkušebním vzorku se vypočte takto: C = 55,36 × ((A2)/(A1)) × ((T1)/(T2)) kde:
9. PŘESNOST METODY Poznámka: Hodnoty pro opakovatelnost byly vypočteny podle Mezinárodní normy IDF (*). 9.1. Opakovatelnost Relativní směrodatná odchylka opakovatelnosti, která vyjadřuje proměnlivost nezávislých analytických výsledků, které u stejného zkušebního vzorku získá stejný pracovník na stejném přístroji v krátkém časovém odstupu, by neměla překročit relativní 2 %. Jestliže se za těchto podmínek získají dvě stanovení, relativní rozdíl mezi jejich výsledky by neměl být větší než 6 % aritmetického průměru výsledků. 9.2. Reprodukovatelnost Jestliže dva pracovníci provedou v různých laboratořích na různých přístrojích a za různých podmínek analýzu téhož zkušebního vzorku a dospějí k různým stanovením, relativní rozdíl mezi oběma výsledky by neměl být větší než 11 % aritmetického průměru výsledků. 10. LITERATURA 10.1. Resmini P., Pellegrino L., Hogenboom J. A., Sadini V., Rampilli M. „Detection of buttermilk solids in skimmilk powder by HPLC quantification of aminophospholipids.“ Sci. Tecn. Latt.-Cas. 39,395[1988]. Obrázek 1 HPLC chromatogram OPA-derivátů fosfatidylserinu (PS) a fosfatidylethanolaminu (PE) v methanolovém extraktu z rekonstituovaného sušeného odstředěného mléka. Je znázorněn integrační režim pro píky PS, PE a tryptamin (vnitřní standard).
Dodatek II ZJIŠŤOVÁNÍ SYŘIDLOVÉ SYROVÁTKY V SUŠENÉM ODSTŘEDĚNÉM MLÉCE URČENÉM PRO VEŘEJNÉ SKLADOVÁNÍ NA ZÁKLADĚ STANOVENÍ KASEINOMAKROPEPTIDŮ VYSOKOÚČINNOU KAPALINOVOU CHROMATOGRAFIÍ (HPLC) 1. PŘEDMĚT A OBLAST POUŽITÍ Tato metoda umožňuje zjistit přítomnost syřidlové syrovátky v sušeném odstředěném mléce určeném k veřejnému skladování na základě stanovení kaseinomakropeptidů. 2. NORMATIVNÍ ODKAZ Mezinárodní norma ISO 707 – Mléko a mléčné výrobky – Směrnice pro odběr vzorků. 3. DEFINICE Obsah sušiny ze syřidlové syrovátky je definován jako hmotnostní procento stanovené popsaným postupem podle obsahu kaseinomakropeptidů. 4. PODSTATA METODY
5. CHEMIKÁLIE Používají se pouze chemikálie čistoty p.a. Použitá voda musí být voda destilovaná nebo voda nejméně rovnocenné čistoty. 5.1. Roztok kyseliny trichloroctové Rozpusťte 240 g kyseliny trichloroctové (CCl3COOH) ve vodě a doplňte na 1 000 ml. Roztok by měl být čirý a bezbarvý. 5.2. Eluční roztok, pH 6,0 Rozpusťte 1,74 g fosforečnanu draselného sekundárního (K2HPO4), 12,37 g hydrogenfosforečnanu draselného (KH2PO4) a 21,41 g síranu sodného (Na2SO4) asi v 700 ml vody. V případě potřeby upravte pH na 6,0 roztokem kyseliny fosforečné nebo hydroxidu draselného. Doplňte vodou na 1 000 ml a homogenizujte. Poznámka: Složení eluentu lze obnovit tak, aby odpovídal osvědčení standardů nebo doporučením výrobce balicích materiálů na kolony. Eluční roztok před použitím přefiltrujte přes membránový filtr o velikosti pórů 0,45 μm. 5.3. Promývací roztok Smíchejte jeden objem acetonitrilu (CH3CN) s devíti objemy vody. Před použitím směs přefiltrujte přes membránový filtr o velikosti pórů 0,45 μm. Poznámka: Lze použít jakýkoliv jiný promývací roztok, který má baktericidní účinek a který nezhoršuje rozlišovací účinnost kolony. 5.4. Srovnávací vzorky 5.4.1. Sušené odstředěné mléko splňující požadavky tohoto nařízení (tj. [0]). 5.4.2. Totéž sušené odstředěné mléko zfalšované přídavkem 5 % hm. sušené syřidlové syrovátky standardního složení (tj. [5]). 6. PŘÍSTROJE A POMŮCKY 6.1. Analytické váhy 6.2. Volitelná odstředivka schopná dosáhnout odstředivé síly 2 200 g a vybavená odstředivkovými zkumavkami se zátkou o objemu asi 50 ml 6.3. Mechanická třepačka 6.4. Magnetická míchačka 6.5. Skleněné nálevky o průměru asi 7 cm 6.6. Filtrační papíry střední hustoty o průměru asi 12,5 cm 6.7. Skleněné filtrační zařízení s membránovým filtrem o velikosti pórů 0,45 μm 6.8. Dělené pipety umožňující dávkovat 10 ml (ISO 648, třída A, nebo ISO/R 835) nebo dávkovací systém schopný dodávat 10,0 ml za dvě minuty 6.9. Dávkovací systém schopný dodávat 20,0 ml vody při teplotě asi 50 °C 6.10. Termostatická vodní lázeň nastavená na 25 ± 0,5 °C 6.11. Zařízení na HPLC, které tvoří:
7. ODBĚR VZORKŮ 7.1. Vzorky musí být odebírány postupem stanoveným v mezinárodní normě ISO 707. Členské státy však mohou používat jinou metodu odběru vzorků za předpokladu, že tato metoda odpovídá zásadám uvedené normy. 7.2. Vzorek skladujte takovým způsobem, aby nemohlo dojít ke znehodnocení nebo změně složení. 8. PRACOVNÍ POSTUP 8.1. Příprava zkušebního vzorku. Sušené mléko převeďte do nádobky, jejíž objem je asi dvakrát větší než objem prášku a která je opatřena vzduchotěsným víčkem. Nádobku ihned uzavřete. Sušené mléko důkladně promíchejte opakovaným převracením nádobky. 8.2. Zkušební dávka Do odstředivkové zkumavky (6.2) nebo vhodné baňky se zátkou na 50 ml navažte 2,000 ± 0,001 g zkušebního vzorku. 8.3. Odstranění tuku a bílkovin 8.3.1. Ke zkušební dávce přidejte 20,0 ml teplé (50 °C) vody. Prášek rozpusťte třepáním po dobu pěti minut na mechanické třepačce (6.3). Zkumavku vložte do vodní lázně (6.10), dokud se její teplota neustálí na 25 °C. 8.3.2. Během dvou minut přidejte za stálého míchání magnetickou míchačkou (6.4) 10,0 ml roztoku kyseliny trichloroctové (5.1) o teplotě asi 25 °C. Zkumavku vložte na 60 minut do vodní lázně (6.10). 8.3.3. Odstřeďujte (6.2) po dobu 10 minut při odstředivé síle 2 200 g, nebo přefiltrujte přes papír (6.6) a prvních 5 ml filtrátu vyhoďte. 8.4. Chromatografické stanovení 8.4.1. Nastříkněte 15 až 30 μl přesně odměřeného supernatantu nebo filtrátu (8.3.3) do zařízení na HPLC (6.11) s průtokem 1,0 ml elučního roztoku (5.2) za minutu. Poznámka 1. V závislosti na vnitřním průměru použitých kolon nebo na pokynech výrobce kolony lze použít i jiný průtok. Poznámka 2. Při každém přerušení promyjte kolony vodou. Nikdy v nich nenechávejte eluční roztok (5.2). Před každým přerušením na více než 24 hodin se kolony propláchnou vodou a potom se promývají roztokem (5.3) nejméně po dobu tří hodin při průtoku 0,2 ml za minutu. 8.4.2. Výsledky chromatografické analýzy zkušebního vzorku [E] se získají ve formě chromatogramu, v němž je každý pík určen svým retenčním časem RT takto:
Retenční časy jednotlivých píků mohou být značně ovlivněny výběrem kolony (kolon). Integrátor (6.11.6) automaticky vypočítává plochu A každého píku:
Před kvantitativní interpretací je nezbytné prozkoumat vzhled každého chromatogramu za účelem zjištění případných anomálií způsobených buď nesprávnou funkcí zařízení, nebo kolon, nebo původem a povahou analyzovaného vzorku. V případě pochybností analýzu zopakujte. 8.5. Kalibrace 8.5.1. U srovnávacích vzorků (5.4) se přesně použije postup popsaný v bodech 8.2 až 8.4.2. Použijte čerstvě připravené roztoky, protože CMP se v prostředí 8 % kyseliny trichloroctové odbourávají. Jejich obsah se při teplotě 30 °C snižuje odhadem o 0,2 % za hodinu. 8.5.2. Před chromatografickým stanovením vzorků stabilizujte kolony opakovanými nástřiky roztoku (8.5.1) srovnávacího vzorku (5.4.2), dokud se plocha a retenční čas píku odpovídajícího CMP neustálí na konstantních hodnotách. 8.5.3. Stanovte odezvové faktory R nastříknutím stejného objemu filtrátů (8.5.1), jakého jste použili pro vzorky. 9. VYJÁDŘENÍ VÝSLEDKŮ 9.1. Metoda výpočtu a vzorce 9.1.1. Výpočet odezvového faktoru R:
kde:
kde:
9.1.2. Výpočet relativní plochy píků ve vzorku [E]
kde:
9.1.3. Výpočet relativního retenčního času píku III ve vzorku [E]: RRTIII[E] = (RTIII[E])/(RTIII[5]) kde:
9.1.4. Pokusy ukazují, že mezi relativním retenčním časem píku III, tj. RRTIII [E] a procentem přidané sušené syrovátky až do přídavku 10 % existuje lineární vztah
Přípustná neurčitost pro hodnoty RRTIII je ± 0,002. Za normálních okolností se hodnota RRTIII [0] jen málo liší od 1,034. Podle stavu kolon se může hodnota blížit 1,000, avšak vždy musí být vyšší než 1,000. 9.2. Výpočet procenta sušené syřidlové syrovátky obsažené ve vzorku: W = SIII[E] – [1, 3 + (SIII[0] – 0, 9)] kde:
9.3. Přesnost metody 9.3.1. Opakovatelnost Rozdíl mezi výsledky dvou stanovení, která u stejného zkušebního vzorku provedl současně nebo v těsném sledu jeden pracovník na stejném přístroji, by neměl překročit 0,2 % hm. 9.3.2. Reprodukovatelnost Rozdíl mezi dvěma jednotlivými a nezávislými výsledky získanými ve dvou různých laboratořích u stejného zkušebního vzorku by neměl překročit 0,4 % hm. 9.4. Interpretace 9.4.1. Lze předpokládat, že syrovátka není přítomna, je-li relativní plocha píku III SIII [E] vyjádřena v gramech syřidlové syrovátky na 100 g produktu, ≤ 2,0 + (SIII[0] – 0,9) kde
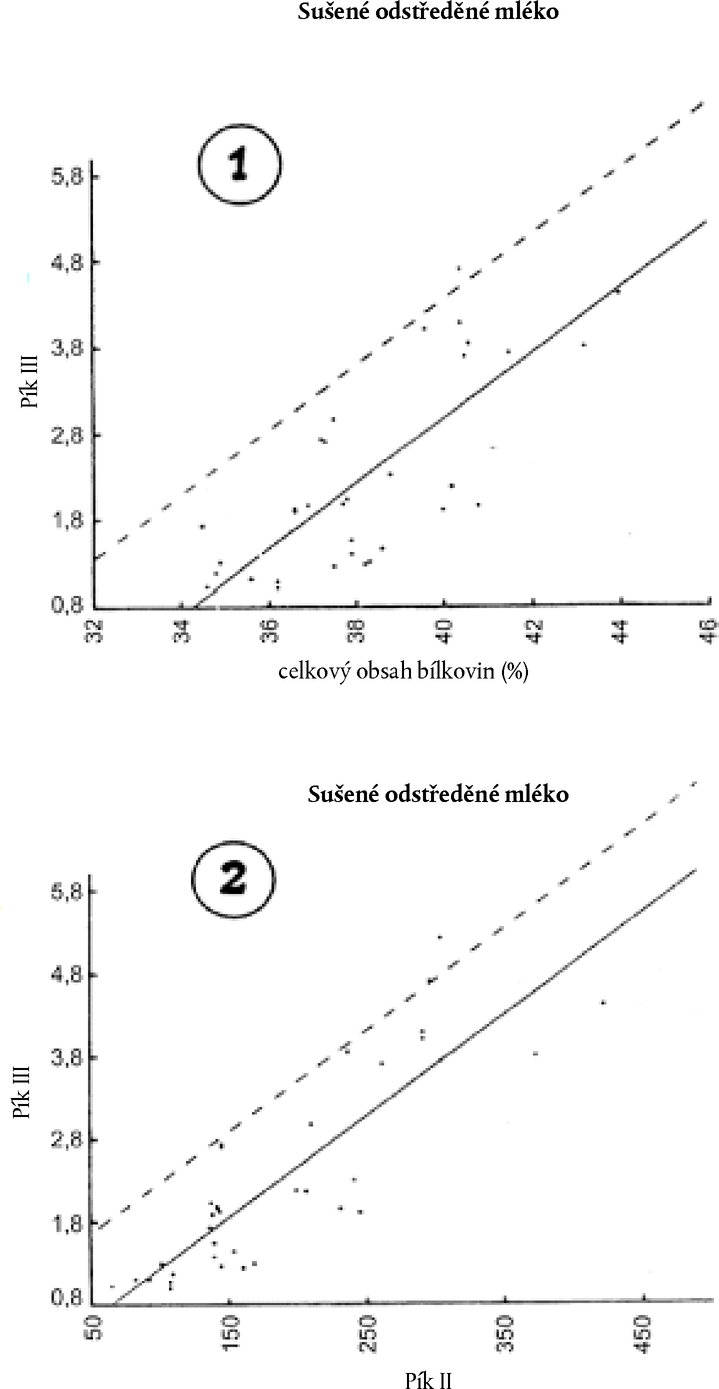
9.4.2. Je-li relativní plocha píku III SIII [E] > 2,0 + (SIII[0] – 0,9) a relativní plocha píku II SII [E] ≤ 160, stanovte obsah syřidlové syrovátky podle bodu 9.2. 9.4.3. Je-li relativní plocha píku III SIII [E] > 2,0 + (SIII[0] – 0,9) a relativní plocha píku II SII [E] ≤ 160, stanovte celkový obsah bílkovin (P %). Potom prostudujte grafy 1 a 2. 9.4.3.1. Data získaná po analýze vzorků nezfalšovaného sušeného odstředěného mléka s vysokým celkovým obsahem bílkovin jsou zanesena do grafů 1 a 2. Plná přímka představuje lineární regresi, jejíž koeficienty se vypočítají metodou nejmenších čtverců. Čárkovaná přímka určuje horní mez relativní plochy píku III, a to s pravděpodobností, že nebude překročena v 90 % případů. Rovnice čárkovaných přímek v grafech 1 a 2 jsou:
kde:
Tyto rovnice jsou rovnocenné číslu 1,3 uvedenému v bodu 9.2. Rozdíl (T1 a T2) mezi zjištěnou relativní plochou SIII [E] a relativní plochou SIII je dán těmito vztahy: T1 = SIII[E] – [(0,376 P% – 10,7) + (SIII[0] – 0,9)]T2 = SIII[E] – [(0,0123 SII[E] + 0,93) + (SIII[0] – 0,9)].
Obsah syřidlové syrovátky se vypočítá podle vzorce: W = T2 + 0,91 kde: 0,91 je vzdálenost na svislé ose mezi plnou a přerušovanou přímkou.
Dodatek III STANOVENÍ SUŠINY ZE SYŘIDLOVÉ SYROVÁTKY V SUŠENÉM ODSTŘEDĚNÉM MLÉCE 1. PŘEDMĚT: STANOVENÍ PŘÍDAVKU SUŠINY ZE SYŘIDLOVÉ SYROVÁTKY DO SUŠENÉHO ODSTŘEDĚNÉHO MLÉKA 2. NORMATIVNÍ ODKAZ: MEZINÁRODNÍ NORMA ISO 707 3. DEFINICE Obsah sušiny ze syřidlové syrovátky je definován jako hmotnostní procento stanovené popsaným postupem podle obsahu kaseinomakropeptidů. 4. PODSTATA METODY Vzorky jsou analyzovány na kaseinomakropeptidy A vysokoúčinnou kapalinovou chromatografií s obrácenými fázemi (metodou HPLC). Vyhodnocení výsledků se provádí porovnáním se srovnávacími vzorky, které tvoří sušené odstředěné mléko bez přídavku nebo s přídavkem známého procenta sušené syrovátky. Pokud je výsledek vyšší než 1 % hm., je přítomnost sušiny ze syřidlové syrovátky prokázána. 5. CHEMIKÁLIE Používají se pouze chemikálie čistoty p.a. Použitá voda musí být voda destilovaná nebo voda nejméně rovnocenné čistoty. Acetonitril musí mít spektroskopickou jakost nebo jakost vhodnou pro HPLC. 5.1. Roztok kyseliny trichloroctové Rozpusťte 240 g kyseliny trichloroctové (CCl3COOH) ve vodě a doplňte na 1 000 ml. Roztok by měl být čirý a bezbarvý. 5.2. Eluenty A a B Eluent A: Vneste 150 ml acetonitrilu (CH3CN), 20 ml isopropanolu (CH3CHOHCH3) a 1,00 ml kyseliny trifluoroctové (TFA, CF3COOH) do odměrné baňky a doplňte na 1 000 ml vodou. Eluent B: Vneste 550 ml acetonitrilu, 20 ml isopropanolu a 1,00 ml TFA do odměrné baňky na 1 000 ml a doplňte na 1 000 ml vodou. Eluční roztok před použitím přefiltrujte přes membránový filtr o velikosti pórů 0,45 μm. 5.3. Uchování kolony Po analýzách se kolona promyje eluentem B (spádem) a potom se propláchne acetonitrilem (spádem po dobu 30 minut). Kolona se uchovává v acetonitrilu. 5.4. Srovnávací vzorky 5.4.1. Sušené odstředěné mléko splňující požadavky stanovené pro veřejné skladování (tj. [0]). 5.4.2. Totéž sušené odstředěné mléko zfalšované přídavkem 5 % hm. sušené syřidlové syrovátky standardního složení (tj. [5]). 5.4.3. Totéž sušené odstředěné mléko zfalšované přídavkem 50 % hm. sušené syřidlové syrovátky standardního složení (tj. [50]). 6. PŘÍSTROJE A POMŮCKY 6.1. Analytické váhy 6.2. Volitelná odstředivka schopná dosáhnout odstředivé síly 2 200 g a vybavená odstředivkovými zkumavkami se zátkou o objemu asi 50 ml 6.3. Mechanická třepačka 6.4. Magnetická míchačka 6.5. Skleněné nálevky o průměru asi 7 cm 6.6. Filtrační papíry střední hustoty o průměru asi 12,5 cm 6.7. Skleněné filtrační zařízení s membránovým filtrem o velikosti pórů 0,45 μm 6.8. Dělené pipety umožňující dávkovat 10 ml (ISO 648, třída A, nebo ISO/R 835), nebo dávkovací systém schopný dodávat 10,0 ml za dvě minuty 6.9. Dávkovací systém schopný dodávat 20,0 ml vody při teplotě asi 50 °C 6.10. Termostatická vodní lázeň nastavená na 25 ± 0,5 °C 6.11. Zařízení na HPLC, které tvoří:
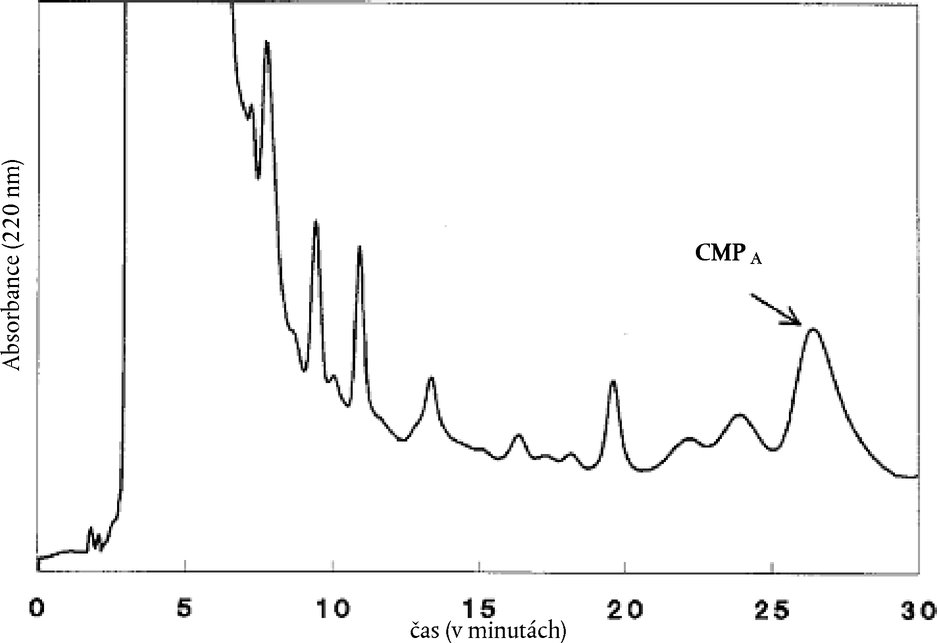
7. ODBĚR VZORKŮ 7.1. Vzorky musí být odebírány postupem stanoveným v mezinárodní normě ISO 707. Členské státy však mohou používat jinou metodu odběru vzorků za předpokladu, že tato metoda odpovídá zásadám uvedené normy. 7.2. Vzorek skladujte takovým způsobem, aby nemohlo dojít ke znehodnocení nebo změně složení. 8. PRACOVNÍ POSTUP 8.1. Příprava zkušebního vzorku Sušené mléko převeďte do nádobky, jejíž objem je asi dvakrát větší než objem prášku a která je opatřena vzduchotěsným víčkem. Nádobku ihned uzavřete. Sušené mléko důkladně promíchejte opakovaným převracením nádobky. 8.2. Zkušební dávka Do odstředivkové zkumavky (6.2) nebo vhodné baňky se zátkou na 50 ml navažte 2,00 ± 0,001 g zkušebního vzorku. Poznámka: V případě směsí navažte takové množství zkušebního vzorku, aby tuku zbavená zkušební dávka odpovídala 2,00 g. 8.3. Odstranění tuku a bílkovin 8.3.1. Ke zkušební dávce přidejte 20,0 ml teplé (50 °C) vody. Pětiminutovým třepáním na mechanické třepačce (6.3) se prášek rozpustí. Zkumavku vložte do vodní lázně (6.10), dokud se její teplota neustálí na 25 °C. 8.3.2. Během dvou minut za stálého míchání magnetickou míchačkou (6.4) přidejte 10,0 ml roztoku kyseliny trichloroctové o teplotě asi 25 °C (5.1). Zkumavku vložte na 60 minut do vodní lázně (6.10). 8.3.3. Odstřeďujte (6.2) po dobu 10 minut při odstředivé síle 2 200 g, nebo přefiltrujte přes papír (6.6) a prvních 5 ml filtrátu vyhoďte. 8.4. Chromatografické stanovení 8.4.1. Metoda HPLC s obrácenými fázemi vylučuje možnost falešně pozitivních výsledků díky přítomnosti sušeného kysaného podmáslí. 8.4.2. Před provedením analýzy HPLC s obrácenými fázemi je třeba optimalizovat podmínky gradientu. Pro gradientové systémy s mrtvým objemem asi 6 ml (objem od bodu, kde se stékají rozpouštědla k objemu nástřikové smyčky včetně) je optimální retenční čas v délce 26 minut ± 2 minuty pro CMPΑ. Pro gradientové systémy s menším mrtvým objemem (např. 2 ml) je optimální retenční čas 22 minut. Vezměte roztoky srovnávacích vzorků (5.4) s obsahem 50 % syřidlové syrovátky a bez obsahu této syrovátky. Nastříkněte 100 μl supernatantu nebo filtrátu (8.3.3) do zařízení na HPLC, které musí fungovat za podmínek referenčního gradientu uvedených v tabulce 1. Tabulka 1 Podmínky referenčního gradientu pro optimalizaci chromatografie
Porovnání obou chromatogramů by mělo ukázat polohu píku CMPΑ. Počáteční složení rozpouštědla, které je nutno použít pro normální gradient (viz 8.4.3), lze vypočítat podle tohoto vzorce: % B = 10 – 2,5 + (13,5 + (RTcmpA – 26) / 6) * 30 / 27 % B = 7,5 + (13,5 + (RTcmpA – 26) / 6) * 1,11 kde
8.4.3. Nástřik roztoků zkušebních vzorků Nastříkněte 100 μl přesně odměřeného supernatantu nebo filtrátu (8.3.3) do zařízení na HPLC s průtokem 1,0 ml elučního roztoku (5.2) za minutu. Složení eluentu na začátku analýzy se získá z 8.4.2. Za normálních okolností se blíží poměru A:B = 76:24 (5.2). Okamžitě po nástřiku se spustí lineární gradient, což způsobí, že procento B se za 27 minut zvětší o 5 %. Potom se spustí lineární gradient, kterým složení eluentu dosáhne v pěti minutách 90 % B. Toto složení se udržuje po dobu pěti minut a po jejím uplynutí se pomocí lineárního gradientu složení opět změní během pěti minut na složení počáteční. V závislosti na vnitřním objemu čerpacího systému může být další nástřik proveden 15 minut po dosažení původních podmínek. Poznámka 1. Retenční čas CMPA by měl být 26 ± 2 minuty. Toho lze dosáhnout změnou počátečních a konečných podmínek prvního gradientu. Rozdíl v % B mezi počátečními a konečnými podmínkami prvního gradientu však zůstává ve výši 5 % B. Poznámka 2. Eluenty je třeba dostatečně odplynit a uchovávat je odplyněné. Je to nezbytně nutné pro to, aby gradientový čerpací systém správně fungoval. Směrodatná odchylka retenčního času píku CMPA musí být menší než 0,1 minuty (n = 10). Poznámka 3. Na každých pět vzorků je třeba nastříknout referenční vzorek [5] a použít jej pro výpočet nového odezvového faktoru R (9.1.1). 8.4.4. Výsledky chromatografických analýz zkušebního vzorku (E) se získávají ve formě chromatogramu, na němž je pík CMPA určen svým retenčním časem přibližně v délce 26 minut. Výšku H píku CMPA automaticky vypočítává integrátor (6.11.6). Polohu základní linie je třeba kontrolovat na každém chromatogramu. Jestliže je základní linie nesprávně umístěna, analýzu nebo integraci je třeba zopakovat. Poznámka: Je-li pík CMPA dostatečně oddělen od ostatních píků, mělo by se použít přiřazení základní linie mezi sedly. V opačném případě použijte svislé kolmice ke společné základní linii, jejichž počáteční bod by měl být v blízkosti píku CMPA (tedy ne v čase t = 0 min!). Pro referenční standard a vzorky použijte stejný typ integrace a u společné základní linie zkontrolujte, zda je se vzorky a referenčním standardem konzistentní. Před kvantitativní interpretací je nezbytné prozkoumat vzhled každého chromatogramu za účelem zjištění případných anomálií způsobených buď nesprávnou funkcí zařízení, nebo kolony, nebo původem a povahou analyzovaného vzorku. V případě pochybností analýzu zopakujte. 8.5. Kalibrace 8.5.1. U srovnávacích vzorků (5.4.1 až 5.4.2) se přesně použije postup popsaný v bodech 8.2 až 8.4.4. Použijte čerstvě připravené roztoky, protože CMP se v prostředí 8 % kyseliny trichloroctové odbourává. Při teplotě 4 °C zůstává roztok stálý po dobu 24 hodin. V případě dlouhých analytických pokusů je žádoucí používat v automatickém nástřikovém zařízení chlazenou misku na vzorek. Poznámka: Fáze 8.4.2 může být vynechána, jestliže je % B v počátečních podmínkách známo z předchozích analýz. Chromatogram referenčního vzorku (5) by měl být obdobný chromatogramu znázorněnému na obrázku 1. Na tomto obrázku jsou před píkem CMPA dva malé píky. Je nezbytně nutné dosáhnout podobné separace. 8.5.2. Před chromatografickým stanovením vzorků nastříkněte 100 μl srovnávacího vzorku bez syřidlové syrovátky [0] (5.4.1). Chromatogram nesmí vykazovat pík v retenčním čase CMPA píku. 8.5.3. Odezvové faktory R stanovte nastříknutím stejného objemu filtrátu (8.5.1), jakého bylo použito pro vzorky. 9. VYJÁDŘENÍ VÝSLEDKŮ 9.1. Metoda výpočtu a vzorce 9.1.1. Výpočet odezvového faktoru R: Pík CMPA: R = W/H kde
9.2. Výpočet procenta sušené syřidlové syrovátky obsažené ve vzorku W(E) = R × H(E) kde
Pokud je W(E) vyšší než 1 % a rozdíl mezi retenčním časem a časem srovnávacího vzorku [5] menší než 0,2 minuty, pak je přítomnost sušiny ze syřidlové syrovátky prokázána. 9.3. Přesnost metody 9.3.1. Opakovatelnost Rozdíl mezi výsledky dvou stanovení, která u stejného zkušebního vzorku provedl současně nebo v těsném sledu jeden pracovník na stejném přístroji, by neměl překročit 0,2 % hm. 9.3.2. Reprodukovatelnost Nestanovena. 9.3.3. Linearita Pro hodnoty od 0 % do 16 % syřidlové syrovátky je třeba získat lineární vztah s korelačním koeficientem > 0,99. 9.4. Interpretace Mezní hodnota 1 % obsahuje nejistotu z důvodu reprodukovatelnosti. Obrázek 1 Ni—4.6 standard
|
|
3) |
Doplňují se nové přílohy, které znějí: „ PŘÍLOHA VI Metody analýzy másla v soukromém skladování
PŘÍLOHA VII Metody analýzy sušeného odstředěného mléka v soukromém skladování
PŘÍLOHA VIII Metody analýzy sýrů v soukromém skladování
Dodatek METODA PRO ZJIŠŤOVÁNÍ KRAVSKÉHO MLÉKA A KASEINÁTŮ V SÝRECH Z OVČÍHO MLÉKA, KOZÍHO MLÉKA NEBO BUVOLÍHO MLÉKA, NEBO SMĚSÍ OVČÍHO, KOZÍHO A BUVOLÍHO MLÉKA 1. PŘEDMĚT Zjišťování kravského mléka a kaseinátů v sýrech vyrobených z ovčího mléka, kozího mléka, buvolího mléka anebo ze směsí ovčího, kozího a buvolího mléka izoelektrickou fokusací γ-kaseinů po plasminolýze. 2. OBLAST POUŽITÍ Metoda je vhodná pro citlivé a specifické zjišťování mléka a kaseinátu, též tepelně ošetřených, v čerstvých a zralých sýrech vyrobených z ovčího mléka, kozího mléka, buvolího mléka nebo ze směsí ovčího, kozího a buvolího mléka. Není vhodná k odhalení falšování mléka a sýrů tepelně ošetřenými bílkovinnými koncentráty z hovězí syrovátky. 3. PODSTATA METODY 3.1. Izolace kaseinů ze sýra a referenčních standardů 3.2. Rozpuštění izolovaných kaseinů a proteolýza plasminem (EC.3.4.21.7) 3.3. Izoelektrická fokusace plasminem ošetřených kaseinů za přítomnosti močoviny a vybarvení bílkovin 3.4. Vyhodnocení vybarvených obrazců kaseinu γ3 a γ2 (důkaz přítomnosti kravského mléka) na základě porovnání obrazce získaného ze vzorku s obrazci získanými u téhož gelu z referenčních standardů obsahujících 0 % a 1 % kravského mléka. 4. CHEMIKÁLIE Není-li stanoveno jinak, používají se pouze chemikálie čistoty p.a. Voda musí být redestilovaná nebo rovnocenné čistoty. Poznámka: Následující údaje se vztahují na laboratorně připravované polyakrylamidové gely s obsahem močoviny o rozměrech 265 × 125 × 0,25 mm. Pokud se použijí jiné rozměry nebo druhy gelu, může být nutné upravit podmínky separace. Izoelektrická fokusace 4.1. Chemikálie pro přípravu polyakrylamidových gelů s obsahem močoviny 4.1.1. Zásobní roztok gelu Ve vodě rozpusťte:
doplňte na 100 ml a uložte do chladničky v hnědé skleněné lahvi. Poznámka: Místo uvedených množství neurotoxického akrylamidu lze použít komerčně dostupný, předem namíchaný roztok akrylamidu a BIS. V případě, že koncentrace komerčního roztoku je 30 % hm./obj. akrylamidu a 0,8 % hm./obj. BIS, pak uvedená množství (4,85 g akrylamidu a 0,15 g BIS) musí být nahrazena komerčním roztokem o objemu 16,2 ml. Zásobní roztok lze skladovat nejdéle 10 dnů. Pokud je jeho vodivost vyšší než 5 μS, je nutné provést jeho deionizaci promícháváním s 2 g Amberlitu MB-3 po dobu 30 minut a poté jej přefiltrovat přes 0,45μm membránu. 4.1.2. Roztok gelu Připravte roztok gelu smícháním přísad a amfolytů (*) se zásobním roztokem gelu (viz 4.1.1).
Gelový roztok promíchejte a odplyňujte po dobu dvou až tří minut v ultrazvukové lázni nebo ve vakuu. Poznámka: Gelový roztok se musí připravit bezprostředně před naléváním (viz 6.2). 4.1.3. Roztoky katalyzátorů 4.1.3.1. N, N, N′, N′-tetramethylendiamin (Temed) 4.1.3.2. 40 % hm./obj. roztoku persíranu amonného (PER) 800 mg PER se rozpustí ve vodě a doplní se do 2 ml. Poznámka: Vždy používejte čerstvě připravený roztok PER. 4.2. Kontaktní tekutina Kerosin nebo kapalný parafin 4.3. Anodový roztok Rozpusťte 5,77 g kyseliny fosforečné (85 % hm.) ve vodě a zřeďte na 100 ml. 4.4. Katodový roztok Rozpusťte 2,00 g hydroxidu sodného ve vodě a zřeďte vodou na 100 ml. Příprava vzorku 4.5. Chemikálie určené k izolaci bílkovin 4.5.1. Zředěná kyselina octová (25,0 ml ledové kyseliny octové doplněné vodou do 100 ml). 4.5.2. Dichlormethan 4.5.3. Aceton 4.6. Tlumivý roztok pro rozpuštění bílkovin Ve vodě rozpusťte:
a doplňte na 50 ml vodou. Poznámka: Skladujte v chladničce; maximální doba uskladnění je jeden týden. 4.7. Chemikálie určené k plasminovému štěpení kaseinů 4.7.1. Tlumivý roztok uhličitanu amonného Titrujte molární roztok hydrogenuhličitanu amonného 0,2 mol/l (1,58 g/100 ml vody) obsahující 0,05 mol/l kyseliny ethylendiamintetraoctové (EDTA, 1,46 g/100 ml) roztokem uhličitanu amonného 0,2 mol/l (1,92 g/100 ml vody) obsahujícího 0,05 mol/l EDTA na pH 8. 4.7.2. Hovězí plasmin (EC. 3.4.21.7), s aktivitou nejméně 5 U/ml. 4.7.3. Roztok kyseliny ε-aminokapronové určené k inhibici enzymu Rozpusťte 2, 624 g kyseliny ε-aminokapronové (6-amino-n-hexankyseliny) ve 100 ml 40 % obj. ethanolu. 4.8. Referenční standardy 4.8.1. Certifikované referenční standardy směsi ovčího a kozího odstředěného mléka vysráženého pomocí syřidla a s obsahem 0 % a 1 % kravského mléka lze získat od Ústavu pro referenční materiály a měření působícího při Komisi, B-2440, Geel, Belgie. 4.8.2. Příprava laboratorních prozatímních standardů buvolího mléka vysráženého pomocí syřidla a s obsahem 0 % a 1 % kravského mléka Odstředěné mléko se připraví odstředěním syrového buvolího nebo kravského mléka při teplotě 37 °C (2 500 g, 20 minut). Po rychlém ochlazení zkumavky a jejího obsahu na 6 až 8 °C se úplně odstraní horní tuková vrstva. Pro přípravu 1 % standardu přidejte 5,00 ml odstředěného kravského mléka do 495 ml odstředěného buvolího mléka v kádince o obsahu 1 l. Hodnotu pH upravte na 6,4 přídavkem zředěné kyseliny mléčné (10 %, hm./obj.). Teplotu upravte na 35 °C, přidejte 100 μl telecího syřidla (aktivita syřidla 1: 10 000, cca 3 000 U/ml) a míchejte po dobu 1 minuty. Poté kádinku zakryjte hliníkovou fólií a nechte hodinu stát při teplotě 35 °C k utvoření sýřeniny. Po utvoření sýřeniny se veškeré mléko vysrážené pomocí syřidla lyofilizuje, aniž by předtím byla provedena homogenizace nebo odebrání syrovátky. Lyofilizované mléko se jemně rozemele na homogenní prášek. K přípravě referenčního standardu 0 % použijte stejný postup u čistého odstředěného buvolího mléka. Referenční standardy musí být skladovány při teplotě – 20 °C. Poznámka: Před přípravou referenčních standardů se doporučuje zkontrolovat čistotu buvolího mléka izoelektrickou fokusací kaseinů vystavených působení plasminu. Chemikálie určené k barvení bílkovin 4.9. Fixativ Rozpusťte 150 g kyseliny trichloroctové ve vodě a doplňte na 1 000 ml. 4.10. Odbarvovací roztok Zřeďte 500 ml methanolu a 200 ml ledové kyseliny octové destilovanou vodou na 2 000 ml. Poznámka: Odbarvovací roztok se připravuje každý den čerstvý; lze jej připravit smícháním stejných objemů zásobního roztoku 50 % (obj.) methanolu a zásobního roztoku 20 % (obj.) ledové kyseliny octové. 4.11. Barvicí roztoky 4.11.1. Barvicí roztok (zásobní roztok 1) V 1 000 ml 90 % obj. methanolu rozpusťte pomocí magnetické míchačky (asi 45 minut) 3,0 g brilantní modři Coomassie G-250 (C. I. 42655) a roztok přefiltrujte přes dva středně husté skládané filtry. 4.11.2. Barvicí roztok (zásobní roztok 2) V 1 000 ml 20 % obj. kyseliny octové rozpusťte 5,0 g síranu měďnatého, pentahydrátu. 4.11.3. Barvicí roztok (pracovní roztok) Bezprostředně před barvením smíchejte 125 ml obou zásobních roztoků (4.11.1 a 4.11.2). Poznámka: Barvicí roztok je třeba použít v den přípravy. 5. PŘÍSTROJE A POMŮCKY 5.1. Skleněné desky (265 x 125 x 4 mm), pryžový váleček (o šířce 15 cm), nivelační stolek. 5.2. Nosná fólie gelu (265 × 125 mm) 5.3. Krycí fólie (280 × 125 mm). Na každý dlouhý okraj nalepte pruh lepicí pásky (280 × 6 × 0,25 mm) (viz obrázek 1). 5.4. Elektrofokusační komora s chladicí deskou (např. 265 × 125 mm) a vhodný napájecí zdroj (≥ 2,5 kV) nebo automatické elektroforetické zařízení 5.5. Cirkulační kryostat s regulací teploty ve výši 12 ± 0,5 °C 5.6. Odstředivka nastavitelná na 3 000 g 5.7. Elektrodové proužky (o délce ≥ 265 mm) 5.8. Plastové kapací nádobky na anodový a katodový roztok 5.9. Aplikátory vzorku (10 × 5 mm, z viskózy nebo filtračního papíru s nízkou adsorpcí bílkovin) 5.10. Odbarvovací a barvicí misky (např. misky na nástroje 280 × 150 mm) z nerezové oceli nebo skleněné 5.12. Nastavitelný tyčový homogenizátor s průměrem tyče 10 mm a rychlostí 8 000 až 20 000 ot/min 5.13. Magnetická míchačka 5.14. Ultrazvuková lázeň 5.15. Svářečka fólií 5.16. Mikropipety na 25 μl 5.17. Vakuová odstředivka nebo lyofilizátor 5.18. Termostatem řízená vodní lázeň, nastavitelná na 35 a 40 ± 1 °C s třepačkou 5.19. Densitometrické zařízení umožňující měření na vlnové délce λ = 634 nm 6. PRACOVNÍ POSTUP 6.1. Příprava vzorku 6.1.1. Izolace kaseinů Do odstředivkové kyvety na 100 ml navažte množství sýra nebo referenčního standardu odpovídající 5 g sušiny, přidejte 60 ml destilované vody a obsah homogenizujte tyčovým homogenizátorem (8 000 až 10 000 ot/min). Upravte pH na 4,6 zředěnou kyselinou octovou (4.5.1) a odstřeďujte (5 minut, 3 000 g). Dekantujte tuk a syrovátku a zbytek homogenizujte při 20 000 ot/min se 40 ml destilované vody s hodnotou pH upravenou na 4,5 zředěnou kyselinou octovou (4.5.1). Přidejte 20 ml dichlormethanu (4.5.2) a znovu homogenizujte a odstřeďuje (5 minut, 3 000 g). Špachtlí vyjměte kaseinovou vrstvu, která se nachází mezi vodnou a organickou fází (viz obrázek 2), a obě fáze dekantujte. Kasein se znovu homogenizuje se 40 ml destilované vody (viz výše) a 20 ml dichlormethanu (4.5.2) a odstředí se. Tento postup opakujte, dokud obě extrakční fáze nejsou bezbarvé (dvakrát až třikrát). Bílkovinný zbytek homogenizujte s 50 ml acetonu (4.5.3) a přefiltrujte přes středně hustý skládaný papírový filtr. Zbytek promyjte dvěma samostatnými 25 ml dávkami acetonu a nechte vysušit na vzduchu nebo v proudu dusíku. Potom jemně rozmělněte v třecí misce. Poznámka: Suché bílkovinné extrakty se musí uchovávat při teplotě – 20 °C. 6.1.2. Přeměna β-kaseinů na γ-kaseiny působením plasminu Rozptylte 25 mg izolovaných kaseinů (6.1.1) v 0,5 ml tlumivého roztoku uhličitanu amonného (4.7.1) a směs 20 minut homogenizujte, např. ultrazvukem. Směs zahřejte na 40 °C a přidejte 10 μl plasminu (4.7.2), promíchejte a inkubujte 1 hodinu při teplotě 40 °C za stálého třepání. Pro inhibici enzymu přidejte 20 μl roztoku kyseliny ε-aminokapronové (4.7.3). Poté přidejte 200 mg pevné močoviny a 2 mg dithiothreitolu. Poznámka: Pro získání lepší symetrie fokusovaných kaseinových pásů se doporučuje po přidání kyseliny ε-aminokapronové roztok lyofilizovat a poté zbytky rozpustit v 0,5 ml tlumivého roztoku pro rozpouštění bílkovin (4.6). 6.2. Příprava polyakrylamidových gelů s obsahem močoviny Na skleněnou desku (5.1) válečkem naneste nosnou fólii gelu (5.2) s pomocí několika kapek vody a přebytečnou vodu odsajte papírovým ubrouskem nebo kapesníčkem. Stejným způsobem válečkem naneste druhou, krycí fólii (5.3) s distančními vložkami (0,25 mm) na jinou skleněnou desku. Tuto druhou desku uložte vodorovně na nivelační stolek. K připravenému odplyněnému roztoku gelu (4.1.2) přidejte 10 μl Temedu (4.1.3.1), protřepejte a přidejte 10 μl roztoku PER (4.1.3.2); důkladně promíchejte a okamžitě nalijte rovnoměrně na střed krycí fólie. Jeden okraj nosné desky gelu (stranou s fólií směrem dolů) přiložte k desce s krycí fólií a pomalu pokládejte tak, aby se mezi fóliemi vytvořila pravidelně rozprostřená tenká vrstva gelu bez bublin (obrázek 3). Pomocí tenké špachtle nosnou desku gelu opatrně přitiskněte po celé délce a položte na ni tři další skleněné desky, aby ji zatížily. Po skončení polymerace (asi po 60 minutách) spolu s krycí fólií sejměte gel zpolymerovaný na nosné fólii gelu tak, že skleněné desky nakloníte. Zadní stranu nosné fólie pečlivě očistěte, aby se odstranily zbytky gelu a močoviny. „Gelový sendvič“ zavařte do tenké fólie a uložte do chladničky (nejdéle na šest týdnů). Poznámka: Krycí fólii s distančními vložkami lze použít znovu. Polyakrylamidový gel může být rozřezán na menší části; je to vhodné v případě omezeného počtu vzorků nebo v případě použití automatického elektroforetického zařízení (dva gely o rozměrech 4,5 × 5 cm). 6.3. Izoelektrická fokusace Nastavte kryostat na 12 °C. Zadní stranu nosné fólie gelu otřete kerosinem a poté na střed chladicího bloku kápněte několik kapek kerosinu (4.2). Opatrně přiložte „gelový sendvič“ nosnou fólií obrácený dolů, aby se vytlačily všechny vzduchové bubliny. Přebytečný kerosin otřete a sejměte krycí fólii. Elektrodové proužky napusťte elektrodovými roztoky (4.3, 4.4), seřízněte na délku gelu a uložte na místo (vzdálenost elektrod 9,5 cm). Podmínky izoelektrické fokusace 6.3.1. Rozměry gelu 265 × 125 × 0,25 mm
Poznámka: Jestliže se změní tloušťka nebo šířka gelů, hodnoty proudu a příkonu musí být odpovídajícím způsobem upraveny (např. hodnoty proudu a příkonu se zdvojnásobí, použije-li se gel o rozměrech 265 × 125 × 0,5 mm). 6.3.2. Příklad programování napětí u automatického elektroforetického zařízení (2 gely 5,0 × 4,5 cm, elektrody bez proužků se přikládají přímo na gel.
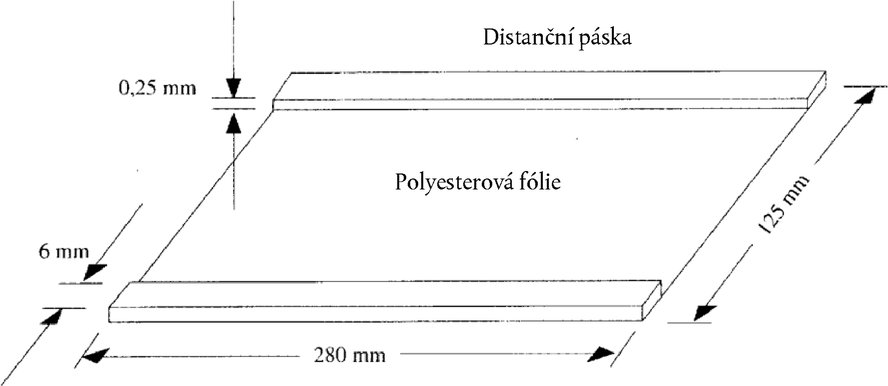
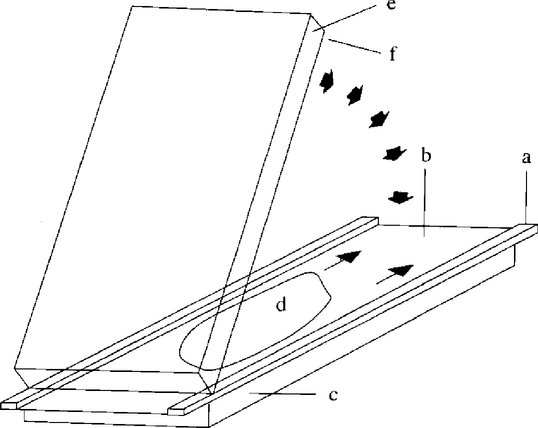
Aplikátor vzorku ve fázi 2 vložte při 0 Vh. Aplikátor vzorku ve fázi 2 odstraňte při 30 Vh. 6.4. Barvení bílkovin 6.4.1. Fixace bílkovin Okamžitě po vypnutí napájení se odstraní elektrodové proužky a gel se ihned vloží do barvicí/odbarvovací misky naplněné 200 ml fixatéru (4.9). Gel v ní ponechte 15 minut za stálého třepání. 6.4.2. Praní a barvení gelové desky Fixatér beze zbytku dekantujte a gelovou desku dvakrát promývejte po dobu 30 sekund vždy ve 100 ml odbarvovacího roztoku (4.10) Odbarvovací roztok dekantujte, misku naplňte 250 ml barvicího roztoku (4.11.3) a gel nechte za mírného třepání po dobu 45 minut zbarvovat. 6.4.3. Odbarvení gelové desky Barvicí roztok dekantujte a gelovou desku dvakrát promývejte po dobu 30 sekund vždy ve 100 ml odbarvovacího roztoku (4.10). Poté 15 minut protřepávejte s 200 ml odbarvovacího roztoku; fázi odbarvování opakujte nejméně dvakrát až třikrát, dokud není pozadí čiré a bezbarvé. Gelovou desku potom promyjte destilovanou vodou (2 × 2 minuty) a nechte uschnout na vzduchu (2 až 3 hodiny) nebo usušte vysoušečem vlasů (10 až 15 minut). Poznámka 1: Fixaci, promývání, barvení a odbarvování provádějte při teplotě 20 °C. Vyšší teploty nepoužívejte. Poznámka 2: Pokud upřednostňujete citlivější obarvení stříbrem (např. Silver Staining Kit, Protein, Pharmacia Biotech, kód č. 17-1150-01), musí být kaseinové vzorky ošetřené plasminem zředěny v poměru 5 mg/ml. 7. HODNOCENÍ Hodnocení se provádí porovnáním proteinových obrazců zkoumaného vzorku s obrazci referenčních standardů u stejného gelu. Zjišťování kravského mléka v sýrech vyrobených z ovčího mléka, kozího mléka, buvolího mléka nebo ze směsí ovčího, kozího a buvolího mléka se provádí prostřednictvím γ3- a γ2-kaseinů, jejichž izoelektrické body se nacházejí v rozmezí mezi pH 6,5 a pH 7,5 (obrázky 4a, b, obrázek 5). Detekční mez je nižší než 0,5 %. 7.1. Vizuální hodnocení Pro vizuální hodnocení množství kravského mléka se doporučuje upravit koncentraci vzorků a referenčních standardů s cílem dosáhnout u hovězích, kozích a/nebo buvolích γ2- a γ3-kaseinů (viz „γ2 E,G,B“ a „γ3 E,G,B“ na obrázcích 4a, b a obrázku 5) stejné úrovně intenzity. Pouze za těchto podmínek lze přímo posoudit množství kravského mléka (menší nebo větší než 1 % nebo ve výši 1 %) v analyzovaném vzorku porovnáním intenzity hovězích γ3- a γ2-kaseinů (viz „γ3 C“ a „γ2 C“ na obrázcích 4 a, b a obrázku 5) s intenzitami kaseinů v referenčních standardech s obsahy 0 % a 1 % (ovčích, kozích), nebo prozatímních laboratorních standardech (buvolích). 7.2. Denzitometrické hodnocení Pokud je to možné, použijte pro stanovení poměru mezi plochami píků hovězích γ2- a γ3-kaseinů a plochami píků ovčích, kozích a/nebo buvolích γ2- a γ3-kaseinů (viz obrázek 5) denzitometrii (5.19). Tuto hodnotu porovnejte s poměrem ploch píků γ2- a γ3-kaseinů v 1 % referenčním standardu (ovčím, kozím) nebo prozatímním laboratorním standardu (buvolím) analyzovaných u téhož gelu. Poznámka: Tato metoda funguje uspokojivě, pokud existuje jasný pozitivní signál obou hovězích γ2- a γ3-kaseinů v 1 % referenčním standardu, avšak nikoli v 0 % referenčním standardu. V opačném případě je nutné postup optimalizovat při přesném dodržení pokynů stanovených v dané metodě. Vzorek je považován za pozitivní, jestliže hodnoty obou hovězích γ2- a γ3-kaseinů nebo příslušných poměrů ploch píků jsou stejné nebo větší než hodnoty odpovídající 1 % referenčnímu standardu. 8. LITERATURA Addeo F., Moio L., Chianese L., Stingo C., Resmini P., Berner I, Krause I., Di Luccia A., Bocca A.: Use of plasmin to increase the sensitivity of the detection of bovine milk in ovine and/or caprine cheese by gel isoelectric focusing of γ2-caseins. Milchwissenschaft 45, 708–711 (1990). Addeo F., Nicolai M.A., Chianese L., Moio L., Spagna Musso S., Bocca A., Del Giovine L.: A control method to detect bovine milk in ewe and water buffalo cheese using immunoblotting. Milchwissenschaft 50, 83–85 (1995). Krause I., Berner I, Klostermeyer H.: Sensitive detection of cow milk in ewe and goat milk and cheese by carrier ampholyte — and carrier ampholyte/immobilized pH gradient — isoelectric focusing of γ-caseins using plasmin as signal amplifier. in: Electrophoresis-Forum 89 (B. J. Radola, ed.) pp 389-393, Bode-Verlag, München (1989). Krause Ι., Belitz H.-D., Kaiser K.-P.: Nachweis von Kuhmilch in Schaf and Ziegenmilch bzw. -käse durch isoelektrische Fokussierung in harnstoffhaltigen Polyacrylamidgelen. Z. Lebensm. Unters. Forsch. 174, 195–199 (1982). Radola B.J.: Ultrathin-layer isoelectric focusing in 50-100 μm polyacrylamide gels on silanised glass plates or polyester films. Electrophoresis 1, 43–56 (1980). Obrázek 1 Schematický výkres krycí fólie
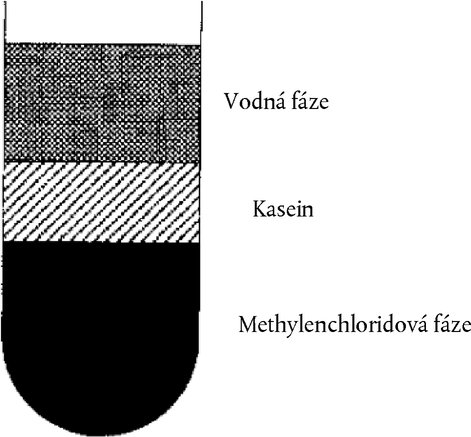
Obrázek 2 Kaseinová vrstva plovoucí mezi vodnou a organickou fází po odstředění
Obrázek 3 Sklápěcí technika pro odlévání ultratenkých polyakrylamidových gelů
a = distanční páska (0,25 mm); b = krycí fólie (5.3); c, e = skleněné desky (5.1); d = gelový roztok (4.1.2); f = nosná fólie gelu (5.2). Obrázek 4a Izoelektrická fokusace kaseinů ze sýrů z ovčího a kozího mléka, ošetřených plasminem, s různým obsahem kravského mléka
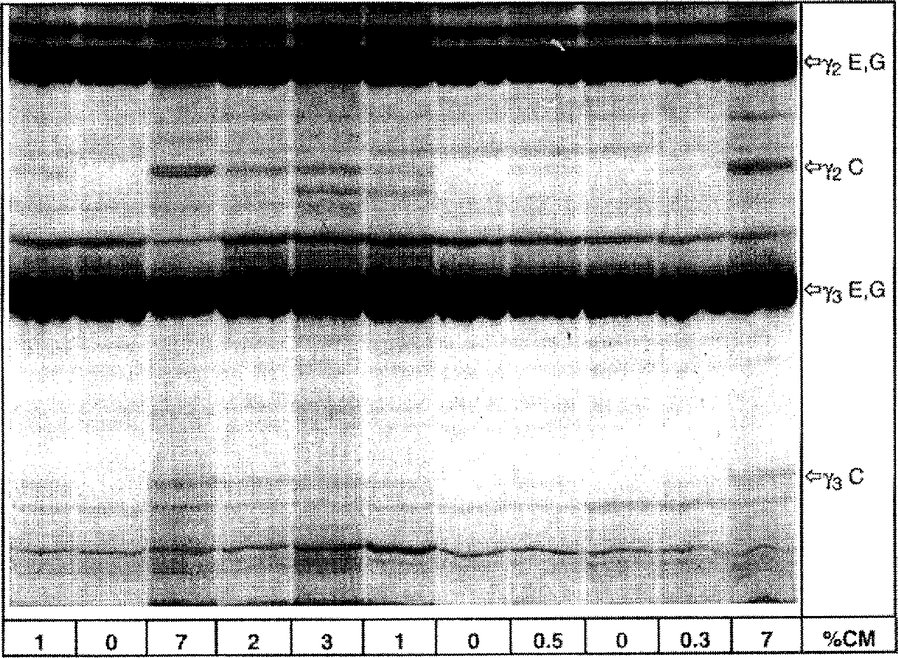
% CM = procento kravského mléka, C = kravské, E = ovčí, G = kozí Na obrázku je znázorněna horní polovina gelu IEF. Obrázek 4b Izoelektrická fokusace kaseinů ze sýrů vyrobených ze směsí ovčího, kozího a buvolího mléka, ošetřených plasminem, s různým obsahem kravského mléka
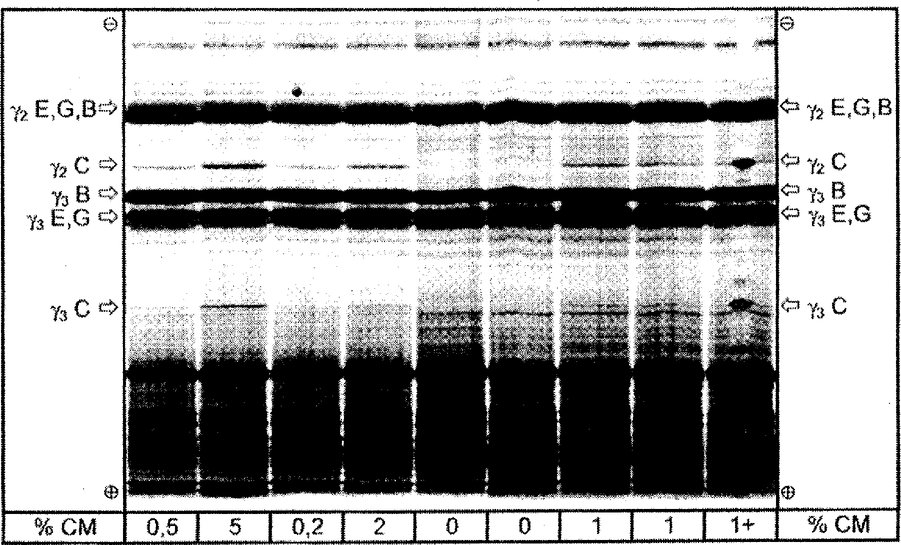
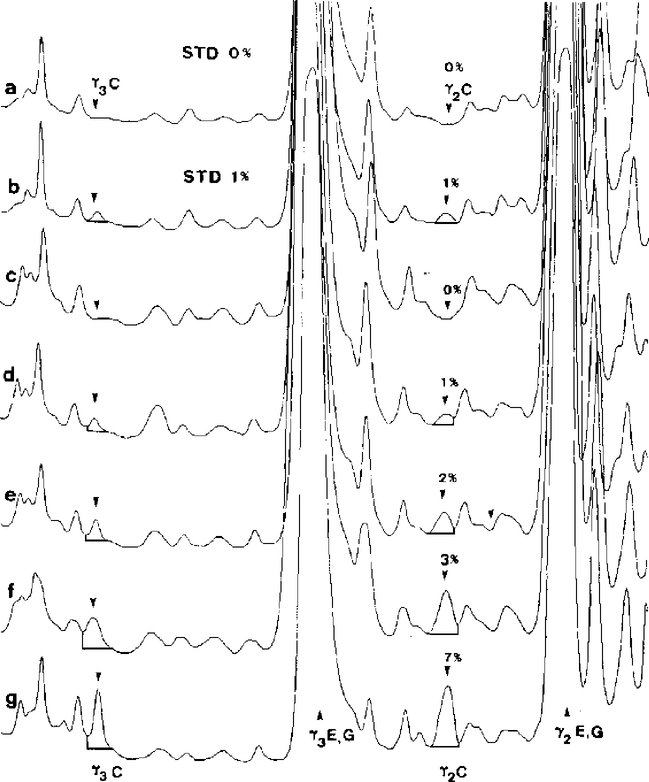
% CM = procento kravského mléka; 1 + = vzorek s obsahem 1 % kravského mléka a s přídavkem čistého hovězího kaseinu ve středu dráhy. C = kravské, E = ovčí, G = kozí, B = buvolí Na obrázku je znázorněna celá separační vzdálenost gelu IEF. Obrázek 5 Superpozice denzitogramů referenčních standardů (STD) a vzorků sýra vyrobeného ze směsi ovčího a kozího mléka, po izoelektrické fokusaci
a, b = referenční standardy obsahující 0 a 1 % kravského mléka; c-g = vzorky sýra obsahujícího 0, 1, 2, 3 a 7 % kravského mléka; C = kravské, E = ovčí, G = kozí. Byla snímána horní polovina gelu IEF při λ = 634 nm. PŘÍLOHA IX Hodnocení analýz 1. Zajištění jakosti Analýzy provádějí laboratoře určené v souladu s článkem 12 nařízení (ES) č. 882/2004 (*) nebo určené příslušnými orgány členského státu. 2. Odběr vzorků a námitky proti výsledkům analýzy
Dodatek Zhodnocení splnění zákonem stanovené mezní hodnoty s ohledem na šarži 1. Podstata metody V případech, kdy právní předpisy týkající se veřejné intervence a soukromého skladování stanoví podrobné postupy pro odběr vzorků, dodržují se tyto postupy. Ve všech ostatních případech se používá vzorek nejméně ze 3 jednotek vzorku odebraných náhodně ze šarže odevzdané ke kontrole. Lze připravit i složený vzorek. Získaný výsledek se porovná se zákonem stanovenými mezními hodnotami na základě výpočtu 95 % intervalu spolehlivosti jakožto dvojnásobku směrodatné odchylky, kde příslušná směrodatná odchylka závisí buď na tom, 1) zda byla metoda validována na základě mezinárodní spolupráce s hodnotami pro σr a σR, nebo v případě vnitrolaboratorní validace na tom, 2) zda byla vypočítána vnitřní reprodukovatelnost. Tento interval spolehlivosti se potom rovná nejistotě měření výsledku. 2. Metoda je validována na základě mezinárodní spolupráce V tomto případě byla směrodatná odchylka opakovatelnosti σr a směrodatná odchylka reprodukovatelnosti σR stanovena a laboratoř může prokázat soulad s pracovními charakteristikami validované metody. Vypočítá se aritmetický průměr Vypočítá se rozšířená nejistota (k = 2) z
Je-li konečný výsledek měření x vypočítán ze vzorce ve tvaru x = y 1 + y 2, x = y 1 – y 2, x = y 1 · y 2 nebo x = y 1/y 2, musí se v takových případech postupovat podle běžných postupů pro kombinování směrodatných odchylek. Šarže se považuje za nevyhovující horní zákonem stanovené mezní hodnotě UL, jestliže
jinak se má za to, že hodnotě UL vyhovuje. Šarže se považuje za nevyhovující dolní zákonem stanovené mezní hodnotě LL, jestliže
jinak se má za to, že hodnotě LL vyhovuje. 3. Vnitrolaboratorní validace s výpočtem směrodatné odchylky vnitřní reprodukovatelnosti V případech, kdy jsou použity metody, které nejsou stanoveny v tomto nařízení, a kdy nebyla stanovena opatření pro zajištění shodnosti, je třeba provést vnitrolaboratorní validaci. Namísto σr a σ R se musí ve vzorci pro výpočet rozšířené nejistoty U použít směrodatná odchylka vnitřní opakovatelnosti sir a směrodatná odchylka vnitřní reprodukovatelnosti si R . Pravidla, jež je třeba dodržovat za účelem splnění zákonem stanovené mezní hodnoty, jsou uvedena v bodě 1. Považuje-li se však šarže za nesplňující zákonem stanovenou mezní hodnotu, měření se zopakují pomocí metody stanovené v tomto nařízení a výsledek se vyhodnotí v souladu s bodem 1.
“ |
 z počtu
z počtu  takto:
takto:
 ;
;(1) Metodu, která má být použita, musí schválit platební agentura.
(2) Analýzy připálených částic mohou být prováděny systematicky. Tyto analýzy se však provádějí vždy, pokud neprobíhají organoleptické kontroly.
(3) Metodu, která má být použita, musí schválit platební agentura (jednu metodu nebo obě).
(4) Metodu, která má být použita, musí schválit platební agentura.
(5) Organoleptické kontroly se provádějí v případě potřeby na základě analýzy rizik schválené platební agenturou.
(6) Metodu, která má být použita, musí schválit platební agentura.
(7) Použití vzorku: Po předběžné fokusaci (krok 1) pipetou odměřte 18 μl vzorku a standardních roztoků do aplikátorů vzorku (10 × 5 mm), vložte na gel ve vzdálenosti 1 mm od sebe a 5 mm podélně od anody a jemně přitlačte. Fokusaci proveďte za výše uvedených podmínek a aplikátory vzorku opatrně vyjměte po 60 minutách fokusace vzorku.