(ES) č. 152/2009Nařízení Komise (ES) č. 152/2009 ze dne 27. ledna 2009 , kterým se stanoví metody odběru vzorků a laboratorního zkoušení pro úřední kontrolu krmiv (Text s významem pro EHP)
| Publikováno: | Úř. věst. L 54, 26.2.2009, s. 1-130 | Druh předpisu: | Nařízení |
| Přijato: | 27. ledna 2009 | Autor předpisu: | Evropská komise |
| Platnost od: | 18. března 2009 | Nabývá účinnosti: | 18. března 2009 |
| Platnost předpisu: | Ano | Pozbývá platnosti: | |
Text předpisu s celou hlavičkou je dostupný pouze pro registrované uživatele.
NAŘÍZENÍ KOMISE (ES) č. 152/2009
ze dne 27. ledna 2009,
kterým se stanoví metody odběru vzorků a laboratorního zkoušení pro úřední kontrolu krmiv
(Text s významem pro EHP)
KOMISE EVROPSKÝCH SPOLEČENSTVÍ,
s ohledem na Smlouvu o založení Evropského společenství,
s ohledem na nařízení Evropského parlamentu a Rady (ES) č. 882/2004 ze dne 29. dubna 2004 o úředních kontrolách za účelem ověření dodržování právních předpisů týkajících se krmiv a potravin a pravidel o zdraví zvířat a dobrých životních podmínkách zvířat (1), a zejména na čl. 11 odst. 4 písm. a), b) a c) uvedeného nařízení,
vzhledem k těmto důvodům:
|
(1) |
Za účelem provedení směrnice 70/373/EHS byly přijaty tyto akty, které v souladu s čl. 61 odst. 2 nařízení (ES) č. 882/2004 zůstávají nadále v platnosti:
|
|
(2) |
Jelikož směrnice 70/373/EHS byla nahrazena nařízením (ES) č. 882/2004, je vhodné nahradit prováděcí akty uvedené směrnice jediným nařízením. Zároveň by měly být přizpůsobeny metody za účelem zohlednění vývoje v oblasti vědeckých a technických poznatků. Metody, které již nejsou vhodné pro zamýšlený účel, by měly být odstraněny. Ve vhodnou dobu se plánuje aktualizace pravidel pro odběr vzorků, aby byl zohledněn nedávný vývoj v oblasti výroby, skladování a přepravy krmiv a jejich uvádění na trh, avšak v současnosti je vhodné zachovat stávající pravidla pro odběr vzorků. |
|
(3) |
Směrnice 71/250/EHS, 71/393/EHS, 72/199/EHS, 73/46/EHS, 76/371/EHS, 76/372/EHS, 78/633/EHS, 81/715/EHS, 84/425/EHS, 86/174/EHS, 93/70/EHS, 93/117/ES, 98/64/ES, 1999/27/ES, 1999/76/ES, 2000/45/ES, 2002/70/ES a 2003/126/ES by proto měly být zrušeny. |
|
(4) |
Opatření stanovená tímto nařízením jsou v souladu se stanoviskem Stálého výboru pro potravinový řetězec a zdraví zvířat, |
PŘIJALA TOTO NAŘÍZENÍ:
Článek 1
Odběr vzorků pro úřední kontrolu krmiv, pokud jde o stanovení jejich složek, doplňkových látek a nežádoucích látek, s výjimkou reziduí pesticidů a mikroorganismů, se provádí podle metod stanovených v příloze I.
Článek 2
Příprava vzorků ke zkoušení a vyjádření výsledků se provádí podle metod stanovených v příloze II.
Článek 3
Zkoušení pro úřední kontrolu krmiv se provádí za použití metod stanovených v příloze III (Metody zkoušení pro kontrolu složení krmných surovin a krmných směsí), v příloze IV (Metody zkoušení pro kontrolu obsahu povolených doplňkových látek v krmivech), v příloze V (Metody zkoušení pro kontrolu nežádoucích látek v krmivech) a v příloze VI (Metody zkoušení pro stanovení složek živočišného původu pro úřední kontrolu krmiv).
Článek 4
Obsah energie u krmných směsí pro drůbež se vypočítá na základě přílohy VII.
Článek 5
Metody zkoušení pro kontrolu nepřípustné přítomnosti již nepovolených doplňkových látek v krmivech, které jsou stanoveny v příloze VIII, se použijí pro účely potvrzování.
Článek 6
Směrnice 71/250/EHS, 71/393/EHS, 72/199/EHS, 73/46/EHS, 76/371/EHS, 76/372/EHS, 78/633/EHS, 81/715/EHS, 84/425/EHS, 86/174/EHS, 93/70/EHS, 93/117/ES, 98/64/ES, 1999/27/ES, 1999/76/ES, 2000/45/ES, 2002/70/ES a 2003/126/ES se zrušují.
Odkazy na zrušené směrnice se považují za odkazy na toto nařízení v souladu se srovnávacími tabulkami v příloze IX.
Článek 7
Toto nařízení vstupuje v platnost dvacátým dnem po vyhlášení v Úředním věstníku Evropské unie.
Použije se od 26. srpna 2009.
Toto nařízení je závazné v celém rozsahu a přímo použitelné ve všech členských státech.
V Bruselu dne 27. ledna 2009.
Za Komisi
Androulla VASSILIOU
členka Komise
(1) Úř. věst. L 165, 30.4.2004, s. 1.
(2) Úř. věst. L 155, 12.7.1971, s. 13.
(3) Úř. věst. L 279, 20.12.1971, s. 7.
(4) Úř. věst. L 123, 29.5.1972, s. 6.
(5) Úř. věst. L 83, 30.3.1973, s. 21.
(6) Úř. věst. L 102, 15.4.1976, s. 1.
(7) Úř. věst. L 102, 15.4.1976, s. 8.
(8) Úř. věst. L 206, 29.7.1978, s. 43.
(9) Úř. věst. L 257, 10.9.1981, s. 38.
(10) Úř. věst. L 238, 6.9.1984, s. 34.
(11) Úř. věst. L 130, 16.5.1986, s. 53.
(12) Úř. věst. L 234, 17.9.1993, s. 17.
(13) Úř. věst. L 329, 30.12.1993, s. 54.
(14) Úř. věst. L 257, 19.9.1998, s. 14.
(15) Úř. věst. L 118, 6.5.1999, s. 36.
(16) Úř. věst. L 207, 6.8.1999, s. 13.
(17) Úř. věst. L 174, 13.7.2000, s. 32.
(18) Úř. věst. L 209, 6.8.2002, s. 15.
(19) Úř. věst. L 339, 24.12.2003, s. 78.
PŘÍLOHA I
METODY ODBĚRU VZORKŮ
1. ÚČEL A ROZSAH POUŽITÍ
Vzorky určené pro úřední kontroly krmiv se odebírají podle níže popsaných metod. Takto získané vzorky jsou považovány za reprezentativní vzorky z dané partie.
2. ZAMĚSTNANCI PROVÁDĚJÍCÍ ODBĚR VZORKŮ
Odběr vzorků provádí osoby pověřené za tímto účelem členskými státy.
3. DEFINICE
Vzorkovaná partie: množství produktů, které tvoří určitou jednotku a u nichž se předpokládají stejné vlastnosti.
Dílčí vzorek: množství odebrané v jednom bodě vzorkované partie.
Souhrnný vzorek: souhrn dílčích vzorků odebraných ze stejné vzorkované partie.
Redukovaný vzorek: reprezentativní část souhrnného vzorku, která se získá redukcí jeho množství.
Konečný vzorek: část redukovaného vzorku nebo homogenizovaného souhrnného vzorku.
4. NÁSTROJE
|
4.1 |
Nástroje určené k odběru vzorků musí být vyrobeny z materiálů, které nemohou kontaminovat produkty, u nichž má být odběr vzorků proveden. Tyto nástroje mohou být oficiálně schváleny členskými státy. |
4.2 Nástroje doporučené pro odběr vzorků tuhých krmiv
4.2.1 Ruční odběr vzorků
|
4.2.1.1 |
Lopatka s rovným dnem a vertikálními okraji. |
|
4.2.1.2 |
Vzorkovač s dlouhým žlábkem nebo vzorkovací krabice. Rozměry vzorkovače musí být přiměřené vlastnostem vzorkované partie (hloubce nádoby, rozměrům pytle apod.) a velikosti částic krmiva. |
4.2.2 Mechanický odběr vzorků
Pro odběr vzorků krmiva v pohybu lze použít schválená mechanická zařízení.
4.2.3 Dělicí nástroj
Nástroje určené k dělení vzorků na přibližně stejné části mohou být použity pro odběr dílčích vzorků i pro úpravu redukovaných či konečných vzorků.
5. KVANTITATIVNÍ POŽADAVKY
|
5.A |
Týkající se kontroly látek nebo produktů obsažených rovnoměrně v krmivech |
|
|
5.A.1 |
Vzorkovaná partie Velikost vzorkované partie musí být taková, aby bylo možné odebrat vzorky ze všech jejích částí. |
|
|
5.A.2 |
Dílčí vzorky |
|
|
5.A.2.1 |
Volně ložené krmivo: |
Minimální počet dílčích vzorků: |
|
5.A.2.1.1 |
vzorkované partie o hmotnosti nepřevyšující 2,5 tuny |
sedm |
|
5.A.2.1.2 |
vzorkované partie o hmotnosti vyšší než 2,5 tuny |
√ 20krát počet tun, který tvoří vzorkovanou partii (1), maximálně 40 dílčích vzorků |
|
5.A.2.2 |
Balené krmivo: |
Minimální počet balení, z nichž má být odebrán vzorek (2): |
|
5.A.2.2.1 |
Balení o hmotnosti vyšší než jeden kilogram: |
|
|
5.A.2.2.1.1 |
vzorkované partie obsahující jedno až čtyři balení |
všechna balení |
|
5.A.2.2.1.2 |
vzorkované partie obsahující pět až 16 balení |
čtyři |
|
5.A.2.2.1.3 |
vzorkované partie obsahující více než 16 balení |
√ počtu balení, který tvoří vzorkovanou partii (1), maximálně 20 balení |
|
5.A.2.2.2 |
Balení o hmotnosti nepřevyšující 1 kg |
čtyři |
|
5.A.2.3 |
Tekuté nebo polotekuté krmivo: |
Minimální počet nádob, z nichž má být odebrán vzorek (2): |
|
5.A.2.3.1 |
Nádoby o obsahu vyšším než jeden litr: |
|
|
5.A.2.3.1.1 |
vzorkované partie obsahující jednu až čtyři nádoby |
všechny nádoby |
|
5.A.2.3.1.2 |
vzorkované partie obsahující pět až 16 nádob |
čtyři |
|
5.A.2.3.1.3 |
vzorkované partie obsahující více než 16 nádob |
√ počtu nádob, které tvoří vzorkovanou partii (1), maximálně 20 nádob |
|
5.A.2.3.2 |
Nádoby o obsahu nepřevyšujícím jeden litr |
čtyři |
|
5.A.2.4 |
Krmivo v blocích a minerální lizy |
Minimální počet bloků nebo lizů, z nichž má být odebrán vzorek (2): jeden blok nebo liz na jednu vzorkovanou partii obsahující 25 jednotek, maximálně čtyři bloky nebo lizy |
|
5.A.3 |
Souhrnný vzorek Požaduje se jediný souhrnný vzorek na jednu vzorkovanou partii. Celkový počet dílčích vzorků, které tvoří souhrnný vzorek, nesmí být nižší než: |
|
|
5.A.3.1 |
Volně ložené krmivo |
4 kg |
|
5.A.3.2 |
Balené krmivo: |
|
|
5.A.3.2.1 |
balení o hmotnosti vyšší než 1 kg |
4 kg |
|
5.A.3.2.2 |
balení o hmotnosti nepřevyšující 1 kg |
hmotnost obsahu čtyř originálních balení |
|
5.A.3.3 |
Tekuté nebo polotekuté krmivo: |
|
|
5.A.3.3.1 |
nádoby o obsahu vyšším než jeden litr |
čtyři litry |
|
5.A.3.3.2 |
nádoby o obsahu nepřevyšujícím jeden litr |
obsah čtyř originálních nádob |
|
5.A.3.4 |
Krmiva v blocích nebo minerální lizy: |
|
|
5.A.3.4.1 |
o hmotnosti vyšší než 1 kg/kus |
4 kg |
|
5.A.3.4.2 |
o hmotnosti nižší než 1 kg/kus |
hmotnost čtyř originálních bloků nebo lizů |
|
5.A.4 |
Konečné vzorky Konečný vzorek se získá ze souhrnného vzorku po případné redukci. Požaduje se provedení zkoušky alespoň u jednoho konečného vzorku. Množství v konečném vzorku pro zkoušku nesmí být nižší než: |
|
|
|
Tuhé krmivo |
500 g |
|
|
Tekuté nebo polotekuté krmivo |
500 ml |
|
5.B |
Týkající se kontroly nežádoucích látek nebo produktů obsažených nerovnoměrně v krmivech, jako jsou aflatoxiny, námel žitný, skočec obecný a crotalaria (3) v krmných surovinách |
|
|
5.B.1 |
Vzorkovaná partie: viz 5.A.1 |
|
|
5.B.2 |
Dílčí vzorky |
|
|
5.B.2.1 |
Volně ložené krmivo: viz 5.A.2.1 |
|
|
5.B.2.2 |
Balené krmivo: |
Minimální počet balení, z nichž má být odebrán vzorek: |
|
5.B.2.2.1 |
vzorkované partie obsahující jedno až čtyři balení |
všechna balení |
|
5.B.2.2.2 |
vzorkované partie obsahující pět až 16 balení |
čtyři |
|
5.B.2.2.3 |
vzorkované partie obsahující více než 16 balení |
√ počtu balení, který tvoří vzorkovanou partii (1), maximálně 40 balení |
|
5.B.3 |
Souhrnné vzorky Počet souhrnných vzorků se bude lišit v závislosti na velikosti vzorkované partie. Minimální počet souhrnných vzorků na jednu vzorkovanou partii je uveden níže. Celková hmotnost dílčích vzorků, které tvoří každý souhrnný vzorek, nesmí být nižší než 4 kg |
|
|
5.B.3.1 |
Volně ložené krmivo |
|
|
|
Hmotnost vzorkované partie v tunách: |
Minimální počet souhrnných vzorků na jednu vzorkovanou partii: |
|
|
méně než 1 |
1 |
|
|
od 1 do 10 |
2 |
|
|
od 10 do 40 |
3 |
|
|
více než 40 |
4 |
|
5.B.3.2 |
Balené krmivo |
|
|
|
Velikost vzorkované partie v počtu balení: |
Minimální počet souhrnných vzorků na jednu vzorkovanou partii: |
|
|
1 až 16 |
1 |
|
|
17 až 200 |
2 |
|
|
201 až 800 |
3 |
|
|
více než 800 |
4 |
|
5.B.4 |
Konečné vzorky Konečný vzorek se získá z každého souhrnného vzorku po redukci. Požaduje se provedení zkoušky alespoň u jednoho konečného vzorku na každý souhrnný vzorek. Hmotnost konečného vzorku pro zkoušku nesmí být nižší než 500 g. |
|
6. POKYNY TÝKAJÍCÍ SE ODBĚRU, PŘÍPRAVY A BALENÍ VZORKŮ
6.1 Obecné pokyny
Vzorky musí být odebrány a upraveny co možná nejrychleji a musí být dodržena nezbytná opatření pro zajištění toho, aby produkt nebyl narušen či kontaminován. Nástroje, plochy i nádoby na vzorky musí být čisté a suché.
6.2 Dílčí vzorky
6.2.A Pokud jde o kontrolu látek nebo produktů obsažených rovnoměrně v krmivech
Odběry dílčích vzorků se provádí náhodně v celé vzorkované partii a jejich hmotnost nebo objem musí být přibližně stejné.
6.2.A.1
Vzorkovaná partie se rozdělí pomyslně na přibližně stejné části. Náhodně se vybere počet částí odpovídající počtu dílčích vzorků požadovanému v bodě 5.A.2 a odebere se nejméně jeden vzorek z každé části.
Případně lze odebrat vzorky v průběhu pohybu vzorkované partie (nakládky či vykládky).
6.2.A.2
Poté, co byl vybrán požadovaný počet balení k odebrání vzorků, který je stanoven v bodě 5.A.2 odebere se část obsahu každého balení pomocí vzorkovače nebo lopatky. V případě potřeby se vzorky odeberou po vyprázdnění každého balení zvlášť. Hrudky se rozdrtí, v případě potřeby se nejprve oddělí a po rozdrcení se vrátí do vzorku, a sice u každého souhrnného vzorku zvlášť.
6.2.A.3
Poté, co byl vybrán požadovaný počet nádob k odebrání vzorků, který je stanoven v bodě 5.A.2, jejich obsah se v případě potřeby homogenizuje a z každé nádoby se odebere určité množství.
Dílčí vzorky mohou být odebrány během stáčení obsahu.
6.2.A.4
Poté, co byl vybrán požadovaný počet nádob k odebrání vzorků, který je stanoven v bodě 5.A.2, provede se odběr v různých vrstvách.
Vzorky mohou být odebrány rovněž během stáčení obsahu, avšak až po odstranění prvních frakcí.
V obou případech nesmí být celkový objem odebraných vzorků nižší než 10 litrů.
6.2.A.5
Poté, co byl vybrán požadovaný počet bloků nebo lizů k odebrání vzorků, který je stanoven v bodě 5.A.2 odebere se část z každého bloku nebo lizu.
6.2.B Pokud jde o kontrolu nežádoucích látek nebo produktů obsažených nerovnoměrně v krmivech, jako jsou aflatoxiny, námel žitný, skočec obecný a crotalaria v krmných surovinách
Vzorkovaná partie se rozdělí pomyslně na přibližně stejné části, jejichž počet odpovídá počtu souhrnných vzorků stanovenému v bodě 5.B.3. Je-li tento počet vyšší než 1, celkový počet dílčích vzorků stanovený v bodě 5.B.2 je rozložen v různých částech přibližně stejně. Odeberou se vzorky přibližně stejné velikosti (4) tak, aby celková hmotnost vzorku z každé části nebyla nižší než minimální hmotnost 4 kg požadovaná pro souhrnný vzorek. Dílčí vzorky odebrané z různých částí nesmí být smíchány.
6.3 Příprava souhrnných vzorků
6.3.A Pokud jde o kontrolu látek nebo produktů obsažených rovnoměrně v krmivech
Dílčí vzorky se smíchají, aby tvořily jeden souhrnný vzorek.
6.3.B Pokud jde o kontrolu nežádoucích látek nebo produktů obsažených nerovnoměrně v krmivech, jako jsou aflatoxiny, námel žitný, skočec obecný a crotalaria v krmných surovinách
Dílčí vzorky z každé části vzorkované partie se smíchají a vytvoří se počet souhrnných vzorků stanovený v bodě 5.B.3 a zaznamená se původ každého souhrnného vzorku.
6.4 Příprava konečných vzorků
Materiál každého souhrnného vzorku se pečlivě smíchá, aby se získal homogenní vzorek (5). Pokud je to nutné, souhrnný vzorek se nejprve redukuje na nejméně 2 kg nebo 2 litry (redukovaný vzorek) buď pomocí mechanického, nebo automatického děliče nebo kvartací.
Potom se připraví nejméně tři konečné vzorky, které mají přibližně stejnou hmotnost nebo stejný objem a odpovídají kvantitativním požadavkům uvedeným v bodech 5.A.4 nebo 5.B.4. Každý vzorek se vloží do vhodné nádoby. Učiní se všechna nutná předběžná opatření pro to, aby nedošlo ke změně složení vzorku nebo kontaminaci či narušení během přepravy nebo skladování.
6.5 Balení konečných vzorků
Nádoby pro balení se zapečetí a opatří štítkem (celý štítek musí být součástí pečeti) tak, aby je nebylo možné otevřít bez porušení pečeti.
7. PROTOKOL O ODBĚRU VZORKU
O každém odběru vzorku musí být veden protokol, který umožňuje jednoznačně identifikovat každou vzorkovanou partii.
8. MÍSTO URČENÍ VZORKŮ
Za každý souhrnný vzorek se společně s údaji nutnými pro laboratorní zkoušení pošle co nejrychleji alespoň jeden konečný vzorek do pověřené zkušební laboratoře.
(1) Pokud výsledné číslo tvoří zlomek, zaokrouhlí se nahoru na nejbližší celé číslo.
(2) U balení nebo nádob, jejichž obsah není vyšší než 1 kg nebo jeden litr, a u bloků nebo lizů, jejichž hmotnost nepřevyšuje 1 kg, tvoří dílčí vzorek obsah jednoho originálního balení nebo nádoby, jednoho bloku nebo jednoho lizu.
(3) Metody stanovené v bodě 5.A se použijí pro kontrolu aflatoxinů, námele žitného, skočce obecného a crotalaria v kompletních a doplňkových krmivech.
(4) U baleného krmiva se pomocí vzorkovače nebo lopatky odebere část obsahu balení, ze kterých má být odebrán vzorek, v případě potřeby po vyprázdnění každého balení zvlášť.
(5) Hrudky se rozdrtí (v případě potřeby se nejprve oddělí a po rozdrcení se vrátí do vzorku), a sice u každého souhrnného vzorku zvlášť.
PŘÍLOHA II
OBECNÁ USTANOVENÍ O METODÁCH LABORATORNÍHO ZKOUŠENÍ KRMIV
A. ÚPRAVA VZORKŮ PRO LABORATORNÍ ZKOUŠENÍ
1. Účel
Postupy popsané níže se týkají úpravy konečných vzorků pro laboratorní zkoušení, které byly zaslány do kontrolních laboratoří po odběru podle ustanovení v příloze I.
Tyto vzorky musí být upraveny tak, aby byly navážky stanovené v metodách laboratorního zkoušení homogenní a reprezentativní pro konečné vzorky.
2. Předběžná opatření
Postup úpravy vzorku, který je třeba dodržet, závisí na použitých metodách zkoušení. Proto je velmi důležité zajistit, aby postup úpravy vzorku odpovídal použité metodě zkoušení.
Veškeré nezbytné úkony musí být prováděny tak, aby se pokud možno zabránilo kontaminaci vzorku a změnám jeho složení.
Rozmělňování, míchání a prosévání se provádí co možná nejrychleji s minimálním vystavením vzorku vlivům vzduchu a světla. Nepoužívají se mlýnky a drtiče, jež by mohly vzorek znatelně zahřát.
Ruční rozmělňování se doporučuje v případě krmiv, která jsou obzvláště citlivá na teplo. Rovněž je třeba zajistit, aby zdrojem kontaminace stopovými prvky nebyl samotný přístroj.
Pokud nelze úpravu provést bez zásadních změn obsahu vlhkosti ve vzorku, stanoví se obsah vlhkosti před úpravou a po úpravě na základě metody uvedené v části A přílohy III.
3. Postup
Vzorek se rozdělí na odpovídající podvzorky pro zkoušku a pro srovnání použitím odpovídající techniky dělení, například střídavým odběrem lopatkou, stacionárním nebo otáčivým proséváním. Dělení vzorku kvartací se nedoporučuje, protože podvzorky vzniklé tímto dělením mohou být zatíženy velkou chybou. Vzorek pro srovnání se uloží do vhodné čisté a suché nádoby se vzduchotěsným uzávěrem a připraví se podvzorky pro zkoušku o minimální hmotnosti 100 g podle níže uvedeného postupu.
3.1 Krmiva, která mohou být rozmělněna ihned
Nestanoví-li metody laboratorního zkoušení jinak, celý vzorek se proseje přes síto s čtvercovými oky o délce strany 1 mm (v souladu s doporučením ISO R565), v případě potřeby až po rozmělnění. Je třeba se vyvarovat nadměrného rozmělnění.
Prosetý vzorek se promíchá a odebere do vhodné čisté a suché nádoby se vzduchotěsným uzávěrem. Těsně před odebráním navážky pro zkoušku se vzorek opět promíchá.
3.2 Krmiva, která mohou být rozmělněna až po usušení
Nestanoví-li metody laboratorního zkoušení jinak, vzorek se suší tak dlouho, dokud se jeho obsah vlhkosti nesníží na úroveň 8 až 12 %, a sice postupem pro předsoušení popsaným v bodě 4.3 metody pro stanovení obsahu vlhkosti uvedené v části A přílohy III. Poté se postupuje podle bodu 3.1.
3.3 Tekutá nebo polotekutá krmiva
Vzorek se odebere do vhodné čisté a suché nádoby se vzduchotěsným uzávěrem. Těsně před odebráním navážky pro zkoušku se vzorek opět důkladně promíchá.
3.4 Ostatní krmiva
Vzorky, které nemohou být upraveny jedním z výše uvedených postupů, se upravují jakýmkoliv jiným postupem, který zajistí, že navážky pro zkoušku budou homogenní a reprezentativní pro konečné vzorky.
4. Skladování vzorků
Vzorky musí být skladovány při takové teplotě, která nezmění jejich složení. Vzorky určené ke zkoušce na obsah vitaminů nebo látek, které jsou obzvláště citlivé na světlo, se skladují v nádobách z hnědého skla.
B. USTANOVENÍ O CHEMIKÁLIÍCH A PŘÍSTROJÍCH A POMŮCKÁCH POUŽÍVANÝCH PŘI METODÁCH LABORATORNÍHO ZKOUŠENÍ
|
1. |
Nestanoví-li metody laboratorního zkoušení jinak, veškeré chemikálie pro zkoušení musí být analyticky čisté. Pokud se provádí zkouška na stopové prvky, čistota chemikálií musí být zkontrolována slepou zkouškou. V závislosti na získaných výsledcích může být požadováno další čištění chemikálií. |
|
2. |
U jakýchkoliv úkonů spojených s úpravou roztoků, ředěním, proplachováním nebo propíráním, jak je uvedeno v metodách laboratorního zkoušení, kdy není specifikována povaha rozpouštědla nebo ředidla, se předpokládá, že musí být použita voda. Obvykle se použije voda demineralizovaná nebo destilovaná. Ve zvláštních případech, které jsou uvedeny v metodách laboratorního zkoušení, se musí podrobit zvláštním čistícím procedurám. |
|
3. |
Pokud jde o vybavení běžně používané v kontrolních laboratořích, uvádějí se v metodách laboratorního zkoušení pouze speciální zařízení a přístroje nebo zařízení a přístroje, které vyžadují zvláštní použití. Musí být čisté, a to zejména v případě, kdy je třeba stanovit velmi malá množství látek. |
C. UPLATŇOVÁNÍ METOD LABORATORNÍHO ZKOUŠENÍ A VYJADŘOVÁNÍ VÝSLEDKŮ
1. Extrakční postup
Některé metody vyžadují zvláštní extrakční postup. Obecně platí, že jiné extrakční postupy, než jsou postupy uvedené v příslušných metodách, mohou být použity jen pod podmínkou, že se prokáže, že použitý extrakční postup má rovnocennou extrakční účinnost na zkoušenou matrici jako postup uvedený v příslušné metodě.
2. Postup čištění
Některé metody vyžadují zvláštní postup čištění. Obecně platí, že jiné postupy čištění, než jsou postupy uvedené v příslušných metodách, mohou být použity jen pod podmínkou, že se prokáže, že použitý postup čištění vykazuje rovnocenné výsledky zkoušky pro zkoušenou matrici jako postup uvedený v příslušné metodě.
3. Zpráva o použité metodě laboratorního zkoušení
Obvykle je pro stanovení každé látky v krmivu určena jediná metoda laboratorního zkoušení. Je-li uvedeno více metod, musí být v protokolu o zkoušce uvedeno, kterou konkrétní metodu kontrolní laboratoř použila.
4. Počet stanovení
Výsledek uvedený v protokolu o zkoušce představuje průměrnou hodnotu získanou nejméně ze dvou stanovení provedených na různých částech vzorku, jejichž opakovatelnost je uspokojivá.
Pokud je však v případě zkoušky na nežádoucí látky výsledek prvního stanovení výrazně (> 50 %) nižší než specifikace, jež má být zkontrolována, není zapotřebí žádných dalších stanovení, jestliže byly dodrženy vhodné postupy kontroly kvality.
Pokud v případě kontroly deklarovaného obsahu látky nebo složky výsledek prvního stanovení potvrdí deklarovaný obsah, tj. jestliže výsledek zkoušky spadá do přijatelného rozsahu odchylky od deklarovaného obsahu, není zapotřebí žádných dalších stanovení, jestliže byly dodrženy vhodné postupy kontroly kvality.
V některých případech je tento přijatelný rozsah odchylky stanoven v právních předpisech, např. ve směrnici Rady 79/373/EHS (1).
5. Zpráva o výsledku zkoušky
Výsledek zkoušky se vyjadřuje způsobem, který je stanoven v metodě laboratorního zkoušení, příslušným počtem platných číslic a v případě potřeby se opraví v závislosti na obsahu vlhkosti v konečném vzorku před úpravou.
6. Nejistota měření a výtěžnost v případě zkoušky na nežádoucí látky
Pokud jde o nežádoucí látky ve smyslu směrnice 2002/32/ES, včetně dioxinů a PCB s dioxinovým efektem, produkt určený ke krmení se považuje za nevyhovující stanovenému maximálnímu obsahu, pokud se na základě výsledku zkoušky pokládá maximální obsah za překročený, přičemž se zohlední rozšířená nejistota měření a korekce na výtěžnost. K posouzení shody se použije zkoušená koncentrace poté, co byla korigovaná na výtěžnost, a poté, co byla odečtena rozšířená nejistota měření. Tento postup se použije pouze v případě, že metoda laboratorního zkoušení umožňuje odhad nejistoty měření a korekci na výtěžnost (nelze jej použít např. v případě mikroskopického zkoušení).
Výsledek zkoušky (pokud použitá metoda laboratorního zkoušení umožňuje odhad nejistoty měření a výtěžnosti) se uvede v této podobě:
|
a) |
korigovaný na výtěžnost s uvedením míry výtěžnosti. Korekce na výtěžnost není nutná v případě, že výtěžnost je v rozmezí 90–110 %; |
|
b) |
ve tvaru „x +/– U“, kde „x“ je výsledek zkoušky a „U“ je rozšířená nejistota měření, přičemž se použije faktor pokrytí 2, který odpovídá hladině spolehlivosti asi 95 %. |
Pokud je však výsledek zkoušky výrazně (> 50 %) nižší než specifikace, jež má být zkontrolována, pokud byly dodrženy vhodné postupy kontroly kvality a pokud zkouška slouží jen pro účely kontroly dodržení ustanovení právních předpisů, může být podána zpráva o výsledku zkoušky bez korekce na výtěžnost a v těchto případech lze vynechat údaje o výtěžnosti a o nejistotě měření.
PŘÍLOHA III
METODY ZKOUŠENÍ PRO KONTROLU SLOŽENÍ KRMNÝCH SUROVIN A KRMNÝCH SMĚSÍ
A. STANOVENÍ OBSAHU VLHKOSTI
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu vlhkosti v krmivech. V případě, že krmivo obsahuje těkavé látky, např. organické kyseliny, je třeba si uvědomit, že spolu s obsahem vlhkosti se stanoví i značné množství těkavých látek.
Nevztahuje se na zkoušení mléčných výrobků jako krmných surovin, zkoušení minerálních látek a směsí, které jsou tvořeny převážně minerálními látkami, zkoušení živočišných a rostlinných tuků a olejů ani na zkoušení olejnatých semen a plodů.
2. Princip
Vzorek se suší za předepsaných podmínek, které závisejí na druhu krmiva. Úbytek hmotnosti se stanoví vážkově. U pevných krmiv s vysokým obsahem vlhkosti je nutné provést předsoušení.
3. Přístroje a pomůcky
|
3.1 |
Laboratorní mlýnek z materiálu, který neabsorbuje vlhkost, snadno se čistí, umožňuje rychlé a rovnoměrné rozemletí, aniž by došlo ke znatelnému ohřevu, pokud možno zabraňuje kontaktu s okolním vzduchem a který odpovídá požadavkům uvedeným v bodech 4.1.1 a 4.1.2 (např. úderové křížové mikromlýnky, vodou chlazené mikromlýnky, rozebíratelné kuželové mlýnky, mlýnky s pomalým chodem a drtiče s ozubenými kolečky). |
|
3.2 |
Analytická váha s přesností na 1 mg. |
|
3.3 |
Vysoušečky z nekorodujícího kovu nebo ze skla, se vzduchotěsně uzavíratelnými víčky; užitková plocha umožňující rozprostření 0,3 g zkoušeného vzorku na 1 cm2. |
|
3.4 |
Elektrická laboratorní sušárna (±2 oC), která zaručuje rychlou regulaci teploty a je vybavena kvalitním větráním (1). |
|
3.5 |
Elektricky vyhřívaná vakuová sušárna s regulací teploty a olejovým čerpadlem, která je vybavena buď zařízením pro přívod teplého a suchého vzduchu nebo vysoušecím prostředkem (např. oxidem vápenatým). |
|
3.6 |
Exsikátor se silnou perforovanou deskou z kovu nebo porcelánu, který obsahuje účinný vysoušecí prostředek. |
4. Postup
|
Poznámka |
: |
Postupy uvedené v tomto bodě musí být provedeny ihned po otevření obalů vzorků. Zkouška musí být provedena nejméně dvakrát. |
4.1 Příprava
4.1.1
Odebere se minimálně 50 g vzorku. Pokud je to nutné, vzorek se rozemele takovým způsobem, aby nedošlo ke změně obsahu vlhkosti (viz bod 6).
4.1.2
Odebere se minimálně 50 g vzorku. Toto množství se rozemele tak, aby alespoň 50 % částic prošlo sítem s oky o velikosti 0,5 mm a zbytek na sítě s kulatými oky velikosti 1 mm byl maximálně 10 %.
4.1.3
Odebere se 25 g vzorku s přesností na 10 mg, smíchá se s odpovídajícím množstvím předem vysušeného písku zváženého s přesností na 10 mg, až vznikne homogenní hmota.
4.2 Sušení
4.2.1
Vysoušečka s víčkem (3.3) se zváží s přesností na 1 mg. Do předem zvážené vysoušečky se naváží asi 5 g vzorku s přesností na 1 mg a rovnoměrně se rozprostře. Vysoušečka se po sejmutí víčka umístí do laboratorní sušárny předehřáté na 103 oC. Aby se zabránilo příliš rychlému poklesu teploty, je třeba vložit vysoušečku do sušárny co nejrychleji. Sušení probíhá 4 hodiny, přičemž doba sušení se počítá od okamžiku, kdy teplota v laboratorní sušárně opět dosáhla 103 oC. Potom se vysoušečka uzavře víčkem, vyjme ze sušárny, nechá vychladnout 30–45 minut v exsikátoru (3.6) a nakonec se zváží s přesností na 1 mg.
U krmiv, která se skládají převážně z olejů a tuků, se suší ještě dalších 30 minut při teplotě 130 oC a znovu se zváží. Rozdíl mezi těmito dvěma váženími nesmí být větší než 0,1 % vlhkosti.
4.2.2
Vysoušečka s víčkem (3.3) se zváží s přesností na 0,5 mg. Do předem zvážené vysoušečky se naváží asi 5 g rozemletého vzorku s přesností na 1 mg a rovnoměrně se rozprostře. Vysoušečka se po sejmutí víčka umístí do laboratorní sušárny předehřáté na 130 oC. Aby se zabránilo příliš rychlému poklesu teploty, je třeba vložit vysoušečku do sušárny co nejrychleji. Sušení probíhá 2 hodiny, přičemž doba sušení se počítá od okamžiku, kdy teplota v laboratorní sušárně opět dosáhla 130 oC. Potom se vysoušečka uzavře víčkem, vyjme ze sušárny, nechá vychladnout 30–45 minut v exsikátoru (3.6) a nakonec se zváží s přesností na 1 mg.
|
4.2.3 |
Krmné směsi s obsahem sacharózy nebo laktózy vyšším než 4 %: krmné suroviny jako šrot ze svatojánského chleba, hydrolyzované vedlejší výrobky z obilovin, sladový květ, cukrovarnické řízky, rybí vývar, cukr solubles; krmné směsi s více než 25 % minerálních látek obsahujících krystalickou vodu. Vysoušečka s víčkem (3.3) se zváží s přesností na 0,5 mg. Do předem zvážené vysoušečky se naváží asi 5 g vzorku s přesností na 1 mg a rovnoměrně se rozprostře. Vysoušečka se po sejmutí víčka umístí do laboratorní vakuové sušárny (3.5) předehřáté na 80–85 oC. Aby se zabránilo příliš rychlému poklesu teploty, je třeba vložit vysoušečku do sušárny co nejrychleji. Tlak se upraví na 100 torrů a vzorek se suší při tomto tlaku 4 hodiny buď pomocí přívodu horkého a suchého vzduchu, nebo pomocí vysoušecího prostředku (přibližně 300 g na 20 vzorků). V tomto druhém případě se přeruší spojení s vakuovou pumpou, jakmile bylo dosaženo předepsaného tlaku. Doba sušení se počítá od okamžiku, kdy bylo v laboratorní sušárně opět dosaženo teploty 80–85 oC. Potom se tlak v sušárně opatrně upraví zpět na atmosférický tlak. Po otevření vakuové laboratorní sušárny se vysoušečka ihned uzavře víčkem, vyjme ze sušárny, nechá vychladnout 30–45 minut v exsikátoru (3.6) a nakonec se zváží s přesností na 1 mg. Vysoušečka se suší dalších 30 minut ve vakuové laboratorní sušárně při teplotě 80–85 oC a znovu se zváží. Rozdíl mezi těmito dvěma váženími nesmí být větší než 0,1 % vlhkosti. |
4.3 Předsoušení
4.3.1
Pevná krmiva s vysokým obsahem vlhkosti, která způsobuje obtíže při mletí, se musí předsoušet takto:
Naváží se 50 g nerozemletého vzorku s přesností na 10 mg (v případě potřeby se provede hrubé rozemletí u lisovaných krmiv nebo u krmiv v kusech) do vhodné nádoby (např. do hliníkové misky 20 × 12 cm s okrajem 0,5 cm). Suší se v laboratorní sušárně při teplotě 60–70 oC až do snížení obsahu vlhkosti na hodnotu 8–12 %. Vysoušečka se vyjme ze sušárny, nechá se 1 hodinu volně chladit a pak se zváží s přesností na 10 mg. Podle druhu krmiva se pak rozemele způsobem uvedeným v bodě 4.1.1 a suší způsobem uvedeným v bodě 4.2.1 nebo 4.2.3.
4.3.2
Zrna, jejichž obsah vlhkosti je vyšší než 17 %, se musí předsoušet takto:
Do vhodné nádoby (např. do hliníkové misky 20 × 12 cm s okrajem 0,5 cm) se naváží 50 g nerozemletých zrn s přesností na 10 mg. Suší se v laboratorní sušárně po dobu 5–7 minut při teplotě 130 oC. Vyjme se ze sušárny, nechá se 2 hodiny volně chladit a pak se zváží s přesností na 10 mg. Pak se ihned rozemele způsobem uvedeným v bodě 4.1.2 a suší způsobem uvedeným v bodě 4.2.2.
5. Výpočet a vyjádření výsledků
Obsah vlhkosti (X) jako procento vzorku se vypočte podle následujícího vzorce:
5.1 Sušení bez předsoušení
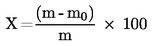
kde:
|
m |
= |
počáteční hmotnost zkoušeného vzorku v gramech |
|
m0 |
= |
hmotnost sušeného zkoušeného vzorku v gramech. |
5.2 Sušení s předsoušením
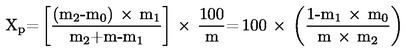
kde:
|
m |
= |
počáteční hmotnost zkoušeného vzorku v gramech |
|
m1 |
= |
hmotnost zkoušeného vzorku po předsoušení v gramech |
|
m2 |
= |
hmotnost zkoušeného vzorku po rozemletí nebo rozmělnění v gramech |
|
m0 |
= |
hmotnost sušeného zkoušeného vzorku v gramech. |
5.3 Opakovatelnost
Rozdíl mezi výsledky dvou paralelních stanovení obsahu vlhkosti provedených na stejném vzorku nesmí překročit absolutní hodnotu vlhkosti 0,2 %.
6. Poznámka
Pokud je nutné provést rozemletí vzorku a pokud se na základě toho musí počítat se změnou obsahu vlhkosti materiálu, je nutno korigovat výsledky zkoušky jednotlivých složek krmiva podle obsahu vlhkosti vzorku v jeho původním stavu.
B. STANOVENÍ OBSAHU VLHKOSTI V ŽIVOČIŠNÝCH A ROSTLINNÝCH TUCÍCH A OLEJÍCH
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu vody a těkavých látek v živočišných a rostlinných tucích a olejích.
2. Princip
Vzorek se vysuší do konstantní hmotnosti při 103 oC (úbytek hmotnosti mezi dvěma po sobě následujícími váženími musí být maximálně 1 mg). Úbytek hmotnosti se stanoví vážkově.
3. Přístroje a pomůcky
|
3.1 |
Miska s plochým dnem z nekorodujícího materiálu, průměr 8–9 cm a výška asi 3 cm. |
|
3.2 |
Rtuťový teploměr se zesílením na vrcholu a expanzní trubicí na horním konci, se stupnicí asi od 80 oC alespoň do 110 oC, asi 10 cm dlouhý. |
|
3.3 |
Písková lázeň nebo elektrická topná deska. |
|
3.4 |
Exsikátor s účinným vysoušecím prostředkem. |
|
3.5 |
Analytické váhy. |
4. Postup
Do misky (3.1), předem vysušené a zvážené, opatřené teploměrem (3.2) se naváží asi 20 g homogenizovaného vzorku s přesností na 1 mg. Zahřívá se na pískové lázni nebo na topné desce (3.3) za stálého promíchávání teploměrem tak, aby teplota během asi 7 minut dosáhla 90 oC.
Zahřívání se sníží a sleduje se intenzita tvorby bublinek, které stoupají ode dna misky. Teplota nesmí přesáhnout 105 oC. V důkladném míchání ode dna misky se pokračuje, dokud se nepřestanou tvořit bubliny.
Během zahřívání se vzorek několikrát zahřeje na 103 ± 2 oC a ochladí na 93 oC, aby se zajistilo úplné odstranění vlhkosti. Poté se nechá vychladnout na laboratorní teplotu v exsikátoru (3.4) a zváží se. Tato operace se opakuje, dokud není dosaženo úbytku hmotnosti mezi dvěma následujícími váženími nižšího než 2 mg.
|
Poznámka |
: |
Zvýšení hmotnosti vzorku po opakovaném zahřívání je způsobeno oxidací tuku. V tomto případě se výsledek počítá z vážení provedeného bezprostředně před tím, než se hmotnost začala zvyšovat. |
5. Výpočet a vyjádření výsledků
Obsah vlhkosti (X) jako procento vzorku se vypočte podle následujícího vzorce:
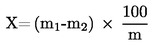
kde:
|
m |
= |
hmotnost zkoušeného vzorku v gramech |
|
m1 |
= |
hmotnost misky s obsahem před ohřevem v gramech |
|
m2 |
= |
hmotnost misky s obsahem po ohřevu v gramech |
Výsledky nižší než 0,05 % musí být uváděny jako „nižší než 0,05 %“.
Opakovatelnost
Rozdíl ve vlhkosti mezi výsledky dvou paralelních stanovení provedených na stejném vzorku nesmí překročit 0,05 % v absolutní hodnotě.
C. STANOVENÍ OBSAHU DUSÍKATÝCH LÁTEK
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu dusíkatých látek v krmivech na základě stanovení obsahu dusíku metodou podle Kjeldahla.
2. Princip
Vzorek se mineralizuje horkou kyselinou sírovou za přítomnosti katalyzátoru. Kyselý roztok se alkalizuje roztokem hydroxidu sodného. Amoniak se vydestiluje a jímá se do odměřeného množství kyseliny sírové a přebytek se titruje standardním roztokem hydroxidu sodného.
Uvolněný amoniak se případně vydestiluje do přebytku roztoku kyseliny borité a následně se titruje roztokem kyseliny chlorovodíkové nebo kyseliny sírové.
3. Chemikálie
|
3.1 |
Síran draselný. |
|
3.2 |
Katalyzátory: oxid měďnatý (CuO) nebo síran měďnatý, pentahydrát (CuSO4 5 H2O). |
|
3.3 |
Granulovaný zinek. |
|
3.4 |
Kyselina sírová, ρ20 = 1,84 g/ml. |
|
3.5 |
Kyselina sírová, standardní odměrný roztok, c(H2SO4) = 0,25 mol/l. |
|
3.6 |
Kyselina sírová, standardní odměrný roztok, c(H2SO4) = 0,10 mol/l. |
|
3.7 |
Kyselina sírová, standardní odměrný roztok, c(H2SO4) = 0,05 mol/l. |
|
3.8 |
Indikátor methylčerveň: 300 mg methylčerveně se rozpustí ve 100 ml ethanolu, σ = 95–96 % (v/v). |
|
3.9 |
Roztok hydroxidu sodného (může být použit technicky čistý), β = 40 g/100 ml (m/v: 40 %). |
|
3.10 |
Hydroxid sodný, standardní odměrný roztok, c(NaOH) = 0,25 mol/l. |
|
3.11 |
Hydroxid sodný, standardní odměrný roztok, c(NaOH) = 0,10 mol/l. |
|
3.12 |
Granulovaná pemza, praná v kyselině chlorovodíkové a vysušená. |
|
3.13 |
Acetanilid (bod tání = 114 oC, obsah N = 10,36 %). |
|
3.14 |
Sacharóza (dusíku prostá). |
|
3.15 |
Kyselina boritá (H3BO3). |
|
3.16 |
Roztok methylčerveně: 100 mg methylčerveně se rozpustí ve 100 ml ethanolu nebo methanolu. |
|
3.17 |
Roztok bromokresolové zeleně: 100 mg bromokresolové zeleně se rozpustí ve 100 ml ethanolu nebo methanolu. |
|
3.18 |
Roztok kyseliny borité (10 g/l až 40 g/l v závislosti na použitém přístroji). Pokud se použije kolorimetrická detekce bodu ekvivalence, do roztoků kyseliny borité se musí přidat methylčerveň a bromokresol. Pokud se připravuje 1 litr roztoku kyseliny borité, před úpravou objemu se přidá 7 ml roztoku methylčerveně (3.16) a 10 ml roztoku bromokresolové zeleně (3.17). V závislosti na použité vodě se může pH jednotlivých roztoků kyseliny borité lišit. Často je nezbytná úprava pomocí malého množství zásadité látky, aby se získalo vhodné pH.
|
|
3.19 |
Kyselina chlorovodíková, standardní odměrný roztok, c(HCl) = 0,10 mol/l.
|
4. Přístroje a pomůcky
Přístroje vhodné pro provedení mineralizace, destilace a titrace podle Kjeldahlovy metody.
5. Postup
5.1 Mineralizace
Do mineralizační baňky se naváží 1 g vzorku s přesností na 0,001 g. Přidá se 15 g síranu draselného (3.1) a vhodné množství katalyzátoru (3.2) (0,3–0,4 g oxidu měďnatého nebo 0,9–1,2 g pentahydrátu síranu měďnatého, 25 ml kyseliny sírové (3.4) a několik granulí pemzy (3.12) a zamíchá se.
Baňka se zahřívá nejprve mírně a podle potřeby se občas zamíchá, dokud nedojde ke zuhelnatění hmoty a neustane pěnění; potom se intenzita zahřívání zvyšuje, dokud se kapalina ustáleně nevaří. Zahřívání je odpovídající, pokud vroucí kyselina kondenzuje na stěnách baňky. Nesmí docházet k přehřívání stěn baňky a k ulpívání organických částic na stěnách baňky.
Jakmile se roztok stane čirým a získá světle zelené zabarvení, pokračuje se v ohřevu ještě 2 hodiny a potom se ponechá vychladnout.
5.2 Destilace
Opatrně se přidá tolik vody, aby došlo k úplnému rozpuštění sulfátů. Nechá se vychladnout a potom se přidá několik granulí zinku (3.3), pokud je třeba. Dále se postupuje podle bodu 5.2.1 nebo 5.2.2.
5.2.1
Do předlohy destilačního zařízení se odměří přesně 25 ml kyseliny sírové (3.5) nebo (3.7) podle předpokládaného obsahu dusíku. Přidá se několik kapek methylčerveně (3.8).
Mineralizační baňka se připojí ke kondenzátoru destilačního přístroje a konec kondenzátoru se ponoří do kapaliny v předloze do hloubky nejméně 1 cm (viz poznámka 8.3). Do mineralizační baňky se zvolna přilije 100 ml hydroxidu sodného (3.9) tak, aby nedošlo ke ztrátám amoniaku (viz poznámka 8.1). Baňka se zahřívá, dokud se nepředestiluje veškerý amoniak.
5.2.2
V případě manuální titrace amoniakového obsahu destilátu se použije níže popsaný postup. Pokud je destilační jednotka plně automatická a zahrnuje titraci amoniakového obsahu destilátu, dodrží se pokyny výrobce pro provoz destilační jednotky.
Předloha s obsahem 25–30 ml roztoku kyseliny borité (3.18) se umístí pod otvor kondenzátoru tak, že jeho vývod je ponořen pod hladinu přebytku roztoku kyseliny borité. Destilační jednotka se upraví na dávkování 50 ml roztoku hydroxidu sodného (3.9). S destilační jednotkou se pracuje podle pokynů výrobce a oddestiluje se amoniak uvolněný přidáním roztoku hydroxidu sodného. Destilát se jímá do roztoku kyseliny borité. Množství destilátu (doba destilace párou) závisí na množství dusíku ve vzorku. Je třeba dodržet pokyny výrobce.
|
Poznámka |
: |
V poloautomatické destilační jednotce se doplnění přebytku hydroxidu sodného a destilace párou provádí automaticky. |
5.3 Titrace
Postupuje se podle bodu 5.3.1 nebo 5.3.2.
5.3.1
Přebytek kyseliny sírové v předloze se titruje roztokem hydroxidu sodného (3.10 nebo 3.11) podle použité koncentrace kyseliny sírové do dosažení bodu ekvivalence.
5.3.2
Obsah předlohy se titruje se standardním odměrným roztokem kyseliny chlorovodíkové (3.19) nebo se standardním odměrným roztokem kyseliny sírové (3.6) s použitím byrety a odečte se množství použitého titračního činidla.
Pokud se použije kolorimetrická detekce bodu ekvivalence, je bodu ekvivalence dosaženo při prvním zbarvení obsahu do růžova. Odečte se objem byretou s přesností na 0,05 ml. Bod ekvivalence bude zřetelnější při použití osvětlené magnetické míchačky nebo fotometrického detektoru.
Lze provést automaticky s použitím parního destilátoru s automatickou titrací.
Je třeba dodržet pokyny výrobce pro provoz konkrétního destilačního přístroje nebo destilačního/titračního přístroje.
|
Poznámka |
: |
Při použití automatického titračního systému začíná titrace ihned po zahájení destilace a použije se 1 % roztok kyseliny borité (3.18). Při použití plně automatické destilační jednotky lze automatickou titraci amoniaku provést rovněž pomocí detekce bodu ekvivalence s použitím potenciometrického systému pH. V tomto případě se použije automatický titrační přístroj s pH metrem. pH metr je správně kalibrován v rozsahu pH 4–7 podle běžných laboratorních postupů pro kalibraci pH. Takto bude pH bodu ekvivalence titrace dosaženo při hodnotě pH 4,6, přičemž se jedná o nejstrmější skok na titrační křivce (inflexní bod). |
5.4 Slepá zkouška
Pro potvrzení, že všechny použité chemikálie jsou prosté dusíku, se provede slepá zkouška (mineralizace, destilace a titrace) s 1 g sacharózy (3.14) místo vzorku.
6. Výpočet a vyjádření výsledků
Výpočet se provede podle bodu 6.1 nebo 6.2.
6.1 Výpočet pro titraci podle bodu 5.3.1
Obsah dusíkatých látek vyjádřený v procentech hmotnosti se vypočítá podle následujícího vzorce:
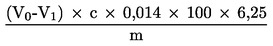
kde:
|
V0 |
= |
spotřeba NaOH (3.10 nebo 3.11) na slepou zkoušku v ml |
|
V1 |
= |
spotřeba NaOH (3.10 nebo 3.11) na titraci vzorku v ml |
|
c |
= |
koncentrace hydroxidu sodného (3.10 nebo 3.11) v mol/l |
|
m |
= |
hmotnost vzorku v g. |
6.2 Výpočet pro titraci podle bodu 5.3.2
6.2.1
Obsah dusíkatých látek vyjádřený v procentech hmotnosti se vypočítá podle následujícího vzorce:
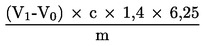
kde:
|
m |
= |
hmotnost zkoušeného vzorku v g |
|
c |
= |
koncentrace standardního odměrného roztoku hydroxidu sodného (3.19) v mol/l |
|
V0 |
= |
spotřeba kyseliny chlorovodíkové na slepou zkoušku v ml |
|
V1 |
= |
spotřeba kyseliny chlorovodíkové na zkušební vzorek v ml. |
6.2.2
Obsah dusíkatých látek vyjádřený v procentech hmotnosti se vypočítá podle následujícího vzorce:
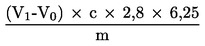
kde:
|
m |
= |
hmotnost zkušebního vzorku v g |
|
c |
= |
koncentrace standardního odměrného roztoku kyseliny sírové (3.6) v mol/l |
|
V0 |
= |
spotřeba kyseliny sírové (3.6) na slepou zkoušku v ml |
|
V1 |
= |
spotřeba kyseliny sírové (3.6) na zkušební vzorek v ml. |
7. Verifikace metody
7.1 Opakovatelnost
Rozdíl mezi výsledky dvou paralelních stanovení provedených u stejného vzorku nesmí překročit:
|
— |
0,2 % v absolutní hodnotě u obsahů dusíkatých látek nižších než 20 %, |
|
— |
1,0 % relativně vzhledem k vyšší hodnotě, u obsahů dusíkatých látek od 20 % do 40 %, |
|
— |
0,4 % v absolutní hodnotě u obsahů dusíkatých látek vyšších než 40 %. |
7.2 Přesnost
Provede se zkouška (mineralizace, destilace a titrace) s 1,5–2,0 g acetanilidu (3.13) a 1 g sacharózy (3.14); spotřeba kyseliny sírové (3.5) na 1 g acetanilidu je 14,80 ml. Výtěžnost musí být nejméně 99 %.
8. Poznámky
|
8.1 |
Přístroje mohou být manuální, poloautomatické nebo automatické. Pokud zařízení vyžaduje převod mezi mineralizačním a destilačním krokem, musí být tento krok proveden bez ztrát. Pokud k baňce destilačního přístroje není připojena přikapávací nálevka, přilévá se hydroxid sodný opatrně po stěně bezprostředně před připojením baňky ke kondenzátoru. |
|
8.2 |
Pokud mineralizát po zchladnutí ztuhne, opakuje se stanovení s větším množstvím kyseliny sírové (3.4), než je uvedeno výše. |
|
8.3 |
U produktů s nízkým obsahem dusíku může být objem kyseliny sírové (3.7) snížen na 10 nebo 15 ml a doplní se vodou na objem 25 ml. |
|
8.4 |
Při rutinní zkoušce lze pro stanovení dusíkatých látek použít alternativní metody, ale referenční metodou je Kjeldahlova metoda uvedená v části C. Ekvivalence výsledků získaných alternativní metodou (např. DUMAS) v porovnání s referenční metodou musí být prokázána samostatně pro každou matrici. Jelikož výsledky získané alternativní metodou se mohou i v případě ověření ekvivalence nepatrně odchylovat od výsledků získaných referenční metodou, je zapotřebí uvést v protokolu o zkoušce metodu laboratorního zkoušení použitou pro stanovení dusíkatých látek. |
D. STANOVENÍ OBSAHU MOČOVINY
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu močoviny v krmivech.
2. Princip
Vzorek s čeřicím činidlem se rozmíchá ve vodě. Suspenze se přefiltruje. Obsah močoviny ve filtrátu se stanoví po přidání 4-dimethylaminobenzaldehydu (4-DMAB) spektrofotometricky při vlnové délce 420 nm.
3. Chemikálie
|
3.1 |
Roztok 4-dimethylaminobenzaldehydu: 1,6 g 4-DMAB se rozpustí v 100 ml 96 % ethanolu a přidá se 10 ml kyseliny chlorovodíkové (ρ201,19 g/ml). Skladovatelnost tohoto roztoku je nejvýše dva týdny. |
|
3.2 |
Carrezovo činidlo I: 21,9 g dihydrátu octanu zinečnatého (CH3CO2)2Zn . 2H2O se rozpustí ve vodě a přidají se 3 ml ledové kyseliny octové. Doplní se vodou na objem 100 ml. |
|
3.3 |
Carrezovo činidlo II: 10,6 g trihydrátu hexakyanoželeznatanu tetradraselného [K4Fe(CN)6] . 3H2O se rozpustí ve vodě. Doplní se vodou na objem 100 ml. |
|
3.4 |
Aktivní uhlí, které neabsorbuje močovinu (musí být ověřeno). |
|
3.5 |
Močovina, 0,1% roztok (w/v). |
4. Přístroje a pomůcky
|
4.1 |
Míchací zařízení: frekvence otáčení asi 35–40 otáček/min. |
|
4.2 |
Zkumavky: 160 mm × 16 mm se zabroušenou zátkou. |
|
4.3 |
Spektrofotometr. |
5. Postup
5.1 Analýza vzorku
Do 500ml odměrné baňky se naváží 2 g vzorku ± 1 mg a přidá se 1 g aktivního uhlí (3.4). Přidá se 400 ml vody a 5 ml Carrezova činidla I (3.2), míchá se po dobu 30 sekund a přidá se 5 ml Carrezova činidla II (3.3). Míchá se po dobu třiceti minut na míchacím zařízení. Doplní se vodou po značku, promíchá a přefiltruje.
Do zkumavek se zabroušenými skleněnými zátkami se odpipetuje 5 ml čirého, bezbarvého filtrátu, přidá se 5 ml roztoku 4-DMAB (3.1) a promíchá. Zkumavky se vloží do vodní lázně nastavené na teplotu 20 oC (+/– 4 oC). Po patnácti minutách se změří absorbance roztoku na spektrofotometru při vlnové délce 420 nm. Porovná se slepou zkouškou roztoku činidel.
5.2 Kalibrační křivka
Do odměrných baněk na 100 ml se napipetuje 1, 2, 4, 5 a 10 ml roztoku močoviny (3.5) a doplní se po značku vodou. Z těchto roztoků se odpipetuje 5 ml, ke každému se přidá 5 ml roztoku 4-DMAB (3.1), homogenizuje se a změří se absorbance, jak je uvedeno výše, a porovná s kontrolním roztokem obsahujícím 5 ml 4-DMAB a 5 ml vody bez močoviny. Sestaví se kalibrační křivka.
6. Výpočet a vyjádření výsledků
Stanoví se množství močoviny ve vzorku s použitím kalibrační křivky.
Výsledek se vyjádří jako procento vzorku.
7. Poznámky
|
7.1 |
V případě, že vzorek obsahuje více než 3 % močoviny, vzorek se redukuje na 1 g nebo se původní roztok zředí tak, aby v něm nebylo více než 50 mg močoviny na 500 ml roztoku. |
|
7.2 |
V případě nízkého obsahu močoviny se vzorek zvětšuje tak dlouho, až je filtrát čirý a bezbarvý. |
|
7.3 |
V případě, že vzorek obsahuje jednoduché dusíkaté sloučeniny, jako například aminokyseliny, absorbance se změří při vlnové délce 435 nm. |
E. STANOVENÍ OBSAHU TĚKAVÝCH DUSÍKATÝCH BÁZÍ
I. MIKRODIFUZE
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu těkavých dusíkatých látek vyjádřených jako amoniak v krmivech.
2. Princip
Vzorek se extrahuje vodou, roztok se vyčeří a přefiltruje. Těkavé dusíkaté báze se po přidání roztoku uhličitanu draselného vytěsní a mikrodifuzí se zachycují v roztoku kyseliny borité a titrují se kyselinou sírovou.
3. Chemikálie
|
3.1 |
Kyselina trichloroctová, roztok 20 % (w/v). |
|
3.2 |
Indikátor: 33 mg bromokresolové zeleně a 65 mg methylčerveně se rozpustí ve 100 ml 95–96 % (v/v) ethanolu. |
|
3.3 |
Roztok kyseliny borité: v odměrné baňce o objemu 1 l se rozpustí 10 g kyseliny borité v 200 ml 95–96 % (v/v) ethanolu a 700 ml vody. Přidá se 10 ml indikátoru (3.2). Roztok se promíchá a v případě potřeby se přidá roztok hydroxidu sodného, dokud roztok nezíská světle červenou barvu. 1 ml tohoto roztoku může vázat až 300 μg NH3. |
|
3.4 |
Nasycený roztok uhličitanu draselného: 100 g uhličitanu draselného se rozpustí ve 100 ml vroucí vody. Po ochlazení se roztok filtruje. |
|
3.5 |
Kyselina sírová, 0,01 mol/l. |
4. Přístroje a pomůcky
|
4.1 |
Míchací zařízení: přibližně 35 až 40 otáček/min. |
|
4.2 |
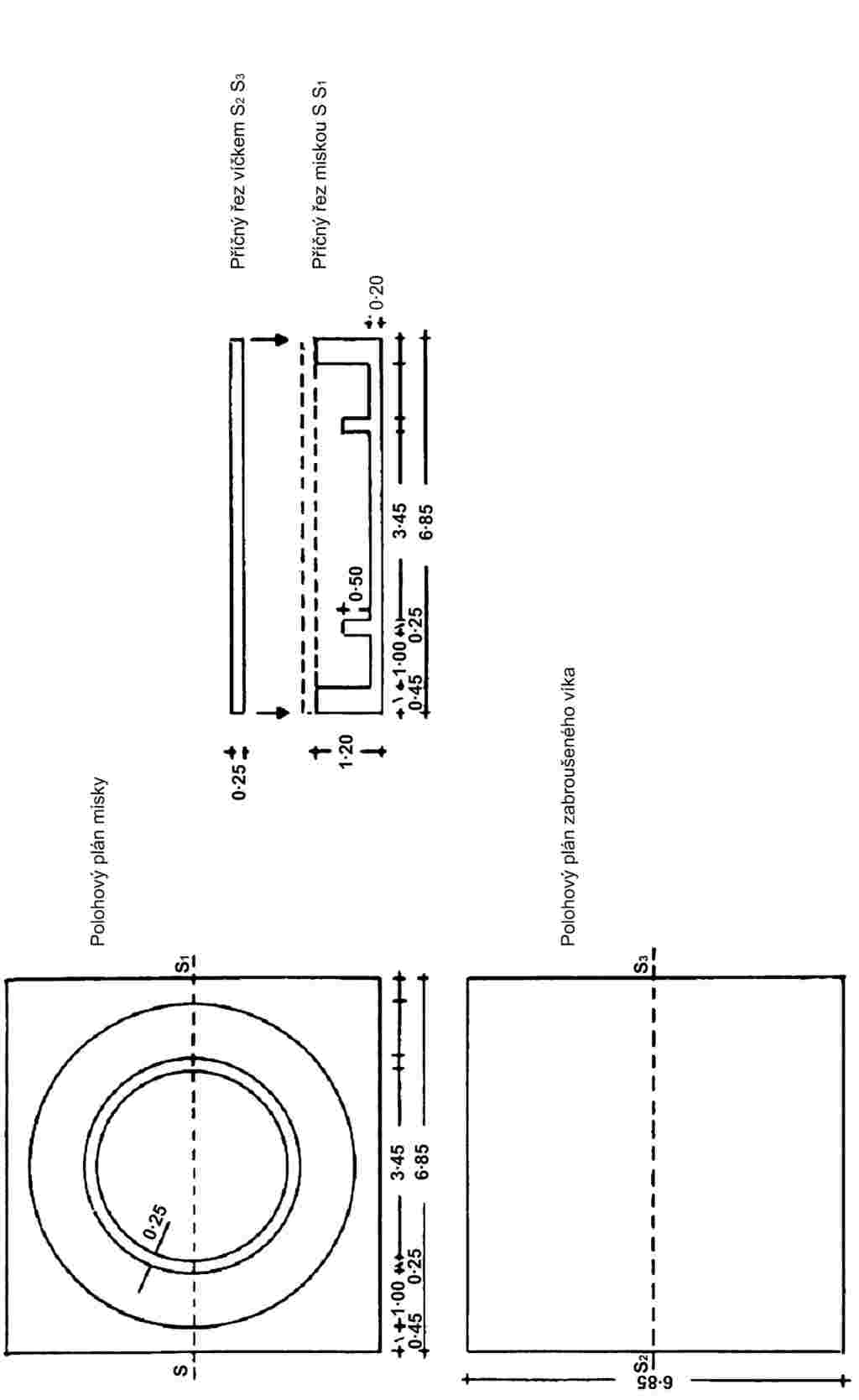
Skleněné nebo plastové Conwayovy misky (viz vyobrazení). |
|
4.3 |
Mikrobyrety s dělením 1/100 ml. |
5. Postup
Do 200 ml odměrné baňky se naváží 10 g vzorku s přesností na 1 mg, přidá se 100 ml vody a míchá se 30 minut v míchacím zařízení. Pak se přidá 50 ml kyseliny trichloroctové (3.1), doplní vodou po značku, důkladně se protřepe a přefiltruje přes skládaný filtr.
Na střed Conwayovy misky se napipetuje 1 ml roztoku kyseliny borité (3.3), okolo ní po obvodu misky se napipetuje 1 ml přefiltrovaného vzorku. Miska se částečné zakryje víkem, potřeným tukem, pak se rychle přidá 1 ml nasyceného roztoku uhličitanu draselného (3.4) na obvod misky a miska se vzduchotěsně uzavře. Aby s jistotou došlo ke smíchání reagenčních činidel, je nutno miskou opatrně otáčet ve vodorovné poloze. Pak se nechá směs stát minimálně 4 hodiny při laboratorní teplotě nebo 1 hodinu při teplotě 40 oC.
Těkavé dusíkaté báze v roztoku kyseliny borité se pak titrují kyselinou sírovou (3.5) pomocí mikrobyrety (4.3).
Stejným způsobem se provede slepá zkouška bez zkoušeného vzorku.
6. Výpočet a vyjádření výsledků
1 ml 0,01 mol/l H2SO4 odpovídá 0,34 mg amoniaku.
Výsledek se vyjádří jako procento vzorku.
Opakovatelnost
Rozdíl mezi výsledky dvou paralelních stanovení provedených na stejném vzorku nesmí přesáhnout:
|
— |
10 % relat. u obsahu amoniaku nižšího než 1,0 %, |
|
— |
0,1 % absol. u obsahu amoniaku 1,0 % nebo vyššího. |
7. Poznámka
Obsahuje-li vzorek více než 0,6 % amoniaku, je nutno původní filtrát zředit.
CONWAYOVA MISKA
Poměr 1/1
II. DESTILACE
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu těkavých dusíkatých bází, vyjádřených jako amoniak, v rybích moučkách, které neobsahují prakticky žádnou močovinu. Metoda je použitelná pouze u obsahu amoniaku nižšího než 0,25 %.
2. Princip
Vzorek se extrahuje vodou, roztok se vyčeří a přefiltruje. Těkavé dusíkaté báze se vytěsní za varu po přidání oxidu hořečnatého a zachycují se v předepsaném množství kyseliny sírové. Přebytek kyseliny sírové se titruje roztokem hydroxidu sodného.
3. Chemikálie
|
3.1 |
Kyselina trichloroctová, roztok 20 % (w/v). |
|
3.2 |
Oxid hořečnatý. |
|
3.3 |
Odpěňovací emulze (např. silikonový olej). |
|
3.4 |
Kyselina sírová, 0,05 mol/l. |
|
3.5 |
Roztok hydroxidu sodného, 0,1 mol/l. |
|
3.6 |
Roztok methylčerveně 0,3 % v 95–96 % (v/v) ethanolu. |
4. Přístroje a pomůcky
|
4.1 |
Míchací zařízení: přibližně 35 až 40 otáček/min. |
|
4.2 |
Kjeldahlova destilační aparatura. |
5. Postup
Do 200 ml odměrné baňky se naváží 10 g vzorku s přesností na 1 mg, přidá se 100 ml vody a míchá se 30 minut v míchacím zařízení. Pak se přidá 50 ml roztoku kyseliny trichloroctové (3.1), doplní se vodou po značku, důkladně se protřepe a přefiltruje přes skládaný filtr.
Podle předpokládaného obsahu těkavých dusíkatých bází se odpipetuje odpovídající množství čirého filtrátu (obvykle 100 ml), zředí se na 200 ml a přidají se 2 g oxidu hořečnatého (3.2) a několik kapek odpěňovací emulze (3.3). Roztok musí na lakmusový papírek reagovat alkalicky; v opačném případě je nutno ještě přidat oxid hořečnatý (3.2). Dále se postupuje podle bodů 5.2 a 5.3 metody laboratorního zkoušení pro stanovení obsahu dusíkatých látek (část C této přílohy).
Stejným způsobem se provede slepá zkouška, ale bez zkoušeného vzorku.
6. Výpočet a vyjádření výsledků
1 ml 0,05 mol/l H2SO4 odpovídá 1,7 mg amoniaku.
Výsledek se vyjádří jako procento vzorku.
Opakovatelnost
Rozdíl mezi výsledky dvou paralelních stanovení provedených na stejném vzorku nesmí překročit hodnotu 10 % amoniaku v relativní hodnotě.
F. STANOVENÍ OBSAHU AMINOKYSELIN (KROMĚ TRYPTOFANU)
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu volných (syntetických a přírodních) a celkových (peptidy vázané a volné) aminokyselin v krmivech za použití analyzátoru aminokyselin. Metoda je použitelná pro tyto aminokyseliny: cyst(e)in, methionin, lysin, threonin, alanin, arginin, kyselinu asparagovou, kyselinu glutamovou, glycin, histidin, isoleucin, leucin, fenylalanin, prolin, serin, tyrosin a valin.
Tato metoda nerozlišuje mezi solemi aminokyselin a nemůže také u aminokyselin rozlišovat mezi formami D a L. Není použitelná pro stanovení tryptofanu nebo hydroxyanalogů aminokyselin.
2. Princip
2.1 Volné aminokyseliny
Volné aminokyseliny se extrahují zředěnou kyselinou chlorovodíkovou. Současně extrahované dusíkaté makromolekuly se vysráží kyselinou sulfosalicylovou a odstraní se filtrací. Přefiltrovaný roztok se upraví na pH 2,20. Aminokyseliny se separují katexovou chromatografií a stanoví se po reakci s ninhydrinem fotometrickou detekcí při 570 nm.
2.2 Celkové aminokyseliny
Zvolený proces závisí na zkoumaných aminokyselinách. Cyst(e)in a methionin musí být před hydrolýzou oxidovány na kyselinu cysteovou a na methioninsulfon. Tyrosin se musí stanovovat v hydrolyzátech neoxidovaných vzorků. Ostatní aminokyseliny uvedené v bodě 1 mohou být stanovovány buď v oxidovaném, nebo v neoxidovaném vzorku.
Oxidace se provádí při 0 oC směsí kyseliny permravenčí a fenolu. Nadbytečné oxidační činidlo se rozloží dvojsíranem sodným. Oxidovaný nebo neoxidovaný vzorek se hydrolyzuje kyselinou chlorovodíkovou (3.20) po 23 hodin. Hydrolyzát se upraví na pH 2,20. Aminokyseliny se separují katexovou chromatografií a stanovují se po reakci s ninhydrinem fotometrickou detekcí při 570 nm (440 nm pro prolin).
3. Chemikálie
Musí být použita dvakrát destilovaná voda nebo voda obdobné jakosti (vodivost < 10 μS).
|
3.1 |
Peroxid vodíku, w (w/w) = 30 %. |
|
3.2 |
Kyselina mravenčí, w (w/w) = 98–100 %. |
|
3.3 |
Fenol. |
|
3.4 |
Disiřičitan sodný. |
|
3.5 |
Hydroxid sodný. |
|
3.6 |
Kyselina 5-sulfosalicylová, dihydrát. |
|
3.7 |
Kyselina chlorovodíková o hustotě přibližně 1,18 g/ml. |
|
3.8 |
Citronan sodný, dihydrát. |
|
3.9 |
2,2'-thiodiethanol (thiodiglykol). |
|
3.10 |
Chlorid sodný. |
|
3.11 |
Ninhydrin. |
|
3.12 |
Petrolether, bod varu 40–60 o C. |
|
3.13 |
Norleucin nebo jiná sloučenina použitelná jako vnitřní standard. |
|
3.14 |
Dusík, plyn (< 10 ppm kyslíku). |
|
3.15 |
1-oktanol. |
|
3.16 |
Aminokyseliny. |
|
3.16.1 |
Standardní látky uvedené v bodě 1. Čisté sloučeniny neobsahující krystalizační vodu. Vysuší se ve vakuu nad P2O5 nebo H2SO4 týden před použitím. |
|
3.16.2 |
Kyselina cysteová. |
|
3.16.3 |
Methioninsulfon. |
|
3.17 |
Roztok hydroxidu sodného, c = 7,5 mol/l: 300 g NaOH (3.5) se rozpustí ve vodě a doplní na 1 litr. |
|
3.18 |
Roztok hydroxidu sodného, c = 1 mol/l: 40 g NaOH (3.5) se rozpustí ve vodě a doplní na 1 litr. |
|
3.19 |
Roztok kyseliny mravenčí a fenolu: smíchá se 889 g kyseliny mravenčí (3.2) se 111 g vody a přidá se 4,73 g fenolu (3.3). |
|
3.20 |
Hydrolyzační směs, c = 6 mol HCl/l s přídavkem 1 g fenolu/l: do 492 ml HCl (3.7) se přidá 1 g fenolu (3.3) a doplní se vodou na 1 litr. |
|
3.21 |
Extrakční směs, c = 0,1 mol HCl/l s přídavkem 2 % thiodiglykolu: 8,2 ml HCl (3.7) se rozředí přibližně 900 ml vody, přidá se 20 ml thiodiglykolu (3.9) a doplní se vodou na 1 litr (nemíchat 3.7 a 3.9 přímo). |
|
3.22 |
Kyselina 5-sulfosalicylová, β = 6 %: 60 g kyseliny 5-sulfosalicylové (3.6) se rozpustí ve vodě a doplní se vodou na 1 litr. |
|
3.23 |
Oxidační směs (kyselina permravenčí – fenol): v malé kádince se smíchá 0,5 ml peroxidu vodíku (3.1) s 4,5 ml roztoku kyseliny mravenčí a fenolu (3.19). Ponechá se v klidu jednu hodinu při 20–30 oC, aby se utvořila kyselina permravenčí, před přidáním ke vzorku se směs ochladí v lázni s ledovou vodou (15 minut). Pozor: je nutno zamezit kontaktu s kůží a používat ochranný oděv. |
|
3.24 |
Citrátový tlumivý roztok, c = 0,2 mol Na+/l, pH 2,20: v přibližně 800 ml vody se rozpustí 19,61 g citronanu sodného (3.8), 5 ml thiodiglykolu (3.9), 1 g fenolu (3.3) a 16,50 ml HCl (3.7). pH se upraví na 2,20. Doplní se vodou na 1 litr. |
|
3.25 |
Eluční tlumivé roztoky připravované podle podmínek pro použitý analyzátor (4.9). |
|
3.26 |
Ninhydrinové činidlo připravované podle podmínek pro použitý analyzátor (4.9). |
|
3.27 |
Standardní roztoky aminokyselin. Tyto roztoky se uchovávají při teplotě pod 5 oC. |
|
3.27.1 |
Zásobní standardní roztok aminokyselin (3.16.1), c = 2,5 μmol/ml pro každou aminokyselinu v kyselině chlorovodíkové. Tento roztok lze obdržet i komerčně připravený. |
|
3.27.2 |
Zásobní standardní roztok kyseliny cysteové a methioninsulfonu, c = 1,25 μmol/ml. Do 1 000 ml odměrné baňky se naváží 0,2115 g kyseliny cysteové (3.16.2) a 0,2265 g methioninsulfonu (3.16.3), rozpustí se v citrátovém tlumivém roztoku (3.24) a doplní se jím po značku. Skladuje se při teplotě pod 5 oC nejdéle 12 měsíců. Tento roztok se nepoužívá, pokud zásobní standardní roztok (3.27.1) obsahuje kyselinu cysteovou a methioninsulfon. |
|
3.27.3 |
Zásobní standardní roztok vnitřního standardu, např. norleucinu, c = 20 μmol/ml. Do 250 ml odměrné baňky se naváží 0,6560 g norleucinu (3.13), rozpustí se v citrátovém tlumivém roztoku (3.24) a doplní se jím po značku. Skladuje se při teplotě pod 5 oC nejdéle 6 měsíců. |
|
3.27.4 |
Kalibrační standardní roztok aminokyselin pro použití s hydrolyzáty, c = 5 nmol/50 μl u kyseliny cysteové a methioninsulfonu a c = 10 nmol/50 μl u ostatních aminokyselin. Ve 100ml kádince se rozpustí 2,2 g chloridu sodného (3.10) v 30 ml citrátového tlumivého roztoku (3.24). Přidají se 4,00 ml zásobního standardního roztoku aminokyselin (3.27.1), 4,00 ml zásobního standardního roztoku kyseliny cysteové a methioninsulfonu (3.27.2) a 0,50 ml zásobního standardního roztoku vnitřního standardu (3.27.3), pokud se používá. pH se upraví na 2,20 hydroxidem sodným (3.18). Roztok se kvantitativně převede do 50ml odměrné baňky, doplní se po značku citrátovým tlumivým roztokem (3.24) a zamíchá. Skladuje se při teplotě pod 5 oC nejdéle 3 měsíce. Viz také poznámka 9.1. |
|
3.27.5 |
Kalibrační standardní roztok aminokyselin pro použití s hydrolyzáty připravenými podle bodu 5.3.3.1 a s extrakty (5.2). Kalibrační roztok se připravuje podle bodu 3.27.4 s vynecháním chloridu sodného. Skladuje se při teplotě pod 5 oC nejdéle 3 měsíce. |
4. Přístroje a pomůcky
|
4.1 |
Baňka s kulatým dnem o obsahu 100 nebo 250 ml opatřená zpětným chladičem. |
|
4.2 |
Láhev z borosilikátového skla o obsahu 100 ml se šroubovým uzávěrem s gumovým nebo teflonovým těsněním (např. Duran, Schott) pro použití v sušárně. |
|
4.3 |
Sušárna s nuceným větráním a tepelnou regulací s přesností lepší než ±2 oC. |
|
4.4 |
pH metr (tři desetinná místa). |
|
4.5 |
Membránový filtr (0,22 μm). |
|
4.6 |
Odstředivka. |
|
4.7 |
Rotační vakuová odparka. |
|
4.8 |
Mechanická třepačka nebo magnetické míchací zařízení. |
|
4.9 |
Analyzátor aminokyselin nebo zařízení HPLC s katexovou kolonou, zařízením pro ninhydrin postkolonovou derivatizaci a fotometrickým detektorem. Kolona se naplní sulfonovanou polystyrenovou pryskyřicí, která umožňuje vzájemné oddělení aminokyselin a oddělení od jiných ninhydrin-pozitivních látek. Průtok tlumivého roztoku a ninhydrinu zajišťují čerpadla se stálostí průtoku ±0,5 % po dobu zahrnující standardní kalibraci i zkoušku vzorku. Některé analyzátory aminokyselin je možno použít také v případě, že hydrolyzát vykazuje koncentraci sodíkových iontů c = 0,8 mol/l a obsahuje zbytkovou kyselinu mravenčí z oxidačního kroku. Jiné analyzátory nezaručují uspokojivé oddělení určitých aminokyselin, obsahuje-li hydrolyzát nadbytečnou kyselinu mravenčí a/nebo vysoké koncentrace sodíkových iontů. V takovém případě se sníží objem kyseliny před úpravou pH odpařením hydrolyzátu na objem asi 5 ml. Odpaření se provádí ve vakuu při teplotě nejvýše 40 oC. |
5. Postup
5.1 Příprava vzorku
Vzorek se upraví mletím tak, aby prošel sítem s oky 0,5 mm. Vzorky s vysokou vlhkostí se musí před mletím vysušit na vzduchu při teplotě nepřesahující 50 oC nebo při zmrazení. Vzorky s vysokým obsahem tuku se před mletím extrahují petroletherem (3.12).
5.2 Stanovení volných aminokyselin v krmivech a v premixech
Do kónické baňky se naváží příslušné množství (1–5 g) upraveného vzorku (5.1) s přesností na 0,2 mg a přidá se 100,0 ml extrakční směsi (3.21). Směs se třepe 60 minut na mechanické třepačce nebo míchá na magnetickém míchacím zařízení (4.8). Po usazení sedimentu se odpipetuje 10,0 ml supernatantu do 100 ml kádinky.
Za stálého míchání se přidá 5,0 ml roztoku sulfosalicylové kyseliny (3.22) a pokračuje se v míchání na magnetickém míchacím zařízení dalších 5 minut. Supernantant se přefiltruje nebo odstředí tak, aby se odstranila veškerá sraženina. 10,0 ml odstředěného roztoku se přenese do 100 ml kádinky, pH se upraví na 2,20 pomocí roztoku hydroxidu sodného (3.18), potom se roztok převede pomocí citrátového tlumivého roztoku (3.24) do odměrné baňky přiměřené velikosti a doplní po značku tlumivým roztokem (3.24).
Použije-li se vnitřní standard, přidá se 1,00 ml roztoku vnitřního standardu (3.27.3) na každých 100 ml odstředěného roztoku a potom se doplní po značku tlumivým roztokem (3.24).
Dále se pokračuje podle bodu 5.4 – chromatografie.
Nejsou-li extrakty zkoušeny týž den, uchovávají se při teplotě pod 5 oC.
5.3 Stanovení celkových aminokyselin
5.3.1
0,1–1 g upraveného vzorku (5.1) se odváží s přesností na 0,2 mg:
|
— |
do 100 ml baňky s kulatým dnem (4.1) pro otevřenou hydrolýzu (5.3.2.3) nebo |
|
— |
do 250 ml baňky s kulatým dnem (4.1), požaduje-li se nízká koncentrace sodíku (5.3.3.1), nebo |
|
— |
do nádoby o obsahu 100 ml se šroubovací uzávěrem (4.2) (pro uzavřenou hydrolýzu 5.3.2.4). |
Navážka vzorku musí mít obsah dusíku asi 10 mg a obsah vlhkosti nepřesahující 100 mg.
Baňka/nádoba se umístí do lázně s ledovou vodou a ochladí se na 0 oC, potom se přidá 5 ml oxidační směsi (3.23) a promíchá se skleněnou lžící se zahnutou špičkou. Baňka/nádoba se uzavře i se lžící vzduchotěsnou fólií a vloží se i s lázní s ledovou vodou do chladničky a ponechá se v ní 16 hodin při 0 oC. Po 16 hodinách se vyjme z chladničky a přebytečné oxidační činidlo se rozloží přidáním 0,84 g disiřičitanu sodného (3.4).
Dále se postupuje podle bodu 5.3.2.1.
5.3.2
5.3.2.1
K oxidovanému vzorku připravenému podle bodu 5.3.1 se přidá 25 ml hydrolyzační směsi (3.20) tak, aby neulpěl žádný vzorek na stěnách nádoby ani na lžíci.
Podle zvoleného postupu hydrolýzy se pokračuje podle bodu 5.3.2.3 nebo 5.3.2.4.
5.3.2.2
Do baňky s kulatým dnem o obsahu 100 ml nebo 250 ml (4.1) nebo do láhve o obsahu 100 ml se šroubovacím uzávěrem (4.2) se naváží 0,1–1 g upraveného vzorku (5.1) s přesností na 0,2 mg. Navážka vzorku musí mít obsah dusíku asi 10 mg. Opatrně se přidá 25 ml hydrolyzační směsi (3.20) a smíchá se vzorkem. Dále se postupuje podle bodu 5.3.2.3 nebo 5.3.2.4.
5.3.2.3
Ke směsi v baňce (připravené podle bodu 5.3.2.1 nebo 5.3.2.2) se přidají 3 skleněné kuličky a směs se uvede do mírného varu pod zpětným chladičem na dobu 23 hodin. Po skončení hydrolýzy se zpětný chladič promyje 5 ml citrátového tlumivého roztoku (3.24). Baňka se odpojí a ochladí se v ledové lázni.
Dále se postupuje podle bodu 5.3.3.
5.3.2.4
Baňka se směsí připravenou podle bodu 5.3.2.1 nebo 5.3.2.2 se umístí do sušárny (4.3) při 110 oC. Aby se během první hodiny zabránilo narůstání tlaku (v důsledku vyvíjení plynných látek) a aby nedošlo k výbuchu, položí se šroubovací uzávěr volně na baňku. Nádoba se neuzavírá. Po uplynutí jedné hodiny se uzávěr zašroubuje a uzavřená baňka se ponechá v sušárně (4.3) 23 hodin. Po skončení hydrolýzy se baňka vyjme ze sušárny, opatrně se odšroubuje uzávěr a baňka se postaví do lázně s ledovou vodou. Nechá se vychladnout.
Podle zvoleného postupu pro úpravu pH (5.3.3) se obsah baňky kvantitativně převede pomocí citrátového tlumivého roztoku (3.24) do 250ml kádinky nebo do 250ml baňky s kulatým dnem.
Dále se postupuje podle bodu 5.3.3.
5.3.3
Podle toho, jakou toleranci k sodíku má analyzátor aminokyselin (4.9), postupuje se při úpravě pH podle bodu 5.3.3.1 nebo 5.3.3.2.
5.3.3.1
Pokud analyzátor aminokyselin vyžaduje nízkou koncentraci sodíku (má-li se snížit obsah kyselin), doporučuje se použít vnitřní zásobní standardní roztok (3.27.3).
V takovém případě se před odpařováním přidají do hydrolyzátu 2,00 ml vnitřního zásobního standardního roztoku (3.27.3).
Do hydrolyzátu připraveného podle bodu 5.3.2.3 nebo 5.3.2.4 se přidají 2 kapky 1-oktanolu (3.15).
Na rotační odparce (4.7) se sníží objem na 5–10 ml za vakua při 40 oC. Pokud by se objem při odpařování snížil pod 5 ml, hydrolyzát je třeba odstranit a zkoušku zahájit znovu.
pH se upraví na 2,20 roztokem hydroxidu sodného (3.18) a dále se postupuje podle bodu 5.3.4.
5.3.3.2
Hydrolyzáty připravené podle bodu 5.3.2.3 nebo 5.3.2.4 se částečně zneutralizují opatrným přidáním 17 ml roztoku hydroxidu sodného (3.17) za stálého míchání a při zaručené teplotě pod 40 oC.
pH se upraví na 2,20 roztokem hydroxidu sodného (3.17) a nakonec roztokem hydroxidu sodného (3.18) při laboratorní teplotě. Dále se postupuje podle bodu 5.3.4.
5.3.4
Hydrolyzát (5.3.3.1 nebo 5.3.3.2) s upraveným pH se kvantitativně převede pomocí citrátového tlumivého roztoku (3.24) do 200 ml odměrné baňky a doplní se po značku tlumivým roztokem (3.24).
Nebyl-li dosud použit vnitřní standard, přidají se 2,00 ml vnitřního standardu (3.27.3) a potom se doplní po značku citrátovým tlumivým roztokem (3.24). Důkladně se promíchá.
Dále se pokračuje v chromatografii (5.4).
Nejsou-li roztoky vzorků zkoušeny týž den, uchovávají se při teplotě pod 5 oC.
5.4 Chromatografie
Před chromatografií musí mít extrakt (5.2) nebo hydrolyzát (5.3.4) laboratorní teplotu. Směs se protřepe a potřebné množství se přefiltruje membránovým filtrem 0,22 μm (4.5). Výsledný čirý roztok se podrobí katexové chromatografii s použitím analyzátoru aminokyselin (4.9).
Nástřik lze provést ručně nebo automaticky. Přitom je důležité, aby se do kolony přidávalo stejné množství roztoku ±0,5 % pro zkoušení standardů a vzorků s výjimkou případu, kdy je použit vnitřní standard. Rovněž poměr sodíku a aminokyselin by měl být v standardních roztocích a v roztocích vzorků co možná nejpodobnější.
Frekvence kalibrace závisí hlavně na stabilitě ninhydrinového činidla a analytického systému. Standard nebo vzorek se ředí citrátovým tlumivým roztokem (3.24) tak, aby plocha píku standardu tvořila 30–200 % plochy píku roztoku vzorků pro jednotlivé aminokyseliny.
Chromatografie aminokyselin se mírně liší v závislosti na typu použitého analyzátoru a použitého ionexu. Zvolený systém musí být schopen oddělit jednotlivé aminokyseliny od sebe navzájem i od ostatních ninhydrin-pozitivních materiálů. V rozsahu svého působení musí chromatografický systém lineárně reagovat na změny objemů aminokyselin přidaných do kolony.
Při zkoušení equimolárního roztoku (zkoušených aminokyselin) by mělo být při chromatografii dosaženo níže uvedených poměrů sedla k vrcholu píku. Tento equimolární roztok musí obsahovat nejméně 30 % maximálního obsahu jednotlivých aminokyselin, což může být přesně měřeno na použitém analyzátoru aminokyselin (4.9).
Při separaci threoninu a serinu nesmí poměr výšek sedla a vrcholu nižšího z píku překrývajících se aminokyselin na chromatogramu přesahovat 2 : 10 (pokud se stanovuje pouze cyst(e)in, methionin, threonin a lysin, je stanovení nepříznivě ovlivněno nedostatečnou separací od přilehlých píků). Pro všechny ostatní aminokyseliny musí být separace lepší než 1 : 10.
Systém musí zaručit, že lysin bude separován od „lysinových náhražek“ a ornithinu.
6. Výpočet a vyjádření výsledků
Plocha píků vzorku a standardu se měří pro každou jednotlivou aminokyselinu a vypočítává se množství (X) aminokyseliny v g na kg vzorku.
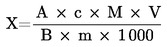
Je-li použit vnitřní standard, násobí se výsledek: 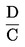
|
A |
= |
plocha píku hydrolyzátu nebo extraktu |
|
B |
= |
plocha píku kalibračního standardního roztoku |
|
C |
= |
plocha píku vnitřního standardu v hydrolyzátu nebo v extraktu |
|
D |
= |
plocha píku vnitřního standardu v kalibračním standardním roztoku |
|
M |
= |
molekulární hmotnost stanovované aminokyseliny |
|
c |
= |
koncentrace standardu v μmol/ml |
|
m |
= |
hmotnost navážky vzorku v g (korigovaná na původní hmotnost, pokud je vzorek vysušen nebo odtučněn) |
|
V |
= |
celkový objem hydrolyzátu (5.3.4) v ml nebo celkový objem zředěného extraktu (6.1) |
Cystin a cystein se v hydrolyzátech oxidovaného vzorku stanovují jako kyselina cysteová, ale vypočítávají se jako cystin (C6H12N2O4S2, MV 240,30 g/mol) s použitím MV 120,15 g/mol (= 0,5 × 240,30 g/mol).
Methionin se v hydrolyzátech oxidovaného vzorku stanovuje jako methioninsulfon, ale vypočítává se jako methionin s použitím MV methioninu: 149,21 g/mol.
Přidaný volný methionin se stanovuje po extrakci jako methionin, pro výpočet se použije stejná MV.
|
6.1 |
Celkový objem zředěného extraktu (F) při stanovení volných aminokyselin (5.2) se vypočítá podle následujícího vzorce: 
|
7. Ověření metody
Tato metoda byla vyzkoušena porovnáním na mezinárodní úrovni v roce 1990 při použití čtyř různých krmiv (krmná směs pro prasata, kombinované krmivo pro brojlery, proteinový koncentrát, premix). Po vyloučení odlehlých hodnot jsou výsledky středních a standardních odchylek uvedeny v následujících tabulkách:
Střední hodnoty v g/kg
|
Referenční materiál |
Aminokyselina |
||||||
|
Threonin |
Cyst(e)in |
Methionin |
Lysin |
||||
|
Krmná směs pro prasata |
6,94 n = 15 |
3,01 n = 17 |
3,27 n = 17 |
9,55 n = 13 |
|||
|
Kombinované krmivo pro brojlery |
9,31 n = 16 |
3,92 n = 18 |
5,08 n = 18 |
13,93 n = 16 |
|||
|
Proteinový koncentrát |
22,32 n = 16 |
5,06 n = 17 |
12,01 n = 17 |
47,74 n = 15 |
|||
|
Premix |
58,42 n = 16 |
— |
90,21 n = 16 |
98,03 n = 16 |
|||
|
|||||||
7.1 Opakovatelnost
Opakovatelnost vyjádřená jako „vnitrolaboratorní standardní odchylka“ je uvedena v následujících tabulkách:
Vnitrolaboratorní standardní odchylka (Sr) v g/kg
|
Referenční materiál |
Aminokyselina |
||||||
|
Threonin |
Cyst(e)in |
Methionin |
Lysin |
||||
|
Krmná směs pro prasata |
0,13 n = 15 |
0,10 n = 17 |
0,11 n = 17 |
0,26 n = 13 |
|||
|
Kombinované krmivo pro brojlery |
0,20 n = 16 |
0,11 n = 18 |
0,16 n = 18 |
0,28 n = 16 |
|||
|
Proteinový koncentrát |
0,48 n = 16 |
0,13 n = 17 |
0,27 n = 17 |
0,99 n = 15 |
|||
|
Premix |
1,30 n = 16 |
— |
2,19 n = 16 |
2,06 n = 16 |
|||
|
|||||||
Variační koeficient (%) vnitrolaboratorní standardní odchylky (Sr)
|
Referenční materiál |
Aminokyselina |
||||||
|
Threonin |
Cyst(e)in |
Methionin |
Lysin |
||||
|
Krmná směs pro prasata |
1,9 n = 15 |
3,3 n = 17 |
3,4 n = 17 |
2,8 n = 13 |
|||
|
Kombinované krmivo pro brojlery |
2,1 n = 16 |
2,8 n = 18 |
3,1 n = 18 |
2,1 n = 16 |
|||
|
Proteinový koncentrát |
2,7 n = 16 |
2,6 n = 17 |
2,2 n = 17 |
2,4 n = 15 |
|||
|
Premix |
2,2 n = 16 |
— |
2,4 n = 16 |
2,1 n = 16 |
|||
|
|||||||
7.2 Reprodukovatelnost
Výsledky pro mezilaboratorní standardní odchylku vyplývající z výše uvedeného srovnání jsou uvedeny v následující tabulce:
Mezilaboratorní standardní odchylka (SR) v g/kg
|
Referenční materiál |
Aminokyselina |
||||||
|
Threonin |
Cyst(e)in |
Methionin |
Lysin |
||||
|
Krmná směs pro prasata |
0,28 n = 15 |
0,30 n = 17 |
0,23 n = 17 |
0,30 n = 13 |
|||
|
Kombinované krmivo pro brojlery |
0,48 n = 16 |
0,34 n = 18 |
0,55 n = 18 |
0,75 n = 16 |
|||
|
Proteinový koncentrát |
0,85 n = 16 |
0,62 n = 17 |
1,57 n = 17 |
1,24 n = 15 |
|||
|
Premix |
2,49 n = 16 |
— |
6,20 n = 16 |
6,62 n = 16 |
|||
|
|||||||
Variační koeficient (%) mezilaboratorní standardní odchylky (SR)
|
Referenční materiál |
Aminokyselina |
||||||
|
Threonin |
Cyst(e)in |
Methionin |
Lysin |
||||
|
Krmná směs pro prasata |
4,1 n = 15 |
9,9 n = 17 |
7,0 n = 17 |
3,2 n = 13 |
|||
|
Kombinované krmivo pro brojlery |
5,2 n = 16 |
8,8 n = 18 |
10,9 n = 18 |
5,4 n = 16 |
|||
|
Proteinový koncentrát |
3,8 n = 16 |
12,3 n = 17 |
13,0 n = 17 |
3,0 n = 15 |
|||
|
Premix |
4,3 n = 16 |
— |
6,9 n = 16 |
6,7 n = 16 |
|||
|
|||||||
8. Použití referenčních materiálů
Správné použití uvedené metody se ověřuje opakovaným měřením certifikovaných referenčních materiálů, jsou-li k dispozici. Doporučuje se kalibrace certifikovanými kalibračními roztoky aminokyselin.
9. Poznámky
|
9.1 |
Vzhledem k rozdílům mezi analyzátory aminokyselin se výsledné koncentrace kalibračních roztoků standardů aminokyselin (viz 3.27.4 a 3.27.5) a hydrolyzátu (viz 5.3.4) považují za určující. Lineární rozsah přístroje je nutno zkontrolovat pro všechny aminokyseliny. Standardní roztok se ředí citrátovým tlumivým roztokem tak, aby se dosáhlo ploch píků uprostřed tohoto rozsahu. |
|
9.2 |
Pokud je k analýze hydrolyzátů použito zařízení pro vysokoúčinnou kapalinovou chromatografii, musí být podmínky zkoušek optimalizovány v souladu s doporučením výrobce. |
|
9.3 |
Při použití této metody u krmiv obsahujících více než 1 % chloridu (koncentráty, minerální krmiva, doplňková krmiva) může dojít k podhodnocení obsahu methioninu, a musí se proto použít speciální postup. |
G. STANOVENÍ OBSAHU TRYPTOFANU
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu celkového a volného tryptofanu v krmivech. Nerozlišuje mezi D- a L- formou.
2. Princip
Pro stanovení celkového tryptofanu se vzorek hydrolyzuje za alkalických podmínek nasyceným roztokem hydroxidu barnatého při 110 oC po dobu 20 hodin. Po hydrolýze se přidává vnitřní standard.
Pro stanovení volného tryptofanu se vzorek extrahuje za mírně kyselých podmínek v přítomnosti vnitřního standardu.
Tryptofan a vnitřní standard se stanoví v hydrolyzátu nebo extraktu metodou HPLC s fluorescenční detekcí.
3. Chemikálie
|
3.1 |
Dvakrát destilovaná voda nebo voda stejné kvality (vodivost < 10 μS/cm). |
|
3.2 |
Standardní látka: tryptofan (čistota/obsah ≥ 99 %), vakuově vysušený nad oxidem fosforečným. |
|
3.3 |
Vnitřní standard: α-methyl-tryptofan (čistota/obsah ≥ 99 %), vakuově vysušený nad oxidem fosforečným. |
|
3.4 |
Hydroxid barnatý, oktahydrát (Ba(OH)2 . 8 H2O), nesmí být nadměrně vystavován vzduchu, aby nedocházelo ke vzniku BaCO3, který by mohl narušovat stanovení (viz poznámka 9.3). |
|
3.5 |
Hydroxid sodný. |
|
3.6 |
Kyselina fosforečná, w (w/w) = 85 %. |
|
3.7 |
Kyselina chlorovodíková, ρ201,19 g/ml. |
|
3.8 |
Methanol, pro HPLC. |
|
3.9 |
Petrolether, bod varu 40–60 oC. |
|
3.10 |
Roztok hydroxidu sodného, c = 1 mol/l: 40,0 g NaOH (3.5) se rozpustí ve vodě a vodou (3.1) se doplní na 1 litr. |
|
3.11 |
Kyselina chlorovodíková, c = 6 mol/l: 492 ml HCl (3.7) se doplní vodou na 1 litr. |
|
3.12 |
Kyselina chlorovodíková, c = 1 mol/l: 82 ml HCl (3.7) se doplní vodou na 1 litr. |
|
3.13 |
Kyselina chlorovodíková, c = 0,1 mol/l: 8,2 ml HCl (3.7) se doplní vodou na 1 litr. |
|
3.14 |
Kyselina fosforečná, c = 0,5 mol/l: 34 ml kyseliny fosforečné (3.6) se doplní vodou (3.1) na 1 litr. |
|
3.15 |
Koncentrovaný roztok tryptofanu (3.2), c = 2,50 μmol/ml: v 500 ml odměrné baňce se rozpustí 0,2553 g tryptofanu (3.2) v kyselině chlorovodíkové (3.13) a doplní se jí po značku. Skladuje se při –18 oC nejdéle 4 týdny. |
|
3.16 |
Koncentrovaný vnitřní standardní roztok, c = 2,50 μmol/ml: v 500 ml odměrné baňce se rozpustí 0,2728 g α-methyl-tryptofanu (3.3) v kyselině chlorovodíkové (3.13) a doplní se jí po značku. Skladuje se při –18 oC nejdéle 4 týdny. |
|
3.17 |
Kalibrační standardní roztok tryptofanu a vnitřního standardu: 2,00 ml koncentrovaného roztoku tryptofanu (3.15) a 2,00 ml koncentrovaného roztoku vnitřního standardu α-methyl-tryptofanu (3.16) se zředí vodou (3.1) a methanolem (3.8) přibližně na stejný objem a stejnou koncentraci methanolu (10–30 %) jako v konečném hydrolyzátu. Roztok musí být připravován čerstvý před použitím. Během přípravy musí být roztok chráněn před slunečním světlem. |
|
3.18 |
Kyselina octová. |
|
3.19 |
1,1,1-trichloro-2-methyl-2-propanol. |
|
3.20 |
Ethanolamin w (w/w) > 98 %. |
|
3.21 |
Roztok 1 g 1,1,1-trichloro-2-methyl-2-propanolu (3.19) ve 100 ml methanolu (3.8). |
|
3.22 |
Mobilní fáze pro HPLC: 3,00 g kyseliny octové (3.18) + 900 ml vody (3.1) +50,0 ml roztoku (3.21) 1,1,1-trichloro-2-methyl-2-propanolu (3.19) v methanolu (3.8) (1 g/100 ml). pH se upraví ethanolaminem (3.20) na hodnotu 5,00. Doplní se vodou (3.1) na 1 000 ml. |
4. Přístroje a pomůcky
|
4.1 |
HPLC vybavení s fluorometrickým detektorem. |
|
4.2 |
Kolona pro kapalinovou chromatografii, 125 mm × 4 mm, C18, náplň 3 μm, nebo obdobná. |
|
4.3 |
pH metr. |
|
4.4 |
Nádoba polypropylenová s širokým hrdlem a šroubovacím uzávěrem o objemu 125 ml. |
|
4.5 |
Membránový filtr, 0,45 μm. |
|
4.6 |
Autokláv, 110 (±2) oC, 1,4 (±0,1) bar. |
|
4.7 |
Mechanická třepačka nebo magnetické míchací zařízení. |
|
4.8 |
Ponorný mixér. |
5. Postup
5.1 Příprava vzorků
Vzorek se upraví mletím tak, aby prošel sítem s oky 0,5 mm. Vzorky s vysokým obsahem vlhkosti musí být před mletím předsušeny buď na vzduchu při teplotě do 50 oC, nebo při zmrazení. Vzorky s vysokým obsahem tuku se před mletím extrahují petroletherem (3.9).
5.2 Stanovení volného tryptofanu (extrakt)
Vhodné množství (1–5 g) upraveného vzorku (5.1) se naváží s přesností na 1 mg do kónické baňky. Přidá se 100,0 ml roztoku kyseliny chlorovodíkové (3.13) a 5,00 ml koncentrovaného roztoku vnitřního standardu (3.16). Směs se třepe nebo míchá 60 minut na mechanické třepačce nebo magnetickém míchacím zařízení (4.7). Sediment se nechá usadit a odpipetuje se 10,0 ml supernatantu do kádinky. Přidá se 5 ml roztoku kyseliny fosforečné (3.14). pH se upraví roztokem hydroxidu sodného (3.10) na 3,0. Přidá se tolik methanolu (3.8), aby jeho konečná koncentrace v roztoku byla 10–30 %. Roztok se převede do odměrné baňky vhodného objemu a zředí se vodou na objem potřebný pro chromatografické stanovení (přibližně stejný objem, jako mají kalibrační standardní roztoky (3.17)).
Před nástřikem na kolonu HPLC se přefiltruje několik ml roztoku přes 0,45 μm membránový filtr (4.5). Dále se postupuje podle bodu 5.4 – chromatografie.
Standardní roztok a extrakty je nutno chránit před přímým slunečním světlem. Pokud není možné provést zkoušku tentýž den, extrakty lze skladovat při 5 oC nejvýše tři dny.
5.3 Stanovení celkového tryptofanu (hydrolyzát)
Do polypropylenové nádoby (4.4) se naváží 0,1–1 g upraveného vzorku (5.1) s přesností na 0,2 mg. Obsah dusíku v navážce vzorku je asi 10 mg. Přidá se 8,4 g oktahydrátu hydroxidu barnatého (3.4) a 10 ml vody. Promíchá se ponorným mixerem (4.8) nebo na magnetickém míchacím zařízení (4.7). Magnet s teflonovým povrchem se ponechá ve směsi. Stěny nádoby se opláchnou 4 ml vody. Nádoba se uzavře a víčko se volně dotáhne. Uzavřená nádoba se umístí na 30–60 minut do autoklávu (4.6) s vroucí vodou a párou. Potom se autokláv uzavře a směs se hydrolyzuje 20 hodin při teplotě 110 (± 2) oC.
Před otevřením autoklávu se sníží teplota pod 100 oC. Aby se zabránilo krystalizaci Ba(OH)2 . 8 H2O, přidá se k horké směsi 30 ml vody o laboratorní teplotě. Mírně se zamíchá nebo protřepe. Přidají se 2,00 ml roztoku koncentrovaného vnitřního standardu α-methyl-tryptofanu (3.16). Nádoba se vychladí 15 minut ve vodní lázni s ledem.
Dále se přidá 5 ml roztoku kyseliny fosforečné (3.14). Nádoba se ponechá v ledové lázni a za stálého míchání se neutralizuje roztokem kyseliny chlorovodíkové (3.11) a potom se pH upraví roztokem kyseliny chlorovodíkové (3.12) na hodnotu 3,0. Přidá se tolik methanolu, aby jeho výsledná koncentrace v konečném objemu byla 10–30 %. Roztok se převede do odměrné baňky vhodného objemu a zředí se vodou na stanovený objem, který je nutný pro chromatografické stanovení (např. 100 ml). Přídavek methanolu nesmí způsobit tvorbu sraženiny.
Před nástřikem na kolonu HPLC se přefiltruje několik ml roztoku přes 0,45 μm membránový filtr (4.5). Dále se postupuje podle bodu 5.4 – chromatografie.
Standardní roztok a hydrolyzáty je nutno chránit před přímým slunečním světlem. Pokud není možné provést zkoušku tentýž den, hydrolyzáty lze skladovat při 5 oC nejvýše tři dny.
5.4 Stanovení HPLC
Následující podmínky izokratické eluce jsou pouze doporučené; mohou být použity jiné podmínky za předpokladu, že poskytují rovnocenné výsledky (viz poznámky 9.1 a 9.2):
|
Kolona pro kapalinovou chromatografii (4.2): |
125 mm × 4 mm, C18, náplň 3 μm, nebo obdobná |
|
Teplota kolony: |
laboratorní teplota |
|
Mobilní fáze (3.22): |
3,00 g kyseliny octové (3.18) + 900 ml vody (3.1) +50,0 ml roztoku (3.21) 1,1,1- trichlor-2-methyl-2-propanolu (3.19) v methanolu (3.8) (1 g/100 ml). pH se upraví na 5,00 ethanolaminem (3.20). Objem se doplní na 1 000 ml vodou (3.1). |
|
Průtok: |
1 ml/min |
|
Celková doba zkoušky: |
asi 34 minut |
|
Detekční vlnová délka: |
excitace: 280 nm, emise: 356 nm |
|
Objem nástřiku: |
20 μl. |
6. Výpočet a vyjádření výsledků
Obsah tryptofanu (X) v g na 100 g vzorku se vypočte podle následujícího vzorce:
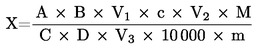
|
A |
= |
plocha píku vnitřního standardu v kalibračním standardním roztoku (3.17) |
|
B |
= |
plocha píku tryptofanu v extraktu (5.2) nebo hydrolyzátu (5.3) |
|
V1 |
= |
objem koncentrovaného roztoku tryptofanu (3.15) přidaného do kalibračního roztoku (3.17) v ml (2 ml) |
|
c |
= |
koncentrace tryptofanu v koncentrovaném roztoku tryptofanu (3.15) přidaném do kalibračního roztoku (3.17) v μmol/ml (= 2,50) |
|
V2 |
= |
objem koncentrovaného roztoku vnitřního standardu (3.16) přidaný při extrakci (5.2) (= 5,00 ml) nebo do hydrolyzátu (5.3) v ml (= 2,00 ml) |
|
C |
= |
plocha píku vnitřního standardu v extraktu (5.2) nebo hydrolyzátu (5.3) |
|
D |
= |
plocha píku tryptofanu v kalibračním standardním roztoku (3.17) |
|
V3 |
= |
objem koncentrovaného roztoku vnitřního standardu (3.16) přidaného do kalibračního standardního roztoku (3.17) v ml (= 2,00 ml) |
|
m |
= |
hmotnost navážky vzorku v g (korigovaná na původní hmotnost, pokud byl vzorek vysušen a/nebo odtučněn) |
|
M |
= |
molekulární hmotnost tryptofanu (= 204,23 g/mol). |
7. Opakovatelnost
Rozdíl mezi výsledky dvou paralelních stanovení provedených na stejném vzorku nesmí překročit 10 % relat. z hodnoty nejvyššího výsledku.
8. Výsledky kruhových testů
ES uspořádala kruhové testy (4. porovnání) pro tři vzorky, které byly zkoušeny ve 12 laboratořích, aby byl ověřen postup stanovení celkového tryptofanu (hydrolýza). Každý vzorek byl zkoušen pětkrát. Výsledky jsou uvedeny v následující tabulce:
|
|
Vzorek 1 Krmivo pro prasata |
Vzorek 2 Krmivo pro prasata s přidaným L-tryptofanem |
Vzorek 3 Koncentrát pro prasata |
|
L |
12 |
12 |
12 |
|
n |
50 |
55 |
50 |
|
Průměr (g/kg) |
2,42 |
3,40 |
4,22 |
|
sr (g/kg) |
0,05 |
0,05 |
0,08 |
|
r (g/kg) |
0,14 |
0,14 |
0,22 |
|
CVr (%) |
1,9 |
1,6 |
1,9 |
|
SR (g/kg) |
0,15 |
0,20 |
0,09 |
|
R (g/kg) |
0,42 |
0,56 |
0,25 |
|
CVR (%) |
6,3 |
6,0 |
2,2 |
|
L |
= |
počet laboratoří, které poskytly výsledky |
|
n |
= |
počet jednotlivých výsledků po vyloučení odlehlých hodnot (určených Cochranovým a Dixonovým testem) |
|
sr |
= |
standardní odchylka opakovatelnosti |
|
SR |
= |
standardní odchylka reprodukovatelnosti |
|
r |
= |
opakovatelnost |
|
R |
= |
reprodukovatelnost |
|
CVr |
= |
variační koeficient opakovatelnosti v % |
|
CVR |
= |
variační koeficient reprodukovatelnosti v % |
Další kruhový test ES (3. porovnání) byl proveden na dvou vzorcích ve 13 laboratořích pro ověření postupu extrakce volného tryptofanu. Každý vzorek byl zkoušen pětkrát. Výsledky jsou uvedeny v následující tabulce:
|
|
Vzorek 4 Směs pšenice a sóji |
Vzorek 5 Směs pšenice a sóji (= vzorek 4) s přidaným tryptofanem (0,457 g/kg) |
|
L |
12 |
12 |
|
n |
55 |
60 |
|
Průměr (g/kg) |
0,391 |
0,931 |
|
sr (g/kg) |
0,005 |
0,012 |
|
r (g/kg) |
0,014 |
0,034 |
|
CVr (%) |
1,34 |
1,34 |
|
SR (g/kg) |
0,018 |
0,048 |
|
R (g/kg) |
0,050 |
0,134 |
|
CVR (%) |
4,71 |
5,11 |
|
L |
= |
počet laboratoří, které poskytly výsledky |
|
n |
= |
počet jednotlivých výsledků po vyloučení odlehlých hodnot (určených Cochranovým a Dixonovým testem) |
|
sr |
= |
standardní odchylka opakovatelnosti |
|
SR |
= |
standardní odchylka reprodukovatelnosti |
|
r |
= |
opakovatelnost |
|
R |
= |
reprodukovatelnost |
|
CVr |
= |
variační koeficient opakovatelnosti v % |
|
CVR |
= |
variační koeficient reprodukovatelnosti v % |
Další kruhový test ES byla proveden na na čtyřech vzorcích v 7 laboratořích pro ověření hydrolýzy tryptofanu. Každý vzorek byl zkoušen pětkrát. Výsledky jsou uvedeny níže.
|
|
Vzorek 1 Krmná směs pro prasata (CRM 117) |
Vzorek 2 Nízkotučná rybí moučka (CRM 118) |
Vzorek 3 Sójová moučka (CRM 119) |
Vzorek 4 Sušené odstředěné mléko (CRM 120) |
|
L |
7 |
7 |
7 |
7 |
|
n |
25 |
30 |
30 |
30 |
|
Průměr (g/kg) |
2,064 |
8,801 |
6,882 |
5,236 |
|
sr (g/kg) |
0,021 |
0,101 |
0,089 |
0,040 |
|
r (g/kg) |
0,059 |
0,283 |
0,249 |
0,112 |
|
CVr (%) |
1,04 |
1,15 |
1,30 |
0,76 |
|
SR (g/kg) |
0,031 |
0,413 |
0,283 |
0,221 |
|
R (g/kg) |
0,087 |
1,156 |
0,792 |
0,619 |
|
CVR (%) |
1,48 |
4,69 |
4,11 |
4,22 |
|
L |
= |
počet laboratoří, které poskytly výsledky |
|
n |
= |
počet jednotlivých výsledků po vyloučení odlehlých hodnot (určených Cochranovým a Dixonovým testem) |
|
sr |
= |
standardní odchylka opakovatelnosti |
|
SR |
= |
standardní odchylka reprodukovatelnosti |
|
r |
= |
opakovatelnost |
|
R |
= |
reprodukovatelnost |
|
CVr |
= |
variační koeficient opakovatelnosti v % |
|
CVR |
= |
variační koeficient reprodukovatelnosti v %. |
9. Poznámky
|
9.1 |
Pro lepší oddělení tryptofanu a α-methyl-tryptofanu jsou doporučeny níže uvedené zvláštní podmínky pro chromatografii. Po izokratické eluci následuje gradientové čištění kolony:
|
||||||||||||||||||||||||||||||||||||||||||||
|
9.2 |
Chromatografie se může měnit podle typu HPLC a náplně kolony. Zvolený systém musí být schopen oddělit tryptofan a vnitřní standard až na základní „baseline“. Navíc je důležité, aby byly degradační produkty dobře odděleny od tryptofanu a vnitřního standardu. Pro kontrolu obsahu nečistot, které by se mohly eluovat ve stejném čase jako vnitřní standard, se provede zkouška hydrolyzátu bez přidaného vnitřního standardu. Je důležité, aby celková doba zkoušky byla dostatečná pro dokonalé vymytí všech degradačních produktů, jinak může docházet v následujících chromatografiích k interferencím. Chromatografický systém musí v rozsahu měření poskytovat lineární odezvu. Lineární odezva se měří s konstantní (normální) koncentrací vnitřního standardu a různými koncentracemi tryptofanu. Je důležité, aby velikost píků tryptofanu i vnitřního standardu byla v lineárním rozsahu HPLC/fluorescenčního systému. Pokud jsou píky tryptofanu a/nebo vnitřního standardu příliš velké nebo příliš malé, zkouška se zopakuje s jinou navážkou vzorku a/nebo s jiným konečným objemem. |
|
9.3 |
Hydroxid barnatý
Stárnutím hydroxidu barnatého se zhoršuje jeho rozpustnost, což způsobuje, že roztok pro stanovení HPLC není čirý a výsledky obsahu tryptofanu mohou být nízké. |
H. STANOVENÍ OBSAHU TUKU
1. Účel a rozsah
Touto metodou se stanoví obsah tuku v krmivech. Metoda se nevztahuje na zkoušení olejnatých semen a plodin.
Podle druhu a složení krmiva a podle účelu, pro který se zkouška provádí, se použije jeden ze dvou níže uvedených postupů.
1.1 Postup A – přímo extrahovatelné tuky
Tato metoda je použitelná pro krmiva rostlinného původu s výjimkou těch, která jsou uvedena v postupu B.
1.2 Postup B – celkové tuky
Tato metoda je použitelná pro krmiva živočišného původu a pro všechny krmné směsi. Používá se pro všechny materiály, z nichž není možné tuk zcela extrahovat bez předchozí hydrolýzy (např. lepky, kvasnice, bramborové proteiny a výrobky, které byly podrobeny zpracování, jako je vytlačování, vločkování a zahřívání).
1.3 Interpretace výsledků
Ve všech případech, kdy se dosáhlo postupem B vyššího výsledku než postupem A, je třeba považovat za platnou hodnotu výsledek dosažený postupem B.
2. Princip
2.1 Postup A
Vzorek se extrahuje petroletherem. Rozpouštědlo se oddestiluje, zbytek se vysuší a zváží.
2.2 Postup B
Vzorek se hydrolyzuje za horka s kyselinou chlorovodíkovou. Směs se ochladí a přefiltruje. Zbytek na filtru se promyje a vysuší a dále se pokračuje ve stanovení podle postupu A.
3. Chemikálie
|
3.1 |
Petrolether, bod varu: 40–60 oC. Bromové číslo musí být menší než 1 a zbytek po odpaření menší než 2 mg/100 ml. |
|
3.2 |
Síran sodný, bezvodý. |
|
3.3 |
Kyselina chlorovodíková, c = 3 mol/l. |
|
3.4 |
Filtrační prostředek, např. Kieselguhr, Hyflo-supercel. |
4. Přístroje a pomůcky
|
4.1 |
Extrakční přístroj. Pokud je opatřen sifonem (typ Soxhlet), rychlost refluxu je taková, aby docházelo k 10 přetokům za hodinu; pokud se jedná o typ bezsifonový, reflux je 10 ml za minutu. |
|
4.2 |
Extrakční patrony neobsahující látky rozpustné v petroletheru a s porozitou odpovídající požadavkům uvedeným v bodu 4.1. |
|
4.3 |
Sušárna, buď vakuová s teplotou 75 ± 3 oC, nebo horkovzdušná s teplotou 100 ± 3 oC. |
5. Postup
5.1 Postup A (viz bod 8.1)
Do extrakční patrony (4.2) se naváží 5 g vzorku s přesností na 1 mg a patrona se uzavře tukuprostou vatou.
Patrona se umístí do extrakčního přístroje (4.1) a extrahuje se šest hodin petroletherem (3.1). Petroletherový extrakt se jímá do suché, předem zvážené baňky s varnými kamínky (2).
Rozpouštědlo se oddestiluje, zbytek v baňce se vysuší 1,5 hodiny v sušárně (4.3). Nechá se vychladnout v exsikátoru a zváží se. Suší se dalších 30 minut, aby se zajistila konstantní hmotnost tuku (rozdíl mezi dvěma následujícími váženími musí být maximálně 1 mg).
5.2 Postup B
Do 400 ml kádinky nebo do 300ml kónické baňky se naváží 2,5 g vzorku s přesností na 1 mg (viz bod 8.2) a přidá se 100 ml kyseliny chlorovodíkové (3.3) a varné kamínky. Kádinka se přikryje hodinovým sklem nebo se kónická baňka napojí na refluxní chladič. Směs se zahřívá k varu na kahanu nebo na topné desce a potom se udržuje v mírném varu jednu hodinu. Během varu se nesmí částečky vzorku usazovat na stěnách nádoby.
Směs se vychladí a přidá se dostatečné množství filtračního prostředku (3.4), aby se zabránilo ztrátám tuku během filtrace. Filtruje se přes zvlhčený dvojitý filtr, který neobsahuje žádný tuk. Zbytek na filtru se promývá studenou vodou, dokud není filtrát neutrální. Provede se kontrola, zda filtrát neobsahuje žádný tuk, jinak je nutné před hydrolýzou provést extrakci petroletherem podle postupu A.
Dvojitý filtr se zbytkem po hydrolýze se umístí na hodinové sklo a suší se 1,5 hodiny v sušárně (4.3) při 100 ± 3 oC.
Dvojitý filtr s vysušeným zbytkem se vloží do extrakční patrony (4.2) a utěsní se tukuprostou vatou. Patrona se umístí do extrakčního přístroje (4.1) a dále se postupuje podle druhého a třetího odstavce bodu 5.1.
6. Vyjádření výsledku
Hmotnost zbytku po extrakci se vyjádří jako % tuku ve vzorku.
7. Opakovatelnost
Rozdíl mezi výsledky dvou paralelních stanovení provedených na stejném vzorku týmž laborantem nesmí překročit:
|
— |
0,2 % v absolutní hodnotě u obsahu tuku do 5 %, |
|
— |
4,0 % relat. z hodnoty nejvyššího výsledku u obsahu 5–10 %, |
|
— |
0,4 % v absolutní hodnotě u obsahu nad 10 %. |
8. Poznámky
|
8.1 |
U výrobků s vysokým obsahem tuků, které se obtížně melou nebo u kterých je obtížné odebrat homogenní redukovaný vzorek, se postupuje následujícím způsobem: Naváží se 20 g vzorku s přesností na 1 mg, promíchá se s 10 nebo více gramy bezvodého síranu sodného (3.2). Extrahuje se petroletherem (3.1) podle bodu 5.1. Získaný extrakt se doplní petroletherem (3.1) na objem 500 ml a promíchá. Do malé, suché, předem zvážené baňky s varnými kamínky se odměří 50 ml tohoto roztoku. Rozpouštědlo se oddestiluje a po vysušení se postupuje podle posledního odstavce bodu 5.1. Ze zbytku po extrakci v patroně se odstraní veškeré rozpouštědlo a zbytek se umele tak, aby prošel sítem s oky 1 mm. Potom se vrátí do extrakční patrony (síran sodný se již nepřidává) a postupuje se podle druhého a třetího odstavce bodu 5.1. Obsah tuku se vypočítá jako procento ze vzorku podle následujícího vzorce: (10m1 + m2) × 5 kde:
|
|
8.2 |
U výrobků s nízkým obsahem tuku se může zvýšit navážka vzorku na 5 g. |
|
8.3 |
Krmiva pro domácí zvířata, která mají vysoký obsah vody, je třeba před hydrolýzou a extrakcí podle postupu B smíchat s bezvodým síranem sodným. |
|
8.4 |
V bodě 5.2 může být účinnější při promývání filtračního zbytku použít horkou vodu místo studené. |
|
8.5 |
U některých krmiv může být zapotřebí prodloužit dobu sušení nad 1,5 hod. Je však třeba zamezit přílišnému vysoušení, které může vést k nižším výsledkům. Je možno použít i mikrovlnnou sušárnu. |
|
8.6 |
Předextrakce podle postupu A před hydrolýzou a další extrakce podle postupu B se doporučuje u krmiv s obsahem tuku nad 15 %. Do jisté míry to závisí na druhu krmiva a druhu tuku v krmivu. |
I. STANOVENÍ OBSAHU VLÁKNINY
1. Účel a rozsah
Tato metoda umožňuje stanovit v krmivu organické látky, které neobsahují tuk, jsou nerozpustné v roztoku kyseliny a louhu a jsou běžně nazývány vláknina.
2. Princip
Vzorek se v případě potřeby odtuční a působí se na něj postupně vroucím roztokem kyseliny sírové a hydroxidu draselného o přesně stanovené koncentraci. Zbytek se oddělí filtrací přes skleněný filtrační kelímek, promyje, vysuší, zváží a spálí při teplotě 475–500 oC. Úbytek váhy po spálení odpovídá obsahu vlákniny ve zkoušeném vzorku.
3. Chemikálie
|
3.1 |
Kyselina sírová, c = 0,13 mol/l. |
|
3.2 |
Odpěňovací činidlo (např. n-oktanol). |
|
3.3 |
Filtrační pomůcka (Celit 545 nebo obdobná) žíhaná při 500 oC po 4 hodiny (8.6). |
|
3.4 |
Aceton. |
|
3.5 |
Petrolether, bod varu 40–60 oC. |
|
3.6 |
Kyselina chlorovodíková, c = 0,5 mol/l. |
|
3.7 |
Roztok hydroxidu draselného, c = 0,23 mol/l. |
4. Přístroje a pomůcky
|
4.1 |
Zahřívací jednotka pro hydrolýzu roztokem kyseliny sírové a hydroxidu draselného, s oporou pro filtrační kelímky (4.2) a vybavená vypouštěcím potrubím s kouhoutem na vypouštěné tekutiny a vakuum, popřípadě stlačený vzduch. Každý den se zařízení před použitím pět minut předehřívá s vroucí vodou. |
|
4.2 |
Skleněný filtrační kelímek s porézní vložkou (fritou) o velikosti pórů 40–90 μm. Před prvním použitím se kelímek několik minut zahřeje na 500 oC a potom vychladí (8.6). |
|
4.3 |
Hydrolyzační nádobka o obsahu nejméně 270 ml s refluxním chladičem, vhodná pro vaření. |
|
4.4 |
Laboratorní sušárna s regulací teploty. |
|
4.5 |
Muflová pec s regulací teploty. |
|
4.6 |
Extrakční jednotka s oporou pro filtrační kelímky (4.2) vybavená vypouštěcím potrubím s kouhoutem na vakuum a vypouštěné tekutiny. |
|
4.7 |
Spojovací kroužky pro spojení zahřívací jednotky (4.1), filtračních kelímků (4.2) a hydrolyzační nádobky (4.3) a pro připojení kelímků ke studené extrakční jednotce (4.6). |
5. Postup
Do filtračního kelímku (4.2) se naváží 1 g upraveného vzorku s přesností na 1 mg (viz poznámky 8.1, 8.2 a 8.3) a přidá se 1 g filtrační pomůcky (3.3).
Filtrační kelímek (4.2) se připojí k zahřívací jednotce (4.1) a poté k hydrolyzační nádobce (4.3). Do spojené hydrolyzační nádobky a filtračního kelímku se nalije 150 ml vroucího roztoku kyseliny sírové (3.1) a v případě potřeby se přidá několik kapek odpěňovacího činidla (3.2).
Zahřeje se během 5 (±2) minut k varu a intenzivně se vaří přesně 30 minut.
Otevře se vypouštěcí ventil (4.1) a kyselina sírová se odsaje za použití vakua přes filtrační kelímek a zbytek se promyje asi třikrát 30 ml horké vody, vždy po úplném odsátí kapaliny z kelímku.
Vypouštěcí ventil se uzavře, přidá se 150 ml vroucího roztoku hydroxidu draselného (3.7) do spojené hydrolyzační nádobky a filtračního kelímku a přidá se několik kapek odpěňovacího činidla (3.2). Kapalina se přivede během 5 (±2) minut k varu a intenzivně se vaří přesně 30 minut. Potom se zopakuje filtrace a promývání jako po varu s kyselinou sírovou.
Po skončení promývání a odsávání se odpojí filtrační kelímek se svým obsahem a připojí se ke studené extrakční jednotce (4.6). Za použití vakua se promyje zbytek ve filtračním kelímku třikrát 25 ml acetonu (3.4), promývá se vždy do úplného odsátí acetonu.
Filtrační kelímek se vysuší do konstantní hmotnosti v sušárně při teplotě 130 oC. Po každém vysušení a ochlazení v exsikátoru se vždy rychle zváží. Filtrační kelímek se vloží do muflové pece a spaluje při 475–500 oC nejméně 30 minut do konstantní hmotnosti (úbytek hmotnosti mezi dvěma váženími nesmí být větší než 2 mg).
Po každém spalování a ochlazení nejprve v peci a potom v exsikátoru se zváží.
Provede se slepá zkouška bez zkoušeného vzorku. Úbytek hmotnosti po spalování nesmí být větší než 4 mg.
6. Výpočet a vyjádření výsledků
Obsah vlákniny jako procento vzorku se vypočítá podle následujícho vzorce:
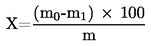
kde:
|
m |
= |
hmotnost vzorku v g |
|
m0 |
= |
úbytek hmotnosti po spálení při vlastním stanovení v g |
|
m1 |
= |
úbytek hmotnosti po spálení při slepé zkoušce v g. |
7. Opakovatelnost
Rozdíl mezi výsledky dvou paralelních stanovení provedených na stejném vzorku nesmí překročit:
|
— |
0,6 % v absolutní hodnotě u obsahu vlákniny nižšího než 10 %, |
|
— |
6 % relat. z hodnoty vyššího výsledku u obsahu vlákniny 10 % a více. |
8. Poznámky
|
8.1 |
Krmiva, která obsahují více než 10 % tuku, musí být před zkouškou odtučněna petroletherem (3.5). Filtrační kelímek (4.2) s obsahem se připojí ke studené extrakční jednotce (4.6) a zbytek se promyje třikrát 30 ml petroletheru za použití vakua až do úplného odsátí. Potom se kelímek připojí k zahřívací jednotce (4.1) a pokračuje se podle bodu 5. |
|
8.2 |
Krmiva obsahující tuk, který nelze přímo extrahovat petroletherem (3.5), musí být odtučněna podle bodu 8.1 a potom ještě jednou po varu s kyselinou. Po varu s kyselinou a následném promytí se filtrační kelímek s obsahem připojí ke studené extrakční jednotce (4.6) a promyje se třikrát 30 ml acetonu a následně třikrát 30 ml petroletheru. Odsaje se do sucha za použití vakua a pokračuje se ve zkoušce podle bodu 5 od přidání hydroxidu draselného. |
|
8.3 |
Pokud krmivo obsahuje více než 5 % uhličitanů vyjádřených jako uhličitan vápenatý, připojí se filtrační kelímek (4.2) s naváženým vzorkem k zahřívací jednotce (4.1). Vzorek se promyje třikrát 30 ml kyseliny chlorovodíkové (3.6). Po každém přidání kyseliny se filtruje po minutové prodlevě. Nakonec se promyje 30 ml vody a pokračuje se podle bodu 5. |
|
8.4 |
Pokud se používá přístroj, u kterého je několik filtračních kelímků připojeno k jedné zahřívací jednotce, neprovádí se obě paralelní stanovení u téhož vzorku ve stejné sérii. |
|
8.5 |
Pokud je po varu s kyselinou nebo louhem obtížné roztok přefiltrovat, použije se stlačený vzduch z vypouštěcího ventilu zahřívací jednotky a potom se pokračuje ve filtraci. |
|
8.6 |
Teplota spalování nesmí přesáhnout 500 oC, aby se prodloužila životnost skleněných filtračních kelímků. Během zahřívání a ochlazování nesmí být kelímky vystaveny nadměrnému teplotnímu šoku. |
J. STANOVENÍ OBSAHU CUKRŮ
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu redukujících cukrů a veškerých cukrů po inverzi vyjádřených jako glukóza nebo sacharóza s použitím faktoru 0,95. Je použitelná pro krmné směsi. Pro ostatní krmiva jsou používány zvláštní metody. Je-li to nutné, může být obsah laktózy zjištěn zvlášť a potom zahrnut do celkového výsledku.
2. Princip
Cukry se vyextrahují zředěným ethanolem. Extrakt se vyčeří Carrezovými činidly I a II. Ethanol se odstraní a množství cukru před a po inverzi se stanoví metodou podle Luff-Schorla.
3. Chemikálie
|
3.1 |
Ethanol, 40% roztok (v/v), hustota: 0,948 g/ml při 20 oC, neutralizovaný na fenolftalein. |
|
3.2 |
Carrezovo činidlo I: ve vodě se rozpustí 21,9 g octanu zinečnatého Zn (CH3COO)2 . 2H2O a 3 g ledové kyseliny octové. Doplní se vodou na 100 ml. |
|
3.3 |
Carrezovo činidlo II: ve vodě se rozpustí 10,6 g kyanoželeznatanu draselného (ferrokyanidu draselného) K4Fe(CN)6 . 3H2O. Doplní se vodou na 100 ml. |
|
3.4 |
Methyloranž, 0,1% roztok (w/v). |
|
3.5 |
Kyselina chlorovodíková 4 mol/l. |
|
3.6 |
Kyselina chlorovodíková 0,1 mol/l. |
|
3.7 |
Roztok hydroxidu sodného, 0,1 mol/l. |
|
3.8 |
Luff-Schoorlovo činidlo: Za opatrného míchání se nalije roztok kyseliny citrónové (3.8.2) do roztoku uhličitanu sodného (3.8.3). Přidá se roztok síranu měďnatého (3.8.1) a doplní se vodou na 1 litr. Nechá se přes noc usadit a pak se přefiltruje. Zkontroluje se koncentrace takto získaného činidla (Cu – 0,05 mol/l; Na2CO3 – 1 mol/l), viz poslední odstavec bodu 5.4. pH tohoto roztoku je přibližně 9,4. |
|
3.8.1 |
Roztok síranu měďnatého: ve 100 ml vody se rozpustí 25 g síranu měďnatého (Cu SO4 . 5H2O) prostého železa. |
|
3.8.2 |
Roztok kyseliny citrónové: v 50 ml vody se rozpustí 50 g kyseliny citrónové (C6H8O7 . H2O). |
|
3.8.3 |
Roztok uhličitanu sodného: v přibližně 300 ml teplé vody se rozpustí 143,8 g bezvodého uhličitanu sodného. Nechá se vychladnout. |
|
3.9 |
Roztok thiosíranu sodného 0,1 mol/l. |
|
3.10 |
Roztok škrobu: ve 30 ml vody se rozpustí 5 g škrobu a tato směs se přidá do jednoho litru vroucí vody. Vaří se tři minuty, nechá se vychladnout a v případě potřeby se přidá 10 mg jodidu rtuťnatého jako stabilizátoru. |
|
3.11 |
Kyselina sírová 3 mol/l. |
|
3.12 |
Roztok jodidu draselného 30 % (w/v). |
|
3.13 |
Granulovaná pemza vyvařená v kyselině chlorovodíkové, propraná ve vodě a vysušená. |
|
3.14 |
3-methylbutan-1-ol (isopenthanol). |
4. Přístroje a pomůcky
Míchací zařízení: přibližně 35–40 otáček/min.
5. Postup
5.1 Extrakce vzorku
Do 250 ml odměrné baňky se naváží 2,5 g vzorku s přesností na 1 mg. Přidá se 200 ml ethanolu (3.1) a míchá se po dobu jedné hodiny na míchacím zařízení. Přidá se 5 ml Carrezova činidla I (3.2) a asi 30 sekund se míchá. Přidá se 5 ml Carrezova činidla II (3.2) a znovu se míchá jednu minutu. Doplní se po značku ethanolem (3.1), homogenizuje a přefiltruje. Odpipetuje se 200 ml filtrátu a odpaří se na polovinu objemu, aby se odstranila většina ethanolu. Zbytek po odpařování se kvantitativně převede do 200 ml odměrné baňky pomocí horké vody. Zchladí se, doplní se vodou po značku, homogenizuje a v případě potřeby přefiltruje. Tento roztok se použije pro stanovení obsahu redukujících cukrů a, po inverzi, pro stanovení obsahu veškerých cukrů.
5.2 Stanovení redukujících cukrů
Odpipetuje se maximálně 25 ml roztoku, který obsahuje nejvýše 60 mg redukujících cukrů vyjádřených jako glukóza. V případě potřeby se odpipetovaný objem doplní destilovanou vodou na 25 ml. Obsah redukujících cukrů se stanoví metodou podle Luff-Schorla. Výsledek se vyjádří jako procentní obsah glukózy ve vzorku.
5.3 Stanovení obsahu veškerých cukrů po inverzi
Do 100 ml odměrné baňky se odpipetuje 50 ml roztoku. Přidá se několik kapek roztoku methyloranže (3.4) a potom se za stálého, opatrného míchání přidá kyselina chlorovodíková (3.5), až se kapalina trvale zbarví do červena. Poté se přidá 15 ml kyseliny chlorovodíkové (3.6) a baňka se rychle ponoří do vařící vodní lázně, kde se ponechá třicet minut. Rychle se vychladí na 20 oC a přidá se 15 ml roztoku hydroxidu sodného (3.7). Doplní se vodou do 100 ml a homogenizuje. Odpipetuje se maximálně 25 ml roztoku, který obsahuje nejvýše 60 mg redukujících cukrů vyjádřených jako glukóza. V případě potřeby se odpipetovaný objem doplní destilovanou vodou na 25 ml. Obsah redukujících cukrů se stanoví metodou podle Luff-Schorla. Výsledek se vyjádří jako procento glukózy nebo, kde je to vhodné, jako sacharózy po vynásobení faktorem 0,95.
5.4 Titrace (metoda podle Luff-Schoorla)
Do 300 ml Erlenmeyerovy baňky se odpipetuje 25 ml Luff-Schoorlova činidla (3.8); přidá se přesně 25 ml vyčeřeného cukerného roztoku. Přidají se dvě granule pemzy (3.13) a při ručním míchání se zahřívá nad otevřeným plamenem střední délky tak, aby se kapalina uvedla do varu přibližně za dvě minuty. Pak se Erlenmeyerova baňka neprodleně umístí na azbestem potaženou drátěnou síťku s otvorem o průměru přibližně 6 cm, pod kterou je zapálen plamen. Tento plamen musí být regulován tak, aby se zahřívalo pouze dno Erlenmeyerovy baňky. K Erlenmeyerově baňce se připojí zpětný chladič a vaří se přesně deset minut. Potom se baňka ihned vychladí ve studené vodě a přibližně po pěti minutách se titruje takto:
Do baňky se přidá 10 ml roztoku jodidu draselného (3.12) a ihned poté (opatrně a pomalu, vzhledem k riziku nadměrného pěnění) se přidá 25 ml kyseliny sírové (3.11). Titruje se roztokem thiosíranu sodného (3.9), až se objeví kalně žlutá barva. Pak se přidá škrobový indikátor (3.10) a dokončí se titrace.
Stejná titrace se provede s přesně odměřenou směsí 25 ml Luff-Schoorlova činidla (3.8) a 25 ml vody, 10 ml roztoku jodidu draselného (3.12) a 25 ml kyseliny sírové (3.11) bez vaření.
6. Výpočet a vyjádření výsledků
S použitím tabulky se stanoví obsah glukózy v mg, který odpovídá rozdílu mezi hodnotami obou titrací vyjádřených v mg thiosíranu sodného 0,1 mol/l. Výsledek se vyjádří jako procento vzorku.
7. Zvláštní postupy
|
7.1 |
V případě krmiv bohatých na melasu nebo jiných krmiv, která nejsou zcela homogenní, se do 1 000 ml odměrné baňky naváží 20 g vzorku a přidá se 500 ml vody. Míchá se jednu hodinu na míchacím zařízení. Vyčeří se Carrezovými činidly I a II (3.2 a 3.3), jak je uvedeno v odstavci 5.1, ale v tomto případě se čtyřnásobným množstvím obou činidel. Doplní se po značku 80% ethanolem (v/v). Homogenizuje se a přefiltruje. Ethanol se odstraní postupem uvedeným v odstavci 5.1. Pokud ve směsi není žádný dextrinovaný škrob, doplní se po značku destilovanou vodou. |
|
7.2 |
V případě melas a krmných surovin, které jsou bohaté na cukry a téměř bez škrobu (svatojánský chléb, sušené řepné řízky), se odváží 5 g, vloží se do 250 ml odměrné baňky, přidá se 200 ml destilované vody a míchá se na míchacím zařízení po dobu jedné hodiny a v případě potřeby i déle. Vyčeří se Carrezovými činidly I a II (3.2 a 3.3), jak uvedeno v odstavci 5.1. Doplní se po značku studenou vodou, homogenizuje a přefiltruje. Pro stanovení obsahu veškerých cukrů se dále postupuje podle bodu 5.3. |
8. Poznámky
|
8.1 |
Pro zamezení pěnění se doporučuje přidat (bez ohledu na množství vzorku) přibližně 1 ml isopenthanolu (3.14) před varem s Luff-Schoorlovým činidlem. |
|
8.2 |
Rozdíl mezi obsahem veškerých cukrů po inverzi vyjádřených jako glukóza a obsahem redukujících cukrů vyjádřených jako glukóza vynásobený koeficientem 0,95 udává procentní obsah sacharózy. |
|
8.3 |
Pro stanovení obsahu redukujících cukrů, s výjimkou laktózy, mohou být použity dvě metody: |
|
8.3.1 |
Pro přibližný výpočet se obsah laktózy stanovený jinou metodou zkoušení vynásobí koeficientem 0,675 a výsledek se odečte od obsahu redukujících cukrů. |
|
8.3.2 |
Pro přesný výpočet obsahu redukujících cukrů, s výjimkou laktózy, se stejný vzorek musí použít pro dvě konečná stanovení. Jedna zkouška se provede s částí roztoku získaného podle bodu 5.1, druhá zkouška se provede s částí roztoku získaného při stanovení laktózy metodou určenou pro tento účel (po fermentaci ostatních druhů cukrů a vyčeření). V obou případech se obsah přítomného cukru stanoví metodou podle Luff-Schoorla a vyjádří v mg glukózy. Jeden údaj se odečte od druhého a rozdíl se vyjádří jako procento ve vzorku. Příklad: Oba objemy odpovídají pro každé stanovení hmotnosti navážky vzorku 250 mg. V prvním případě je spotřebováno 17 ml roztoku thiosíranu sodného 0,1 mol/l, což odpovídá 44,2 mg glukózy. V druhém případě je spotřebováno 11 ml téhož roztoku, což odpovídá 27,6 mg glukózy. Rozdíl činí 16,6 mg glukózy. Obsah redukujících cukrů (s výjimkou laktózy) vyjádřených jako glukóza je tedy:
|
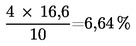
Tabulka hodnot pro 25 ml Luff-Schoorlova činidla
ml roztoku thiosíranu sodného (Na2S2O3) 0,1 mol/l, dvě minuty zahřívání k varu a deset minut varu
|
Na2S2O3 0,1 mol/l |
Glukóza, fruktóza, invertní cukry C6H12O6 |
Laktóza C12H22O11 |
Maltóza C12H22O11 |
Na2S2O3 0,1 mol/l |
|||
|
ml |
mg |
rozdíl |
mg |
rozdíl |
mg |
rozdíl |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
K. STANOVENÍ OBSAHU LAKTÓZY
1. Účel a rozsah
Tato metoda umožňuje stanovení množství laktózy v krmivech obsahujících více než 0,5 % laktózy.
2. Princip
Cukry se vyextrahují ze vzorku vodou. Extrakt se vystaví fermentačnímu účinku kvasinek Saccharomyces cerevisiae, které ponechají laktózu v nezměněném stavu. Po vyčeření a filtraci se obsah laktózy ve filtrátu stanoví metodou podle Luff-Schoorla.
3. Chemikálie
|
3.1 |
Kvasinky Saccharomyces cerevisiae, suspenze: ve 100 ml vody se suspenduje 25 g čerstvého droždí. Suspenze vydrží v chladničce maximálně jeden týden. |
|
3.2 |
Carrezovo činidlo I: ve vodě se rozpustí 21,9 g octanu zinečnatého, Zn (CH3COO)2 . 2H2O a 3 g ledové kyseliny octové. Doplní se vodou na 100 ml. |
|
3.3 |
Carrezovo činidlo II: ve vodě se rozpustí 10,6 g kyanoželeznatanu draselného (ferrokyanidu draselného) K4Fe(CN)6 . 3H2O. Doplní se vodou na 100 ml. |
|
3.4 |
Luff-Schoorlovo činidlo: Za opatrného míchání se nalije roztok kyseliny citrónové (3.4.2) do roztoku uhličitanu sodného (3.4.3). Přidá se roztok síranu měďnatého (3.4.1) a doplní se vodou na 1 litr. Nechá se přes noc usadit a potom se přefiltruje. Zkontroluje se koncentrace takto získaného činidla (Cu 0,05 mol/l; Na2 CO3 1 mol/l). pH tohoto roztoku je přibližně 9,4. |
|
3.4.1 |
Roztok síranu měďnatého: ve 100 ml vody se rozpustí 25 g síranu měďnatého (Cu SO4 . 5H2O) prostého železa. |
|
3.4.2 |
Roztok kyseliny citrónové: v 50 ml vody se rozpustí 50 g kyseliny citrónové (C6H8O7 · H2O). |
|
3.4.3 |
Roztok uhličitanu sodného: v přibližně 300 ml teplé vody se rozpustí 143,8 g bezvodého uhličitanu sodného. Nechá se vychladnout. |
|
3.5 |
Granulovaná pemza vyvařená v kyselině chlorovodíkové, promytá vodou a vysušená. |
|
3.6 |
Roztok jodidu draselného 30 % (w/v). |
|
3.7 |
Kyselina sírová 3 mol/l. |
|
3.8 |
Roztok thiosíranu sodného 0,1 mol/l. |
|
3.9 |
Roztok škrobu: ve 30 ml vody se rozpustí 5 g škrobu a směs se přidá do jednoho litru vroucí vody. Vaří se tři minuty, nechá se vychladnout a bude-li třeba, přidá se 10 g jodidu rtuťnatého jako stabilizátoru. |
4. Přístroje a pomůcky
Vodní lázeň s termostatem nastaveným na 38–40 oC.
5. Postup
Do 100 ml odměrné baňky se naváží 1 g vzorku s přesností na 1 mg. Přidá se 25–30 ml vody. Baňka se umístí do vroucí vodní lázně na třicet minut a pak se ochladí přibližně na 35 oC. Přidá se 5 ml suspenze kvasinek (3.1) a homogenizuje. Obsah baňky se nechá stát po dobu dvou hodin ve vodní lázni o teplotě 38–40 oC a potom se vychladí se na teplotu přibližně 20 oC.
Přidá se 2,5 ml Carrezova činidla I (3.2) a třicet vteřin se míchá. Potom se přidá 2,5 ml Carrezova činidla II (3.3) a znovu se míchá třicet vteřin. Doplní se vodou na 100 ml, zamíchá se a přefiltruje. Do 300 ml Erlenmeyerovy baňky se odpipetuje část filtrátu, nejvýše 25 ml, která obsahuje asi 40–80 mg laktózy. Pokud je třeba, doplní se vodou na 25 ml.
Stejným způsobem se provede slepá zkouška s 5 ml suspenze kvasinek (3.1). Stanoví se obsah laktózy metodou podle Luff-Schoorla tímto způsobem: přidá se přesně 25 ml Luff-Schoorlova činidla (3.4) a dvě granule pemzy (3.5). Ručně se zamíchá při zahřívání na volném plameni střední výšky a kapalina se přivede do varu přibližně během dvou minut. Pak se Erlenmeyerova baňka neprodleně umístí na azbestem potaženou drátěnou síťku s otvorem o průměru přibližně 6 cm, pod kterou byl zapálen plamen. Tento plamen musí být regulován tak, aby se zahřívalo pouze dno Erlenmeyerovy baňky. K Erlenmeyerově baňce se připevní zpětný chladič. Vaří se přesně deset minut. Potom se baňka ihned ochladí ve studené vodě a přibližně po pěti minutách se titruje takto:
Přidá se 10 ml roztoku jodidu sodného (3.6) a ihned poté (opatrně a pomalu, vzhledem k riziku nadměrného pěnění) se přidá 25 ml kyseliny sírové (3.7). Titruje se roztokem thiosíranu sodného (3.8), až se objeví kalně žlutá barva. Pak se přidá škrobový indikátor (3.9) a dokončí se titrace.
Stejná titrace se provede s přesně odměřenou směsí 25 ml Luff-Schoorlova činidla (3.4) a 25 ml vody, po přidání 10 ml roztoku jodidu sodného (3.6) a 25 ml kyseliny sírové (3.7) bez vaření.
6. Výpočet a vyjádření výsledků
S použitím přiložené tabulky se stanoví množství laktózy v mg, které odpovídá rozdílu mezi výsledky dvou titrací, vyjádřených v ml roztoku thiosíranu sodného 0,1 mol/l.
Výsledek se vyjádří jako bezvodá laktóza vyjádřená jako procento ve vzorku.
7. Poznámka
U látek obsahujících více než 40% zkvasitelného cukru se použije více než 5 ml kvasinkové suspenze (3.1).
Tabulka hodnot pro 25 ml Luff-Schoorlova činidla
ml roztoku thiosíranu sodného (Na2S2O3) 0,1 mol/l, dvě minuty zahřívání k varu a deset minut varu
|
Na2S2O3 0,1 mol/l |
Glukóza, fruktóza, invertní cukry C6H12O6 |
Laktóza C12H22O11 |
Maltóza C12H22O11 |
Na2S2O3 0,1 mol/l |
|||
|
ml |
mg |
rozdíl |
mg |
rozdíl |
mg |
rozdíl |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
L. STANOVENÍ OBSAHU ŠKROBU
– POLARIMETRICKÁ METODA –
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu škrobu a degradačních produktů škrobu s vysokou molekulární hmotností v krmivech za účelem kontroly shody s obsahem metabolizovatelné energie (ustanovení v příloze VII) a se směrnicí Rady 96/25/ES (3).
2. Princip
Metoda zahrnuje dvě stanovení. Nejprve je vzorek podroben působení zředěné kyseliny chlorovodíkové. Po vyčeření a filtraci je optická otáčivost roztoku měřena polarimetricky.
Potom je vzorek extrahován 40% ethanolem. Po okyselení filtrátu kyselinou chlorovodíkovou, vyčeření a filtraci je optická otáčivost roztoku měřena stejně jako u prvního stanovení.
Rozdíl mezi těmito dvěma měřeními násobený známým faktorem udává obsah škrobu ve vzorku.
3. Chemikálie
|
3.1 |
Kyselina chlorovodíková, roztok 25 % (w/w), hustota: 1,126 g/ml. |
|
3.2 |
Kyselina chlorovodíková, roztok 1,13 % (w/v). Koncentrace se musí zkontrolovat titrací za použití roztoku 0,1 mol/l hydroxidu sodného a přítomnosti 0,1 % (w/v) methylčerveně v 94% (v/v) ethanolu. Pro neutralizaci 10 ml je zapotřebí 30,94 ml NaOH 0,1 mol/l. |
|
3.3 |
Carrezovo činidlo I: ve vodě se rozpustí 21,9 g octanu zinečnatého Zn (CH3COO)2 . 2H2O a 3 g ledové kyseliny octové. Doplní se vodou na 100 ml. |
|
3.4 |
Carrezovo činidlo II: 10,6 g kyanoželeznatanu draselného (ferrokyanidu draselného K4Fe(CN)6 . 3H2O) se rozpustí ve vodě. Doplní se vodou na 100 ml. |
|
3.5 |
Ethanol, roztok 40 % (v/v), hustota: 0,948 g/ml při 20 oC. |
4. Přístroje a pomůcky
|
4.1 |
250 ml Erlenmeyerova baňka se standardním skleněným zábrusem a zpětným chladičem. |
|
4.2 |
Polarimetr nebo sacharimetr. |
5. Postup
5.1 Příprava vzorku
Vzorek se upraví mletím tak, aby prošel sítem s kruhovými otvory o velikosti 0,5 mm.
5.2 Stanovení celkové optické otáčivosti (P nebo S) (viz poznámka 7.1)
Do 100 ml odměrné baňky se naváží 2,5 g umletého vzorku s přesností na 1 mg. Přidá se 25 ml kyseliny chlorovodíkové (3.2), protřepe se, aby se vzorek rovnoměrně rozprostřel, a potom se přidá dalších 25 ml kyseliny chlorovodíkové (3.2). Baňka se ponoří do vroucí vodní lázně a první tři minuty se s ní intenzivně třepe, aby nedošlo k tvorbě shluků. Množství vody ve vodní lázni musí být takové, aby při ponoření baňky do lázně nedošlo k přerušení varu. Během protřepávání nesmí být baňka vyjmuta z lázně. Přesně po 15 minutách se baňka vyjme z lázně, přidá se 30 ml studené vody a ihned se ochladí na 20 oC.
Přidá se 5 ml Carrezova činidla I (3.3) a asi 30 sekund se protřepává. Přidá se 5 ml Carrezova činidla II (3.4) a opět se protřepává asi 30 sekund. Baňka se doplní po značku vodou, promíchá a přefiltruje. Pokud není filtrát zcela čirý (k tomu dochází pouze výjimečně), stanovení se zopakuje s použitím většího množství Carrezových činidel I a II, například 10 ml.
Optická otáčivost roztoku se měří na polarimetru nebo sacharimetru v 200 mm trubici.
5.3 Stanovení optické otáčivosti (P' nebo S') látek rozpustných ve 40% ethanolu
Do 100 ml odměrné baňky se naváží 5 g vzorku s přesností na 1 mg a přidá se asi 80 ml ethanolu (3.5) (viz poznámka 7.2). Baňka se nechá stát 1 hodinu při laboratorní teplotě; během této doby se šestkrát důkladně protřepe, aby se zkoušený vzorek zcela promísil s ethanolem. Doplní se ethanolem (3.5) po značku, promíchá a přefitruje.
Odpipetuje se 50 ml filtrátu (= 2,5 g vzorku) do 250 ml Erlenmeyerovy baňky, přidá se 2,1 ml kyseliny chlorovodíkové (3.1) a důkladně se protřepe. Na Erlenmeyerovu baňku se připojí zpětný chladič a baňka se ponoří do vroucí vodní lázně. Přesně po 15 minutách se baňka vyjme z lázně, její obsah se převede do 100 ml odměrné baňky, varná baňka se vypláchne malým množstvím studené vody a ochladí se na 20 oC.
Potom se vyčeří Carrezovými činidly I a II (3.3 a 3.4), doplní se vodou po značku, promíchá a přefiltruje. Při měření optické otáčivosti se postupuje podle druhého a třetího odstavce bodu 5.2.
6. Výpočet a vyjádření výsledků
Obsah škrobu (v %) se vypočte podle následujících vzorců:
6.1 Měření na polarimetru
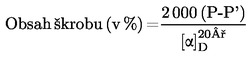
|
P |
= |
celková optická otáčivost v úhlových stupních |
||||||||||||||
|
P' |
= |
optická otáčivost v úhlových stupních látek rozpustných ve 40% (v/v) ethanolu |
||||||||||||||
|
|
= |
specifická optická otáčivost čistého škrobu. Konvenční hodnoty D přijaté pro tento faktor jsou následující:
|
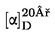
6.2 Měření na sacharimetru
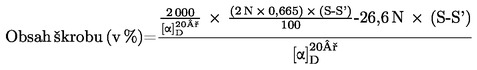
|
S |
= |
celková optická otáčivost ve stupních sacharizace |
|
S' |
= |
optická otáčivost ve stupních sacharizace látek rozpustných ve 40% (v/v) ethanolu |
|
N |
= |
navážka sacharózy v g ve 100 ml vody, která poskytne ve 200mm trubici optickou otáčivost rovnou 100o sacharizace 16,29 g pro francouzské sacharimetry 26,00 g pro německé sacharimetry 20,00 g pro směsné sacharimetry |
|
|
= |
specifická optická otáčivost čistého škrobu (viz 6.1). |
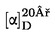
6.3 Opakovatelnost
Rozdíl mezi výsledky dvou paralelních stanovení provedených na stejném vzorku nesmí překročit 0,4 % v absolutní hodnotě u obsahu škrobu do 40 % a 1 % relat. u obsahu škrobu 40 % a více.
7. Poznámky
|
7.1 |
Pokud vzorek obsahuje více než 6 % uhličitanů, vyjádřených jako uhličitan vápenatý, musí být přítomné uhličitany před stanovením celkové optické otáčivosti rozloženy přesně odpovídajícím množstvím zředěné kyseliny sírové. |
|
7.2 |
U výrobků s vysokým obsahem laktózy, jako např. sušené mléčné sérum nebo sušené odstředěné mléko, postupuje se po přidání 80 ml ethanolu (3.5) níže uvedeným způsobem. Na baňku se upevní zpětný chladič a baňka se ponoří do vodní lázně o teplotě 50 oC na 30 minut. Nechá se vychladnout a dále se pokračuje podle bodu 5.3. |
|
7.3 |
Pokud jsou v krmivu ve významném množství přítomny níže uvedené krmné suroviny, dochází k interferencím při stanovení škrobu polarimetrickou metodou, což může způsobit nesprávné výsledky:
|
M. STANOVENÍ OBSAHU POPELA
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu popela v krmivech.
2. Princip
Vzorek se zpopelní při 550 oC; zbytek se zváží.
3. Chemikálie
Dusičnan amonný, roztok 20 % (w/v).
4. Přístroje a pomůcky
|
4.1 |
Topná deska. |
|
4.2 |
Elektrická muflová pec s termostatem. |
|
4.3 |
Spalovací kelímky z křemíku, porcelánu nebo platiny, buď pravoúhlé (asi 60 × 40 × 25 mm) nebo kulaté (průměr 60–70 mm, výška 20–40 mm). |
5. Postup
Do spalovacího kelímku, který byl předem zahřát na teplotu 550 oC, ochlazen a zvážen, se s přesností na 1 mg naváží 5 g vzorku (2,5 g v případě látek, které mají během spalování tendenci zvětšovat svůj objem). Potom se kelímek vloží na topnou desku a postupně se zahřívá, až látka zuhelnatí. Spaluje se podle bodu 5.1 nebo 5.2.
|
5.1 |
Spalovací kelímek se vloží do muflové pece zahřáté na 550 oC. Při této teplotě se spaluje, dokud nevznikne bílý, lehce šedý nebo načervenalý popel, který se zdá být prostý zuhelnatělých částic. Kelímek se vloží do exsikátoru, nechá se vychladnout a ihned se zváží. |
|
5.2 |
Spalovací kelímek se vloží do muflové pece zahřáté na 550 oC. Spaluje se 3 hodiny. Kelímek se vloží do exsikátoru, nechá se vychladnout a ihned se zváží. Spaluje se dalších 30 minut, aby se zajistilo, že je dosažená hmotnost popela konstantní (úbytek hmotnosti mezi dvěma váženími nesmí být větší než 1 mg). |
6. Výpočet a vyjádření výsledků
Vypočítá se hmotnost zbytku po odečtení hmotnosti kelímku (táry).
Výsledek se vyjádří jako procento vzorku.
7. Poznámky
|
7.1 |
Popel látek, které se obtížně zpopelňují, se musí spalovat nejméně po dobu tří hodin. Po vychladnutí se přidá několik kapek 20% roztoku dusičnanu amonného nebo vody (opatrně, aby nedošlo k rozptylu popela anebo ke tvoření hrudek). Po vysušení v sušárně se pokračuje v žíhání. Postup se opakuje podle potřeby tak dlouho, až je zpopelnění úplné. |
|
7.2 |
V případě látek, které jsou rezistentní na proces popsaný v bodě 7.1, se postupuje takto: po spalování po dobu tří hodin se popel rozmíchá v horké vodě a přefiltruje se přes malý bezpopelný filtr. Filtr s popelem se dále spaluje v původním kelímku. Filtrát se převede do vychlazeného kelímku, odpaří do sucha, opět se spálí a popel se zváží. |
|
7.3 |
V případě olejů a tuků se přesně naváží 25 g vzorku do kelímku vhodné velikosti. Zuhelnatí se zapálením látky proužky bezpopelného papírového filtru. Po spálení se zvlhčí co nejmenším množstvím vody. Vysuší se a spálí podle popisu v bodu 5. |
N. STANOVENÍ OBSAHU POPELA NEROZPUSTNÉHO V KYSELINĚ CHLOROVODÍKOVÉ
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu minerálních látek nerozpustných v kyselině chlorovodíkové v krmivech. Podle druhu vzorku mohou být použity dvě různé metody.
|
1.1 |
Metoda A: použije se pro krmné suroviny organického původu a pro většinu krmných směsí. |
|
1.2 |
Metoda B: použije se pro minerální suroviny a minerální krmné směsi a pro krmné směsi, u nichž je obsah látek nerozpustných v kyselině chlorovodíkové, stanovený metodou A, vyšší než 1 %. |
2. Princip
|
2.1 |
Metoda A: vzorek se zpopelní, popel se povaří s kyselinou chlorovodíkovou a nerozpuštěný zbytek se přefiltruje a zváží. |
|
2.2 |
Metoda B: vzorek se rozpustí v kyselině chlorovodíkové. Roztok se přefiltruje, zbytek na filtru se zpopelní a dále se postupuje podle metody A. |
3. Chemikálie
|
3.1 |
Kyselina chlorovodíková 3 mol/l. |
|
3.2 |
Kyselina trichloroctová, 20% roztok (w/v). |
|
3.3 |
Kyselina trichloroctová, 1% roztok (w/v). |
4. Přístroje a pomůcky
|
4.1 |
Topná deska. |
|
4.2 |
Elektrická muflová pec s termostatem. |
|
4.3 |
Spalovací kelímky z křemíku, porcelánu nebo platiny, buď pravoúhlé (asi 60 × 40 × 25 mm) nebo kulaté (průměr 60–75 mm, výška 20–40 mm). |
5. Postup
5.1 Metoda A
Vzorek se zpopelní podle postupu uvedeného v metodě pro stanovení obsahu popela. Může se použít rovněž popel získaný při uvedeném stanovení.
Popel se pomocí 75 ml kyseliny chlorovodíkové (3.1) převede do 250–400 ml kádinky. Uvede se pomalu do varu a mírně se vaří patnáct minut. Teplý roztok se přefiltruje přes bezpopelný papírový filtr a zbytek na filtru se promyje teplou vodou do vymizení kyselé reakce. Filtr s promytým zbytkem se vysuší a potom zpopelní v předem zváženém kelímku při teplotě minimálně 550 oC a maximálně 700 oC. Nechá se vychladnout v exsikátoru a zváží se.
5.2 Metoda B
Do 250–400 ml kádinky se naváží 5 g vzorku s přesností na 1 mg. Přidá se postupně 25 ml vody a 25 ml kyseliny chlorovodíkové (3.1), promíchá se a nechá se stát, dokud neustane pěnění. Přidá se dalších 50 ml kyseliny chlorovodíkové (3.1). Po ukončení vývoje plynu se kádinka umístí na vroucí vodní lázeň, kde se ponechá 30 minut nebo v případě potřeby déle, aby mohlo dojít k hydrolýze veškerého přítomného škrobu. Přefiltruje se za horka přes bezpopelný papírový filtr a filtr se promyje 50 ml horké vody (viz poznámka 7). Filtr se zbytkem se vloží do spalovacího kelímku, vysuší se a potom zpopelní při teplotě minimálně 550 oC a maximálně 700 oC. Po spálení se popel převede pomocí 75 ml kyseliny chlorovodíkové (3.1) do 250–400 ml kádinky; pokračuje se podle druhého odstavce bodu 5.1.
6. Výpočet a vyjádření výsledků
Vypočítá se hmotnost zbytku po odečtení hmotnosti kelímku (táry). Výsledek se vyjádří jako procento vzorku.
7. Poznámka
Jestliže je filtrace obtížná, je možno nahradit 50 ml kyseliny chlorovodíkové (3.1) 50 ml 20% kyseliny trichloroctové (3.2) a zbytek na filtru promývat teplým roztokem 1% kyseliny trichloroctové (3.3).
O. STANOVENÍ OBSAHU UHLIČITANŮ
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu uhličitanů vyjádřených obvykle jako uhličitan vápenatý ve většině krmiv.
Nicméně v některých případech (např. u uhličitanu železnatého) musí být použita zvláštní metoda.
2. Princip
Uhličitany se rozloží v kyselině chlorovodíkové; vzniklý oxid uhličitý se změří v kalibrované trubici a jeho objem se porovná s objemem plynu uvolněného za stejných podmínek ze známého množství uhličitanu vápenatého.
3. Chemikálie
|
3.1 |
Kyselina chlorovodíková, hustota 1,10 g/ml. |
|
3.2 |
Uhličitan vápenatý. |
|
3.3 |
Kyselina sírová, přibližně 0,05 mol/l, obarvená methylčervení. |
4. Přístroje a pomůcky
Scheibler-Dietrichův přístroj (viz obrázek) nebo obdobný přístroj.
5. Postup
Podle obsahu uhličitanů ve vzorku se naváží:
|
— |
0,5 g u produktů obsahujících 50–100 % uhličitanů vyjádřených jako uhličitan vápenatý, |
|
— |
1 g u produktů obsahujících 40–50 % uhličitanů vyjádřených jako uhličitan vápenatý, |
|
— |
2 až 3 g u ostatních produktů. |
Navážka vzorku se umístí do zvláštní nádobky (4) přístroje, vybavené malou trubicí z nerozbitného materiálu, která obsahuje 10 ml kyseliny chlorovodíkové (3.1), a nádobka se připojí k přístroji. Trojcestný kohout (5) se otočí tak, aby trubice (1) byla spojena s vnějším prostorem. Pomocí mobilní trubice (2), která je naplněna obarvenou kyselinou sírovou (3.3) a spojena s kalibrovanou trubicí (1), se vyrovnají hladiny na nulu. Kohout (5) se otočí tak, aby byly spojeny trubice (1) a (3), a zkontroluje se, zda je hladina na nule.
Pomalým nakláněním nádobky (4) se kyselina chlorovodíková (3.1) nalije na navážku vzorku. Tlak se vyrovná snížením trubice (2). Nádobkou (4) se třepe tak dlouho, dokud zcela neustane uvolňování oxidu uhličitého.
Tlak se obnoví vyrovnáním kapalin v trubicích (1) a (2) na stejnou hladinu. Po několika minutách, až se ustálí tlak plynu, se odečte jeho hodnota.
Provede se kontrolní zkouška za stejných podmínek s navážkou 0,5 g uhličitanu vápenatého (3.2).
6. Výpočet a vyjádření výsledků
Obsah uhličitanů vyjádřených jako uhličitan vápenatý se vypočítá podle následujícho vzorce:
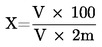
kde:
|
X |
= |
% (w/w) uhličitanů vyjádřených jako uhličitan vápenatý ve vzorku |
|
V |
= |
ml CO2 uvolněné z navážky vzorku |
|
V1 |
= |
ml CO2 uvolněné z 0,5 g CaCO3 |
|
m |
= |
hmotnost navážky vzorku v gramech. |
7. Poznámky
|
7.1 |
Pokud je hmotnost navážky vzorku větší než 2 g, přidá se ke vzorku v nádobce (4) nejprve 15 ml destilované vody a před zahájením zkoušky se promíchá. Při kontrolní zkoušce se použije stejný objem vody. |
|
7.2 |
Pokud se používá přístroj s jiným objemem než u Schleibler-Dietrichova přístroje, musí se upravit velikost navážky vzorku a kontrolní látky a musí se upravit výpočet výsledků.
|
P. STANOVENÍ OBSAHU CELKOVÉHO FOSFORU
FOTOMETRICKÁ METODA
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu celkového fosforu v krmivech. Je vhodná především pro zkoušení krmiv s nízkým obsahem fosforu. V určitých případech (u produktů s vysokým obsahem fosforu) je možno použít gravimetrickou metodu.
2. Princip
Vzorek se mineralizuje buď suchou cestou (zejména u organických krmiv), nebo mokrou cestou (zejména u minerálních sloučenin a tekutých krmiv), a převede se do kyselého roztoku. K tomuto roztoku se přidá molybdátovanadátové činidlo. Absorbance žlutě zbarveného roztoku se měří při 430 nm pomocí spektrofotometru.
3. Chemikálie
|
3.1 |
Uhličitan vápenatý. |
|
3.2 |
Kyselina chlorovodíková, ρ20 = 1,10 g/ml (přibližně 6 mol/l). |
|
3.3 |
Kyselina dusičná, ρ20 = 1,045 g/ml. |
|
3.4 |
Kyselina dusičná, ρ20 = 1,38–1,42 g/ml. |
|
3.5 |
Kyselina sírová, ρ20 = 1,84 g/ml. |
|
3.6 |
Molybdátovanadátové činidlo: v odměrné baňce o objemu 1 l se smíchá 200 ml roztoku heptamolybdenanu amonného (3.6.1) s 200 ml roztoku vanadičnanu amonného (3.6.2) a se 134 ml kyseliny dusičné (3.4) a doplní se vodou po značku. |
|
3.6.1 |
Roztok heptamolybdenanu amonného: 100 g heptamolybdenanu amonného (NH4)6Mo7O24 . 4H2O se rozpustí v horké vodě. Přidá se 10 ml amoniaku (hustota 0,91g/ml) a doplní se vodou na 1 litr. |
|
3.6.2 |
Roztok vanadičnanu amonného: 2,35 g vanadičnanu amonného NH4VO3 se rozpustí ve 400 ml horké vody. Za stálého míchání se pomalu přilévá 20 ml zředěné kyseliny dusičné (7 ml HNO3 (3.4) + 13 ml H2O) a doplní se vodou na 1 litr. |
|
3.7 |
Standardní roztok 1 mg fosforu na ml: 4,387 g dihydrogenfosforečnanu draselného KH2PO4 se rozpustí ve vodě. Doplní se vodou na 1 litr. |
4. Přístroje a pomůcky
|
4.1 |
Křemenné, porcelánové nebo platinové spalovací kelímky. |
|
4.2 |
Elektrická muflová pec s termostatem, regulovaná na teplotu 550 oC. |
|
4.3 |
Kjeldahlova baňka, 250 ml. |
|
4.4 |
Odměrné baňky a přesné pipety. |
|
4.5 |
Spektrofotometr. |
|
4.6 |
Zkumavky, průměr asi 16 mm, se zátkou o průměru 14,5 mm; objem: 25–30 ml. |
5. Postup
5.1 Příprava roztoku
Podle druhu vzorku se připraví roztok způsobem, který je uveden v bodě 5.1.1 nebo 5.1.2.
5.1.1
Do Kjeldahlovy baňky se naváží 1 g nebo větší množství vzorku s přesností na 1 mg. Přidá se 20 ml kyseliny sírové (3.5), baňka se důkladně protřepe, aby se vzorek a kyselina dobře spojily a aby se zabránilo usazování vzorku na stěnách baňky; pak se roztok zahřeje a 10 minut vaří. Po mírném ochlazení se přidají 2 ml kyseliny dusičné (3.4); slabě se zahřeje a opět mírně ochladí. Pak se opět přidá trochu kyseliny dusičné (3.4) a uvede se opět do varu. Postup se opakuje tolikrát, až se získá čirý roztok. Potom se ochladí, přidá se trochu vody, kapalina se převede do 500 ml odměrné baňky a Kjeldahlova baňka se vypláchne horkou vodou. Po ochlazení se baňka doplní vodou po značku, homogenizuje a přefiltruje.
5.1.2
Do spalovacího kelímku se naváží přibližně 2,5 g vzorku s přesností na 1 mg a dokonale se promíchá s 1 g uhličitanu vápenatého (3.1). V muflové peci se spaluje při teplotě 550 oC tak dlouho, dokud se nezíská popel bílé nebo šedé barvy (malé množství uhlíkatých částic není na závadu). Popel se převede do 250 ml kádinky, přidá se 20 ml vody a potom se přidává kyselina chlorovodíková (3.2), dokud neustane pěnění; nakonec se přidá dalších 10 ml kyseliny chlorovodíkové (3.2). Kádinka se postaví na pískovou lázeň a odpaří se do sucha, aby se vyloučily křemičitany. Zbytek se rozpustí v 10 ml kyseliny dusičné (3.3) a 5 minut vaří na pískové lázni nebo na topné desce tak, aby nedošlo k úplnému vysušení. Kapalina se převede do 500 ml odměrné baňky a kádinka se několikrát vymyje horkou vodou. Po ochlazení se doplní vodou po značku, homogenizuje a přefiltruje.
5.2 Vývoj zbarvení a měření absorbance
Alikvotní množství filtrátu připraveného podle bodu 5.1.1 nebo 5.1.2 se zředí tak, aby koncentrace fosforu byla maximálně 40 μg/ml. 10 ml tohoto roztoku se odpipetuje do zkumavky (4.6) a přidá se 10 ml molybdátovanadátového činidla (3.6). Homogenizuje se a nechá se stát minimálně po dobu 10 minut při teplotě 20 oC. Pak se měří absorbance ve spektrofotometru při 430 nm proti roztoku připravenému z 10 ml vody a 10 ml molybdátovanadátového činidla (3.6).
5.3 Kalibrační křivka
Ze standardního roztoku (3.7) se připraví roztoky, které obsahují 5, 10, 20, 30 a 40 μg fosforu na jeden ml. Ke každým 10 ml těchto roztoků se přidá 10 ml molybdátovanadátového činidla (3.6). Homogenizuje se a nechá se stát minimálně po dobu 10 minut při teplotě 20 oC. Pak se měří absorbance, jak je uvedeno v bodě 5.2. Kalibrační křivka se vytvoří vynesením hodnot absorbancí proti odpovídajícím množstvím fosforu. Křivka je lineární při koncentraci fosforu 0–40 μg/ml.
6. Výpočet a vyjádření výsledků
Obsah fosforu ve zkoušeném vzorku se stanoví pomocí kalibrační křivky.
Výsledek se vyjádří jako procento vzorku.
Opakovatelnost
Rozdíl mezi výsledky dvou paralelních stanovení provedených na stejném vzorku nesmí přesáhnout:
|
— |
3 % relat. z hodnoty vyššího výsledku u vzorků s obsahem fosforu nižším než 5 %, |
|
— |
0,15 % v absolutní hodnotě u vzorků s obsahem fosforu 5 % a více. |
Q. STANOVENÍ OBSAHU VE VODĚ ROZPUSTNÝCH CHLORIDŮ
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu ve vodě rozpustných chloridů obvykle vyjadřovaných jako chlorid sodný. Tato metoda je vhodná pro všechna krmiva.
2. Princip
Chloridy se rozpustí ve vodě. Jestliže produkt obsahuje organickou hmotu, roztok se vyčeří. Roztok se mírně okyselí kyselinou dusičnou a chloridy se vysrážejí pomocí roztoku dusičnanu stříbrného jako chlorid stříbrný. Přebytek dusičnanu stříbrného se titruje roztokem thiokyanatanu amonného podle Volhardovy metody.
3. Chemikálie
|
3.1 |
Roztok thiokyanatanu amonného 0,1 mol/l. |
|
3.2 |
Roztok dusičnanu stříbrného 0,1 mol/l. |
|
3.3 |
Nasycený roztok síranu železitoamonného (NH4)Fe(SO4)2. |
|
3.4 |
Kyselina dusičná, hustota: 1,38 g/ml. |
|
3.5 |
Diethylether. |
|
3.6 |
Aceton. |
|
3.7 |
Carrezovo činidlo I: ve vodě se rozpustí 21,9 g octanu zinečnatého Zn (CH3COO)2 . 2H2O a 3 g ledové kyseliny octové. Doplní se vodou na 100 ml. |
|
3.8 |
Carrezovo činidlo II: ve vodě se rozpustí 10,6 g kyanoželeznatanu draselného (ferrokyanidu draselného) K4Fe(CN)6 . 3H2O. Doplní se vodou na 100 ml. |
|
3.9 |
Aktivní uhlí, chloridů prosté a neabsorbující chloridy. |
4. Přístroje a pomůcky
Míchací zařízení: přibližně 35–40 otáček/min.
5. Postup
5.1 Příprava roztoku
Příprava vzorku závisí na druhu vzorku a roztok se připraví podle odstavce 5.1.1, 5.1.2 nebo 5.1.3.
Současně se provede slepá zkouška bez zkoušeného vzorku.
5.1.1
Do 500 ml odměrné baňky se naváží maximálně 10 g vzorku s přesností na 1 mg tak, aby navážka obsahovala nejvýše 3 g ve vodě rozpustných chloridů, přidá se asi 400 ml vody a třicet minut se míchá na míchacím zařízení při teplotě asi 20 oC. Potom se doplní po značku, homogenizuje a přefiltruje.
5.1.2
Do 500 ml odměrné baňky se naváží asi 5 g vzorku s přesností na 1 mg a 1 g aktivního uhlí. Přidá se 400 ml vody o teplotě přibližně 20 oC a 5 ml Carrezova činidla I (3.7), asi 30 sekund se míchá, pak se přidá 5 ml Carrezova činidla II (3.8). Třicet minut se míchá na míchacím zařízení, potom se doplní po značku, homogenizuje a přefiltruje.
5.1.3
Roztok se připraví podle bodu 5.1.2, ale nefiltruje se. Místo toho se dekantuje nebo v případě potřeby odstředí. Do 200 ml odměrné baňky se odpipetuje 100 ml supernatantu, smíchá se s acetonem (3.6) a doplní se tímto rozpouštědlem po značku. Homogenizuje se a přefiltruje.
5.2 Titrace
Do Erlenmayerovy baňky se odpipetuje 25–100 ml filtrátu (podle předpokládaného obsahu chlóru) získaného podle bodu 5.1.1, 5.1.2 nebo 5.1.3. Alikvotní část nesmí obsahovat více než 150 ml chlóru (Cl). V případě potřeby se doplní vodou na minimálně 50 ml, přidá se 5 ml kyseliny dusičné (3.4), 20 ml nasyceného roztoku síranu železitoamonného (3.3) a dvě kapky roztoku thiokyanatanu amonného (3.1) z byrety naplněné ke značce nula. Jinou byretou se přidá roztok dusičnanu stříbrného (3.2) tak, aby se získal přebytek 5 ml. Dále se přidá 5 ml diethyletheru (3.5) a silně se protřepe, aby sraženina dobře koagulovala. Přebytek dusičnanu stříbrného se titruje s roztokem thiokyanatanu amonného (3.1), dokud není dosaženo červenohnědého zabarvení, které vydrží jednu minutu.
6. Výpočet a vyjádření výsledků
Obsah ve vodě rozpustných chloridů (X) vyjádřených jako procento chloridu sodného se vypočítá podle následujícího vzorce:
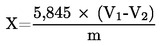
kde:
|
V1 |
= |
přidaný roztok dusičnanu stříbrného 0,1 mol/l v ml |
|
V2 |
= |
roztok thiokyanatanu amonného 0,1 mol/l použitý pro titraci v ml |
|
m |
= |
hmotnost vzorku |
Jestliže byl při slepé zkoušce roztok dusičnanu stříbrného 0,1 mol/l spotřebován, odečte se tato hodnota z objemu (V1 - V2).
7. Poznámky
|
7.1 |
Titraci lze provádět také potenciometricky. |
|
7.2 |
V případě produktů velmi bohatých na oleje a tuky se vzorek nejprve odtuční diethyletherem nebo petroletherem. |
|
7.3 |
V případě rybích mouček lze titraci provést Mohrovou metodou. |
(1) Laboratorní sušárna určená pro sušení obilí, mouky, krup a krupice musí mít takovou tepelnou kapacitu, aby, pokud je nastavena na teplotu 131 oC, této teploty opět dosáhla v době kratší než 45 minut poté, co byl do sušárny umístěn maximální počet zkoušených vzorků, které se mají sušit současně. Větrání musí být takové, že když se všechny vzorky pšenice obecné, které může sušárna pojmout, současně suší po dobu dvou hodin, vykazují s ohledem na výsledky čtyřhodinového sušení rozdíl, který je nižší než 0,15 %.
(2) Pokud má být vyextrahovaný tuk použit pro další zkoušky kvality, nahradí se varné kamínky skleněnými kuličkami.
PŘÍLOHA IV
METODY ZKOUŠENÍ PRO KONTROLU OBSAHU POVOLENÝCH DOPLŇKOVÝCH LÁTEK V KRMIVECH
A. STANOVENÍ OBSAHU VITAMINU A
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu vitaminu A (retinolu) v krmivech a premixech. Vitamin A zahrnuje all-trans-retinyl alkohol a jeho cis izomery, které se stanovují touto metodou. Obsah vitaminu A se vyjadřuje v mezinárodních jednotkách (m. j.) na kilogram. Jedna mezinárodní jednotka odpovídá aktivitě 0,300 μg all-trans vitaminu A (alkohol) nebo 0,344 μg all-trans vitaminu A (acetát) nebo 0,550 μg all-trans vitaminu A (palmitát).
Mez kvantifikace je 2 000 m. j. vitaminu A/kg.
2. Princip
Vzorek se hydrolyzuje ethanolickým roztokem hydroxidu draselného a vitamin A se extrahuje petroletherem. Rozpouštědlo se odpaří a zbytek se rozpustí v methanolu a, pokud je to nutné, naředí se na vhodnou koncentraci. Obsah vitaminu A se stanoví metodou vysokoúčinné kapalinové chromatografie na reverzní fázi (RP-HPLC) s UV nebo fluorescenční detekcí. Chromatografické parametry jsou vybrány tak, aby nedocházelo k separaci all-trans vitaminu A (alkohol) a jeho cis izomerů.
3. Chemikálie
|
3.1 |
Ethanol, σ = 96 %. |
|
3.2 |
Petrolether, bod varu 40–60 oC. |
|
3.3 |
Methanol. |
|
3.4 |
Roztok hydroxidu draselného, c = 50 g/100 ml. |
|
3.5 |
Roztok askorbátu sodného, c = 10 g/100 ml (viz poznámky 7.7). |
|
3.6 |
Sulfid sodný, Na2S · x H2O (x = 7–9). |
|
3.6.1. |
Roztok sulfidu sodného, c = 0,5 mol/l v glycerolu, β = 120 g/l (pro x = 9) (viz poznámky 7.8). |
|
3.7 |
Roztok fenolftaleinu, c = 2 g/100 ml v ethanolu (3.1). |
|
3.8 |
2-propanol. |
|
3.9 |
Mobilní fáze pro HPLC: směs methanolu (3,3) a vody, např. 980 + 20 (v + v). Přesný poměr bude upraven podle charakteristiky použité kolony. |
|
3.10 |
Dusík, prostý kyslíku. |
|
3.11 |
All-trans-vitamin A (acetát), extra čistý, s ověřenou aktivitou, např. 2,80 × 106 m. j./g. |
|
3.11.1 |
Zásobní roztok all-trans-vitaminu A (acetát): do 100 ml odměrné baňky se s přesností na 0,1 mg naváží 50 mg vitaminu A (acetát) (3.11). Rozpustí se v 2-propanolu (3.8) a doplní se jím po značku. Nominální koncentrace vitaminu A v roztoku je 1 400 m. j./ml. Přesný obsah se stanoví podle bodu 5.6.3.1. |
|
3.12 |
All-trans-vitamin A (palmitát), extra čistý, s ověřenou aktivitou, např. 1,80 × 106 m. j./g. |
|
3.12.1 |
Zásobní roztok all-trans-vitaminu A (palmitát): do 100 ml odměrné baňky se s přesností na 0,1 mg naváží 80 mg vitaminu A (palmitát) (3.12). Rozpustí se v 2-propanolu (3.8) a doplní se jím po značku. Nominální koncentrace vitaminu A v roztoku je 1 400 m. j./ml. Přesný obsah se stanoví podle bodu 5.6.3.2. |
|
3.13 |
2,6-di-terc-butyl-4-methylfenol (BHT) (viz poznámky 7.5). |
4. Přístroje a pomůcky
|
4.1 |
Vakuová rotační odparka. |
|
4.2 |
Tmavé sklo. |
|
4.2.1 |
Baňky s plochým dnem nebo kónické baňky, 500 ml, se zábrusovým hrdlem. |
|
4.2.2 |
Odměrné baňky s úzkým hrdlem a zábrusovou zátkou, objem 10, 25, 100 a 500 ml. |
|
4.2.3 |
Dělicí nálevky, kónické, 1 000 ml, se zábrusovou zátkou. |
|
4.2.4 |
Hruškovité baňky, 250 ml, se zábrusovým hrdlem. |
|
4.3 |
Allihnův chladič, délka pláště 300 mm, se zábrusem a s adaptérem na zavádění plynu. |
|
4.4 |
Skládaný filtrační papír pro oddělování fází, průměr 185 mm (např. Schleicher & Schuell 597 HY 1/2). |
|
4.5 |
Zařízení pro HPLC s dávkovacím systémem. |
|
4.5.1 |
Kolona pro kapalinovou chromatografii, 250 mm × 4 mm, C18, náplň 5 nebo 10 μm nebo obdobná (výkonnostní parametry: pouze jediný pík pro všechny izomery retinolu za podmínek HPLC). |
|
4.5.2 |
UV nebo fluorescenční detektor s nastavitelnou vlnovou délkou. |
|
4.6 |
Spektrofotometr s 10 mm křemennými kyvetami. |
|
4.7 |
Vodní lázeň s magnetickým mícháním. |
|
4.8 |
Extrakční zařízení (viz obrázek 1) sestávající ze: |
|
4.8.1 |
Skleněného válce o objemu 1 l vybaveného zábrusovým hrdlem a zátkou. |
|
4.8.2 |
Vkladatelného zařízení se zábrusem, s postranním raménkem a nastavitelnou trubičkou, která prochází středem. Nastavitelná trubička má konec vytvarovaný do tvaru U a zúženou výpust na opačné straně tak, aby bylo možné převést horní vrstvu z válce do dělicí nálevky. |
5. Postup
|
Poznámka |
: |
Vitamin A je citlivý na (UV) světlo a oxidaci. Všechny operace se provádí bez přístupu světla (používá se tmavé sklo nebo se chrání před světlem hliníkovou fólií) a kyslíku (pod dusíkem). Vzduch nad kapalinou se během extrakce vytěsní dusíkem (aby nedošlo k přetlakování, je nutno občas uvolnit zátku). |
5.1 Příprava vzorku
Vzorek se rozmělní tak, aby prošel sítem o velikosti oka 1 mm a aby během úpravy nedošlo k jeho zahřátí. Vzorek se musí rozmělňovat bezprostředně před vážením a saponifikací, jinak může docházet ke ztrátám vitaminu A.
5.2 Saponifikace
Podle obsahu vitaminu A ve vzorku se s přesností na 1 mg naváží 2–25 g vzorku do 500 ml baňky s plochým dnem nebo kónické baňky (4.2.1). Za intenzivního míchání se přidá 130 ml ethanolu (3.1), asi 100 mg BHT (3.13), 2 ml roztoku askorbátu sodného (3.5) a 2 ml roztoku sulfidu sodného (3.6). Baňka se připojí k chladiči (4.3) a ponoří se do vodní lázně s magnetickým mícháním (4.7). Zahřeje se k varu a vaří se pod refluxem 5 minut. Potom se přes chladič (4.3) přidá 25 ml roztoku hydroxidu draselného (3.4) a vaří se pod refluxem za míchání pod pomalým proudem dusíku dalších 25 minut. Potom se chladič opláchne asi 20 ml vody a obsah baňky se nechá vychladnout na laboratorní teplotu.
5.3 Extrakce
Saponifikovaný roztok se dekantací a propláchnutím celkem 250 ml vody převede kvantitativně do 1 000 ml dělicí nálevky (4.2.3) nebo do extrakčního zařízení (4.8). Saponifikační baňka se opláchne 25 ml ethanolu (3.1) a poté 100 ml petroletheru (3.2) a oba oplachy se převedou do dělicí nálevky nebo do extrakčního zařízení. Konečný poměr vody a ethanolu v kombinovaných roztocích musí být asi 2 : 1. Roztok se 2 minuty intenzivně třepe a potom se nechá 2 minuty stát.
5.3.1
Po rozdělení vrstev (poznámka 7.3) se petroletherová vrstva převede do další dělicí nálevky (4.2.3). Extrakce se opakuje ještě dvakrát se 100 ml petroletheru (3.2) a dvakrát s 50 ml petroletheru (3.2).
Spojené extrakty v dělicí nálevce se dvakrát promyjí mírným kroužením (aby nevznikala emulze) 100 ml vody a potom se opakovaně protřepávají se 100 ml vody, dokud voda nezůstane po přidání roztoku fenolftaleinu (3.7) bezbarvá (obvykle stačí 4 promytí). Promytý extrakt se přefiltruje do 500 ml odměrné baňky (4.2.2) přes suchý skládaný filtr pro oddělování fází (4.4), aby se odstranila suspendovaná voda. Dělicí nálevka se opláchne 50 ml petroletheru (3.2) a ten se přidá přes filtr do téže odměrné baňky. Baňka se petroletherem (3.2) doplní po značku a dobře promíchá.
5.3.2
Po rozdělení vrstev (poznámka 7.3) se odstraní zátka z válce (4.8.1) a místo ní se vloží vnitřní zařízení (4.8.2). Spodní strana U-trubice se umístí tak, aby byla právě nad rozhraním. Za použití tlaku dusíku (přiváděného postranním raménkem) se převede horní petroletherová vrstva do 1 000 ml dělicí nálevky (4.2.3). Přidá se 100 ml petroletheru (3.2), válec se zazátkuje a důkladně protřepe. Po oddělení vrstev se horní vrstva opět převede stejným způsobem do dělicí nálevky. Extrakce se opakuje s dalšími 100 ml petroletheru (3.2) a pak ještě dvakrát s 50 ml petroletheru (3.2). Po rozdělení fází se vždy horní petroletherová vrstva přidá k předchozí do dělicí nálevky.
Spojené extrakty petroletheru v dělicí nálevce se promyjí, jak je popsáno v bodě 5.3.1, a postupuje se dále podle bodu 5.3.1.
5.4 Příprava roztoku vzorku pro stanovení HPLC
Alikvotní část roztoku petroletheru (podle bodu 5.3.1 nebo 5.3.2) se odpipetuje do 250 ml hruškovité baňky (4.2.4). Rozpouštědlo se na vakuové rotační odparce (4.1) odpaří za sníženého tlaku při teplotě do 40 oC téměř do sucha. Atmosférický tlak se obnoví zavedením dusíku (3.10) do baňky a ta se odpojí od rotační odparky. Zbytek rozpouštědla se odpaří pod proudem dusíku (3.10) a odparek se ihned rozpustí ve známém objemu (10–100 ml) methanolu (3.3) (výsledná koncentrace vitaminu A musí být v rozsahu 5–30 m. j./ml).
5.5 Stanovení HPLC
Vitamin A se separuje na koloně C18 s reverzní fází (4.5.1) a jeho koncentrace se měří UV detektorem (325 nm) nebo fluorescenčním detektorem (excitace: 325 nm, emise: 475 nm) (4.5.2).
Do přístroje se nastřikuje alikvotní část (např. 20 μl) methanolického roztoku získaného podle bodu 5.4 a eluuje se mobilní fází (3.9). Průměrná hodnota výšek (ploch) píků se vypočte z několika opakovaných nástřiků téhož vzorku nebo kalibračních roztoků (5.6.2).
Následující podmínky jsou doporučené; mohou být použity jiné podmínky za předpokladu, že poskytují rovnocenné výsledky.
|
Kolona pro kapalinovou chromatografii (4.5.1): |
250 mm × 4 mm, C18, náplň 5 nebo 10 μm nebo obdobná |
|
Mobilní fáze (3.9): |
směs methanolu (3.3) a vody, např. 980 + 20 (v + v) |
|
Průtok: |
1–2 ml/min |
|
Detektor (4.5.2): |
UV detektor (325 nm) nebo fluorescenční detektor (excitace: 325 nm / emise: 475 nm). |
5.6 Kalibrace
5.6.1
Do 500 ml baňky s plochým dnem nebo kónické baňky (4.2.1) se odpipetuje 20 ml zásobního roztoku vitaminu A (acetát) (3.11.1) nebo 20 ml zásobního roztoku vitaminu A (palmitát) (3.12.1) a hydrolyzuje se podle bodu 5.2, ale bez přidání BHT. Potom se provede extrakce petroletherem (3.2) podle bodu 5.3 a doplní se na 500 ml petroletherem (3.2). 100 ml tohoto extraktu se odpaří na rotační odparce téměř do sucha (viz 5.4), zbylé rozpouštědlo se odpaří pod proudem dusíku (3.10) a odparek se rozpustí v 10,0 ml methanolu (3.3). Nominální koncentrace vitaminu A v tomto roztoku je 560 m. j./ml. Přesný obsah se stanoví podle bodu 5.6.3.3. Pracovní standardní roztok musí být připraven čerstvý před použitím.
Do 20 ml odměrné baňky se odpipetují 2,0 ml tohoto pracovního standardního roztoku, baňka se doplní po značku methanolem (3.3) a promíchá. Nominální koncentrace vitaminu A v tomto zředěném pracovním standardním roztoku je 56 m. j./ml.
5.6.2
Do 20 ml odměrných baněk se odpipetuje postupně 1,0, 2,0, 5,0 a 10,0 ml zředěných pracovních standardních roztoků, doplní se methanolem (3.3) po značku a promíchá. Nominální koncentrace vitaminu A v těchto roztocích jsou 2,8, 5,6, 14,0 a 28,0 m. j./ml.
Do přístroje se opakovaně nastřikuje 20 μl jednotlivých kalibračních roztoků a stanoví se průměrné hodnoty výšek (ploch) píků. Z průměrných hodnot výšek (ploch) píků se sestrojí kalibrační křivka, přičemž se použijí výsledky UV kontroly (5.6.3.3).
5.6.3
5.6.3.1
Do 50 ml odměrné baňky (4.2.2) se odpipetují 2,0 ml zásobního roztoku vitaminu A (acetát) (3.11.1) a doplní se po značku 2-propanolem (3.8). Nominální koncentrace vitaminu A v tomto roztoku je 56 m. j./ml. Do 25 ml odměrné baňky se odpipetuje 3,0 ml tohoto zředěného roztoku vitaminu A (acetát) a doplní se po značku 2-propanolem (3.8). Nominální koncentrace vitaminu A v tomto roztoku je 6,72 m. j./ml. Na spektrofotometru (4.6) se proměří UV spektrum tohoto roztoku proti 2-propanolu (3.8) v rozmezí 300–400 nm. Extinkční maximum musí ležet mezi 325 a 327 nm.
Výpočet obsahu vitaminu A:
m. j. vitaminu A / ml = E326 × 19,0
(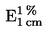 pro vitamin A acetát = 1 530 při 326 nm v 2-propanolu).
pro vitamin A acetát = 1 530 při 326 nm v 2-propanolu).
5.6.3.2
Do 50 ml odměrné baňky (4.2.2) se odpipetují 2,0 ml zásobního roztoku vitaminu A (palmitát) (3.12.1) a doplní se po značku 2-propanolem (3.8). Nominální koncentrace vitaminu A v tomto roztoku je 56 m. j./ml. Do 25 ml odměrné baňky se odpipetuje 3,0 ml tohoto zředěného roztoku vitaminu A (palmitát) a doplní se po značku 2-propanolem (3.8). Nominální koncentrace vitaminu A v tomto roztoku je 6,72 m. j./ml. Na spektrofotometru (4.6) se proměří UV spektrum tohoto roztoku proti 2-propanolu (3.8) v rozmezí 300–400 nm. Extinkční maximum musí ležet mezi 325 a 327 nm.
Výpočet obsahu vitaminu A:
m. j. vitaminu A / ml = E326 × 19,0
(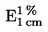 pro vitamin A palmitát = 957 při 326 nm v 2-propanolu).
pro vitamin A palmitát = 957 při 326 nm v 2-propanolu).
5.6.3.3
Do 50 ml odměrné baňky (4.2.2) se odpipetují 3,0 ml nezředěného pracovního standardního roztoku vitaminu A připraveného podle bodu 5.6.1 a doplní se po značku 2-propanolem (3.8). Do 25 ml odměrné baňky se odpipetuje 5,0 ml tohoto roztoku a doplní se po značku 2-propanolem (3.8). Nominální koncentrace vitaminu A v tomto roztoku je 6,72 m. j./ml. Na spektrofotometru (4.6) se proměří UV spektrum tohoto roztoku proti 2-propanolu (3.8) v rozmezí 300–400 nm. Extinkční maximum musí ležet mezi 325 a 327 nm.
Výpočet obsahu vitaminu A:
m. j. vitaminu A / ml = E325 × 18,3
(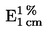 pro vitamin A alkohol = 1 821 při 325 nm v 2-propanolu).
pro vitamin A alkohol = 1 821 při 325 nm v 2-propanolu).
6. Výpočet a vyjádření výsledků
Koncentrace vitaminu A v extraktu vzorku v m. j./ml se stanoví porovnáním s kalibrační křivkou podle průměrných hodnot výšek (ploch) píků (5.6.2).
Obsah vitaminu A (w) v m. j./kg ve vzorku se vypočte podle následujícího vzorce:
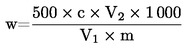 (m. j./kg)
(m. j./kg)
kde:
|
c |
= |
koncentrace vitaminu A v extraktu vzorku (5.4) v m. j./ml |
|
V1 |
= |
objem extraktu vzorku (5.4) v ml |
|
V2 |
= |
objem alikvotní části podle bodu 5.4 v ml |
|
m |
= |
hmotnost zkušebního vzorku v gramech. |
7. Poznámky
|
7.1 |
U vzorků s nízkou koncentrací vitaminu A může být užitečné spojit extrakty petroletheru ze dvou saponifikovaných vzorků (navážka: 25 g) do jednoho roztoku vzorku pro stanovení HPLC. |
|
7.2 |
Navážka vzorku pro zkoušku nesmí obsahovat více než 2 g tuku. |
|
7.3 |
Pokud nedojde k rozdělení fází, přidá se asi 10 ml ethanolu (3.1), aby se odstranila emulze. |
|
7.4 |
U oleje z tresčích jater a ostatních čistých tuků se doba saponifikace prodlouží na 45–60 minut. |
|
7.5 |
Místo BHT je možno použít hydrochinon. |
|
7.6 |
Pro separaci izomerů retinolu je možné použít kolonu s normální fází. V tomto případě je však nutné sečíst pro výpočet výšky (plochy) píků všech cis a trans izomerů. |
|
7.7 |
Místo roztoku askorbátu sodného lze použít asi 150 mg kyseliny askorbové. |
|
7.8 |
Místo roztoku sulfidu sodného lze použít asi 50 mg EDTA. |
|
7.9 |
V případě zkoušky na obsah vitaminu A v mléčných krmných směsích je třeba věnovat zvláštní pozornost:
Pro kontrolu toho, zda použitá metoda zkoušení poskytuje spolehlivé výsledky, pokud jde o tuto specifickou matrici (mléčné krmné směsi), se provede zkouška na výtěžnost u dodatečné navážky vzorku. Pokud je výtěžnost nižší než 80 %, výsledek zkoušky je třeba korigovat na výtěžnost. |
8. Opakovatelnost
Rozdíl mezi výsledky dvou paralelních stanovení provedených na stejném vzorku nesmí překročit 15 % relat. z hodnoty vyššího výsledku.
9. Výsledky kruhových testů (1)
|
|
Premix |
Premix krmiva |
Minerální koncentrát |
Proteinové krmivo |
Krmivo pro selata |
|
L |
13 |
12 |
13 |
12 |
13 |
|
n |
48 |
45 |
47 |
46 |
49 |
|
průměr (m. j./kg) |
17,02 × 106 |
1,21 × 106 |
537 100 |
151 800 |
18 070 |
|
sr (m. j./kg) |
0,51 × 106 |
0,039 × 106 |
22 080 |
12 280 |
682 |
|
r (m. j./kg) |
1,43 × 106 |
0,109 × 106 |
61 824 |
34 384 |
1 910 |
|
CVr (%) |
3,0 |
3,5 |
4,1 |
8,1 |
3,8 |
|
sR (m. j./kg) |
1,36 × 106 |
0,069 × 106 |
46 300 |
23 060 |
3 614 |
|
R (m. j./kg) |
3,81 × 106 |
0,193 × 106 |
129 640 |
64 568 |
10 119 |
|
CVR (%) |
8,0 |
6,2 |
8,6 |
15 |
20 |
|
L |
= |
počet laboratoří |
|
n |
= |
počet jednotlivých hodnot |
|
sr |
= |
standardní odchylka opakovatelnosti |
|
sR |
= |
standardní odchylka reprodukovatelnosti |
|
r |
= |
opakovatelnost |
|
R |
= |
reprodukovatelnost |
|
CVr |
= |
variační koeficient opakovatelnosti |
|
CVR |
= |
variační koeficient reprodukovatelnosti |
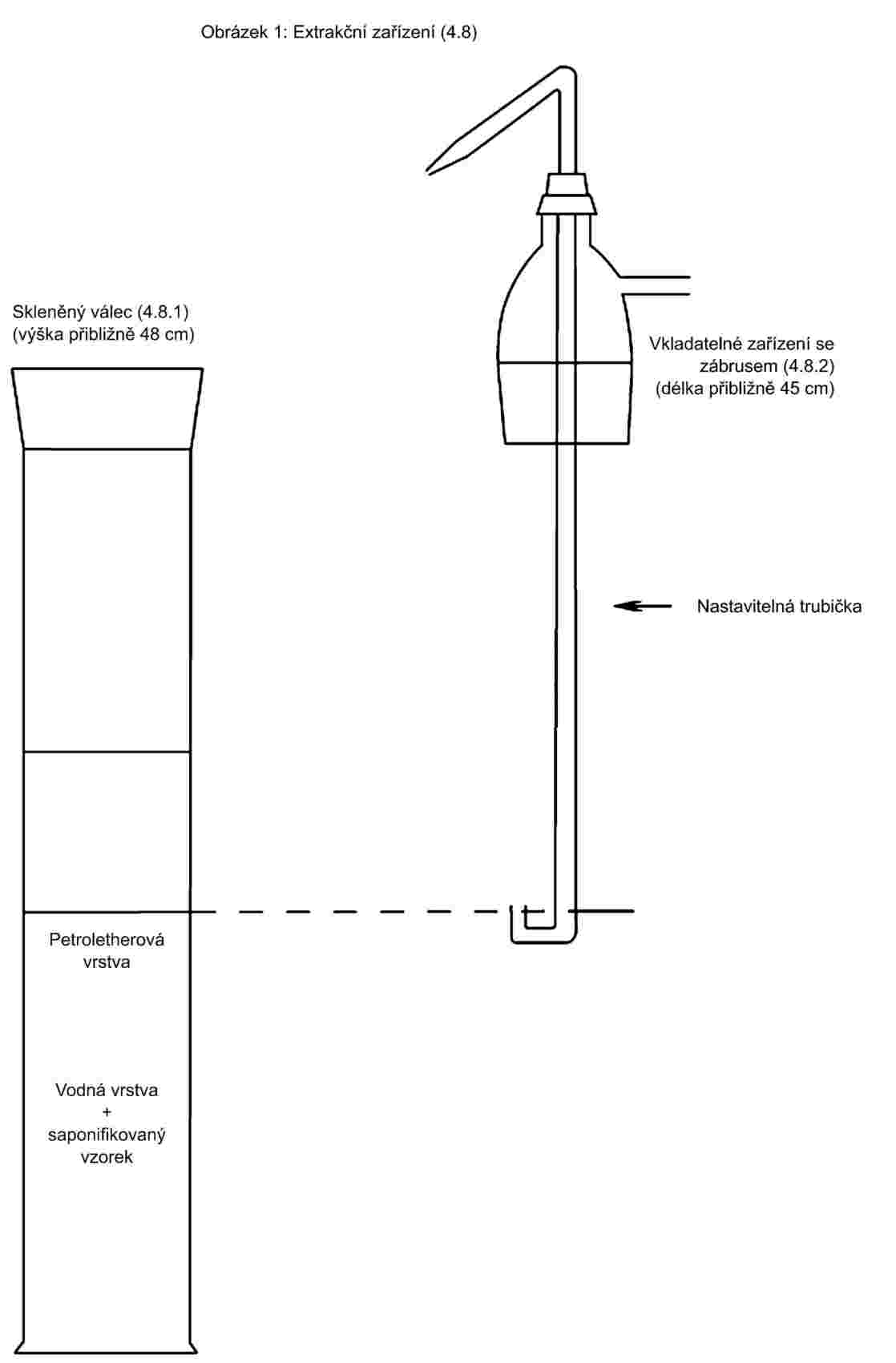
B. STANOVENÍ OBSAHU VITAMINU E
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu vitaminu E v krmivech a premixech. Obsah vitaminu E se vyjadřuje jako mg DL-α-tokoferol acetátu na kilogram. 1 mg DL-α-tokoferol acetátu odpovídá 0,91 mg DL-α-tokoferolu (vitamin E).
Mez kvantifikace je 2 mg vitaminu E/kg. Tato mez kvantifikace je dosažitelná pouze s fluorescenčním detektorem. Při použití UV detektoru je mez kvantifikace 10 mg/kg.
2. Princip
Vzorek se hydrolyzuje ethanolickým roztokem hydroxidu draselného a vitamin E se extrahuje petroletherem. Rozpouštědlo se odpaří a zbytek se rozpustí v methanolu a, pokud je to nutné, naředí se na vhodnou koncentraci. Obsah vitaminu E se stanoví metodou vysokoúčinné kapalinové chromatografie na reverzní fázi (RP-HPLC) s fluorescenční nebo UV detekcí.
3. Chemikálie
|
3.1 |
Ethanol, σ = 96 %. |
|
3.2 |
Petrolether, bod varu 40–60 oC. |
|
3.3 |
Methanol. |
|
3.4 |
Roztok hydroxidu draselného, c = 50 g/100 ml. |
|
3.5 |
Roztok askorbátu sodného, c = 10 g/100 ml (viz poznámky 7.7). |
|
3.6 |
Sulfid sodný, Na2S · x H2O (x = 7–9). |
|
3.6.1 |
Roztok sulfidu sodného, c = 0,5 mol/l v glycerolu, β = 120 g/l (pro x = 9) (viz poznámky 7.8). |
|
3.7 |
Roztok fenolftaleinu, c = 2 g/100 ml v ethanolu (3.1). |
|
3.8 |
Mobilní fáze pro HPLC: směs methanolu (3.3) a vody, např. 980 + 20 (v + v). Přesný poměr bude upraven podle charakteristiky použité kolony. |
|
3.9 |
Dusík, prostý kyslíku. |
|
3.10 |
DL-α-tokoferol acetát, extra čistý, s ověřenou aktivitou. |
|
3.10.1 |
Zásobní roztok DL-α-tokoferol acetátu: do 100 ml odměrné baňky se s přesností na 0,1 mg naváží 100 mg DL-α-tokoferol acetátu (3.10). Rozpustí se v ethanolu (3.1) a doplní se jím po značku. 1 ml tohoto roztoku obsahuje 1 mg DL-α-tokoferol acetátu. (UV kontrola – viz 5.6.1.3; stabilizace – viz poznámky 7.4). |
|
3.11 |
DL-α-tokoferol, extra čistý, s ověřenou aktivitou. |
|
3.11.1 |
Zásobní roztok DL-α-tokoferolu: do 100 ml odměrné baňky se s přesností na 0,1 mg naváží 100 mg DL-α-tokoferolu (3.11). Rozpustí se v ethanolu (3.1) a doplní se jím po značku. 1 ml tohoto roztoku obsahuje 1 mg DL-α-tokoferolu. (UV kontrola – viz 5.6.2.3; stabilizace – viz poznámky 7.4). |
|
3.12 |
2,6-di-terc-butyl-4-methylfenol (BHT) (viz poznámky 7.5). |
4. Přístroje a pomůcky
|
4.1 |
Rotační odparka. |
|
4.2 |
Tmavé sklo. |
|
4.2.1 |
Baňky s plochým dnem nebo kónické baňky, 500 ml, se zábrusovým hrdlem. |
|
4.2.2 |
Odměrné baňky s úzkým hrdlem a zábrusovou zátkou, objem 10, 25, 100 a 500 ml. |
|
4.2.3 |
Dělicí nálevky, kónické, 1 000 ml, se zábrusovou zátkou. |
|
4.2.4 |
Hruškovité baňky, 250 ml, se zábrusovým hrdlem. |
|
4.3 |
Allihnův chladič, délka pláště 300 mm, se zábrusem a s adaptérem na zavádění plynu. |
|
4.4 |
Skládaný filtrační papír pro oddělování fází, průměr 185 mm (např. Schleicher & Schuell 597 HY 1/2). |
|
4.5 |
Zařízení HPLC s dávkovacím systémem. |
|
4.5.1 |
Kolona pro kapalinovou chromatografii, 250 mm × 4 mm, C18, náplň 5 nebo 10 μm nebo obdobná. |
|
4.5.2 |
UV nebo fluorescenční detektor s nastavitelnou vlnovou délkou. |
|
4.6 |
Spektrofotometr s 10 mm křemennými kyvetami. |
|
4.7 |
Vodní lázeň s magnetickým mícháním. |
|
4.8 |
Extrakční zařízení (viz obrázek 1) sestávající z: |
|
4.8.1 |
Skleněného válce o objemu 1 l vybaveného zábrusovým hrdlem a zátkou. |
|
4.8.2 |
Vkladatelného zařízení se zábrusem, s postranním raménkem a nastavitelnou trubičkou, která prochází středem. Nastavitelná trubička má konec vytvarovaný do tvaru U a zúženou výpust na opačné straně tak, aby bylo možné převést horní vrstvu z válce do dělicí nálevky. |
5. Postup
|
Poznámka |
: |
Vitamin E je citlivý na (UV) světlo a oxidaci. Všechny operace se provádí bez přístupu světla (používá se tmavé sklo nebo se chrání před světlem hliníkovou fólií) a kyslíku (pod dusíkem). Vzduch nad kapalinou se během extrakce vytěsní dusíkem (aby nedošlo k přetlakování, je nutno občas uvolnit zátku). |
5.1 Příprava vzorku
Vzorek se rozmělní tak, aby prošel sítem o velikosti oka 1 mm a aby během úpravy nedošlo k jeho zahřátí. Rozmělnění musí být provedeno bezprostředně před vážením a saponifikací, jinak dochází ke ztrátám vitaminu E.
5.2 Saponifikace
Podle obsahu vitaminu E ve vzorku se s přesností na 0,01 g naváží 2–25 g vzorku do 500 ml baňky s plochým dnem nebo kónické baňky (4.2.1). Za intenzivního míchání se postupně přidá 130 ml ethanolu (3.1), asi 100 mg BHT (3.12), 2 ml roztoku askorbátu sodného (3.5) a 2 ml roztoku sulfidu sodného (3.6). Baňka se připojí k chladiči (4.3) a ponoří se do vodní lázně s magnetickým mícháním (4.7). Zahřeje se k varu a vaří se pod refluxem 5 minut. Potom se přes chladič (4.3) přidá 25 ml roztoku hydroxidu draselného (3.4) a vaří se pod refluxem za míchání pod pomalým proudem dusíku dalších 25 minut. Potom se chladič opláchne asi 20 ml vody a obsah baňky se nechá vychladnout na laboratorní teplotu.
5.3 Extrakce
Saponifikovaný roztok se dekantací a propláchnutím celkem 250 ml vody převede kvantitativně do 1 000 ml dělicí nálevky (4.2.3) nebo do extrakčního zařízení (4.8). Saponifikační baňka se postupně opláchne 25 ml ethanolu (3.1) a 100 ml petroletheru (3.2) a oba oplachy se převedou do dělicí nálevky nebo do extrakčního zařízení. Konečný poměr vody a ethanolu v kombinovaných roztocích musí být asi 2 : 1. Roztok se 2 minuty intenzivně třepe a potom se nechá 2 minuty stát.
5.3.1
Po rozdělení vrstev (viz poznámka 7.3) se petroletherová vrstva převede do další dělicí nálevky (4.2.3). Extrakce se opakuje dvakrát se 100 ml petroletheru (3.2) a dvakrát s 50 ml petroletheru (3.2).
Spojené extrakty v dělicí nálevce se dvakrát promyjí mírným kroužením (aby nevznikala emulze) 100 ml vody a potom se opakovaně protřepávají se 100 ml vody, dokud voda nezůstane po přidání roztoku fenolftaleinu (3.7) bezbarvá (obvykle stačí 4 promytí). Promytý extrakt se přefiltruje do 500 ml odměrné baňky (4.2.2) přes suchý skládaný filtr pro oddělování fází (4.4), aby se odstranila suspendovaná voda. Dělicí nálevka se opláchne 50 ml petroletheru (3.2) a ten se přidá přes filtr do téže odměrné baňky. Baňka se petroletherem (3.2) doplní po značku a důkladně promíchá.
5.3.2
Po rozdělení vrstev (poznámka 7.3) se odstraní zátka z válce (4.8.1) a místo ní se vloží vnitřní zařízení (4.8.2). Spodní strana U-trubice se umístí tak, aby byla právě nad rozhraním. Za použití tlaku dusíku (přiváděného postranním raménkem) se převede horní petroletherová vrstva do 1 000 ml dělicí nálevky (4.2.3). Přidá se 100 ml petroletheru (3.2), válec se zazátkuje a důkladně protřepe. Po oddělení vrstev se horní vrstva opět převede stejným způsobem do dělicí nálevky. Extrakce se opakuje s dalšími 100 ml petroletheru (3.2) a pak ještě dvakrát s 50 ml petroletheru (3.2). Po rozdělení fází se vždy horní petroletherová vrstva přidá k předchozí do dělicí nálevky.
Spojené extrakty petroletheru v dělicí nálevce se promyjí, jak je popsáno v bodě 5.3.1, a postupuje se dále podle bodu 5.3.1.
5.4 Příprava roztoku vzorku pro stanovení HPLC
Alikvotní část roztoku petroletheru (podle bodu 5.3.1 nebo 5.3.2) se odpipetuje do 250 ml hruškovité baňky (4.2.4). Roztok se na vakuové rotační odparce (4.1) odpaří za sníženého tlaku při teplotě do 40 oC téměř do sucha. Atmosférický tlak se obnoví zavedením dusíku (3.9) do baňky a ta se odpojí od rotační odparky. Zbytek rozpouštědla se odpaří pod proudem dusíku (3.9) a odparek se ihned rozpustí ve známém objemu (10–100 ml) methanolu. (3.3). Výsledná koncentrace DL-α-tokoferolu musí být 5–30 μg/ml.
5.5 Stanovení HPLC
Vitamin E se separuje na koloně C18 s reverzní fází (4.5.1) a jeho koncentrace se měří fluorescenčním detektorem (excitace: 295 nm, emise: 330 nm) nebo UV detektorem (292 nm) (4.5.2).
Do přístroje se nastřikuje alikvotní část (např. 20 μl) methanolického roztoku získaného podle bodu 5.4 a eluuje se mobilní fází (3.8). Průměrná hodnota výšek (ploch) píků se vypočte z několika opakovaných nástřiků téhož vzorku nebo kalibračních roztoků (5.6.2).
Následující podmínky jsou doporučené; mohou být použity jiné podmínky za předpokladu, že poskytují rovnocenné výsledky.
|
Kolona pro kapalinovou chromatografii (4.5.1): |
250 mm × 4 mm, C18, náplň 5 nebo 10 μm nebo obdobná |
|
Mobilní fáze (3.8): |
směs methanolu (3.3) a vody např. 980 + 20 (v + v) |
|
Průtok: |
1–2 ml/min |
|
Detektor (4.5.2): |
fluorescenční detektor (excitace: 295 nm/emise: 330 nm) nebo UV detektor (292 nm). |
5.6 Kalibrace (DL-α-tokoferol acetát nebo DL-α-tokoferol)
5.6.1
5.6.1.1
Do 500 ml baňky s plochým dnem nebo kónické baňky (4.2.1) se odpipetuje 25 ml zásobního roztoku DL-α-tokoferol acetátu (3.10.1) a hydrolyzuje se podle bodu 5.2. Potom se provede extrakce petroletherem (3.2) podle bodu 5.3 a doplní se na 500 ml petroletherem. 25 ml tohoto extraktu se odpaří na rotační odparce (viz. 5.4) téměř do sucha, zbylé rozpouštědlo se odpaří pod proudem dusíku (3.9) a odparek se rozpustí v 25,0 ml methanolu (3.3). Nominální koncentrace tohoto roztoku je 45,5 μg DL-α-tokoferolu/ml, což odpovídá 50 μg DL-α-tokoferol acetátu/ml. Pracovní standardní roztok se připravuje čerstvý před použitím.
5.6.1.2
Do odměrných baněk na 20 ml se odpipetuje postupně 1,0, 2,0, 4,0 a 10,0 ml pracovních standardních roztoků, doplní se methanolem (3.3) po značku a promíchá. Nominální koncentrace těchto roztoků jsou 2,5, 5,0, 10,0 a 25,0 μg/ml DL-α-tokoferol acetátu, tj. 2,28, 4,55, 9,10 a 22,8 μg/ml DL-α-tokoferolu.
Do přístroje se opakovaně nastřikuje 20 μl jednotlivých kalibračních roztoků a stanoví se průměrné hodnoty výšek (ploch) píků. Z průměrných hodnot výšek (ploch) píků se sestrojí kalibrační křivka.
5.6.1.3
Do 25 ml odměrné baňky se odpipetuje 5,0 ml zásobního roztoku DL-α-tokoferol acetátu (3.10.1) a doplní se po značku ethanolem. Na spektrofotometru (4.6) se proměří UV spektrum tohoto roztoku proti ethanolu (3.1) v rozmezí 250–320 nm.
Absorpční maximum je při 284 nm:
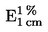 = 43,6 při 284 nm v ethanolu
= 43,6 při 284 nm v ethanolu
Při tomto ředění musí být získaná hodnota extinkce 0,84–0,88.
5.6.2
5.6.2.1
Do 50 ml odměrné baňky se odpipetují 2 ml zásobního roztoku DL-α-tokoferolu (3.11.1), zředí se methanolem (3.3) a doplní se jím po značku. Nominální koncentrace tohoto roztoku je 40 μg/ml DL-α-tokoferolu, což odpovídá 44,0 μg/ml DL-α-tokoferol acetátu. Pracovní standardní roztok se připravuje čerstvý před použitím.
5.6.2.2
Do odměrných baněk na 20 ml se odpipetuje postupně 1,0, 2,0, 4,0 a 10,0 ml pracovních standardních roztoků, doplní se methanolem (3.3) po značku a promíchá. Nominální koncentrace těchto roztoků jsou 2,0, 4,0, 8,0 a 20,0 μg/ml DL-α-tokoferolu, tj. 2,20, 4,40, 8,79 a 22,0 μg/ml DL-α-tokoferol acetátu.
Do přístroje se opakovaně nastřikuje 20 μl jednotlivých kalibračních roztoků a stanoví se průměrné hodnoty výšek (ploch) píků. Z průměrných hodnot výšek (ploch) píků se sestrojí kalibrační křivka.
5.6.2.3
Do 25 ml odměrné baňky se odpipetuje 2,0 ml zásobního roztoku DL-α-tokoferolu (3.11.1) a doplní se po značku ethanolem. Na spektrofotometru (4.6) se proměří UV spektrum tohoto roztoku proti ethanolu (3.1) v rozmezí 250–320 nm. Absorpční maximum je při 292 nm:
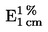 = 75,8 při 292 nm v ethanolu
= 75,8 při 292 nm v ethanolu
Při tomto ředění musí být získána hodnota extinkce 0,6.
6. Výpočet a vyjádření výsledků
Ze průměrné výšky (plochy) píků vitaminu E pro roztok vzorku se stanoví koncentrace roztoku vzorku v μg/ml (počítáno jako α-tokoferol acetát) porovnáním s kalibrační křivkou (5.6.1.2 nebo 5.6.2.2).
Obsah vitaminu E (w) v mg/kg ve vzorku se vypočte podle následujícího vzorce:
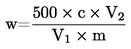 (mg/kg)
(mg/kg)
kde:
|
c |
= |
koncentrace vitaminu E (jako α-tokoferol acetátu) v extraktu vzorku (5.4) v μg/ml |
|
V1 |
= |
objem extraktu vzorku (5.4) v ml |
|
V2 |
= |
objem alikvotní části (5.4) v ml |
|
m |
= |
navážka zkušebního vzorku v g. |
7. Poznámky
|
7.1 |
Pro vzorky s nízkým obsahem vitaminu E může být užitečné spojit dva petroletherové extrakty ze dvou saponifikovaných vzorků (navážka: 25 g) do jednoho roztoku vzorku pro stanovení HPLC. |
|
7.2 |
Navážka vzorku pro zkoušku nesmí obsahovat více než 2 g tuku. |
|
7.3 |
Pokud nedojde k rozdělení fází, přidá se asi 10 ml ethanolu (3.1), aby se odstranila emulze. |
|
7.4 |
Po spektrofotometrickém měření roztoku DL-α-tokoferol acetátu nebo DL-α-tokoferolu podle bodů 5.6.1.3 nebo 5.6.2.3 se přidá do roztoku (3.10.1 nebo 3.10.2) přibližně 10 mg BHT (3.12) a roztok se skladuje v chladničce (maximální doba skladovatelnosti je čtyři týdny). |
|
7.5 |
Místo BHT je možno použít hydrochinon. |
|
7.6 |
Oddělení α-, β-, γ- a δ-tokoferolů je možné s použitím normální fázové kolony. |
|
7.7 |
Místo roztoku askorbátu sodného lze použít asi 150 mg kyseliny askorbové. |
|
7.8 |
Místo roztoku sulfidu sodného lze použít asi 50 mg EDTA. |
|
7.9 |
Vitamin E acetát hydrolyzuje v alkalickém prostředí velmi rychle, a proto je velmi citlivý na oxidaci, zejména v přítomnosti stopových prvků, jako je železo nebo měď. V případě stanovení vitaminu E v premixech s obsahem vyšším než 5 000 mg/kg by důsledkem mohl být rozklad vitaminu E. Proto se pro potvrzení doporučuje metoda HPLC včetně enzymatického rozkladu vitaminu E bez alkalické saponifikace. |
8. Opakovatelnost
Rozdíl mezi výsledky dvou paralelních stanovení provedených na stejném vzorku nesmí překročit 15 % relat. z hodnoty vyššího výsledku.
9. Výsledky kruhových testů (2)
|
|
Premix |
Premix krmiva |
Minerální koncentrát |
Proteinové krmivo |
Krmivo pro selata |
|
L |
12 |
12 |
12 |
12 |
12 |
|
n |
48 |
48 |
48 |
48 |
48 |
|
průměr (mg/kg) |
17 380 |
1 187 |
926 |
315 |
61,3 |
|
Sr (mg/kg) |
384 |
45,3 |
25,2 |
13,0 |
2,3 |
|
r (mg/kg) |
1 075 |
126,8 |
70,6 |
36,4 |
6,4 |
|
CVr (%) |
2,2 |
3,8 |
2,7 |
4,1 |
3,8 |
|
sR (mg/kg) |
830 |
65,0 |
55,5 |
18,9 |
7,8 |
|
R (mg/kg) |
2 324 |
182,0 |
155,4 |
52,9 |
21,8 |
|
CVR (%) |
4,8 |
5,5 |
6,0 |
6,0 |
12,7 |
|
L |
= |
počet laboratoří |
|
n |
= |
počet jednotlivých hodnot |
|
sr |
= |
standardní odchylka opakovatelnosti |
|
sR |
= |
standardní odchylka reprodukovatelnosti |
|
r |
= |
opakovatelnost |
|
R |
= |
reprodukovatelnost |
|
CVr |
= |
variační koeficient opakovatelnosti |
|
CVR |
= |
variační koeficient reprodukovatelnosti |
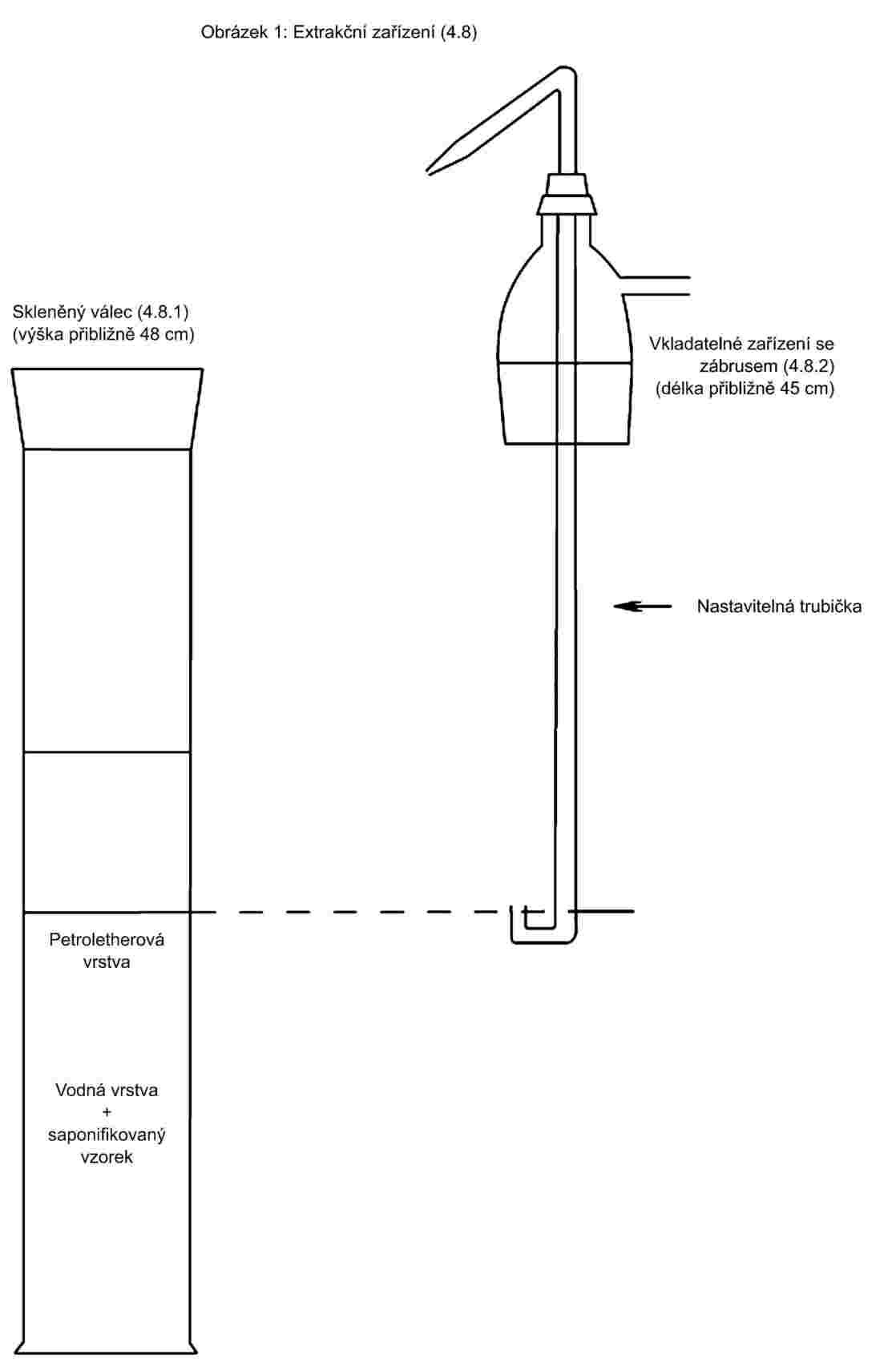
C. STANOVENÍ OBSAHU STOPOVÝCH PRVKŮ ŽELEZA, MĚDI, MANGANU A ZINKU
1. Účel a rozsah
Tato metoda umožňuje stanovení stopových prvků železa, mědi, manganu a zinku v krmivech. Meze stanovitelnosti jsou:
|
— |
železo (Fe): 20 mg/kg, |
|
— |
měď (Cu): 10 mg/kg, |
|
— |
mangan (Mn): 20 mg/kg, |
|
— |
zinek (Zn): 20 mg/kg. |
2. Princip
Vzorek se převede po destrukci organické hmoty (pokud je obsažena) do roztoku kyselinou chlorovodíkovou. Prvky železo, měď, mangan a zinek se stanovují po příslušném zředění atomovou absorpční spektrofotometrií.
3. Chemikálie
Úvodní poznámky
Pro přípravu chemikálií a analytických roztoků se používá voda, dvakrát destilovaná ve skleněném destilačním přístroji (borosilikátové nebo křemičité sklo) nebo po dvojím průchodu přes ionex, která prokazatelně neobsahuje kationty.
Chemikálie musí být nejméně analytické čistoty. Nepřítomnost prvků, které mají být stanoveny, musí být kontrolována slepou zkouškou. Pokud je to nutné, musí být chemikálie přečištěny.
Místo standardních roztoků popsaných níže mohou být použity komerční standardní roztoky za předpokladu, že mají deklarovaný obsah a před použitím byly překontrolovány.
|
3.1 |
Kyselina chlorovodíková (d: 1,19 g/ml). |
|
3.2 |
Kyselina chlorovodíková (6 mol/l). |
|
3.3 |
Kyselina chlorovodíková (0,5 mol/l). |
|
3.4 |
Kyselina fluorovodíková 38–40 % (v/v) s obsahem železa (Fe) pod 1 mg/l a zbytkem po odpaření menším než 10 mg (jako sulfát)/l. |
|
3.5 |
Kyselina sírová (d: 1,84 g/ml). |
|
3.6 |
Peroxid vodíku (přibližně 100 objemů kyslíku (30 % hmotnostních)). |
|
3.7 |
Standardní roztok železa (1 000 μg Fe/ml) připravený podle níže uvedeného postupu nebo obdobný komerčně dostupný roztok: v odměrné baňce na 1 000 ml se ve 200 ml kyseliny chlorovodíkové 6 mol/l (3.2) rozpustí 1 g železného drátu, přidá se 16 ml peroxidu vodíku (3.6) a doplní se po značku vodou. |
|
3.7.1 |
Pracovní standardní roztok železa (100 μg Fe/ml) připravený zředěním standardního roztoku (3.7) vodou v poměru 1 + 9. |
|
3.8 |
Standardní roztok mědi (1 000 μg Cu/ml) připravený podle níže uvedeného postupu nebo obdobný komerčně dostupný roztok:
|
|
3.8.1 |
Pracovní standardní roztok mědi (10 μg Cu/ml) připravený zředěním standardního roztoku (3.8) vodou v poměru 1 + 9 a potom zředěním výsledného roztoku vodou v poměru 1 + 9. |
|
3.9 |
Standardní roztok manganu (1 000 μg Mn/ml) připravený podle níže uvedeného postupu nebo obdobný komerčně dostupný roztok:
|
|
3.9.1 |
Pracovní standardní roztok manganu (10 μg Mn/ml) připravený zředěním standardního roztoku (3.9) vodou v poměru 1 + 9 a potom zředěním výsledného roztoku vodou v poměru 1 + 9. |
|
3.10 |
Standardní roztok zinku (1 000 μg Zn/ml) připravený podle níže uvedeného postupu nebo obdobný komerčně dostupný roztok:
|
|
3.10.1 |
Pracovní standardní roztok zinku (10 μg Zn/ml) připravený zředěním standardního roztoku (3.10) vodou v poměru 1 + 9 a potom zředěním výsledného roztoku vodou v poměru 1 + 9. |
|
3.11 |
Roztok chloridu lanthanitého: v odměrné baňce na 1 000 ml se ve 150 ml vody a 100 ml kyseliny chlorovodíkové 6 mol/l (3.2) rozpustí 12 g kysličníku lanthanitého a doplní se po značku vodou. |
4. Přístroje a pomůcky
|
4.1 |
Muflová pec s regulací teploty a záznamníkem (pokud možno). |
|
4.2 |
Skleněné nádobí rezistentního borosilikátového typu. Doporučuje se používat přístroje, které jsou používány výlučně pro stanovení stopových prvků. |
|
4.3 |
Atomový absorpční spektrofotometr splňující požadavky metody na citlivost a přesnost v požadovaném rozsahu. |
5. Postup (3)
5.1 Vzorky obsahující organickou hmotu
5.1.1 (4)
|
5.1.1.1 |
Do platinového nebo křemenného kelímku (4.3) (viz poznámka b)), předem zváženého, se naváží 5–10 g vzorku s přesností na 0,2 mg, vysuší se v sušárně při 105 oC a potom se vloží do chladné muflové pece (4.1). Pec se uzavře (viz poznámka c)) a teplota se postupně zvyšuje na 450–475 oC během asi 90 minut. Tato teplota se udržuje 4 až 16 hodin (např. přes noc), aby se odstranily uhlíkaté částice. Potom se pec otevře a nechá vychladnout (viz poznámka d)). Popel se zvlhčí vodou a přenese se do 250 ml kádinky. Kelímek se vypláchne celkem asi 5 ml kyseliny chlorovodíkové (3.1) a další kyselina se přidává pomalu a opatrně do kádinky (může proběhnout prudká reakce v důsledku tvorby CO2). Kyselina chlorovodíková (3.1) se přidává po kapkách a za míchání, dokud neustane šumění. Odpaří se do sucha za občasného zamíchání skleněnou tyčinkou. K odparku se přidá 15 ml kyseliny chlorovodíkové 6 mol/l (3.2) a poté asi 120 ml vody. Promíchá se skleněnou tyčinkou, která se ponechá v kádince, a poté se kádinka zakryje hodinovým sklem. Obsah kádinky se uvede do mírného varu a udržuje se na bodu varu, dokud se popel nerozpustí. Roztok se přefiltruje přes bezpopelný papírový filtr a filtrát se jímá do 250 ml odměrné baňky. Kádinka a filtr se promyjí 5 ml horké kyseliny chlorovodíkové 6 mol/l (3.2) a dvakrát vroucí vodou. Odměrná baňka se doplní po značku vodou (koncentrace HCl je asi 0,5 mol/l). |
|
5.1.1.2 |
Pokud je zbytek na filtru zbarven černě (uhlík), vrátí se do pece a spaluje se další tři až pět hodin při 450–475 oC. Spalování je dokončeno, když je popel bílý nebo téměř bílý. Popel se rozpustí v asi 2 ml kyseliny chlorovodíkové (3.1), odpaří se do sucha a přidá se 5 ml kyseliny chlorovodíkové 6 mol/l (3.2). Roztok se zahřeje k varu a potom se přefiltruje do odměrné baňky a doplní po značku vodou (koncentrace HCl je asi 0,5 mol/l). Poznámky:
|
5.1.2
5.1.2.1
Pro každý stanovovaný prvek se připraví z pracovních standardních roztoků (3.7.1, 3.8.1, 3.9.1 a 3.10.1) řada kalibračních roztoků. Kalibrační roztoky mají koncentraci HCl asi 0,5 mol/l a (v případě železa, manganu a zinku) koncentraci chloridu lanthanitého asi 0,1 % La (w/v).
Koncentrace vybraných stopových prvků musí ležet v rozsahu citlivosti použitého spektrofotometru. Níže uvedené tabulky ukazují příklad složení typických řad kalibračních roztoků; v závislosti na typu a citlivosti použitého spektrofotometru však může být nutno zvolit jiné koncentrace.
Železo
|
μg Fe/ml |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
pracovní standardní roztok (3.7.1) (1 ml = 100 μg Fe) v ml |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
HCl (3.2) v ml |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml roztoku chloridu lanthanitého (3.11) a doplní se vodou po značku. |
|||||||
Měď
|
μg Cu/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
pracovní standardní roztok (3.8.1) (1 ml = 10 μg Cu) v ml |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
HCl (3.2) v ml |
8 |
8 |
8 |
8 |
8 |
8 |
8 |
Mangan
|
μg Mn/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
pracovní standardní roztok (3.9.1) (1 ml = 10 μg Mn) v ml |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
HCl (3.2) v ml |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml roztoku chloridu lanthanitého (3.11) a doplní se vodou po značku. |
|||||||
Zinek
|
μg Zn/ml |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
|
pracovní standardní roztok (3.10.1) (1 ml = 10 μg Zn) v ml |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
|
HCl (3.2) v ml |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml roztoku chloridu lanthanitého (3.11) a doplní se vodou po značku. |
|||||||
5.1.2.2
Při stanovení mědi lze použít přímo roztok připravený podle bodu 5.1.1. Pokud je nutné jeho koncentraci naředit na úroveň kalibračních roztoků, odpipetuje se alikvotní část roztoku do 100 ml odměrné baňky a doplní se po značku kyselinou chlorovodíkovou 0,5 mol/l (3.3).
Při stanovení železa, manganu a zinku se odpipetujte alikvotní část roztoku připraveného podle bodu 5.1.1 do 100 ml odměrné baňky, přidá se 10 ml roztoku chloridu lanthanitého (3.11) a doplní se po značku kyselinou chlorovodíkovou 0,5 mol/l (3.3) (viz rovněž bod 8 Poznámka).
5.1.2.3
Slepá zkouška musí zahrnovat všechny předepsané kroky postupu, ale vlastní vzorek se nepoužije. „0“ kalibrační roztok nemůže být použit jako slepá zkouška.
5.1.2.4
Atomová absorpce kalibračních roztoků a zkoušeného roztoku se změří s použitím oxidačního plamene vzduch-acetylen při následujících vlnových délkách:
|
|
Fe: 248,3 nm |
|
|
Cu: 324,8 nm |
|
|
Mn: 279,5 nm |
|
|
Zn: 213,8 nm |
Každé měření se provede čtyřikrát.
5.2 Minerální krmiva
Pokud vzorek neobsahuje organickou hmotu, nemusí se spalovat. Příprava se provádí podle druhého odstavce bodu 5.1.1.1. Nemusí se provádět ani odpařování s kyselinou fluorovodíkovou.
6. Výpočet a vyjádření výsledků
Koncentrace příslušného stopového prvku v roztoku se vypočte z kalibrační křivky a výsledek se vyjádří v miligramech stopového prvku na kilogram vzorku (ppm).
7. Opakovatelnost
Rozdíl mezi výsledky dvou paralelních stanovení provedených na stejném vzorku týmž laborantem nesmí překročit:
|
— |
5 mg/kg v absolutní hodnotě u obsahů příslušného stopového prvku do 50 mg/kg, |
|
— |
10 % z vyššího výsledku u obsahů příslušného stopového prvku od 50 do 100 mg/kg, |
|
— |
10 mg/kg v absolutní hodnotě u obsahů příslušného stopového prvku od 100 do 200 mg/kg, |
|
— |
5 % z vyššího výsledku u obsahů příslušného stopového prvku nad 200 mg/kg. |
8. Poznámka
Přítomnost velkého množství fosfátů může ovlivňovat stanovení železa, manganu a zinku. Proto musí být přidáván roztoku chloridu lanthanitého (3.11). Pokud je váhový poměr (Ca + Mg)/P ve vzorku > 2, nemusí se do roztoku pro zkoušku ani do kalibračních roztoků přidávat roztok chloridu lanthanitého (3.11).
D. STANOVENÍ OBSAHU HALOFUGINONU
DL-trans-7-brom-6-chlor-3-[3-(3-hydroxy-2-piperidyl)acetonyl]-quinazolin-4-(3H)-1-hydrobromid
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu halofuginonu v krmivech. Mez kvantifikace je 1 mg/kg.
2. Princip
Obsah halofuginonu se stanoví jako volná báze po extrakci horkou vodou do ethylacetátu a následně se rozdělí jako hydrochlorid do vodného kyselého roztoku. Extrakt se přečistí na ionexové chromatografické koloně. Obsah halofuginonu se stanoví metodou vysokoúčinné kapalinové chromatografie (HPLC) na reverzní fázi s UV detekcí.
3. Chemikálie
|
3.1 |
Acetonitril, pro HPLC. |
|
3.2 |
Amberlit XAD-2 ionexová pryskyřice. |
|
3.3 |
Octan amonný. |
|
3.4 |
Ethylacetát. |
|
3.5 |
Kyselina octová, ledová. |
|
3.6 |
Halofuginon, standardní látka (DL-trans-7-brom-6-chlor-3-[3-(3-hydroxy-2-piperidyl)acetonyl]-quinazolin-4-(3H)-1-hydrobromid, E 764). |
|
3.6.1 |
Do 500 ml odměrné baňky se naváží 50 mg halofuginonu (3.6) s přesností na 0,1 mg, rozpustí se v tlumivém roztoku octanu amonného (3.18), doplní po značku tlumivým roztokem a promíchá. Tento roztok je stálý tři týdny při teplotě 5 oC, je-li skladován ve tmě. |
|
3.6.2 |
Do řady 100 ml odměrných baněk se odpipetuje 1,0, 2,0, 3,0, 4,0 a 6,0 ml standardního zásobního roztoku (3.6.1). Doplní se po značku mobilní fází (3.21) a promíchá. Roztoky mají koncentrace halofuginonu 1,0, 2,0, 3,0, 4,0 a 6,0 μg/ml. Tyto roztoky se musí připravovat čerstvé před použitím. |
|
3.7 |
Kyselina chlorovodíková (ρ20 přibližně 1,16 g/ml). |
|
3.8 |
Methanol. |
|
3.9 |
Dusičnan stříbrný. |
|
3.10 |
Askorbát sodný. |
|
3.11 |
Uhličitan sodný. |
|
3.12 |
Chlorid sodný. |
|
3.13 |
EDTA (disodná sůl kyseliny ethylendiaminotetraoctové). |
|
3.14 |
Voda, pro HPLC. |
|
3.15 |
Uhličitan sodný, roztok, c = 10 g/100 ml. |
|
3.16 |
Uhličitan sodný, roztok, saturovaný chloridem sodným, c = 5 g/100 ml. 50 g uhličitanu sodného (3.11) se rozpustí ve vodě, doplní se na 1 000 ml a přidává se chlorid sodný (3.12), až k nasycení roztoku. |
|
3.17 |
Kyselina chlorovodíková, přibližně 0,1 mol/l. 10 ml HCl (3.7) se zředí vodou na 1 000 ml. |
|
3.18 |
Tlumivý roztok octanu amonného, přibližně 0,25 mol/l. 19,3 g octanu amonného (3.3) a 30 ml kyseliny octové (3.5) se rozpustí ve vodě (3.14) a zředí se na 1 000 ml. |
|
3.19 |
Ionexová pryskyřice Amberlit XAD-2 – příprava. Přiměřené množství amberlitu (3.2) se promývá ve vodě, až se odstraní všechny chloridové ionty, což se prokáže zkouškou pomocí dusičnanu stříbrného (3.20) na odstraňované vodní fázi. Pak se pryskyřice promyje 50 ml methanolu (3.8), který se odstraní, a pryskyřice se uloží pod čerstvý methanol. |
|
3.20 |
Dusičnan stříbrný, roztok přibližně 0,1 mol/l. 0,17 g dusičnanu stříbrného (3.9) se rozpustí v 10 ml vody. |
|
3.21 |
Mobilní fáze HPLC. Smíchá se 500 ml acetonitrilu (3.1), 300 ml tlumivého roztoku octanu amonného (3.18) a 1 200 ml vody (3.14). pH se upraví na 4,3 za použití kyseliny octové (3.5). Přefiltruje se filtrem 0,22 μm (4.8) a roztok se odplyní (např. tím, že se vystaví ultrazvuku po dobu 10 minut). Tento roztok je stálý jeden měsíc, je-li skladován ve tmě a v uzavřené nádobě. |
4. Přístroje a pomůcky
|
4.1 |
Ultrazvuková lázeň. |
|
4.2 |
Rotační odparka. |
|
4.3 |
Odstředivka. |
|
4.4 |
Zařízení HPLC s ultrafialovým detektorem s nastavitelnou vlnovou délkou nebo s detektorem diodového pole. |
|
4.4.1 |
Kolona pro kapalinovou chromatografii, 300 mm × 4 mm, C18, náplň 10 μm, nebo obdobná kolona. |
|
4.5 |
Skleněné kolony (300 mm × 10 mm) opatřené filtrem z křemičitého skla a uzavíracím kohoutem. |
|
4.6 |
Filtry ze skleněných vláken, průměr 150 mm. |
|
4.7 |
Membránové filtry, 0,45 μm. |
|
4.8 |
Membránové filtry, 0,22 μm. |
5. Postup
|
Poznámka |
: |
Halofuginon jako volná báze je nestálý v alkalických roztocích a v roztocích ethylacetátu. Nesmí zůstat v roztoku ethylacetátu déle než 30 minut. |
5.1 Obecné pokyny
|
5.1.1 |
Slepý vzorek krmiva se předem podrobí zkoušce pro kontrolu, zda neobsahuje halofuginon nebo jiné rušivé látky. |
|
5.1.2 |
Provede se zkouška na výtěžnost u slepého vzorku krmiva, které bylo obohaceno přidáním takového množství halofuginonu, které odpovídá jeho obsahu ve vzorku. Pro obohacení na úroveň 3 mg/kg se přidá 300 μl standardního zásobního roztoku (3.6.1) k 10 g slepého vzorku krmiva, promíchá se a vyčká 10 minut, potom se pokračuje v extrakčním postupu (5.2).
|
5.2 Extrakce
Do odstředivkové zkumavky o obsahu 200 ml se naváží 10 g upraveného vzorku s přesností na 0,1 g, přidá se 0,5 g askorbátu sodného (3.10), 0,5 g EDTA (3.13) a 20 ml vody a promíchá se. Zkumavka se umístí na 5 minut do vodní lázně (80 oC). Po vychladnutí na laboratorní teplotu se přidá 20 ml roztoku uhličitanu sodného (3.15) a promíchá se. Ihned se přidá 100 ml ethylacetátu (3.4) a ručně se 15 sekund intenzivně třepe. Pak se zkumavka s uvolněnou zátkou umístí na tři minuty do ultrazvukové lázně (4.1). Odstředí se dvě minuty a ethylacetátová fáze se odlije přes filtr ze skleněných vláken (4.6) do 500 ml separační nálevky. Extrakce vzorku se opakuje s dalšími 100 ml ethylacetátu. Kombinované extrakty se promývají jednu minutu 50 ml roztoku uhličitanu sodného saturovaného chloridem sodným (3.16) a poté se vodná vrstva odstraní.
Organická vrstva se extrahuje po dobu jedné minuty 50 ml kyseliny chlorovodíkové (3.17). Spodní kyselinová vrstva se vypustí do 250 ml separační nálevky. Organická vrstva se znovu extrahuje 1,5 minuty s dalšími 50 ml kyseliny chlorovodíkové a kyselinová vrstva se spojí s prvním extraktem. Spojené kyselinové extrakty se třepou přibližně 10 sekund s 10 ml ethylacetátu (3.4).
Vodná vrstva se kvantitativně převede do 250 ml baňky s kulatým dnem a organická fáze se odstraní. Pomocí rotační odparky (4.2) se z kyselého roztoku odpaří všechen zbývající ethylacetát. Teplota vodní lázně nesmí přesáhnout 40 oC. Ve vakuu přibližně 25 mbar se všechen zbývající ethylacetát odstraní během 5 minut při teplotě 38 oC.
5.3 Přečištění
5.3.1
Pro každý extrakt vzorku se připraví kolona s XAD-2. Do skleněné kolony (4.5) se přenese 10 g připraveného amberlitu (3.19) spolu s methanolem (3.8). Na horní konec pryskyřicové kolony se vloží malá zátka ze skelné vaty. Methanol z kolony se odstraní a kolona se promyje 100 ml vody. Přívod vody se zastaví, jakmile tekutina dosáhne horního konce pryskyřicové kolony. Před použitím se kolona nechá 10 minut ekvilibrovat. Kolona nesmí nikdy vyschnout.
5.3.2
Extrakt (5.2) se kvantitativně převede na horní část připravené amberlitové kolony (5.3.1) a eluuje se, přičemž se eluát odstraňuje. Rychlost promývání nesmí přesáhnout 20 ml/min. Baňka s kulatým dnem se vypláchne 20 ml kyseliny chlorovodíkové (3.17) a tato tekutina se použije k promytí pryskyřicové kolony. Zbytek kyselého roztoku se z kolony odstraní proudem vzduchu. Promývací roztok se také odstraní. Na kolonu se přidá 100 ml methanolu (3.8) a vypustí se asi 5–10 ml eluátu do 250 ml baňky s kulatým dnem. Zbývající methanol se nechá 10 minut ustát na koloně a potom se pokračuje v promývání rychlostí, která nepřesáhne 20 ml/min. Eluát se shromažďuje v téže baňce s kulatým dnem. Methanol se odpaří na rotační odparce (4.2), teplota vodní lázně nesmí přesáhnout 40 oC. Odparek se pomocí mobilní fáze (3.21) kvantitativně převede do 10 ml odměrné baňky. Doplní se mobilní fází po značku a promíchá. Alikvotní část se filtruje membránovým filtrem (4.7). Tento roztok se uschová pro stanovení HPLC (5.4).
5.4 Stanovení HPLC
5.4.1
Následující podmínky jsou doporučené, lze použít i jiné podmínky, pokud zajišťují srovnatelné výsledky:
Kolona pro kapalinovou chromatografii (4.4.1).
HPLC mobilní fáze (3.21).
Průtok: 1,5–2 ml/min.
Detekční vlnová délka: 243 nm.
Objem nástřiku: 40–100 μl.
Stálost chromatografického systému se kontroluje opakovaným nástřikem kalibračního roztoku (3.6.2), který obsahuje 3,0 μg/ml halofuginonu. Opakuje se několikrát, až se dosáhne konstantní výšky (plochy) píků a konstantních retenčních časů.
5.4.2
Provede se několikrát nástřik každého kalibračního roztoku (3.6.2) a změří se výšky (plochy) píků pro každou koncentraci. Kalibrační křivka se vytvoří vynesením průměrných hodnot výšek nebo ploch píků kalibračních roztoků proti příslušným koncentracím v μg/ml.
5.4.3
Provede se několikrát nástřik roztoku vzorku (5.3.2), přičemž se použije stejný objem jako pro kalibrační roztoky a zjistí se průměrná hodnota výšky (plochy) halofuginonových píků.
6. Výpočet a vyjádření výsledků
Koncentrace halofunginonu v roztoku vzorku v μg/ml se stanoví podle průměrné hodnoty výšky (plochy) halofuginonových píků v roztoku vzorku porovnáním s kalibrační křivkou (5.4.2).
Obsah halofuginonu (w) v mg/kg ve vzorku se vypočítá podle následujícího vzorce:
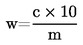
kde:
|
c |
: |
koncentrace halofuginonu v roztoku vzorku v μg/ml |
|
m |
: |
hmotnost navážky vzorku v gramech. |
7. Ověření výsledků
7.1 Identita
Identita analytu může být potvrzena opakovaným stanovením s přídavkem (co-chromatography) nebo využitím detektoru diodového pole, přičemž se porovnávají spektra extraktu vzorku a kalibračního roztoku (3.6.2) obsahujícího 6,0 μg/ml halofuginonu.
7.1.1
K extraktu vzorku se přidá vhodné množství kalibračního roztoku (3.6.2). Množství přidaného halofuginonu musí odpovídat odhadovanému obsahu halofuginonu v extraktu vzorku.
Přídavek se projeví pouze zvýšením píku halofuginonu, a to úměrně k přidanému množství a ředění extraktu. Šířka píku v polovině maximální výšky se může odchylovat od šířky původního píku nejvýše o ±10 %.
7.1.2
Výsledky jsou posuzovány podle následujících kritérií:
|
a) |
při vlnové délce maximální absorpce vzorku i standardu musí být záznam spektra zaznamenaný ve vrcholu píku na chromatogramu stejný s nepřesností danou rozlišovací schopností detekčního systému. Pro detektor diodového pole se udává ±2 nm; |
|
b) |
při vlnové délce mezi 225 a 300 nm se nesmí záznam spektra ve zkoušeném vzorku a standardu zaznamenaný ve vrcholu píku chromatogramu lišit od ostatních částí spektra v rozsahu 10–100 % relativní absorbance. Tohoto kritéria je dosaženo, jestliže jsou vykázána stejná maxima a jestliže odchylka mezi dvěma spektry nikde nepřesahuje 15 % absorbance standardního analytu; |
|
c) |
při vlnové délce mezi 225 a 300 nm se nesmí spektrum vzestupné části, vrcholu a sestupné části píku zkoušeného vzorku lišit od ostatních částí spektra v rozsahu 10 až 100 % relativní absorbance. Tohoto kritéria je dosaženo, jestliže jsou vykázána stejná maxima a jestliže ve všech sledovaných bodech odchylka mezi spektry nepřesahuje 15 % absorbance spektra ve vrcholu píku. |
Jestliže některé z těchto kritérií není splněno, přítomnost analytu není potvrzena.
7.2 Opakovatelnost
Rozdíl mezi výsledky dvou paralelních stanovení provedených na stejném vzorku nesmí překročit 0,5 mg/kg při obsahu halofuginonu do 3 mg/kg.
7.3 Výtěžnost
Výtěžnost pro obohacený slepý vzorek krmiva musí být nejméně 80 %.
8. Výsledky kruhových testů
Při kruhových testech (5) byly osmi laboratořemi zkoušeny tři vzorky.
Výsledky
|
|
Vzorek A (slepý) při převzetí |
Vzorek B (mouka) |
Vzorek C (pelety) |
||
|
|
|
při převzetí |
po dvou měsících |
při převzetí |
po dvou měsících |
|
průměr (mg/kg) |
ND |
2,80 |
2,42 |
2,89 |
2,45 |
|
SR (mg/kg) |
— |
0,45 |
0,43 |
0,40 |
0,42 |
|
CVR (%) |
— |
16 |
18 |
14 |
17 |
|
Rec. (%) |
|
86 |
74 |
88 |
75 |
|
ND |
= |
nezjištěno |
|
SR |
= |
standardní odchylka reprodukovatelnosti |
|
CVR |
= |
variační koeficient reprodukovatelnosti (%) |
|
Rec. |
= |
výtěžnost (%). |
E. STANOVENÍ OBSAHU ROBENIDINU
1,3-bis[(4-chlorobenzyliden) amino]guanidin hydrochlorid
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu robenidinu v krmivech. Mez kvantifikace je 5 mg/kg.
2. Princip
Vzorek se extrahuje okyseleným methanolem. Extrakt se vysuší a jeho alikvotní část se přečistí na koloně s oxidem hlinitým. Robenidin se eluuje z kolony methanolem, koncentruje se a na vhodný objem se upraví pomocí mobilní fáze. Obsah robenidinu se stanoví vysokoúčinnou kapalinovou chromatografií (HPLC) na reverzní fázi pomocí UV detektoru.
3. Chemikálie
|
3.1 |
Methanol. |
|
3.2 |
Okyselený methanol. Do 500 ml odměrné baňky se odměří 4,0 ml kyseliny chlorovodíkové (ρ20 = 1,18 g/ml), doplní se po značku methanolem (3.1) a promíchá. Tento roztok se připravuje čerstvý před použitím. |
|
3.3 |
Acetonitril, pro HPLC. |
|
3.4 |
Molekulární síto. Typ 3A, 8–12 mesh (1,6–2,5 mm perličky krystalického hlinitokřemičitanu, průměr pórů 0,3 mm). |
|
3.5 |
Oxid hlinitý, kyselý, stupeň aktivity I pro kolonovou chromatografii. 100 g oxidu hlinitého se naváží do vhodné nádoby a přidají se 2,0 ml vody. Zazátkuje se a protřepává přibližně 20 minut. Skladuje se v dobře zazátkované nádobě. |
|
3.6 |
Dihydrogenfosforečnan draselný, roztok, c = 0,025 mol/l. 3,4 g dihydrogenfosforečnanu draselného se rozpustí ve vodě (pro HPLC) v 1 000 ml odměrné baňce, doplní se po značku a promíchá. |
|
3.7 |
Hydrogenfosforečnan sodný, roztok, c = 0,025 mol/l. 3,55 g bezvodého (nebo 4,45 g dihydrátu, nebo 8,95 g dodekahydrátu) hydrogenfosforečnanu sodného se rozpustí ve vodě (pro HPLC) v 1 000 ml odměrné baňce, doplní se po značku a promíchá. |
|
3.8 |
Mobilní fáze HPLC Smíchá se:
Přefiltruje se filtrem 0,22 μm (4.6) a roztok se odplyní (např. ultrazvukem po dobu 10 minut). |
|
3.9 |
Standardní látka. Čistý robenidin: 1,3-bis[(4-chlorobenzyliden) amino]guanidin hydrochlorid. |
|
3.9.1 |
Naváží se 30 mg standardní látky robenidinu (3.9) s přesností na 0,1 mg. Rozpustí se v okyseleném methanolu (3.2) ve 100 ml odměrné baňce, doplní se po značku týmž rozpouštědlem a promíchá se. Baňka se obalí aluminiovou fólií a uloží ve tmě. |
|
3.9.2 |
Odměří se 10,0 ml základního standardního roztoku (3.9.1) do 250 ml odměrné baňky, doplní se po značku mobilní fází (3.8) a promíchá. Baňka se obalí aluminiovou fólií a uloží ve tmě. |
|
3.9.3 |
Do sady 50 ml odměrných baněk se odměří 5,0, 10,0, 15,0, 20,0 a 25,0 ml pracovního standardního roztoku (3.9.2). Doplní se po značku mobilní fází (3.8) a promíchá. Tyto roztoky odpovídají 1,2, 2,4, 3,6, 4,8 a 6,0 μg/ml robenidinu. Tyto roztoky se připravují čerstvé před použitím. |
|
3.10 |
Voda, pro HPLC. |
4. Přístroje a pomůcky
|
4.1 |
Skleněná kolona. Vyrobená z tmavého skla, vybavená uzavíracím kohoutem a nádržkou o obsahu přibližně 150 ml. Vnitřní průměr 10–15 mm, délka 250 mm. |
|
4.2 |
Mechanická třepačka nebo magnetické míchadlo. |
|
4.3 |
Rotační odparka. |
|
4.4 |
Zařízení na vysokoúčinnou kapalinovou chromatografii (HPLC) vybavené ultrafialovým detektorem s nastavitelnou vlnovou délkou nebo detektorem diodového pole, pracujícím v pásmu 250–400 nm. |
|
4.4.1 |
Kolona pro kapalinovou chromatografii: 300 mm × 4 mm, C18, náplň 10 μm nebo obdobná. |
|
4.5 |
Filtr ze skleněných vláken (Whatman GF/A nebo podobný). |
|
4.6 |
Membránové filtry, 0,22 μm. |
|
4.7 |
Membránové filtry, 0,45 μm. |
5. Postup
|
Poznámka |
: |
Robenidin je citlivý na světlo. Proto se při všech operacích použije vždy tmavé sklo. |
5.1 Obecné pokyny
|
5.1.1 |
Provede se zkouška slepého vzorku krmiva pro kontrolu, zda neobsahuje robenidin ani jiné rušivé látky. |
|
5.1.2 |
Provede se zkouška na výtěžnost u slepého vzorku krmiva (5.1.1), do kterého bylo přidáno stejné množství robenidinu, jako je ve vzorku. K dosažení obsahu 60 mg/kg se do 250 ml kónické baňky přidají 3,0 ml základního standardního roztoku (3.9.1). Roztok se odpaří asi na objem 0,5 ml v proudu dusíku. Přidá se 15 g slepého vzorku krmiva, promíchá se a po deseti minutách se může začít s extrakcí (5.2).
|
5.2 Extrakce
Do 250 ml kónické baňky se naváží přibližně 15 g upraveného vzorku s přesností na 0,01 g, přidá se 100,0 ml okyseleného methanolu (3.2), zazátkuje a hodinu třepe na třepačce (4.2). Roztok se přefiltruje přes filtr ze skleněných vláken (4.5) a celý objem filtrátu se jímá do 150 ml kónické baňky. Přidá se 7,5 g molekulárního síta (3.4), zazátkuje a pět minut se třepe. Potom se ihned přefiltruje přes filtr ze skleněných vláken. Tento roztok se použije k čištění (5.3).
5.3 Čištění
5.3.1
Do spodního konce skleněné kolony (4.1) se vloží malá zátka ze skelné vaty a utěsní se skleněnou tyčinkou. Naváží se 11,0 g připraveného kysličníku hlinitého (3.5) a nasype se do kolony, pokud možno s minimální časovou prodlevou v okolním prostředí. Oxid hlinitý se v naplněné koloně setřese lehkým poklepem na dolní konec kolony.
5.3.2
Na kolonu se napipetuje 5,0 ml upraveného extraktu vzorku (5.2). Špička pipety se opře o stěnu kolony a roztok se nechá absorbovat oxidem hlinitým. Robenidin se z kolony eluuje pomocí 100 ml methanolu (3.1) při rychlosti průtoku 2–3 ml za minutu a eluát se jímá do 250 ml baňky s kulatým dnem. Methanolový roztok se za redukovaného tlaku a při teplotě 40 oC odpaří do sucha v rotační odparce (4.3). Odparek se znovu rozpustí ve 3–4 ml mobilní fáze (3.8) a kvantitativně se převede do 10 ml odměrné baňky. Baňka se několikrát vypláchne 1–2 ml mobilní fáze a tyto roztoky se přidají do odměrné baňky. Baňka se doplní po značku týmž rozpouštědlem a promíchá. Alikvotní část se přefiltruje přes 0,45 μm membránový filtr (4.7). Tento roztok se použije pro stanovení HPLC (5.4).
5.4 Stanovení HPLC
5.4.1
Následující podmínky jsou doporučené, mohou být použity jiné podmínky za předpokladu, že poskytují rovnocenné výsledky.
Kolona pro kapalinovou chromatografii (4.4.1)
Mobilní fáze HPLC (3.8)
Průtok: 1,5–2 ml/min
Vlnová délka detektoru: 317 nm
Objem nástřiku: 20–50 μl
Stabilita chromatografického systému se kontroluje opakovaným nástřikem kalibračního roztoku (3.9.3), který obsahuje 3,6 μg/ml, dokud se nedosáhne konstantních výšek píků a konstantních retenčních časů.
5.4.2
Pro každý kalibrační roztok (3.9.3) se provede několik nástřiků a změří se výšky (plochy) píků pro každou koncentraci. Kalibrační křivka se vytvoří vynesením průměrných hodnot výšek nebo ploch píků kalibračních roztoků proti příslušným koncentracím v μg/ml.
5.4.3
Opakovaně se nastříkne stejný objem extraktu vzorku (5.3.2) jako při kalibraci a určí se průměrná hodnota výšky (plochy) píků robenidinu.
6. Výpočet a vyjádření výsledků
Z průměrných hodnot výšky (plochy) robenidinových píků v roztoku vzorku se určí koncentrace robenidinu v roztoku vzorku v μg/ml porovnáním s kalibrační křivkou (5.4.2).
Obsah robenidinu (w) v mg/kg ve vzorku se vypočítá podle následujícího vzorce:
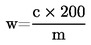
kde:
|
c |
= |
koncentrace robenidinu v roztoku vzorku v μg/ml |
|
m |
= |
hmotnost navážky vzorku v gramech. |
7. Ověření výsledků
7.1 Identita
Identitu analytů lze potvrdit opakovaným stanovením s přídavkem anebo využitím detektoru diodového pole, který umožňuje porovnání spektra extraktu vzorku a kalibračního roztoku (3.9.3) obsahujícího 6 μg/ml robenidinu.
7.1.1
K extraktu vzorku se přidá vhodné množství kalibračního roztoku (3.9.3). Množství přidaného robenidinu musí odpovídat předpokládanému obsahu robenidinu v extraktu vzorku.
Přídavek se projeví pouze zvýšením píku robenidinu, a to úměrně k přidanému množství a ředění. Šířka píku v polovině jeho maximální výšky se může odchylovat od šířky původního píku nejvýše o ±10 %.
7.1.2
Výsledky jsou posuzovány podle následujících kritérií:
|
a) |
při vlnové délce maximální absorpce vzorku i standardu musí být záznam spektra zaznamenaný ve vrcholu píku na chromatogramu stejný s nepřesností danou rozlišovací schopností detekčního systému. Pro detektor diodového pole se udává ±2 nm; |
|
b) |
při vlnové délce mezi 220 a 400 nm se nesmí záznam spektra ve zkoušeném vzorku a standardu zaznamenaný ve vrcholu píku chromatogramu lišit od ostatních částí spektra v rozsahu 10–100 % relativní absorbance. Tohoto kritéria je dosaženo, jestliže jsou vykázána stejná maxima a jestliže odchylka mezi dvěma spektry nikde nepřesahuje 15 % absorbance standardního analytu; |
|
c) |
při vlnové délce mezi 220 a 400 nm se nesmí spektrum vzestupné části, vrcholu a sestupné části píku zkoušeného vzorku lišit od ostatních částí spektra v rozsahu 10–100 % relativní absorbance. Tohoto kritéria je dosaženo, jestliže jsou vykázána stejná maxima a jestliže ve všech sledovaných bodech odchylka mezi spektry nepřesahuje 15 % absorbance spektra ve vrcholu píku. |
Jestliže některé z těchto kritérií není splněno, přítomnost analytu není potvrzena.
7.2 Opakovatelnost
Rozdíl mezi výsledky dvou paralelních stanovení provedených na stejném vzorku nesmí přesáhnout 10 % z hodnoty nejvyššího výsledku při obsahu robenidinu nad 15 mg/kg.
7.3 Výtěžnost
Výtěžnost pro obohacený slepý vzorek krmiva musí být nejméně 85 %.
8. Výsledky kruhových testů
Byl proveden kruhový test ES, ve kterém dvanáct laboratoří analyzovalo čtyři vzorky krmiv pro drůbež a králíky, sypkých nebo peletovaných. U každého vzorku byly provedeny dvě zkoušky. Výsledky jsou uvedeny v následující tabulce:
|
|
Drůbež |
Králíci |
||
|
|
Sypké krmivo |
Pelety |
Sypké krmivo |
Pelety |
|
průměr (mg/kg) |
27,00 |
27,99 |
43,6 |
40,1 |
|
sr (mg/kg) |
1,46 |
1,26 |
1,44 |
1,66 |
|
CVr (%) |
5,4 |
4,5 |
3,3 |
4,1 |
|
SR (mg/kg) |
4,36 |
3,36 |
4,61 |
3,91 |
|
CVR (%) |
16,1 |
12,0 |
10,6 |
9,7 |
|
Výtěžnost (%) |
90,0 |
93,3 |
87,2 |
80,2 |
|
sr |
= |
standardní odchylka opakovatelnosti |
|
CVr |
= |
variační koeficient opakovatelnosti v % |
|
SR |
= |
standardní odchylka reprodukovatelnosti |
|
CVR |
= |
variační koeficient reprodukovatelnosti v %. |
F. STANOVENÍ DICLAZURILU
2,6-dichlor-alfa-(4-chlorofenyl)-4-(4,5-dihydro-3,5-dioxo-1,2,4-triazin-2-(3-H)yl)benzenacetonitril
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu diclazurilu v krmivech a premixech. Mez detekce je 0,1 mg/kg, mez kvantifikace 0,5 mg/kg.
2. Princip
Po přidání interního standardu se vzorek extrahuje okyseleným methanolem. U krmiv se alikvotní část extraktu přečistí na SPE patroně C18. Diclazuril se z patrony eluuje směsí okyseleného methanolu a vody. Po odpaření se zbytek rozpustí v roztoku DMF/voda. U premixů se extrakt odpaří a zbytek se rozpustí v roztoku DMF/voda. Obsah diclazurilu se stanoví vysokoúčinnou kapalinovou chromatografií (HPLC) na reverzní fázi s ternárním gradientem s UV detekcí.
3. Chemikálie
|
3.1 |
Voda, pro HPLC. |
|
3.2 |
Octan amonný. |
|
3.3 |
Tetrabutylamonium hydrogensíran (TBHS). |
|
3.4 |
Acetonitril, pro HPLC. |
|
3.5 |
Methanol, pro HPLC. |
|
3.6 |
N,N-dimethylformamid (DMF). |
|
3.7 |
Kyselina chlorovodíková, ρ20 = 1,19 g/ml. |
|
3.8 |
Standardní látka: diclazuril II-24: 2,6-dichlor-alfa-(4-chlorofenyl)-4-(4,5-dihydro-3,5-dioxo-1,2,4-triazin-2-(3-H)yl)benzenacetonitril, deklarované čistoty, E 771. |
|
3.8.1 |
Do 50 ml odměrné baňky se naváží 25 mg diclazurilu (3.8) s přesností na 0,1 mg, rozpustí se v DMF (3.6), doplní se DMF (3.6) po značku a promíchá. Odměrná baňka se obalí hliníkovou fólií nebo se použije odměrná baňka z tmavého skla a uchovává se v chladničce. Roztok je při teplotě ≤ 4 oC stálý jeden měsíc. |
|
3.8.2 |
Do 50 ml odměrné baňky se převede 5,00 ml základního standardního roztoku (3.8.1), doplní se DMF (3.6) po značku a promíchá. Odměrná baňka se obalí hliníkovou fólií nebo se použije odměrná baňka z tmavého skla a uchovává se v chladničce. Roztok je při teplotě ≤ 4 oC stálý jeden měsíc. |
|
3.9 |
Interní standardní substance: 2,6-dichloro-α-(4-chlorofenyl)-4-(4,5-dihydro-3,5-dioxo-1,2,4-triazin-2-(3-H)-yl)-α-metylbenzen-acetonitril. |
|
3.9.1 |
Do 50 ml odměrné baňky se naváží 25 mg interní standardní substance (3.9) s přesností na 0,1 mg, rozpustí se v DMF (3.6), doplní se DMF (3.6) po značku a promíchá. Odměrná baňka se obalí hliníkovou fólií nebo se použije odměrná baňka z tmavého skla a uchovává se v chladničce. Roztok je při teplotě ≤ 4 oC stálý jeden měsíc. |
|
3.9.2 |
Do 50 ml odměrné baňky se převede 5,00 ml interního základního standardního roztoku (3.9.1), doplní se DMF (3.6) po značku a promíchá. Odměrná baňka se obalí hliníkovou fólií nebo se použije odměrná baňka z tmavého skla a uchovává se v chladničce. Roztok je při teplotě ≤ 4 oC stálý jeden měsíc. |
|
3.9.3 |
(p = deklarovaný obsah diclazurilu v premixu v mg/kg) Do 100 ml odměrné baňky se naváží p/10 mg interní standardní substance s přesností na 0,1 mg, rozpustí se v DMF (3.6) v ultrazvukové lázni (4.6), doplní DMF po značku a promíchá. Odměrná baňka se obalí hliníkovou fólií nebo se použije odměrná baňka z tmavého skla a uchovává se v chladničce. Roztok je při teplotě ≤ 4 oC stálý jeden měsíc. |
|
3.10 |
Kalibrační roztok, 2 μg/ml Do 50 ml odměrné baňky se odpipetuje 2,00 ml standardního roztoku diclazurilu (3.8.2) a 2,00 ml interního standardního roztoku (3.9.2). Přidá se 16 ml DMF (3.6), doplní se vodou po značku a promíchá. Tento roztok se připravuje čerstvý před použitím. |
|
3.11 |
SPE patrona C18, např. Bond Elut, velikost: 1 ml, sorpční hmotnost: 100 mg. |
|
3.12 |
Extrakční směs: okyselený methanol. Do 1 000 ml methanolu (3.5) se napipetuje 5,0 ml kyseliny chlorovodíkové (3.7) a promíchá. |
|
3.13 |
Mobilní fáze pro HPLC. |
|
3.13.1 |
Složka (eluent) A: roztok octanu amonného a tetrabutylamonium hydrogensíranu. 5 g octanu amonného (3.2) a 3,4 g TBHS (3.3) se rozpustí v 1 000 ml vody a promíchá se. |
|
3.13.2 |
Složka (eluent) B: acetonitril (3.4). |
|
3.13.3 |
Složka (eluent) C: methanol (3.5). |
4. Přístroje a pomůcky
|
4.1 |
Mechanické míchací zařízení. |
|
4.2 |
Zařízení pro HPLC s ternárním gradientem. |
|
4.2.1 |
Kolona pro kapalinovou chromatografii, Hypersil ODS, náplň 3 μm, 100 mm × 4,6 mm nebo obdobná. |
|
4.2.2 |
UV detektor s nastavitelnou vlnovou délkou nebo detektor diodového pole. |
|
4.3 |
Rotační odparka. |
|
4.4 |
Membránový filtr, 0,45 μm. |
|
4.5 |
Vakuové zařízení. |
|
4.6 |
Ultrazvuková lázeň. |
5. Postup
5.1 Obecné pokyny
5.1.1
Za účelem kontroly toho, že slepý vzorek krmiva neobsahuje ani diclazuril, ani jiné rušivé látky, se provede slepá zkouška. Slepý vzorek krmiva má podobné složení jako zkoušený vzorek a není zjištěn diclazuril ani jiné rušivé látky.
5.1.2
Zkouška na výtěžnost se provede na slepém vzorku krmiva, ke kterému bylo přidáno takové množství diclazurilu, které odpovídá množství diclazurilu ve zkoušeném vzorku. Pro obohacení na obsah 1 mg/kg se k 50 g slepého vzorku krmiva přidá 0,1 ml základního standardního roztoku (3.8.1). Důkladně se promíchá, nechá se 10 minut stát a před zahájením extrakce (5.2) se znovu několikrát promíchá.
Není-li k dispozici slepý vzorek krmiva podobného složení jako zkoušený vzorek (viz 5.1.1), lze provést zkoušku na výtěžnost metodou standardního přídavku. V tom případě se ke zkoušenému vzorku přidá přibližně stejné množství diclazurilu, jaké je ve vzorku již obsaženo. Obohacený vzorek se potom zkouší společně se vzorkem neobohaceným a výtěžnost se vypočte odečtením.
5.2 Extrakce
5.2.1
Do 500 ml kónické baňky se naváží 50 g vzorku s přesností na 0,01 g. Přidá se 1,00 ml interního standardního roztoku (3.9.2) a 200 ml extrakční směsi (3.12) a baňka se uzavře. Směs se přes noc třepe na míchacím zařízení (4.1). Poté se nechá 10 minut usadit. 20 ml supernatantu se převede do vhodné skleněné nádoby a zředí se 20 ml vody. Tento roztok se nanese na extrakční patronu (3.11) a prosaje se pomocí vakua (4.5). Patrona se promyje 25 ml směsi složené z extrakční směsi (3.12) a vody, 65 + 35 (V + V). Spojené frakce se odstraní, účinná látka a interní standard se eluují 25 ml směsi složené z extrakční směsi (3.12) a vody, 80 + 20 (V + V). Tato frakce se v rotační odparce (4.3) odpaří při 60 oC do sucha. Odparek se rozpustí v 1,0 ml DMF (3.6), přidá se 1,5 ml vody (3.1) a promíchá. Roztok se přefiltruje přes membránový filtr (4.4). Dále se pokračuje se stanovením HPLC (5.3).
5.2.2
Do 500 ml kónické baňky se naváží 1 g vzorku s přesností na 0,001 g. Přidá se 1,00 ml interního standardního roztoku (3.9.3) a 200 ml extrakční směsi (3.12) a baňka se uzavře. Směs se přes noc třepe na třepačce (4.1). Poté se nechá 10 minut usadit. 10 000/p ml (p = deklarovaný obsah diclazurilu v premixu v mg/kg) supernatantu se převede do baňky s kulatým dnem vhodné velikosti. V rotační odparce (4.3) se odpaří za sníženého tlaku při 60 oC do sucha. Odparek se rozpustí v 10,0 ml DMF (3.6), přidá se 15,0 ml vody (3.1) a promíchá. Dále se pokračuje se stanovením HPLC (5.3).
5.3 Stanovení HPLC
5.3.1
Níže uvedené podmínky jsou doporučené, lze použít jiné podmínky, pokud poskytují rovnocenné výsledky.
|
Kolona pro kapalinovou chromatografii (4.2.1): |
100 mm × 4,6 mm, Hypersil ODS, náplň 3 μm nebo obdobná |
|
||||||
|
Mobilní fáze: |
Složka A (3.13.1): |
vodný roztok octanu amonného a tetrabutylamonium hydrogensíranu |
||||||
|
|
Složka B (3.13.2): |
acetonitril |
||||||
|
|
Složka C (3.13.3): |
methanol |
||||||
|
Druh eluce: |
10 minut proplachovat složkou B |
|||||||
|
Průtok: |
1,5–2 ml/min |
|
||||||
|
Objem nástřiku: |
20 μl |
|
||||||
|
Vlnová délka detektoru: |
280 nm |
|
||||||
Stabilita chromatografického systému se kontroluje opakovaným nástřikem kalibračního roztoku (3.10), který obsahuje 2 μg/ml, dokud se nedosáhne konstantních výšek píků a konstantních retenčních časů.
5.3.2
Několikrát se nastřikuje 20 μl kalibračního roztoku (3.10) a stanoví se průměrná hodnota výšky (plochy) píku diclazurilu a interního standardu.
5.3.3
Několikrát se nastřikuje 20 μl roztoku vzorku (5.2.1 nebo 5.2.2) a stanoví se průměrná hodnota výšky (plochy) píku diclazurilu a interního standardu.
6. Výpočet výsledků
6.1 Krmiva
Obsah diclazurilu (w) v mg/kg ve vzorku se vypočte podle následujícího vzorce:
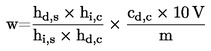 (mg/kg)
(mg/kg)
kde:
|
hd,s |
= |
výška (plocha) píku diclazurilu v roztoku vzorku (5.2.1) |
|
hi,s |
= |
výška (plocha) píku interního standardu v roztoku vzorku (5.2.1) |
|
hd,c |
= |
výška (plocha) píku diclazurilu v kalibračním roztoku (3.10) |
|
hi,c |
= |
výška (plocha) píku interního standardu v kalibračním roztoku (3.10) |
|
cd,c |
= |
koncentrace diclazurilu v kalibračním roztoku v μg/ml (3.10) |
|
m |
= |
hmotnost navážky vzorku v g |
|
V |
= |
objem extrakční směsi podle 5.2.1 (tj. 2,5 ml). |
6.2 Premixy
Obsah diclazurilu (w) v mg/kg ve vzorku se vypočte podle následujícího vzorce:
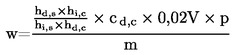 (mg/kg)
(mg/kg)
kde:
|
hd,c |
= |
výška (plocha) píku diclazurilu v kalibračním roztoku (3.10) |
|
hi,c |
= |
výška (plocha) píku interního standardu v kalibračním roztoku (3.10) |
|
hd,s |
= |
výška (plocha) píku diclazurilu v roztoku vzorku (5.2.2) |
|
hi,s |
= |
výška (plocha) píku interního standardu v roztoku vzorku (5.2.2) |
|
cd,c |
= |
koncentrace diclazurilu v kalibračním roztoku v μg/ml (3.10) |
|
m |
= |
hmotnost navážky vzorku v g |
|
V |
= |
objem extrakční směsi podle 5.2.2 (tj. 25 ml) |
|
p |
= |
deklarovaný obsah diclazurilu v premixu v mg/kg. |
7. Ověření výsledků
7.1 Identita
Identitu analytů lze potvrdit opakovaným stanovením s přídavkem (co-chromatography) nebo využitím detektoru diodového pole, přičemž se srovnávají spektra extraktu vzorku (5.2.1 nebo 5.2.2) a kalibračního roztoku (3.10).
7.1.1
K extraktu vzorku (5.2.1 nebo 5.2.2) se přidá vhodné množství kalibračního roztoku (3.10). Přidané množství diclazurilu musí odpovídat obsahu diclazurilu v extraktu vzorku.
Přídavek se projeví pouze zvýšením píku diclazurilu a píku interního standardu, a to úměrně přidanému množství a ředění. Šířka píku v polovině maximální výšky se může odchylovat od šířky původního píku diclazurilu nebo píku interního standardu v neobohaceném extraktu vzorku nejvýše o ± 10 %.
7.1.2
Výsledky jsou posuzovány podle následujících kritérií:
|
a) |
při vlnové délce maximální absorpce vzorku i standardu musí být záznam spektra zaznamenaný ve vrcholu píku na chromatogramu stejný s přesností danou rozlišovací schopností detekčního systému. Pro detektor diodového pole se udává obvykle ±2 nm; |
|
b) |
při vlnové délce mezi 230 a 320 nm se nesmí záznam spektra zkoušeného vzorku a standardu zaznamenaný ve vrcholu píku chromatogramu lišit od ostatních částí spektra v rozsahu 10–100 % relativní absorbance. Tohoto kritéria je dosaženo, jestliže jsou vykázána stejná maxima a jestliže odchylka mezi dvěma spektry nikde nepřesahuje 15 % absorbance standardního analytu; |
|
c) |
při vlnové délce mezi 230 a 320 nm se nesmí spektrum vzestupné části, vrcholu a sestupné části píku zkoušeného vzorku lišit od ostatních částí spektra v rozsahu 10–100 % relativní absorbance. Tohoto kritéria je dosaženo, jestliže jsou vykázána stejná maxima a jestliže ve všech sledovaných bodech odchylka mezi spektry nepřesahuje nikde 15 % absorbance spektra ve vrcholu píku. |
Jestliže některé z těchto kritérií není splněno, přítomnost analytu není potvrzena.
7.2 Opakovatelnost
Rozdíl mezi výsledky dvou paralelních stanovení provedených u stejného vzorku nesmí překročit:
|
— |
30 % relat. z hodnoty vyššího výsledku u obsahu diclazurilu mezi 0,5 mg/kg a 2,5 mg/kg, |
|
— |
0,75 mg/kg u obsahu diclazurilu mezi 2,5 mg/kg a 5 mg/kg, |
|
— |
15 % relat. z hodnoty vyššího výsledku u obsahu diclazurilu nad 5 mg/kg. |
7.3 Výtěžnost
Pro obohacený (slepý) vzorek musí být výtěžnost nejméně 80 %.
8. Výsledky kruhových testů
Při mezilaboratorních zkouškách bylo zkoušeno 5 vzorků 11 laboratořemi. Tyto vzorky sestávaly ze dvou premixů, z nichž jeden byl smísen s organickou (O 100) a druhý s anorganickou matricí (A 100). Teoretický obsah diclazurilu je 100 mg/kg. Tři krmné směsi pro drůbež pocházely od 3 různých výrobců (NL) (L1/Z1/K1). Teoretický obsah diclazurilu je 1 mg/kg. Laboratoře byly pověřeny, aby každý ze vzorků podrobily jedné nebo dvěma zkouškám. (Přesnější údaje o těchto kruhových testech viz Journal of AOAC International, Volume 77, No6, 1994, s. 1 359–1 361). Výsledky jsou shrnuty v níže uvedené tabulce.
|
|
Vzorek 1 A 100 |
Vzorek 2 O 100 |
Vzorek 3 L1 |
Vzorek 4 Z1 |
Vzorek 5 K1 |
|
L |
11 |
11 |
11 |
11 |
6 |
|
n |
19 |
18 |
19 |
19 |
12 |
|
Průměr |
100,8 |
103,5 |
0,89 |
1,15 |
0,89 |
|
Sr (mg/kg) |
5,88 |
7,64 |
0,15 |
0,02 |
0,03 |
|
CVr (%) |
5,83 |
7,38 |
17,32 |
1,92 |
3,34 |
|
SR (mg/kg) |
7,59 |
7,64 |
0,17 |
0,11 |
0,12 |
|
CVR (%) |
7,53 |
7,38 |
18,61 |
9,67 |
13,65 |
|
Deklarovaný obsah (mg/kg) |
100 |
100 |
1 |
1 |
1 |
|
L |
= |
počet laboratoří |
|
n |
= |
počet jednotlivých hodnot |
|
Sr |
= |
standardní odchylka opakovatelnosti |
|
CVr |
= |
variační koeficient opakovatelnosti |
|
SR |
= |
standardní odchylka reprodukovatelnosti |
|
CVR |
= |
variační koeficient reprodukovatelnosti. |
9. Poznámky
Musí být předem prokázáno, že odezva diclazurilu je měřena v lineární oblasti koncentrace.
G. STANOVENÍ OBSAHU LASALOCIDU
Sodná sůl polyetherové karboxylové kyseliny produkované Streptomyces lasaliensis
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu lasalocidu v krmivech a premixech. Mez detekce je 5 mg/kg, mez kvantifikace 10 mg/kg.
2. Princip
Lasalocid se extrahuje ze vzorku okyseleným methanolem a stanoví se vysokoúčinnou kapalinovou chromatografií (HPLC) na reverzní fázi s použitím spektrofluorimetru.
3. Chemikálie
|
3.1 |
Dihydrogenfosforečnan draselný (KH2PO4). |
|
3.2 |
Kyselina fosforečná, w (w/w) = 85 %. |
|
3.3 |
Roztok kyseliny fosforečné, c = 20 %. 23,5 ml kyseliny fosforečné (3.2) se doplní vodou na 100 ml. |
|
3.4 |
6-methyl-2-heptylamin (1,5-dimethylhexylamin), w (w/w) = 99 %. |
|
3.5 |
Methanol, pro HPLC. |
|
3.6 |
Kyselina chlorovodíková, hustota = 1,19 g/ml. |
|
3.7 |
Fosfátový tlumivý roztok, c = 0,01 mol/l. V 500 ml vody (3.11) se rozpustí 1,36 g KH2PO4 (3.1), přidá se 3,5 ml kyseliny fosforečné (3.2) a 10,0 ml 6-methyl-2-heptylaminu (3.4). Hodnota pH se upraví roztokem kyseliny fosforečné (3.3) na 4,0 a doplní se vodou (3.11) na 1 000 ml. |
|
3.8 |
Okyselený methanol. Do 1 000 ml odměrné baňky se odměří 5,0 ml kyseliny chlorovodíkové (3.6), doplní se methanolem (3.5) po značku a promíchá. Tento roztok se připravuje čerstvý před použitím. |
|
3.9 |
HPLC mobilní fáze, roztok fosfátového tlumivého roztoku a methanolu 5 + 95 (V + V). Smíchá se 5 ml fosfátového tlumivého roztoku (3.7) a 95 ml methanolu (3.5). |
|
3.10 |
Standardní látka lasalocid, sodná sůl s deklarovanou čistotou, C34H53O8Na (sodná sůl polyetherové karboxylové kyseliny produkované Streptomyces lasaliensis), E 763. |
|
3.10.1 |
Do 100 ml odměrné baňky se naváží 50 mg lasalocidu (3.10) s přesností na 0,1 mg a rozpustí se v okyseleném methanolu (3.8), doplní se jím po značku a promíchá. Tento roztok se musí připravovat čerstvý před použitím. |
|
3.10.2 |
10,0 ml základního standardního roztoku (3.10.1) se odpipetuje do 100 ml odměrné baňky, doplní se po značku okyseleným methanolem (3.8) a promíchá. Tento roztok se musí připravovat čerstvý před použitím. |
|
3.10.3 |
Do řady 50 ml odměrných baněk se odpipetuje 1,0, 2,0, 4,0, 5,0 a 10,0 ml pracovního standardního roztoku (3.10.2). Doplní se po značku okyseleným methanolem (3.8) a promíchá. Tyto roztoky odpovídají 1,0, 2,0, 4,0, 5,0 a 10,0 μg lasalocidu na ml. Tyto roztoky se musí připravovat čerstvé před použitím. |
|
3.11 |
Voda, pro HPLC. |
4. Přístroje a pomůcky
|
4.1 |
Ultrazvuková lázeň (nebo vodní třepací lázeň) s kontrolou teploty. |
|
4.2 |
Membránové filtry, 0,45 μm. |
|
4.3 |
Zařízení HPLC s dávkovacím systémem na 20 μl. |
|
4.3.1 |
Kolona pro kapalinovou chromatografii 125 mm × 4 mm, reverzní fáze C18, náplň 5 μm nebo obdobná. |
|
4.3.2 |
Spektrofluorimetr s nastavitelnou excitační i emisní vlnovou délkou. |
5. Postup
5.1 Obecné pokyny
5.1.1
Pro provedení zkoušky na výtěžnost (5.1.2) se provede zkouška slepého vzorku krmiva pro kontrolu, že není přítomen ani lasalocid ani jiné rušivé látky. Slepý vzorek krmiva má podobné složení jako zkoušený vzorek a není zjištěn lasalocid ani jiné rušivé látky.
5.1.2
Zkouška na výtěžnost se provede na slepém vzorku krmiva, ke kterému bylo přidáno takové množství lasalocidu, které odpovídá množství lasalocidu ve zkoušeném vzorku. Pro obohacení na obsah 100 mg/kg se přidá 10,0 ml základního standardního roztoku (3.10.1) do 250 ml kónické baňky a roztok se odpaří na objem asi 0,5 ml. Potom se přidá 50 g slepého vzorku krmiva. Důkladně se promíchá, nechá se 10 minut stát a před zahájením extrakce (5.2) se znovu několikrát promíchá.
Není-li k dispozici slepý vzorek krmiva podobného složení jako zkoušený vzorek (viz 5.1.1), lze provést zkoušku na výtěžnost metodou standardního přídavku. V tom případě se ke zkoušenému vzorku přidá přibližně stejné množství lasalocidu, jaké je ve vzorku již obsaženo. Obohacený vzorek se potom zkouší společně se vzorkem neobohaceným a výtěžnost se vypočte odečtením.
5.2 Extrakce
5.2.1
Do 250 ml kónické baňky se zátkou se naváží 5–10 g vzorku s přesností na 0,01 g. Pipetou se přidá 100,0 ml okyseleného methanolu (3.8). Baňka se volně uzavře zátkou a promíchá kroužením. Potom se vloží na 20 minut do ultrazvukové lázně (4.1) při teplotě asi 40 oC. Pak se baňka vyjme a ochladí na laboratorní teplotu. Nechá se stát asi hodinu, aby se usadily suspedované částice, a potom se alikvotní část přefiltruje přes membránový filtr 0,45 μm (4.2) do vhodné nádoby. Dále se pokračuje se stanovením HPLC (5.3).
5.2.2
Do 250 ml odměrné baňky se naváží asi 2 g nemletého vzorku premixu s přesností na 0,001 g. Přidá se 100,0 ml okyseleného methanolu (3.8) a promíchá kroužením. Potom se vloží na 20 minut do ultrazvukové lázně (4.1) při teplotě asi 40 oC. Pak se baňka vyjme a ochladí na laboratorní teplotu. Doplní se po značku okyseleným methanolem (3.8) a důkladně promíchá. Nechá se stát asi hodinu, aby se usadily suspendované částice, a potom se alikvotní část přefiltruje přes membránový filtr 0,45 μm (4.2). Vhodný objem čirého filtrátu se naředí okyseleným methanolem (3.8) na konečnou koncentraci asi 4 μg/ml lasalocidu. Dále se pokračuje se stanovením HPLC (5.3).
5.3 Stanovení HPLC
5.3.1
Následující podmínky jsou doporučené; mohou být použity jiné podmínky za předpokladu, že poskytují rovnocenné výsledky:
|
Kolona pro kapalinovou chromatografii (4.3.1): |
125 mm × 4 mm, reverzní fáze C18, náplň 5 μm nebo obdobná |
|
Mobilní fáze (3.9): |
směs fosfátového tlumivého roztoku (3.7) a methanolu (3.5), 5 + 95 (V + V) |
|
Průtok: |
1,2 ml/min |
|
Detekční vlnová délka: |
|
|
excitace: |
310 nm |
|
emise: |
419 nm |
|
Objem nástřiku: |
20 μl |
Stabilita chromatografického systému se kontroluje opakovaným nástřikem kalibračního roztoku (3.10.3), který obsahuje 4,0 μg/ml, dokud se nedosáhne konstantních výšek (nebo ploch) píků a konstantních retenčních časů.
5.3.2
Každý kalibrační roztok (3.10.3) se nastřikuje několikrát a změří se výšky (plochy) píků pro každou koncentraci. Kalibrační křivka se vytvoří vynesením průměrných hodnot výšek nebo ploch píků kalibračních roztoků proti příslušným koncentracím v μg/ml.
5.3.3
Extrakt ze vzorku 5.2.1 nebo 5.2.2 se nastřikuje několikrát za použití stejného objemu jako v případě kalibračního roztoku a zjišťuje se průměrná hodnota výšek (ploch) píků lasalocidu.
6. Výpočet a vyjádření výsledků
Z průměrné hodnoty výšky (plochy) píku v roztoku vzorku (5.3.3) se porovnáním s kalibrační křivkou stanoví koncentrace lasalocidu v μg/ml.
6.1 Krmiva
Obsah lasalocidu (w) v mg/kg ve vzorku se vypočítá podle následujícího vzorce:
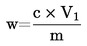 (mg/kg)
(mg/kg)
kde:
|
c |
= |
koncentrace lasalocidu v roztoku vzorku (5.2.1) v μg/ml |
|
V1 |
= |
objem extraktu vzorku podle 5.2.1 v ml (tj. 100 ml) |
|
m |
= |
hmotnost navážky vzorku v g. |
6.2 Premixy
Obsah lasalocidu (w) v mg/kg ve vzorku se vypočítá podle následujícího vzorce:
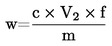 (mg/kg)
(mg/kg)
kde:
|
c |
= |
koncentrace lasalocidu v roztoku vzorku (5.2.2) v μg/ml |
|
V2 |
= |
objem extraktu vzorku podle 5.2.2 v ml (tj. 250 ml) |
|
f |
= |
faktor ředění podle 5.2.2 |
|
m |
= |
hmotnost navážky vzorku v g. |
7. Ověření výsledků
7.1 Identita
Metody založené na spektrofluorimetrické detekci jsou méně náchylné na interference než metody s UV detekcí. Identita analytu může být potvrzena opakovaným stanovením s přídavkem (co-chromatography).
7.1.1
K extraktu vzorku (5.2.1 nebo 5.2.2) se přidá vhodné množství kalibračního roztoku (3.10.3). Přidané množství lasalocidu musí odpovídat obsahu lasalocidu v extraktu vzorku. Přídavek se projeví pouze zvýšením píku lasalocidu, a to úměrně k přidanému množství lasalocidu a ředění extraktu. Šířka píku v polovině maximální výšky se může odchylovat od šířky původního píku v neobohaceném extraktu vzorku nejvýše o ±10 %.
7.2 Opakovatelnost
Rozdíl mezi výsledky dvou paralelních stanovení provedených u stejného vzorku nesmí překročit:
|
— |
15 % relat. z hodnoty vyššího výsledku pro obsah lasalocidu v rozmezí od 30 mg/kg do 100 mg/kg, |
|
— |
15 mg/kg pro obsah lasalocidu v rozmezí od 100 mg/kg do 200 mg/kg, |
|
— |
7,5 % relat. z hodnoty vyššího výsledku pro obsah lasalocidu nad 200 mg/kg. |
7.3 Výtěžnost
Pro obohacený (slepý) vzorek krmiva je výtěžnost nejméně 80 %. Pro obohacené vzorky premixů je výtěžnost nejméně 90 %.
8. Výsledky kruhových testů
Kruhové testy (6) byly provedeny se 2 vzorky premixů (vzorky 1 a 2) a 5 vzorky krmiva (vzorky 3–7) ve 12 laboratořích. U každého vzorku byly provedeny dvě zkoušky. Výsledky jsou uvedeny v následující tabulce:
|
|
Vzorek 1 Premix pro kuřata |
Vzorek 2 Premix pro krůty |
Vzorek 3 Krmná směs pro krůty peletovaná |
Vzorek 4 Krmná směs pro kuřata sypká |
Vzorek 5 Krmná směs pro krůty |
Vzorek 6 Krmná směs pro drůbež A |
Vzorek 7 Krmná směs pro drůbež B |
|
L |
12 |
12 |
12 |
12 |
12 |
12 |
12 |
|
n |
23 |
23 |
23 |
23 |
23 |
23 |
23 |
|
průměr (mg/kg) |
5 050 |
16 200 |
76,5 |
78,4 |
92,9 |
48,3 |
32,6 |
|
sr (mg/kg) |
107 |
408 |
1,71 |
2,23 |
2,27 |
1,93 |
1,75 |
|
CVr (%) |
2,12 |
2,52 |
2,24 |
2,84 |
2,44 |
4,00 |
5,37 |
|
sR (mg/kg) |
286 |
883 |
3,85 |
7,32 |
5,29 |
3,47 |
3,49 |
|
CVR (%) |
5,66 |
5,45 |
5,03 |
9,34 |
5,69 |
7,18 |
10,70 |
|
Deklarovaný obsah (mg/kg) |
5 000 (7) |
16 000 (7) |
80 (7) |
105 (7) |
120 (7) |
50 (8) |
35 (8) |
|
L |
= |
počet laboratoří |
|
n |
= |
počet jednotlivých hodnot |
|
sr |
= |
standardní odchylka opakovatelnosti |
|
sR |
= |
standardní odchylka reprodukovatelnosti |
|
CVr |
= |
variační koeficient opakovatelnosti v % |
|
CVR |
= |
variační koeficient reprodukovatelnosti v %. |
(1) Provedla pracovní skupina pro krmiva Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
(2) Provedla pracovní skupina pro krmiva Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
(3) Lze použít i jiné metody mineralizace, pokud prokazatelně poskytují podobné výsledky (např. mikrovlnný tlakový rozklad).
(4) Zelené pícniny (čerstvé nebo suché) často obsahují velké množství rostlinného křemíku, který může zadržovat stopové prvky, a proto musí být odstraněn. U vzorků těchto krmiv musí být proto dodržen následující modifikovaný postup. Postupuje se podle bodu 5.1.1.1 až k filtraci. Filtrační papír obsahující nerozpustný zbytek se promyje dvakrát vroucí vodou a vloží se zpět do křemíkového nebo platinového kelímku. Spaluje se v muflové peci (4.1) při teplotě pod 550 oC, dokud všechny uhlíkaté částice zcela nezmizí. Nechá se vychladnout, přidá se několik kapek vody a poté 10–15 ml kyseliny fluorovodíkové (3.4) a odpaří se do sucha při asi 150 oC. Pokud zůstane nějaký křemičitý zbytek, rozpustí se znovu v několika mililitrech kyseliny fluorovodíkové (3.4) a odpaří se do sucha. Přidá se pět kapek kyseliny sírové (3.5) a zahřívá se, dokud se nepřestane uvolňovat bílý kouř. Po přidání 5 ml kyseliny chlorovodíkové 6 mol/l (3.2) a asi 30 ml vody se roztok zahřeje, přefiltruje do 250 ml odměrné baňky a doplní se po značku vodou (koncentrace HCl je asi 0,5 mol/l). Pak se pokračuje podle bodu 5.1.2.
(5) The Analyst 108, 1983, s. 1 252–1 256.
(6) Analyst, 1995, 120, s. 2 175–2 180.
(7) Obsah deklarovaný výrobcem.
(8) Krmivo připravené v laboratoři.
PŘÍLOHA V
METODY ZKOUŠENÍ PRO KONTROLU NEŽÁDOUCÍCH LÁTEK V KRMIVECH
A. STANOVENÍ VOLNÉHO A CELKOVÉHO GOSSYPOLU
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu volného a celkového gossypolu a chemicky příbuzných látek v bavlníkových semenech, bavlníkové mouce a bavlníkových pokrutinách a v krmných směsích, které obsahují krmné suroviny, v nichž je obsažen volný a celkový gossypol a chemicky příbuzné látky v koncentraci nad 20 mg/kg.
2. Princip
Gossypol se extrahuje buď za přítomnosti aminopropanolu (3-aminopropan-1-ol) směsí 2-propanolu a hexanu při stanovení volného gossypolu, nebo dimethylformamidem při stanovení celkového gossypolu. Gossypol je reakcí s anilinem převeden na gossypol-dianilin a absorbance roztoku je měřena při 440 nm.
3. Chemikálie
|
3.1 |
Směs 2-propanolu a hexanu: směs 2-propanolu a n-hexanu v poměru 60 + 40. |
|
3.2 |
Rozpouštědlo A: do 1 000 ml odměrné baňky se nalije asi 500 ml směsi 2-propanolu a hexanu (3.1), 2 ml aminopropanolu, 8 ml ledové kyseliny octové a 50 ml vody. Doplní se po značku směsí 2-propanolu a hexanu (3.1) a promíchá. Rozpouštědlo je stálé jeden týden. |
|
3.3 |
Rozpouštědlo B: do 100 ml odměrné baňky se napipetují 2 ml aminopropanolu a 10 ml ledové kyseliny octové. Vychladí se na laboratorní teplotu a doplní se po značku N, N-dimethylformamidem. Rozpouštědlo je stálé jeden týden. |
|
3.4 |
Anilin: pokud absorbance anilinu (slepá zkouška) přesáhne 0,022, musí se anilin destilovat přes zinkový prach, přičemž prvních a posledních 10 % frakcí destilátu se odstraní. Při skladování v chladničce v tmavé zábrusové láhvi vydrží tato chemikálie několik měsíců. |
|
3.5 |
Standardní roztok gossypolu A: do 250 ml odměrné baňky se naváží 27,9 mg octanu gossypolu. Rozpustí se a doplní po značku rozpouštědlem A (3.2). Do 250 ml odměrné baňky se odpipetuje 50 ml tohoto roztoku a doplní se rozpouštědlem A po značku. 1 ml tohoto roztoku obsahuje 0,02 mg gossypolu. Před použitím se nechá hodinu stát při laboratorní teplotě. |
|
3.6 |
Standardní roztok gossypolu B: do 50 ml odměrné baňky se naváží 27,9 mg octanu gossypolu, rozpustí se a doplní po značku rozpouštědlem B (3.3). 1 ml tohoto roztoku obsahuje 0,5 mg gossypolu. Standardní roztoky gossypolu A a B jsou stálé 24 hodin, pokud jsou chráněny před světlem. |
4. Přístroje a pomůcky
|
4.1 |
Míchací zařízení: přibližně 35 ot./min. |
|
4.2 |
Spektrofotometr. |
5. Postup
5.1 Zkoušený vzorek
Množství zkoušeného vzorku závisí na předpokládaném obsahu gossypolu ve vzorku. Je vhodnější pracovat s malým množstvím zkoušeného vzorku a relativně velkou alikvotní částí filtrátu, aby obsah gossypolu byl dostatečný pro přesné spektrofotometrické měření. Pro stanovení volného gossypolu v bavlníku, bavlníkové mouce a bavlníkových pokrutinách navážka vzorku nepřesáhne 1 g; v krmných směsích může být 5 g. Ve většině případů je vhodný alikvotní podíl filtrátu 10 ml; obsahuje 50–100 μg gossypolu. Pro stanovení celkového gossypolu je navážka 0,5–5 g, tedy 2 ml alikvotní části filtrátu budou obsahovat 40–200 μg gossypolu.
Stanovení se provádí při laboratorní teplotě asi 20 oC.
5.2 Stanovení volného gossypolu
Do širokohrdlé 250 ml baňky, na jejíž dno bylo přidáno drcené sklo, se naváží zkušební vzorek. Do baňky se odpipetuje 50 ml rozpouštědla A (3.2), baňka se uzavře a mixuje po dobu jedné hodiny v míchacím zařízení. Pak se obsah baňky filtruje suchým filtrem do malé širokohrdlé baňky. Během filtrace se filtrační nálevka přikryje hodinovým krycím sklem.
Do dvou 25 ml odměrných baněk (označí se A a B) se odpipetuje stejná alikvotní část filtrátu obsahující 50–100 μg gossypolu. Pokud je třeba, doplní se rozpouštědlem A (3.2) na objem 10 ml. Roztok v baňce A se pak doplní po značku směsí 2-propanolu a hexanu (3.1). Tento roztok se použije jako referenční roztok, proti kterému se měří roztok vzorku.
Do dalších dvou 25 ml odměrných baněk (označí se C a D) se odpipetuje 10 ml rozpouštědla A (3.2). Baňka C se doplní po značku směsí 2-propanolu a hexanu (3.1). Tento roztok se použije jako referenční roztok, proti kterému se měří roztok slepé zkoušky.
Do odměrných baněk B a D se odpipetuje po 2 ml anilinu (3.4). Baňky se zahřívají po dobu 30 minut na vroucí vodní lázni, aby došlo k vybarvení roztoků. Potom se ochladí na laboratorní teplotu, doplní směsí 2-propanolu a hexanu (3.1) po značku a homogenizují. Nechají se asi jednu hodinu stát.
Na spektrofotometru se měří absorbance roztoku slepé zkoušky (D) proti referenčnímu roztoku (C) a absorbance roztoku vzorku (B) proti referenčnímu roztoku (A) v kyvetách optické délky 10 mm při vlnové délce 440 nm.
Od absorbance vzorku se odečte absorbance slepé zkoušky a rozdíl absorbancí (korigovaná absorbance) je základem výpočtu obsahu volného gossypolu podle odstavce 6.
5.3 Stanovení celkového gossypolu
Do 50 ml odměrné baňky se naváží zkoušený vzorek, který obsahuje 1–5 mg gossypolu a přidá se 10 ml rozpouštědla B (3.3). Do další 50 ml odměrné baňky se přidá přesně 10 ml rozpouštědla B (3.3) pro slepou zkoušku. Obě baňky se zahřívají na vroucí vodní lázni po dobu 30 minut. Potom se ochladí na laboratorní teplotu a obě baňky se doplní po značku směsí 2-propanolu a hexanu (3.1). Homogenizují se a nechají 10–15 minut usadit, poté se obsah filtruje do širokohrdlých baněk.
Do obou 25 ml odměrných baněk se z filtrátu vzorku odpipetuje po 2 ml a z filtrátu slepé zkoušky do obou 25 ml odměrných baněk rovněž po 2 ml. Jedna baňka se vzorkem a jedna se slepou zkouškou se doplní směsí 2-propanolu a hexanu (3.1) po značku. Tyto roztoky se použijí jako referenční roztoky.
Do druhých dvou baněk se přidají 2 ml anilinu (3.4). Baňky se zahřívají po dobu 30 minut na vroucí vodní lázni, aby došlo k vybarvení roztoků. Potom se ochladí na laboratorní teplotu, doplní směsí 2-propanolu a hexanu (3.1) po značku a homogenizují. Nechají se jednu hodinu stát.
Absorbance se stanoví podle bodu 5.2 pro volný gossypol. Z těchto hodnot se vypočítá obsah celkového gossypolu podle odstavce 6.
6. Výpočet a vyjádření výsledků
Výsledky mohou být vypočítány ze specifických absorbančních koeficientů (6.1) nebo podle kalibrační křivky (6.2).
6.1 Ze specifických absorbančních koeficientů
Specifické absorbanční koeficienty jsou na základě uvedených podmínek tyto:
|
Volný gossypol: |
|
|
Celkový gossypol: |
|
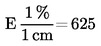
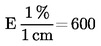
Obsah volného nebo celkového gossypolu ve vzorku se vypočítá podle následujícího vzorce:
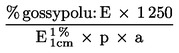
kde:
|
E |
= |
korigovaná absorbance stanovená podle 5.2 |
|
p |
= |
navážka zkušebního vzorku v g |
|
a |
= |
alikvotní podíl filtrátu v ml. |
6.2 Z kalibrační křivky
6.2.1
Připraví se 2 sady pěti 25 ml odměrných baněk. Do každé sady baněk se odpipetuje 2,0, 4,0, 6,0, 8,0 a 10,0 ml standardního roztoku gossypolu A (3.5). Doplní se na 10 ml rozpouštědlem A (3.2). Každá sada se doplní o 25 ml odměrnou baňku obsahující pouze 10 ml rozpouštědla A (3.2) (slepá zkouška).
Obsah baněk první sady (včetně baňky pro slepou zkoušku) se doplní na 25 ml směsí 2-propanolu a hexanu (3.1) (referenční sada).
Do každé baňky druhé sady (včetně baňky pro slepou zkoušku) se přidají 2 ml anilinu (3.4). Baňky se zahřívají po dobu 30 minut na vroucí vodní lázni, aby došlo k vybarvení roztoků. Potom se ochladí na laboratorní teplotu, doplní směsí 2-propanolu a hexanu (3.1) po značku a homogenizují. Nechají se asi jednu hodinu stát (standardní sada).
Absorbance roztoků standardní sady se stanoví podle bodu 5.2 porovnáním s odpovídajícími roztoky v referenční sadě. Kalibrační křivka se vytvoří vynesením hodnot absorbancí proti odpovídajícím množstvím gossypolu (v μg).
6.2.2
Připraví se šest 50 ml odměrných baněk. Do první baňky se napipetuje 10 ml rozpouštědla B (3.3) a do ostatních 2,0, 4,0, 6,0, 8,0 a 10,0 ml standardního roztoku gossypolu B (3.6). Obsah každé baňky se doplní na 10 ml rozpouštědlem B (3.3). Baňky se zahřívají na vroucí vodní lázni po dobu 30 minut. Potom se ochladí na laboratorní teplotu, doplní směsí 2-propanolu a hexanu (3.1) po značku a homogenizují.
2,0 ml těchto roztoků se přenese do každé ze dvou sad šesti 25 ml odměrných baněk. Baňky první sady se doplní směsí 2-propanolu a hexanu (3.1) na 25 ml (referenční sada).
Do každé baňky druhé sady se přidají 2 ml anilinu (3.4). Baňky se zahřívají na vroucí vodní lázni po dobu 30 minut. Potom se ochladí na laboratorní teplotu, doplní se směsí 2-propanolu a hexanu (3.1) po značku a homogenizují. Nechají se asi jednu hodinu stát (standardní sada).
Absorbance roztoků standardní sady se stanoví podle bodu 5.2 porovnáním s odpovídajícími roztoky v referenční sadě. Kalibrační křivka se vytvoří vynesením hodnot absorbancí proti odpovídajícím množstvím gossypolu (v μg).
6.3 Opakovatelnost
Rozdíl mezi výsledky dvou paralelních stanovení provedených u stejného vzorku nesmí překročit:
|
— |
15 % relat. z hodnoty vyššího výsledku u obsahu gossypolu nižšího než 500 ppm, |
|
— |
75 ppm absol. u obsahu od 500 ppm do 750 ppm, |
|
— |
10 % relat. z hodnoty vyššího výsledku u obsahu nad 750 ppm. |
B. STANOVENÍ OBSAHU DIOXINŮ (PCDD/PCDF) A PCB S DIOXINOVÝM EFEKTEM
I. METODY ODBĚRU VZORKŮ A INTERPRETACE VÝSLEDKŮ ZKOUŠEK
1. Účel a rozsah
Vzorky určené pro úřední kontrolu obsahu dioxinů (polychlorovaných dibenzo-p-dioxinů (PCDD) a polychlorovaných dibenzofuranů (PCDF)) a polychlorovaných bifenylů (PCB) (1) s dioxinovým efektem v krmivech se odebírají v souladu s pravidly v příloze I. Musí být použity kvantitativní požadavky týkající se kontroly látek nebo produktů obsažených rovnoměrně v krmivech, jak je stanoveno v bodě 5.A přílohy I. Takto získané souhrnné vzorky se považují za reprezentativní pro vzorkované partie nebo části vzorkované partie. Dodržení maximálních hodnot stanovených ve směrnici Evropského parlamentu a Rady 2002/32/ES (2) se posuzuje na základě obsahů určených v laboratorních vzorcích.
2. Dodržení maximálního obsahu ve vzorkované partii nebo v části vzorkované partie
Vzorkovaná partie se přijme, jestliže výsledek jediné zkoušky nepřekročí příslušný maximální obsah stanovený ve směrnici 2002/32/ES při zohlednění nejistoty měření.
Vzorkovaná partie nevyhovuje maximálnímu obsahu stanovenému ve směrnici 2002/32/ES, pokud horní (3) výsledek zkoušky potvrzený opakovanou zkouškou (4) při současném zohlednění nejistoty měření nepochybně překračuje maximální obsah.
Nejistotu měření lze zohlednit jedním z těchto způsobů:
|
— |
započítáním rozšířené nejistoty při použití faktoru pokrytí 2, který odpovídá hladině spolehlivosti asi 95 %. Vzorkovaná partie nesplňuje podmínky, pokud měřená hodnota, od níž se odečte U, přesahuje maximální obsah. V případě samostatného stanovení dioxinů a PCB s dioxinovým efektem se součet odhadované rozšířené nejistoty měření samostatných výsledků zkoušek u dioxinů a PCB s dioxinovým efektem musí použít pro součet dioxinů a PCB s dioxinovým efektem, |
|
— |
stanovením rozhodovací meze (CCα) v souladu s rozhodnutím Komise 2002/657/ES (5) (bod 3.1.2.5 přílohy – pro látky, pro něž byla stanovena přípustná hodnota). Vzorkovaná partie nesplňuje podmínky, pokud se naměřená hodnota rovná CCα, nebo je vyšší. |
Uvedená pravidla interpretace se použijí na výsledky zkoušek získané u vzorků pro úřední kontrolu. Není jimi dotčeno právo členských států použít vnitrostátní pravidla pro zkoušky pro účely obhajoby nebo rozhodčího řízení.
II. PŘÍPRAVA VZORKŮ A POŽADAVKY NA METODY ZKOUŠENÍ POUŽITÉ PŘI ÚŘEDNÍ KONTROLE OBSAHU DIOXINŮ (PCDD/PCDF) A PCB S DIOXINOVÝM EFEKTEM
1. Cíl a oblast použití
Tyto požadavky se použijí tam, kde jsou zkoušeny krmné suroviny a krmiva za účelem stanovení dioxinů (polychlorovaných dibenzo-p-dioxinů (PCDD) a polychlorovaných dibenzofuranů (PCDF)) a polychlorovaných bifenylů (PCB) s dioxinovým efektem.
Pro sledování přítomnosti dioxinů v krmivech může být využita screeningová metoda k vyhledání vzorků s obsahem dioxinů a PBC s dioxinovým efektem, který je o méně než 25 % nižší nebo vyšší, než je sledovaná úroveň. Koncentrace dioxinů ve vzorcích s významným množstvím se stanoví nebo potvrdí potvrzovací metodou.
Screeningové metody jsou metody, které slouží ke zjišťování přítomnosti dioxinů a PCB s dioxinovým efektem na sledované úrovni. Tyto metody mají vysokou kapacitu, pokud jde o množství vzorků, a jsou používány k vytřídění potenciálně pozitivních vzorků z velkého množství vzorků. Jsou speciálně vyvinuty tak, aby neposkytovaly nesprávně negativní výsledky.
Potvrzovací metody jsou metody, jež poskytují úplnou nebo doplňující informaci, která na sledované úrovni umožní jednoznačnou identifikaci a jednoznačnou kvantifikaci dioxinů a PCB s dioxinovým efektem.
2. Souvislosti
Protože vzorky z životního prostředí a biologické vzorky (včetně vzorků krmných surovin/krmiv) obvykle obsahují složité směsi různých dioxinových kongenerů, byla vyvinuta koncepce koeficientů toxické ekvivalence (TEF), aby se usnadnilo hodnocení nebezpečí. Tyto TEF jsou určeny k tomu, aby vyjádřily koncentrace směsí 2,3,7,8-substituovaných PCDD a PCDF a některých non-orto a mono-orto-chlorovaných PCB (PCB s jedním nebo žádným atomem chlóru v orto-poloze), které vykazují aktivitu podobnou dioxinům v toxických ekvivalentech (TEQ) 2,3,7,8-TCDD. Koncentrace jednotlivých látek v daném vzorku se násobí jejich příslušným TEF a následně sčítají, aby se dospělo k celkové koncentraci sloučenin s dioxinovým efektem vyjádřené v TEQ.
Pouze pro účely tohoto nařízení je schválenou specifickou mezí kvantifikace jednotlivého kongeneru koncentrace analytu v extraktu vzorku, jenž vytváří instrumentální odezvu na dva různé ionty, která má být monitorována v poměru signál-šum 3 : 1 pro méně citlivé signály a při splnění základních požadavků, jako je např. retenční čas a poměr izotopu podle postupu stanovení popsaného v metodě EPA 1613 revize B.
3. Požadavky na zabezpečení kvality, které musí být splněny při úpravě vzorku
Použijí se obecná ustanovení o úpravě vzorků pro laboratorní zkoušení uvedená v příloze II.
Kromě toho musí být splněny tyto požadavky:
|
— |
vzorky musí být uchovávány a přepravovány v nádobách ze skla, hliníku, polypropylenu nebo polyethylenu. Z nádoby na vzorky musí být odstraněny stopy papírového prachu. Skleněné nádoby se vypláchnou rozpouštědly, u nichž byla předem provedena kontrola, zda neobsahují dioxiny, |
|
— |
provede se slepá zkouška, při níž se provede celý postup laboratorního zkoušení bez vzorku, |
|
— |
hmotnost vzorku použitého pro extrakci musí být dostatečná na to, aby byly splněny požadavky na citlivost stanovení. |
4. Požadavky na laboratoře
|
— |
laboratoře prokazují funkčnost metody v rozsahu kolem sledované úrovně, např. poloviny, jednonásobku nebo dvojnásobku sledované úrovně, a to s přijatelným variačním koeficientem pro opakovanou zkoušku. Podrobnosti o kritériích přijatelnosti jsou uvedeny v bodě 5, |
|
— |
mez kvantifikace pro potvrzovací metodu musí být v rozsahu asi jedné pětiny sledované úrovně, aby bylo zajištěno, že v rozsahu sledované úrovně se dosahuje přijatelných variačních koeficientů, |
|
— |
jako opatření v rámci vnitřní kontroly kvality se provádějí pravidelné slepé zkoušky, stanovení s přídavkem nebo zkoušky kontrolních vzorků (nejlépe certifikovaného referenčního materiálu, je-li k dispozici), |
|
— |
úspěšné výsledky mezilaboratorních srovnávacích zkoušek, při nichž se hodnotí odbornost laboratoře, jsou nejlepším způsobem ověření odborné způsobilosti pro specifické zkoušky. Úspěšné výsledky kruhových testů, např. pro vzorky půd nebo kalů, nejsou nezbytně důkazem odborné způsobilosti, pokud jde o vzorky potravin nebo krmiv, v nichž se vyskytují nižší úrovně kontaminace. Proto je povinná stálá účast při kruhových testech pro stanovení dioxinů a PCB s dioxinovým efektem v relevantních matricích potravin nebo krmiv, |
|
— |
laboratoře musí být akreditovány uznávaným subjektem působícím v souladu s ISO Guide 58, aby bylo zajištěno, že uplatňují zabezpečování kvality při zkouškách. Laboratoře musí být akreditovány podle normy ISO/IEC/17025. |
5. Požadavky na postupy laboratorního zkoušení pro dioxiny a PCB s dioxinovým efektem
Základní požadavky na přijatelnost postupů laboratorního zkoušení:
|
— |
vysoká citlivost a nízké meze detekce. V případě PCDD a PCDF musí být zjistitelné množství z důvodu extrémní toxicity některých těchto sloučenin na úrovni pikogramů TEQ (10-12 g). Je známo, že PCB se vyskytují ve vyšších koncentracích než PCDD a PCDF. Pro většinu kongenerů PCB již postačuje citlivost v řádu nanogramů (10-9 g). Pro měření toxičtějších kongenerů PCB s dioxinovým efektem (zejména non-orto substituovaných kongenerů) musí být dosaženo stejné citlivosti měření jako pro PCDD a PCDF, |
|
— |
vysoká selektivita/specifičnost. Je třeba rozlišovat PCDD, PCDF a PCB s dioxinovým efektem od ostatních sloučenin, které se extrahují společně s těmito látkami, mohou rušit při jejich stanovení a jsou přítomny v koncentracích až o několik řádů vyšších než koncentrace sledovaných analytů. Pro plynovou chromatografii/hmotnostní spektrometrii (GC/MS) je nezbytné rozlišení mezi různými kongenery, tj. mezi toxickými (např. sedmnácti 2,3,7,8-substituovanými PCDD a PCDF a PCB s dioxinovým efektem) a ostatními kongenery. Biotesty musí umožnit selektivně určit hodnoty TEQ jako součet PCDD, PCDF a PCB s dioxinovým efektem, |
|
— |
vysoká správnost (pravdivost a přesnost). Stanovení poskytne platný a spolehlivý odhad skutečné koncentrace ve vzorku. Vysoká správnost (správnost měření: stupeň shody mezi výsledkem měření a skutečnou nebo přidělenou hodnotou) je nezbytná k tomu, aby nedošlo k zamítnutí výsledku zkoušky vzorku na základě malé spolehlivosti odhadu TEQ. Správnost je vyjádřena pravdivostí (rozdílem mezi střední naměřenou hodnotou analytu v certifikovaném materiálu a jeho certifikovanou hodnotou, vyjádřeným v procentech této hodnoty) a přesností (RSDR je relativní směrodatná odchylka vypočtená z výsledků získaných za podmínek reprodukovatelnosti). |
Screeningové metody mohou zahrnovat metody biotestů sloučenin a metody GC/MS; potvrzovací metody jsou metodami plynové chromatografie s vysokým rozlišením/hmotnostní spektrometrie s vysokým rozlišením (HRGC/HRMS).
Hodnota celkového TEQ musí splňovat následující kritéria:
|
|
Screeningové metody |
Potvrzovací metody |
|
Nesprávné negativní výsledky |
< 1 % |
|
|
Pravdivost |
|
-20 % až +20 % |
|
Přesnost RSDR |
< 30 % |
< 15 % |
6. Zvláštní požadavky, které musí splňovat metody GC/MS, aby vyhovovaly pro účely screeningu nebo ověřování
|
— |
vnitřní standardy 2,3,7,8-chlor-substituovaných PCDD/F značené izotopem 13C a standardy PCB s dioxinovým efektem značené izotopem 13C musí být přimíšeny na samém začátku nebo při zahájení zkoušky, např. před extrakcí, aby bylo možné ověřit postup laboratorního zkoušení. Alespoň jeden kongener musí být přidán pro každou z tetra až okta-chlorovaných homologických skupin PCDD/F a alespoň jeden kongener pro každou z homologických skupin PCB s dioxinovým efektem (nebo alespoň jeden kongener pro každou skupinu vybraných iontů při použití hmotnostní spektrometrie v režimu registrace vybraných iontů, použitou pro sledování PCDD/F a PCB s dioxinovým efektem). Jednoznačně se dává přednost, zejména v případě potvrzovacích metod, použití všech 17 vnitřních standardů 2,3,7,8-substituovaných PCDD/F značených izotopem 13C a všech 12 vnitřních standardů PCB s dioxinovým efektem značených izotopem 13C, |
|
— |
relativní odezvové faktory se s pomocí vhodných kalibračních roztoků stanoví také pro kongenery, pro něž nebyly přimíšeny sloučeniny značené izotopem 13C, |
|
— |
u krmiv rostlinného původu nebo krmiv živočišného původu s obsahem tuku nižším než 10 % je příměs vnitřních standardů před extrakcí povinná. U krmiv živočišného původu s obsahem tuku vyšším než 10 % lze vnitřní standardy přimísit buď před extrakcí, nebo po extrakci tuku. Provede se vhodné ověření účinnosti extrakce v závislosti na tom, kdy je vnitřní standard přidáván, a na tom, zda jsou výsledky uváděny v produktu nebo v tuku, |
|
— |
před zkouškou metodou GC/MS musí být přimíšeny 1 nebo 2 náhradní standardy pro stanovení výtěžnosti, |
|
— |
kontrola výtěžnosti je nezbytná. U potvrzovacích metod se výtěžnost jednotlivých vnitřních standardů musí pohybovat v rozsahu 60–120 %. Nižší nebo vyšší hodnota výtěžnosti jednotlivých kongenerů, zejména některých hepta- a okta-chlorovaných dibenzodioxinů a dibenzofuranů, je přípustná pod podmínkou, že jejich příspěvek k hodnotě TEQ nepřesáhne 10 % celkové hodnoty TEQ (na základě součtu PCDD/F a PCB s dioxinovým efektem). Hodnota výtěžnosti u screeningových metod se musí pohybovat mezi 30–140 %, |
|
— |
oddělení dioxinů od rušivých chlorovaných sloučenin, jako jsou jiné PCB než PCB s dioxinovým efektem a chlorované difenylethery, se provádí vhodnými chromatografickými technikami (nejlépe na florisilové, aluminiové a/nebo uhlíkové koloně), |
|
— |
oddělení izomerů pomocí plynové chromatografie musí být dostatečné (< 25 % mezi píky mezi 1,2,3,4,7,8-HxCDF a 1,2,3,6,7,8-HxCDF), |
|
— |
stanovení se provádí metodou EPA 1613 revize B: „Tetra- through octa-chlorinated dioxins and furans by isotope dilution HRGC/HRMS“ nebo jinou metodou s rovnocennými pracovními kritérii, |
|
— |
u krmiv kontaminovaných dioxiny v rozsahu maximálního obsahu nebo nad ním nesmí rozdíl mezi horní a dolní mezní úrovní překročit 20 %. U krmiv s úrovněmi kontaminace výrazně pod maximálním obsahem by se měl rozdíl pohybovat mezi 25–40 %. |
7. Screeningové metody laboratorního zkoušení
7.1 Úvod
Při zkoušce mohou být použity různé přístupy ke screeningové metodě: čistý screening a kvantitativní zkoušení.
Odezva vzorků je porovnávána s odezvou referenčního vzorku na sledované úrovni. Vzorky s odezvou nižší, než má referenční vzorek, se prohlásí za negativní, vzorky s vyšší odezvou se považují za pozitivní. Požadavky:
|
— |
do každé zkušební sady se zařadí slepý a referenční vzorek, které jsou extrahovány a zkoušeny současně a za stejných podmínek. Referenční vzorek musí ve srovnání se slepým vzorkem vykazovat zřetelně vyšší odezvu, |
|
— |
kromě toho se zařadí referenční vzorky o poloviční a dvojnásobné koncentraci, než je sledovaná úroveň, aby bylo prokázáno správné provádění zkoušky v rozsahu odpovídajícím sledované úrovni, |
|
— |
při zkoušení jiných matric musí být prokázána vhodnost referenčního vzorku/referenčních vzorků, a to přednostně zařazením vzorků, u nichž byla metodou HRGC/HRMS prokázána úroveň TEQ blízká úrovni v referenčním vzorku nebo také slepého vzorku uměle obohaceného na tuto úroveň, |
|
— |
vzhledem k tomu, že v biotestech nelze použít žádné vnitřní standardy, jsou zkoušky opakovatelnosti velmi důležité pro získání informací o směrodatné odchylce v rámci zkoušené sady. Variační koeficient musí být nižší než 30 %, |
|
— |
u biotestů se určí cílové sloučeniny, možné interference a nejvyšší přípustný obsah ve slepém vzorku. |
Kvantitativní zkoušení vyžaduje sérii ředění standardního roztoku, dvakrát nebo třikrát opakované čištění a měření a rovněž zařazení slepých vzorků a kontroly výtěžnosti. Výsledky mohou být vyjádřeny v TEQ, přičemž se vychází z toho, že sloučeniny, jež způsobily signál, vyhovují principu TEQ. To lze provést pomocí TCDD (nebo standardní směsi dioxin/furan/PCB s dioxinovým efektem), přičemž se sestrojí kalibrační křivka pro výpočet TEQ extraktu, a tedy i vzorku. Poté se provede korekce na hodnotu TEQ, která je spočítána pro slepý vzorek (aby se zohlednily nečistoty v použitých rozpouštědlech a chemikáliích), a na výtěžnost (vypočítanou z hodnoty TEQ, která je u vzorku určeného pro kontrolu kvality na sledované úrovni). Je nezbytné upozornit na to, že zjevné snížení výtěžnosti může být částečně způsobeno matricovými jevy a/nebo rozdíly mezi hodnotami TEF v biotestech a úředními hodnotami TEF stanovenými WHO.
7.2 Požadavky na metody laboratorního zkoušení používané pro screening
|
— |
pro screening mohou být používány metody laboratorního zkoušení GC/MS a biotesty. V případě metod GC/MS platí požadavky uvedené v bodě 6. Pro biotesty sloučenin platí zvláštní požadavky stanovené v bodu 7.3 a pro biotesty sloučenin prováděné pomocí souprav platí zvláštní požadavky stanovené v bodu 7.4, |
|
— |
nezbytné jsou informace o počtu nesprávných pozitivních a nesprávných negativních výsledků velkého souboru vzorků s hodnotami ležícími nad a pod maximálním obsahem nebo akční hodnotou ve srovnání s obsahem TEQ určeným potvrzovací metodou zkoušení. Skutečný podíl nesprávných negativních výsledků musí být pod 1 %. Podíl nesprávných pozitivních výsledků je natolik nízký, aby se použití screeningového nástroje stalo výhodným, |
|
— |
pozitivní výsledky musí být vždy potvrzeny potvrzovací metodou zkoušení (HRGC/HRMS). Kromě toho se pomocí HRGC/HRMS potvrdí vzorky z širokého rozmezí hodnot TEQ (přibližně 2–10 % negativních vzorků). Jsou dány k dispozici informace o souladu výsledků biotestu a HRGC/HRMS. |
7.3 Zvláštní požadavky na biotesty s intaktními buňkami
|
— |
při provádění biotestů vyžaduje každá zkouška sérii referenčních koncentrací TCDD nebo směsi dioxinu a furanu (kalibrační křivka v plném rozsahu s R2 > 0,95). Avšak pro účely screeningu může být ke zkoušce vzorků s nízkým obsahem použita kalibrační křivka s rozšířením pro nízké koncentrace, |
|
— |
pro vyjádření výsledků biotestů za určité časové období se použije referenční koncentrace TCDD (asi trojnásobek meze kvantifikace) uvedená v záznamech o kontrole kvality. Jinou možností může být porovnání relativní odezvy referenčního vzorku vzhledem ke kalibrační křivce TCDD, protože odezva buněk může záviset na mnoha faktorech, |
|
— |
pro každý typ referenčního materiálu se zaznamenávají a ověřují grafy kontrol kvality, aby bylo zajištěno, že výsledky jsou v souladu se stanovenými pokyny, |
|
— |
zejména při kvantitativních výpočtech musí být použita taková indukce ředění vzorku, aby ležela v lineárním úseku křivky závislosti odezvy. Vzorky ležící nad lineárním úsekem křivky závislosti odezvy se musí zředit a zkoušet znovu. Proto se doporučuje zkoušet alespoň tři nebo více různě ředěných vzorků najednou, |
|
— |
procentní standardní odchylka nesmí být vyšší než 15 % při trojnásobném stanovení pro každé ředění vzorku a pro tři nezávislé pokusy by neměla přesáhnout 30 %, |
|
— |
mez detekce může být stanovena na úrovni trojnásobku standardní odchylky slepého vzorku rozpouštědla nebo odezvy pozadí. Další možností je použít odezvu, která leží nad odezvou pozadí a vypočte se z kalibrační křivky sestrojené v daný den (pětinásobek odezvy slepého vzorku rozpouštědla). Mez kvantifikace může být stanovena na úrovni pěti- až šestinásobku standardní odchylky odezvy slepého vzorku rozpouštědla nebo odezvy pozadí nebo se použije odezva, která je nad odezvou pozadí a která se vypočte z kalibrační křivky sestrojené v daný den (desetinásobek odezvy slepého vzorku rozpouštědla). |
7.4 Specifické požadavky na biotesty s intaktními buňkami prováděné pomocí souprav
|
— |
musí být zaručeno, že biotesty prováděné pomocí souprav jsou dostatečně citlivé a spolehlivé pro použití u krmiv, |
|
— |
musí být dodržovány pokyny výrobce pro úpravu a zkoušení vzorků, |
|
— |
nesmí se používat zkušební soupravy, u nichž uplynula doba použitelnosti, |
|
— |
nesmí se používat materiály nebo součásti navržené pro použití s jinou soupravou, |
|
— |
zkušební soupravy se uchovávají v předepsaném rozsahu skladovacích teplot a používají se při předepsané pracovní teplotě, |
|
— |
mez detekce imunologických zkoušek se stanoví jako podíl hodnoty součtu střední odchylky a trojnásobku směrodatné odchylky odezvy na základě deseti opakovaných zkoušek slepého vzorku a hodnoty směrnice přímky získané lineární regresí, |
|
— |
pro kontrolu, zda odezva leží v přijatelném rozmezí, se při laboratorních zkouškách používají referenční standardy. |
8. Zpráva o výsledcích
Pokud to postup laboratorního zkoušení umožňuje, výsledky obsahují hodnoty jednotlivých kongenerů PCDD/F a PCB a výsledky zkoušky se musí oznamovat jako dolní, horní a střední odhad, aby zpráva o výsledcích obsahovala co nejvíce informací a aby tak výsledky mohly být vykládány v souladu se zvláštními požadavky.
Ve zprávě se také uvede obsah lipidů ve vzorku a použitá metoda extrakce lipidů.
Pokud výtěžnost leží mimo rozsah uvedený v bodě 6, je-li překročen maximální obsah nebo v ostatních případech na žádost, musí být dány k dispozici hodnoty výtěžnosti pro jednotlivé vnitřní standardy.
Protože má být při rozhodování o souladu vzorku přihlédnuto také k nejistotě měření, je dán k dispozici také tento parametr. Výsledek zkoušky se proto oznámí ve tvaru „x +/– U“, kde x je výsledek zkoušky a U je rozšířená nejistota měření, přičemž se použije faktor pokrytí 2, který odpovídá hladině spolehlivosti přibližně 95 %. V případě samostatného stanovení dioxinů a PCB s dioxinovým efektem se součet odhadované rozšířené nejistoty měření samostatných výsledků zkoušek u dioxinů a PCB s dioxinovým efektem musí použít pro součet dioxinů a PCB s dioxinovým efektem.
Je-li nejistota měření zohledněna uplatněním rozhodovací meze (CCα) (jak je popsáno v bodě I.2 části B), tento parametr se oznámí.
(1) Tabulka koeficientů toxické ekvivalence (TEF) pro dioxiny, furany a PCB s dioxinovým efektem:
|
Kongener |
Hodnota TEF |
Kongener |
Hodnota TEF |
|
Dibenzo-p-dioxiny („PCDD“) |
|
PCB „s dioxinovým efektem“: |
|
|
2,3,7,8-TCDD |
1 |
|
|
|
1,2,3,7,8-PeCDD |
1 |
Non-orto PCB |
|
|
1,2,3,4,7,8-HxCDD |
0,1 |
PCB 77 |
0,0001 |
|
1,2,3,6,7,8-HxCDD |
0,1 |
PCB 81 |
0,0001 |
|
1,2,3,7,8,9-HxCDD |
0,1 |
PCB 126 |
0,1 |
|
1,2,3,4,6,7,8-HpCDD |
0,01 |
PCB 169 |
0,01 |
|
OCDD |
0,0001 |
Mono-orto PCB |
|
|
|
|
PCB 105 |
0,0001 |
|
Dibenzofurany („PCDF“) |
|
PCB 114 |
0,0005 |
|
2,3,7,8-TCDF |
0,1 |
PCB 118 |
0,0001 |
|
1,2,3,7,8-PeCDF |
0,05 |
PCB 123 |
0,0001 |
|
2,3,4,7,8-PeCDF |
0,5 |
PCB 156 |
0,0005 |
|
1,2,3,4,7,8-HxCDF |
0,1 |
PCB 157 |
0,0005 |
|
1,2,3,6,7,8-HxCDF |
0,1 |
PCB 167 |
0,00001 |
|
1,2,3,7,8,9-HxCDF |
0,1 |
PCB 189 |
0,0001 |
|
2,3,4,6,7,8-HxCDF |
0,1 |
|
|
|
1,2,3,4,6,7,8-HpCDF |
0,01 |
|
|
|
1,2,3,4,7,8,9-HpCDF |
0,01 |
|
|
|
OCDF |
0,0001 |
|
|
|
Použité zkratky: „T“ = tetra; „Pe“ = penta; „Hx“ = hexa; „Hp“ = hepta; „O“ = okta; „CDD“ = chlorodibenzo-p-dioxin; „CDF“ = chlorodibenzofuran; „CB“ = chlorobifenyl. |
|||
(2) Úř. věst. L 140, 30.5.2002, s. 10.
(3) Koncepce „horního odhadu“ vyžaduje použití mezní hodnoty kvantifikace jakožto příspěvku každého nekvantifikovaného kongeneru do TEQ.
Pro výpočet „dolního odhadu“ se pro velikost každého příspěvku množstevně nestanoveného kongeneru k TEQ zvolí hodnota nula.
Koncepce „středního odhadu“ vyžaduje použití poloviny mezní hodnoty kvantifikace pro výpočet příspěvku každého nekvantifikovaného kongeneru do TEQ.
(4) Opakovaná zkouška je nutná k vyloučení možnosti vnitřní křížové kontaminace nebo náhodného promíchání vzorků. První zkouška zohledňující nejistotu měření se použije pro ověření souladu.
Je-li zkouška prováděna v rámci kontaminace dioxiny, lze od potvrzení opakovanou zkouškou upustit, pokud jsou vzorky vybrané pro zkoušku spojeny zpětnou sledovatelností s daným případem kontaminace dioxiny.
PŘÍLOHA VI
METODY ZKOUŠENÍ PRO STANOVENÍ SLOŽEK ŽIVOČIŠNÉHO PŮVODU PRO ÚŘEDNÍ KONTROLU KRMIV
Podmínky pro mikroskopické zjišťování, identifikaci nebo určování složek živočišného původu v krmivech
1. Cíl a oblast použití
Tyto podmínky se použijí, provádí-li se zjišťování složek živočišného původu (definovaných jako produkty ze zpracování těl a částí těl savců, drůbeže a ryb) v krmivech mikroskopickou zkouškou v rámci koordinovaného programu kontroly v oblasti výživy zvířat v souladu s nařízením Evropského parlamentu a Rady (ES) č. 882/2004 (1). Pokud se metody popsané v této příloze používají při všech úředně stanovených zkouškách, lze rovněž provést druhou zkoušku pomocí odlišných nebo alternativních metod, aby se lépe zjistily některé druhy složek živočišného původu nebo více zpřesnil původ těchto složek. Kromě toho lze použít odlišný protokol, pokud se zkoumají některé zvláštní složky živočišného původu, jako například plazma nebo kosti v loji (viz též bod 9), za předpokladu, že se tyto zkoušky provádějí jako doplněk ke zkouškám stanoveným v koordinovaném programu kontroly.
2. Citlivost
Podle druhu složek živočišného původu lze v krmivech zjistit velmi malá množství (< 0,1 %).
3. Princip
K identifikaci se používá reprezentativní vzorek odebraný v souladu s ustanoveními uvedenými v příloze I, jenž byl vhodným způsobem upraven. Pro manipulaci s krmivem o nízkém obsahu vlhkosti je vhodný následující protokol. Krmivo s obsahem vlhkosti vyšším než 14 % se před manipulací suší (zahušťuje). Některá krmiva a některé krmné suroviny (např. tuky a oleje) vyžadují speciální ošetření (viz bod 9). Složky živočišného původu jsou identifikovány na základě charakteristických, mikroskopicky rozeznatelných znaků (tj. svalových vláken a jiných masitých částic, chrupavek, kostí, rohů, chlupů, štětin, krve, peří, vaječných skořápek, rybích kostí, šupin). Identifikace musí být provedena jak v prosívané frakci (6.1), tak v koncentrovaném sedimentu (6.2) vzorku.
4. Chemikálie
4.1 Zalévací přípravek
|
4.1.1 |
Chloralhydrát (vodný, 60 % w/v). |
|
4.1.2 |
Louh (NaOH 2,5 % w/v nebo KOH 2,5 % w/v) pro prosívané frakce. |
|
4.1.3 |
Parafínový olej nebo glycerol (viskozita: 68–81) pro mikroskopická pozorování v sedimentu. |
4.2 Oplachovací činidla
|
4.2.1 |
Alkohol, 96 %. |
|
4.2.2 |
Aceton. |
4.3 Koncentrační přípravek
|
4.3.1 |
Tetrachloroethylen (hustota 1,62). |
4.4 Činidla k barvení
|
4.4.1 |
Roztok jódu v jodidu draselném (2 g jodidu draselného se rozpustí ve 100 ml vody a přidá se 1 g jódu za častého protřepávání). |
|
4.4.2 |
Alizarinová červeň (2,5 ml 1M kyseliny chlorovodíkové se rozpustí ve 100 ml vody a do tohoto roztoku se přidá 200 mg alizarinové červeně). |
|
4.4.3 |
Činidlo na cystin (2 g octanu olovnatého, 10 g NaOH/100 ml H2O). |
|
4.4.4 |
Roztok jódu v jodidu draselném (rozředěný v 70 % ethanolu). |
4.5 Bělicí činidlo
|
4.5.1 |
Komerční roztok hypochloritu sodného (9,6 % aktivního chlóru). |
5. Přístroje a pomůcky
|
5.1 |
Analytické váhy (přesnost 0,01 g, kromě vážení koncentrovaného sedimentu: 0,001 g). |
|
5.2 |
Rozmělňovací zařízení (mlýnek nebo hmoždíř, zejména pro zkoušení krmiv obsahujících více než 15 % tuku). |
|
5.3 |
Síto se čtvercovými oky o velikosti nejvýše 0,50 mm. |
|
5.4 |
Dělicí nálevka nebo usazovací kádinka s kónickým dnem. |
|
5.5 |
Stereoskopický mikroskop (s alespoň 40násobným zvětšením). |
|
5.6 |
Kombinovaný mikroskop (s alespoň 400násobným zvětšením), pro pozorování v procházejícím světle nebo s polarizačním zařízením. |
|
5.7 |
Standardní laboratorní sklo. Všechny přístroje a pomůcky musí být důkladně vyčištěny. Dělicí nálevky a sklo musí být umyty v myčce. Síta je třeba vyčistit kartáčem s tuhými štětinami. |
6. Postup
Peletované krmivo lze předběžně přesít, pokud se obě frakce zkoušejí jako samostatné vzorky.
Upraví se alespoň 50 g vzorku (v případě nutnosti se opatrně rozmělní pomocí vhodného rozmělňovacího zařízení (5.2), aby se dosáhlo vhodné struktury). Z rozmělněného materiálu se odeberou dvě reprezentativní části, jedna pro prosévané frakce (nejméně 5 g) (6.1) a jedna pro koncentrovaný sediment (nejméně 5 g) (6.2). Z důvodu identifikace lze dodatečně použít činidla k barvení (6.3).
Aby se zjistila povaha živočišných bílkovin a původ částeček, lze použít systém pro podporu rozhodování, například ARIES, a referenční vzorky.
6.1 Identifikace složek živočišného původu v prosévaných frakcích
Nejméně 5 g vzorku se proseje přes síto (5.3) na dvě frakce.
Prosetá (proseté) frakce s velkými částečkami (nebo reprezentativní část frakce) se nanese (nanesou) v tenké vrstvě na vhodnou podložku a pod stereoskopickým mikroskopem (5.5) se systematicky zkoumá (zkoumají) při různém zvětšení za účelem zjištění přítomnosti složek živočišného původu.
Podložní sklíčka s prosetou frakcí (prosetými frakcemi) obsahující jemné částečky se systematicky zkoumají pod kombinovaným mikroskopem (5.6) při různém zvětšení za účelem zjištění přítomnosti složek živočišného původu.
6.2 Identifikace složek živočišného původu v koncentrovaném sedimentu
Do dělicí nálevky nebo do usazovací kádinky s kónickým dnem se naváží nejméně 5 g (s přesností na 0,01 g) vzorku a přidá se alespoň 50 ml tetrachlorethylenu (4.3.1). Tato směs se opakovaně protřepe nebo promíchá.
|
— |
Je-li použita uzavřená dělicí nálevka, sediment se nechá dostatečně dlouho stát (alespoň tři minuty), než se oddělí. Opakuje se protřepání a sediment se opět nechá stát alespoň tři minuty. Sediment se znovu oddělí. |
|
— |
Je-li použita otevřená kádinka, sediment se před oddělením nechá stát alespoň pět minut. |
Veškerý sediment se vysuší a potom zváží (s přesností na 0,001 g). Vážení je nezbytné pouze tehdy, pokud je požadován odhad. Pokud sediment obsahuje mnoho velkých částeček, může se prosít sítem (5.3) na dvě frakce. Vysušený sediment se zkoumá pod stereoskopickým (5.5) a kombinovaným (5.6) mikroskopem za účelem zjištění přítomnosti částic kostí.
6.3 Použití zalévacích přípravků a činidel k barvení
Mikroskopická identifikace složek živočišného původu může být podpořena použitím speciálních zalévacích přípravků a činidel k barvení.
Chloralhydrát (4.1.1): při opatrném ohřevu lze buněčné struktury vidět zřetelněji, protože zrna škrobu rosolovatí a nežádoucí obsah buněk je odstraněn.
Louh (4.1.2): hydroxid sodný nebo hydroxid draselný čistí materiál krmiva a napomáhá detekci svalových vláken, chlupů a jiných keratinových struktur.
Parafínový olej a glycerol (4.1.3): částice kostí lze tímto zalévacím přípravkem dobře identifikovat, protože většina lakun zůstane naplněna vzduchem a jeví se jako černé otvory o velikosti 5–15 μm.
Roztok jódu v jodidu draselném (4.4.1): používá se pro detekci škrobu (modrofialová barva) a bílkoviny (žlutooranžová barva). Roztoky mohou být zředěny, pokud je třeba.
Roztok alizarinové červeně (4.4.2): červené/růžové zabarvení kostí, rybích kostí a šupin. Před vysušením sedimentu (viz bod 6.2) se veškerý sediment převede do skleněné zkumavky a dvakrát vypláchne asi 5 ml alkoholu (4.2.1) (vždy se použije ponorný mixér, rozpouštědlo se nechá usadit asi jednu minutu a odlije se). Před použitím tohoto činidla k barvení se sediment vybělí přidáním nejméně 1 ml roztoku hypochloritu sodného (4.5.1). Nechá se reagovat 10 minut. Zkumavka se naplní vodou, sediment se nechá usadit 2–3 minuty a voda se suspendovanými částečkami se odlije. Sediment se ještě dvakrát propláchne asi 10 ml vody (vždy se použije ponorný mixér, nechá se usadit a voda se odlije). Přidá se 2–10 kapek či více roztoku alizarinové červeně (v závislosti na množství zbytku). Směs se protřepe a nechá se několik sekund reagovat. Zbarvený sediment se dvakrát propláchne asi 5 ml alkoholu (4.2.1) a pak jednou acetonem (4.2.2) (vždy se použije ponorný mixér, rozpouštědlo se nechá usadit asi jednu minutu a odlije se). Poté je sediment připraven k vysušení.
Činidlo na cystin (4.4.3): při opatrném ohřevu se částečky obsahující cystin (chlupy, peří atd.) zbarví do černohněda.
6.4 Zkoumání možné přítomnosti rybí moučky v krmivu
Pod kombinovaným mikroskopem se zkoumá alespoň jedno podložní sklíčko s jemnou prosévanou frakcí a jemnou frakcí sedimentu (viz body 6.1 a 6.2).
Pokud je na etiketě uvedeno, že složky zahrnují rybí moučku, nebo v případě podezření na přítomnost rybí moučky nebo jejího zjištění při prvním zkoumání, zkoumají se alespoň dvě další podložní sklíčka s jemnou prosévanou frakcí z původního vzorku a s celkovou frakcí sedimentu.
7. Výpočet a vyhodnocení
Členské státy zajistí, aby se postupy popsané v tomto bodě používaly vždy, když se provádí úřední zkouška za účelem určení množství (nikoli jen přítomnosti) složek živočišného původu.
Výpočet lze provést pouze tehdy, jsou-li ve složkách živočišného původu obsaženy částice kostí.
Částice kostí teplokrevných suchozemských druhů (tj. savců a ptáků) se v mikroskopickém preparátu odlišují od rybích kostí charakteristickými lakunami. Určení podílu složek živočišného původu ve vzorku se provádí s přihlédnutím k:
|
— |
odhadovanému podílu ( % hmotnosti) částic kostí v koncentrovaném sedimentu a |
|
— |
podílu ( % hmotnosti) kostí ve složkách živočišného původu. |
Určování se provádí na základě (pokud možno) nejméně tří preparátů a nejméně pěti polí na preparát. Koncentrovaný sediment u krmných směsí zpravidla obsahuje kromě částic kostí suchozemských zvířat a částic rybích kostí i jiné částice vysoké specifické hmotnosti, například minerály, písek, zdřevnatělé úlomky rostlin apod.
7.1 Určení procentuálního podílu kostních částic
částice kostí suchozemských zvířat (%) = (S × c)/W
částice rybích kostí a šupin (%) = (S × d)/W
|
(S = |
hmotnost sedimentu (mg), c = opravný koeficient (%) pro odhadovaný podíl kostí suchozemských živočichů v sedimentu, d = opravný koeficient (%) pro odhadovaný podíl částic rybích kostí a šupin v sedimentu, W = navážka vzorku pro sedimentaci (mg)). |
7.2 Určení podílu složek živočišného původu
Podíl kostí v produktech živočišného původu se může značně lišit (procentuální podíl kostí je v případě kostní moučky řádově 50–60 % a v případě masových mouček řádově 20–30 %; v případě rybích mouček se obsah kostí a šupin liší podle kategorie a původu rybí moučky a obvykle se pohybuje v řádu 10–20 %).
Je-li znám druh živočišné moučky ve vzorku, lze podíl odhadnout:
Odhadovaný podíl složek produktů ze suchozemských živočichů (%) = (S × c)/(W × f) × 100
Odhadovaný podíl složek rybích produktů (%) = (S × d)/(W × f) × 100
|
(S = |
hmotnost sedimentu (mg), c = opravný koeficient (%) pro odhadovaný podíl kostí suchozemských živočichů v sedimentu, d = opravný koeficient (%) pro odhadovaný podíl částic rybích kostí a šupin v sedimentu, f = opravný koeficient pro podíl kostí ve složkách živočišného původu ve zkoušeném vzorku, W = navážka vzorku pro sedimentaci (mg)). |
8. Vyjádření výsledku zkoušky
Zpráva musí obsahovat přinejmenším informace o přítomnosti složek ze suchozemských živočichů a z rybí moučky. Různé nálezy lze vyjádřit takto:
|
8.1 |
Pokud jde o přítomnost složek ze suchozemských živočichů:
|
|
8.2 |
A pokud jde o přítomnost rybí moučky:
V případě zjištění složek z ryb nebo ze suchozemských živočichů může být v případě nutnosti ve zprávě o zkoušce uveden odhad množství zjištěných složek (x %, < 0,1 %, 0,1–0,5 %, 0,5–5 % nebo > 5 %) a upřesněn druh suchozemských živočichů (je-li to možné) a zjištěné živočišné složky (svalová vlákna, chrupavky, kosti, rohovina, chlupy, štětiny, peří, krev, vaječné skořápky, rybí kosti, šupiny). Je-li uveden odhad množství složek živočišného původu, uvede se rovněž použitý opravný koeficient f. Pokud byly prokázány částice kostí suchozemských živočichů, obsahuje zpráva tento dodatek: „Nelze vyloučit možnost, že výše uvedené složky pocházejí ze savců.“ Tento dodatek není nutný v případech, kdy byly částice kostí suchozemských živočichů specifikovány jako částice kostí drůbeže nebo savců. |
9. Nepovinný protokol pro zkoušení tuků nebo olejů
Pro zkoušení tuků nebo olejů lze použít tento protokol:
|
— |
je-li tuk v pevném stavu, ohřívá se například v mikrovlnné troubě, dokud není kapalný, |
|
— |
pomocí pipety se přenese 40 ml tuku ze spodní části vzorku do odstředivkové zkumavky, |
|
— |
odstřeďuje se po dobu 10 minut při 4 000 ot./min, |
|
— |
pokud tuk při odstřeďování ztuhl, znovu se ohřeje v troubě, dokud není kapalný. Opakuje se odstřeďování po dobu 5 minut při 4 000 ot./min, |
|
— |
pomocí lžičky nebo stěrky se polovina dekantovaných nečistot přenese na malou Petriho misku nebo na podložní sklíčko pro mikroskopické zkoumání za účelem zjištění možného obsahu živočišných složek (svalových vláken, peří, částic kostí atd.). Jako zalévací přípravek pro mikroskopické zkoumání se doporučuje použít parafínový olej nebo glycerol, |
|
— |
zbylé nečistoty se použijí pro sedimentaci podle bodu 6.2. |
PŘÍLOHA VII
METODA VÝPOČTU OBSAHU ENERGIE U KRMNÝCH SMĚSÍ PRO DRŮBEŽ
1. Metoda výpočtu a vyjádření obsahu energie
Obsah energie krmných směsí pro drůbež se vypočítá podle níže uvedeného vzorce na základě procentního podílu určitých analytických složek krmiva. Tato hodnota se vyjadřuje v megajoulech (MJ) metabolizovatelné energie (ME) s korekcí na dusík na jeden kilogram krmné směsi:
MJ/kg ME = 0,1551 × % hrubého proteinu +0,3431 × % tuku +0,1669 × % škrobu +0,1301 × % veškerého cukru (vyjádřeného jako sacharóza).
2. Tolerance pro deklarované obsahy
Jestliže úřední kontroly zjistí odchylku mezi výsledkem kontroly a deklarovaným obsahem energie znamenající zvýšení nebo snížení obsahu energie krmiva, povoluje se minimální přípustná odchylka 0,4 MJ/kg metabolizovatelné energie.
3. Vyjádření výsledku
Výsledek vypočítaný podle výše uvedeného vzorce se uvádí s přesností na jedno desetinné místo.
4. Metody odběru vzorků a metody laboratorního zkoušení
Odběr vzorku krmné směsi a stanovení obsahu analytických složek uvedených ve výpočetní metodě se provádí podle metod odběru vzorků a metod laboratorního zkoušení Společenství pro úřední kontrolu krmiv.
Použije se:
|
— |
ke stanovení obsahu tuku: postup B metody pro stanovení obsahu tuku uvedené v části H přílohy III, |
|
— |
ke stanovení obsahu škrobu: polarimetrická metoda uvedená v části L přílohy III. |
PŘÍLOHA VIII
METODY ZKOUŠENÍ PRO KONTROLU NEPŘÍPUSTNÉ PŘÍTOMNOSTI JIŽ NEPOVOLENÝCH DOPLŇKOVÝCH LÁTEK V KRMIVECH
DŮLEŽITÉ UPOZORNĚNÍ:Pro zjištění nepřípustné přítomnosti již nepovolených doplňkových látek v krmivech lze použít citlivější metody zkoušení, než jsou metody zkoušení uvedené v této příloze.Metody zkoušení uvedené v této příloze se použijí pro účely potvrzování.
A. STANOVENÍ OBSAHU METHYLBENZOCHÁTU
2-methyl-7-benzyl-oxy-6-butyl-1,4-dihydro-4-oxychinolin-3-karboxylát
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu methylbenzochátu v krmivech. Mez kvantifikace je 1 mg/kg.
2. Princip
Methylbenzochát se extrahuje ze vzorku pomocí methanolového roztoku kyseliny methansulfonové. Extrakt se přečistí dichlormethanem, ionexovou chromatografií, a následně pak znovu dichlormethanem. Obsah methylbenzochátu se stanoví metodou vysokoúčinné kapalinové chromatografie (HPLC) na reverzní fázi s UV detekcí.
3. Chemikálie
|
3.1 |
Dichlormethan. |
|
3.2 |
Methanol, pro HPLC. |
|
3.3 |
Mobilní fáze HPLC. Směs methanolu (3.2) a vody (pro HPLC) 75 + 25 (v + v). Přefiltruje se filtrem 0,22 μm (4.5) a roztok se odplyní (např. vystaví se ultrazvuku po dobu 10 minut). |
|
3.4 |
Roztok kyseliny methansulfonové, c = 2 %. 20 ml kyseliny methansulfonové se zředí v 1 000 ml methanolu (3.2). |
|
3.5 |
Roztok kyseliny chlorovodíkové, c = 10 %. 100 ml kyseliny chlorovodíkové (ρ201,18 g/ml) se zředí v 1 000 ml vody. |
|
3.6 |
Ionex Amberlit CG – 120 (Na), 100–200 mesh. 100 g ionexu se rozmíchá v 500 ml roztoku kyseliny chlorovodíkové (3.5) a za stálého míchání se zahřeje na topné desce k varu. Po ochlazení se kyselina dekantuje. Filtruje se přes papírový filtr za vakua. Ionex se propláchne dvakrát 500 ml vody a potom 250 ml methanolu (3.2). Potom se ionex propláchne dalšími 250 ml methanolu a vysuší se prosáváním vzduchu přes filtrační koláč. Vysušený ionex se uchovává v uzavřené láhvi. |
|
3.7 |
Standardní látka: čistý methylbenzochát (2-methyl-7-benzyl-oxy-6-butyl-1,4-dihydro-4-oxychinolin-3-karboxylát). |
|
3.7.1 |
Naváží se 50 mg standardní látky (3.7) s přesností na 0,1 mg, rozpustí se ve 100 ml odměrné baňce v roztoku kyseliny methansulfonové (3.4), doplní se jí po značku a promíchá. |
|
3.7.2 |
Do 50 ml odměrné baňky se odpipetuje 5,0 ml základního standardního roztoku methylbenzochátu (3.7.1), doplní se methanolem (3.2) po značku a promíchá. |
|
3.7.3 |
Do sady 25 ml odměrných baněk se odpipetuje 1,0, 2,0, 3,0, 4,0 a 5,0 ml pracovního standardního roztoku methylbenzochátu (3.7.2). Doplní se po značku mobilní fází (3.3) a promíchá. Tyto roztoky mají koncentrace 2,0, 4,0, 6,0, 8,0 a 10,0 μg/ml methylbenzochátu. Tyto roztoky se připravují čerstvé před použitím. |
4. Přístroje a pomůcky
|
4.1 |
Laboratorní míchací zařízení. |
|
4.2 |
Rotační odparka. |
|
4.3 |
Skleněná kolona (250 mm × 15 mm) vybavená uzavíracím kohoutkem a nádržkou s kapacitou asi 200 ml. |
|
4.4 |
Zařízení HPLC s ultrafialovým detektorem s nastavitelnou vlnovou délkou nebo s detektorem diodového pole. |
|
4.4.1 |
Kolona pro kapalinovou chromatografii: 300 mm × 4 mm, C18, náplň 10 μm nebo obdobná. |
|
4.5 |
Membránové filtry, 0,22 μm. |
|
4.6 |
Membránové filtry, 0,45 μm. |
5. Postup
5.1 Obecné pokyny
|
5.1.1 |
Provede se zkouška slepého vzorku krmiva, aby se prokázalo, že v něm není přítomen ani methylbenzochát, ani jiné rušivé látky. |
|
5.1.2 |
Provede se zkouška na výtěžnost u slepého vzorku krmiva, ke kterému bylo přidáno takové množství methylbenzochátu, které odpovídá množství ve vzorku. Pro obohacení na úroveň 15 mg/kg se přidá ke 20 g slepého vzorku krmiva 600 μl základního standardního roztoku (3.7.1), promíchá se a po 10 minutách se začne s extrakcí (5.2). Poznámka: slepý vzorek krmiva pro účel této metody je podobného typu jako vzorek a při jeho zkoušce nesmí být zjištěn methylbenzochát. |
5.2 Extrakce
Do 250 ml kónické baňky se naváží s přesností na 0,01 g asi 20 g upraveného vzorku. Přidá se 100 ml roztoku kyseliny methansulfonové (3.4) a mechanicky se třepe (4.1) po dobu 30 minut. Roztok se přefiltruje přes papírový filtr a filtrát se použije na extrakci kapalina-kapalina (5.3).
5.3 Extrakce kapalina-kapalina
Do 500 ml dělicí nálevky obsahující 100 ml roztoku kyseliny chlorovodíkové (3.5) se přidá 25,0 ml filtrátu, který byl získán postupem uvedeným v bodě 5.2. Dále se do nálevky přidá 100 ml dichlormethanu (3.1) a třepe po dobu jedné minuty. Potom se nechají vrstvy separovat a spodní vrstva (dichlormethanu) se odpustí do 500 ml baňky s kulatým dnem. Extrakce vodné fáze se opakuje se dvěma dalšími 40 ml dávkami dichlormethanu a ty se spojí s prvním extraktem v baňce s kulatým dnem. Extrakt dichlormethanu se odpaří do sucha na rotační odparce (4.2) při sníženém tlaku a teplotě 40 oC. Odparek se rozpustí v 20–25 ml methanolu (3.2), baňka se zazátkuje a celý extrakt se použije pro chromatografii s iontovou výměnou (5.4).
5.4 Chromatografie s iontovou výměnou
5.4.1
Do dolního konce skleněné kolony (4.3) se vloží tampon ze skelné vaty. Připraví se suspenze z 5,0 g upraveného ionexu (3.6) a z 50 ml kyseliny chlorovodíkové (3.5). Tato směs se nalije do skleněné kolony a nechá se usadit. Přebytečná kyselina se odpustí až těsně nad hladinu ionexu a kolona se promývá vodou, dokud proteklý eluát není neutrální na lakmusový papír. Potom se do kolony se nalije 50 ml methanolu (3.2) a nechá se stéci až k povrchu ionexu.
5.4.2
Na kolonu se opatrně napipetuje získaný extrakt (5.3). Baňka s kulatým dnem se vypláchne dvěma dávkami 5–10 ml methanolu (3.2) a výplachy se také převedou na kolonu. Kolona se promyje 50 ml methanolu tak, aby průtok nepřesahoval 5 ml za minutu. Proteklá kapalina se odstraní. Methylbenzochát se eluuje z kolony 150 ml roztoku kyseliny methansulfonové (3.4) a eluát se jímá do 250 ml kónické baňky.
5.5 Extrakce kapalina-kapalina
Získaný eluát (5.4.2) se převede do 1 000 ml dělicí nálevky. Kónická baňka se propláchne 5–10 ml methanolu (3.2) a výplach se přidá do dělicí nálevky. Dále se do dělicí nálevky přidá 300 ml roztoku kyseliny chlorovodíkové (3.5) a 130 ml dichlormethanu (3.1). Třepe se po dobu jedné minuty a potom se nechají fáze separovat. Spodní vrstva (dichlormethanu) se odpustí do 500 ml baňky s kulatým dnem. Extrakce vodné fáze se opakuje se dvěma dalšími 70 ml dávkami dichlormethanu a tyto extrakty se smísí s prvním extraktem v baňce s kulatým dnem.
Extrakt dichlormethanu se odpaří do sucha v rotační odparce (4.2) při sníženém tlaku a teplotě 40 oC. Odparek v baňce se rozpustí v přibližně 5 ml methanolu (3.2) a tento roztok se kvantitativně převede do 10 ml odměrné baňky. Baňka s kulatým dnem se vypláchne dvěma dalšími dávkami 1–2 ml methanolu a opět se převede do odměrné baňky. Baňka se doplní po značku methanolem a promíchá. Alikvotní část se přefiltruje přes membránový filtr (4.6). Tento roztok se použije pro stanovení HPLC (5.6).
5.6 Stanovení HPLC
5.6.1
Následující podmínky jsou doporučené, mohou být použity jiné podmínky za předpokladu, že poskytují rovnocenné výsledky:
|
— |
kolona pro kapalinovou chromatografii (4.4.1), |
|
— |
mobilní fáze HPLC: směs methanolu a vody (3.3), |
|
— |
průtok: 1–1,5 ml/min, |
|
— |
vlnová délka detektoru: 265 nm, |
|
— |
objem nástřiku: 20–50 μl. |
Stabilita chromatografického systému se kontroluje opakovaným nástřikem kalibračního roztoku (3.7.3) obsahujícího 4 μg/ml, dokud se nedosáhne konstantních výšek nebo ploch píků a konstantních retenčních časů.
5.6.2
Provede se několikrát nástřik každého kalibračního roztoku (3.7.3) a změří výšky (plochy) píků pro každou koncentraci. Kalibrační křivka se vytvoří vynesením průměrných hodnot výšek nebo ploch píků kalibračních roztoků proti příslušným koncentracím v μg/ml.
5.6.3
Opakovaně se nastřikuje extrakt vzorku (5.5) s použitím stejného objemu, jaký byl použit pro kalibrační roztoky, a určí se průměrná hodnota výšky (plochy) píků methylbenzochátu.
6. Výpočet a vyjádření výsledků
Z průměrných hodnot výšek (ploch) píků methylbenzochátu v roztoku vzorku se stanoví koncentrace v roztoku vzorku v μg/ml porovnáním s kalibrační křivkou (5.6.2).
Obsah methylbenzochátu (w) ve vzorku v mg/kg se vypočítá podle následujícího vzorce:
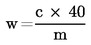
kde:
|
c |
= |
koncentrace methylbenzochátu v roztoku vzorku v g/ml |
|
m |
= |
hmotnost navážky vzorku v gramech. |
7. Ověření výsledků
7.1 Identita
Identita analytu může být potvrzena opakovaným stanovením s přídavkem nebo pomocí detektoru diodového pole, který umožňuje porovnání spektra extraktu vzorku se spektrem kalibračního roztoku (3.7.3) obsahujícího 10 μg/ml methylbenzochátu.
7.1.1
Extrakt vzorku se obohatí přidáním vhodného množství pracovního standardního roztoku (3.7.2). Množství přidaného methylbenzochátu musí být podobné jako odhadovaný obsah methylbenzochátu v extraktu vzorku.
Přídavek se projeví pouze zvýšením píku methylbenzochátu, a to úměrně k přidanému množství a ředění extraktu. Šířka píku v polovině maximální výšky se může odchylovat od šířky původního píku nejvýše o ±10 %.
7.1.2
Výsledky jsou posuzovány podle následujících kritérií:
|
a) |
při vlnové délce maximální absorpce vzorku i standardu musí být záznam spektra zaznamenaný ve vrcholu píku na chromatogramu stejný s přesností danou rozlišovací schopností detekčního systému. Pro detektor diodového pole se udává obvykle ±2 nm; |
|
b) |
při vlnové délce mezi 220 a 350 nm se nesmí záznam spektra ve zkoušeném vzorku a standardu zaznamenaný ve vrcholu píku chromatogramu lišit od ostatních částí spektra v rozsahu 10–100 % relativní absorbance. Tohoto kritéria je dosaženo, jestliže jsou vykázána stejná maxima a jestliže odchylka mezi dvěma spektry nikde nepřesahuje 15 % absorbance standardního analytu; |
|
c) |
při vlnové délce mezi 220 a 350 nm se nesmí spektrum vzestupné části, vrcholu a sestupné části píku zkoušeného vzorku lišit od ostatních v této částí spektra v rozsahu 10–100 % relativní absorbance. Tohoto kritéria je dosaženo, jestliže jsou vykázána stejná maxima a jestliže ve všech sledovaných bodech odchylka mezi spektry nikde nepřesahuje 15 % absorbance spektra ve vrcholu píku. |
Jestliže některé z těchto kritérií není splněno, přítomnost analytu není potvrzena.
7.2 Opakovatelnost
Rozdíl mezi výsledky dvou paralelních stanovení provedených u stejného vzorku nesmí překročit: 10 % relat. z hodnoty vyššího výsledku při obsahu methylbenzochátu od 4,0 do 20 mg/kg.
7.3 Výtěžnost
Výtěžnost pro obohacený vzorek musí být nejméně 90 %.
8. Výsledky kruhových testů
V deseti laboratořích bylo zkoušeno pět vzorků. U každého vzorku byly provedeny dvě zkoušky.
|
|
Slepý vzorek krmiva |
Sypké krmivo 1 |
Pelety 1 |
Sypké krmivo 2 |
Pelety 2 |
|
průměr (mg/kg) |
ND |
4,50 |
4,50 |
8,90 |
8,70 |
|
sr (mg/kg) |
— |
0,30 |
0,20 |
0,60 |
0,50 |
|
CVr (%) |
— |
6,70 |
4,40 |
6,70 |
5,70 |
|
sR (mg/kg) |
— |
0,40 |
0,50 |
0,90 |
1,00 |
|
CVR (%) |
— |
8,90 |
11,10 |
10,10 |
11,50 |
|
Výtěžnost (%) |
— |
92,00 |
93,00 |
92,00 |
89,00 |
|
ND |
= |
nezjištěno |
|
sr |
= |
standardní odchylka opakovatelnosti |
|
CVr |
= |
variační koeficient opakovatelnosti v % |
|
sR |
= |
standardní odchylka reprodukovatelnosti |
|
CVR |
= |
variační koeficient reprodukovatelnosti v %. |
B. STANOVENÍ OLACHINDOXU
2-[N-2'-(hydroxyethyl)karbamoyl]-3-methylchinoxalin-N1,N4-dioxid
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu olachindoxu v krmivech. Mez kvantifikace je 5 mg/kg.
2. Princip
Vzorek se extrahuje směsí vody a methanolu. Obsah olachindoxu se stanoví vysokoúčinnou kapalinovou chromatografií (HPLC) na reverzní fázi za použití UV detektoru.
3. Chemikálie
|
3.1 |
Methanol. |
|
3.2 |
Methanol, pro HPLC. |
|
3.3 |
Voda, pro HPLC. |
|
3.4 |
Mobilní fáze pro HPLC. Směs vody (3.3) a methanolu (3.2), 900 + 100 (V + V). |
|
3.5 |
Standardní látka: čistý olachindox 2-[N-2'-(hydroxyethyl)karbomoyl]-3-methylchinoxalin-N1,N4-dioxid, E 851. |
|
3.5.1 |
Do 200 ml odměrné baňky se naváží 50 mg olachindoxu (3.5) s přesností na 0,1 mg a přidá se asi 190 ml vody. Baňka se postaví na 20 minut do ultrazvukové lázně (4.1). Po vyjmutí z ultrazvukové lázně se roztok ochladí na laboratorní teplotu, doplní vodou po značku a promíchá. Baňka se obalí hliníkovou fólií a uschová v chladničce. Tento roztok se musí připravovat čerstvý každý měsíc. |
|
3.5.2 |
10,0 ml základního standardního roztoku (3.5.1) se převede do 100 ml odměrné baňky, doplní se po značku mobilní fází (3.4) a promíchá. Baňka se obalí hliníkovou fólií a uschová v chladničce. Tento roztok se musí připravovat denně čerstvý. |
|
3.5.3 |
Do sady 50 ml odměrných baněk se odpipetuje 1,0, 2,0, 5,0, 10,0, 15,0 a 20,0 ml pracovního standardního roztoku (3.5.2). Doplní se po značku mobilní fází (3.4) a promíchá. Baňky se obalí hliníkovou fólií. Tyto roztoky odpovídají 0,5, 1,0, 2,5, 5,0, 7,5 a 10,0 μg/ml olachindoxu. Tyto roztoky se musí připravovat denně čerstvé. |
4. Přístroje a pomůcky
|
4.1 |
Ultrazvuková lázeň. |
|
4.2 |
Mechanické míchací zařízení. |
|
4.3 |
Zařízení HPLC s UV detektorem s nastavitelnou vlnovou délkou nebo s detektorem diodového pole. |
|
4.3.1 |
Kolona pro kapalinovou chromatografii, 250 mm × 4 mm, C18, náplň 10 μm nebo obdobná. |
|
4.4 |
Membránové filtry, 0,45 μm. |
5. Postup
|
Poznámka |
: |
Olachindox je citlivý na světlo. Všechny postupy se musí provádět při tlumeném světle nebo v nádobách z tmavého skla. |
5.1 Obecné pokyny
|
5.1.1 |
Provede se zkouška slepého vzorku krmiva pro kontrolu, zda neobsahuje olachindox nebo jiné rušivé látky. |
|
5.1.2 |
Zkouška na výtěžnost se provede na slepém vzorku krmiva, ke kterému bylo přidáno takové množství olachindoxu, které odpovídá obsahu ve vzorku. Pro obohacení na úroveň 50 mg/kg se odpipetuje 10,0 ml základního standardního roztoku (3.5.1) do 250 ml kónické baňky a roztok se odpaří na objem asi 0,5 ml. Přidá se 50 g slepého vzorku krmiva, řádně se promíchá a nechá se 10 minut odstát, přičemž se ještě několikrát promíchá. Dále se pokračuje s extrakcí podle bodu 5.2.
|
5.2 Extrakce
Naváží se přibližně 50 g vzorku s přesností na 0,01 g. Přenese se do kónické baňky o obsahu 1 000 ml, přidá se 100 ml methanolu (3.1) a baňka se umístí na pět minut do ultrazvukové lázně (4.1). Přidá se 410 ml vody a ponechá se v ultrazvukové lázni dalších 15 minut. Baňka se vyjme z ultrazvukové lázně, dále se třepe 30 minut na míchacím zařízení (4.2) a potom se přefiltruje složeným filtrem. 10,0 ml filtrátu se přenese do 20 ml odměrné baňky, doplní se vodou po značku a promíchá. Poměrná část se přefiltruje membránovým filtrem (4.4). (viz bod 9 Poznámka). Dále se pokračuje se stanovením HPLC (5.3).
5.3 Stanovení HPLC
5.3.1
Následující podmínky jsou doporučené, mohou být použity jiné podmínky za předpokladu, že poskytují rovnocenné výsledky:
|
Analytická kolona (4.3.1) |
směs vody (3.3) a methanolu (3.2), 900 + 100 (V + V) |
|
Mobilní fáze (3.4): |
|
|
Průtok: |
1,5–2 ml/min |
|
Detekční vlnová délka: |
380 nm |
|
Objem nástřiku: |
20 μl–100 μl |
Stabilita chromatografického systému se kontroluje opakovaným nástřikem kalibračního roztoku (3.5.3), který obsahuje 2,5 μg/ml, dokud se nedosáhne konstantních výšek píků a konstantních retenčních časů.
5.3.2
Provede se několikrát nástřik každého kalibračního roztoku (3.5.3) a změří se průměrná hodnota výšky (plochy) píků pro každou koncentraci. Kalibrační křivka se vytvoří vynesením průměrných hodnot výšek (ploch) píků kalibračních roztoků proti příslušným koncentracím v μg/ml.
5.3.3
Provede se několik nástřiků extraktu vzorku (5.2) při použití stejného objemu jako u kalibračních roztoků a změří se průměrná hodnota výšky (plochy) píků olachindoxu.
6. Výpočet a vyjádření výsledků
Z průměrné hodnoty výšky (plochy) píků olachindoxu v roztoku vzorku se stanoví koncentrace v roztoku vzorku v μg/ml porovnáním s kalibrační křivkou (5.3.2).
Obsah olachindoxu ve vzorku (w) v mg/kg se vypočítá podle následujícího vzorce:
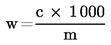
kde:
|
c |
= |
koncentrace olachindoxu v extraktu vzorku (5.2) v μg/ml |
|
m |
= |
hmotnost navážky vzorku v g (5.2). |
7. Ověření výsledků
7.1 Identita
Identita analytu může být potvrzena opakovaným stanovením s přídavkem (co-chromatography) nebo využitím detektoru diodového pole, přičemž se porovnávají spektra extraktu vzorku (5.2) a kalibračního roztoku (3.5.3), obsahujícího 5,0 μg/ml.
7.1.1
Extrakt vzorku (5.2) se obohatí přídavkem přiměřeného množství kalibračního roztoku (3.5.3). Množství přidaného olachindoxu musí odpovídat množství olachindoxu nalezeného v extraktu vzorku.
Přídavek se projeví pouze zvýšením píku olachindoxu, a to úměrně k přidanému množství a ředění extraktu. Šířka píku v polovině jeho výšky musí být v rozmezí ±10 % původní šířky píku olachindoxu v neobohaceném extraktu vzorku.
7.1.2
Výsledky jsou posuzovány podle následujících kritérií:
|
a) |
při vlnové délce maximální absorpce vzorku i standardu musí být záznam spektra zaznamenaný ve vrcholu píku na chromatogramu stejný s nepřesností danou rozlišovací schopností detekčního systému. Pro detektor diodového pole se udává obvykle ±2 nm; |
|
b) |
při vlnové délce mezi 220 a 400 nm se nesmí záznam spektra zkoušeného vzorku a standardu zaznamenaný ve vrcholu píku chromatogramu lišit od ostatních částí spektra v rozsahu 10–100 % relativní absorbance. Tohoto kritéria je dosaženo, jestliže jsou vykázána stejná maxima a jestliže odchylka mezi dvěma spektry nikde nepřesahuje 15 % absorbance standardního analytu; |
|
c) |
při vlnové délce mezi 220 a 400 nm se nesmí spektrum vzestupné části, vrcholu a sestupné části píku zkoušeného vzorku lišit od ostatních částí spektra v rozsahu 10–100 % relativní absorbance. Tohoto kritéria je dosaženo, jestliže jsou vykázána stejná maxima a jestliže ve všech sledovaných bodech odchylka mezi spektry nepřesahuje nikde 15 % absorbance spektra ve vrcholu píku. |
Jestliže některé z těchto kritérií není splněno, přítomnost analytu není potvrzena.
7.2 Opakovatelnost
Rozdíl mezi výsledky dvou paralelních stanovení provedených na stejném vzorku nesmí přesáhnout 15 % relat. z vyššího výsledku při obsahu olachindoxu mezi 10 a 200 mg/kg.
7.3 Výtěžnost
Výtěžnost pro obohacený slepý vzorek krmiva musí být nejméně 90 %.
8. Výsledky kruhových testů
V rámci ES byly provedeny kruhové testy, při nichž až 13 laboratoří zkoušelo čtyři vzorky krmiva pro selata, včetně jednoho slepého vzorku. Výsledky jsou uvedeny v následující tabulce:
|
|
Vzorek 1 |
Vzorek 2 |
Vzorek 3 |
Vzorek 4 |
|
L |
13 |
10 |
11 |
11 |
|
n |
40 |
40 |
44 |
44 |
|
průměr (mg/kg) |
— |
14,6 |
48,0 |
95,4 |
|
Sr (mg/kg) |
— |
0,82 |
2,05 |
6,36 |
|
SR (mg/kg) |
— |
1,62 |
4,28 |
8,42 |
|
CVr (%) |
— |
5,6 |
4,3 |
6,7 |
|
CVR (%) |
— |
11,1 |
8,9 |
8,8 |
|
Deklarovaný obsah |
|
|
|
|
|
(mg/kg) |
— |
15 |
50 |
100 |
|
výtěžnost % |
— |
97,3 |
96,0 |
95,4 |
|
L |
= |
počet laboratoří |
|
n |
= |
počet jednotlivých hodnot |
|
Sr |
= |
standardní odchylka opakovatelnosti |
|
SR |
= |
standardní odchylka reprodukovatelnosti |
|
CVr |
= |
variační koeficient opakovatelnosti |
|
CVR |
= |
variační koeficient reprodukovatelnosti. |
9. Poznámka
Ačkoliv tato metoda nebyla ověřena pro krmiva obsahující více než 100 mg/kg olachindoxu, je možné obdržet uspokojivé výsledky menší navážkou vzorku a/nebo zředěním extraktu (5.2) tak, aby se dosáhlo koncentrace v rozsahu kalibrační křivky (5.3.2).
C. STANOVENÍ OBSAHU AMPROLIA
1-[(4-amino-2-propyl-pyrimidinyl)-metyl]-2-picoliniumchlorid hydrochlorid
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu amprolia v krmivech a premixech. Mez detekce je 1 mg/kg, mez kvantifikace je 5 mg/kg.
2. Princip
Vzorek se extrahuje směsí methanolu a vody. Po zředění mobilní fází a membránové filtraci se obsah amprolia stanoví vysokoúčinnou kapalinovou chromatografií (HPLC) s výměnou kationtů a UV detekcí.
3. Chemikálie
|
3.1 |
Methanol. |
|
3.2 |
Acetonitril, pro HPLC. |
|
3.3 |
Voda, pro HPLC. |
|
3.4 |
Roztok dihydrogenfosforečnanu sodného, c = 0,1 mol/l. 13,80 g dihydrogenfosforečnanu sodného se rozpustí ve vodě (3.3) v odměrné baňce o objemu 1 000 ml, doplní se vodou (3.3) po značku a promíchá. |
|
3.5 |
Roztok chloristanu sodného, c = 1,6 mol/l. 224,74 g chloristanu sodného se rozpustí ve vodě (3.3) v odměrné baňce o objemu 1 000 ml, doplní se vodou (3.3) po značku a promíchá. |
|
3.6 |
Mobilní fáze pro HPLC (viz poznámka 9.1). Směs acetonitrilu (3.2), roztoku dihydrogenfosforečnanu sodného (3.4) a roztoku chloristanu sodného (3.5), 450 + 450 + 100 (v + v + v). Roztok se před použitím přefiltruje přes membránový filtr 0,22 μm (4.3) a odplyní (např. v ultrazvukové lázni (4.4) po dobu nejméně 15 minut). |
|
3.7 |
Standardní látka: amprolium, čisté, 1-[(4-amino-2-propyl-5-pyrimidinyl)-methyl]-2-picoliniumchlorid hydrochlorid, E 750 (viz 9.2). |
|
3.7.1 |
Do 100 ml odměrné baňky se s přesností na 0,1 mg naváží 50 mg amprolia (3.7), rozpustí se v 80 ml methanolu (3.1) a vloží se na 10 minut do ultrazvukové lázně (4.4). Po vyjmutí z ultrazvukové lázně se roztok ochladí na laboratorní teplotu, doplní vodou po značku a promíchá. Roztok je při teplotě ≤ 4 oC stálý jeden měsíc. |
|
3.7.2 |
Do 50 ml odměrné baňky se napipetuje 5,0 ml základního standardního roztoku (3.7.1), doplní se po značku extrakční směsí (3.8) a promíchá. Roztok je při teplotě ≤ 4 oC stálý jeden měsíc. |
|
3.7.3 |
Do sady 50 ml odměrných baněk se napipetuje 0,5, 1,0 a 2,0 ml pracovního standardního roztoku (3.7.2). Doplní se po značku mobilní fází (3.6) a promíchá. Tyto roztoky obsahují 0,5, 1,0 a 2,0 μg amprolia v 1 ml. Tyto roztoky se musí připravovat čerstvé před použitím. |
|
3.8 |
Extrakční směs. Směs methanolu (3.1) a vody, 2 + 1 (v + v). |
4. Přístroje a pomůcky
|
4.1 |
Zařízení HPLC s dávkovacím systémem pro injektované objemy 100 μl. |
|
4.1.1 |
Kolona pro kapalinovou chromatografii, 125 mm × 4 mm, katex Nucleosil 10 SA, náplň 5 nebo 10 μm, nebo obdobná. |
|
4.1.2 |
UV detektor s nastavitelnou vlnovou délkou nebo detektor diodového pole. |
|
4.2 |
Membránový filtr, materiál PTFE, velikost pórů 0,45 μm. |
|
4.3 |
Membránový filtr, 0,22 μm. |
|
4.4 |
Ultrazvuková lázeň. |
|
4.5 |
Mechanické nebo magnetické míchací zařízení. |
5. Postup
5.1 Obecné pokyny
5.1.1
Pro provedení zkoušky na výtěžnost (5.1.2) se provede zkouška slepého vzorku krmiva za účelem kontroly, že ve vzorku není přítomno amprolium ani jiné rušivé látky. Slepý vzorek krmiva je podobného složení jako zkoušený vzorek a nesmí být zjištěno amprolium ani rušivé látky.
5.1.2
Zkouška na výtěžnost se provede na slepém vzorku krmiva, ke kterému bylo přidáno takové množství amprolia, které odpovídá množství amprolia ve zkoušeném vzorku. Pro obohacení na obsah 100 mg/kg se převede 10,0 ml základního standardního roztoku (3.7.1) do 250 ml kónické baňky a roztok se odpaří na přibližně 0,5 ml. Poté se přidá 50 g slepého vzorku krmiva. Důkladně se promíchá, nechá se 10 minut stát a před zahájením extrakce (5.2) se znovu několikrát promíchá.
Není-li k dispozici slepý vzorek krmiva podobného složení jako zkoušený vzorek (viz 5.1.1), lze provést zkoušku na výtěžnost metodou standardního přídavku. V tom případě se ke zkoušenému vzorku přidá přibližně stejné množství amprolia, jaké je ve vzorku již obsaženo. Obohacený vzorek se potom zkouší společně se vzorkem neobohaceným a výtěžnost se vypočte odečtením.
5.2 Extrakce
5.2.1
Podle obsahu amprolia se do 500 ml kónické baňky naváží 5–40 g vzorku s přesností na 0,01 g a přidá se 200 ml extrakční směsi (3.8). Baňka se vloží na 15 minut do ultrazvukové lázně (4.4). Poté se vyjme a 1 hodinu třepe nebo míchá na magnetickém míchacím zařízení (4.5). Alikvotní část extraktu se zředí mobilní fází (3.6) na obsah amprolia 0,5–2 μg/ml a promíchá se (viz poznámka 9.3). 5–10 ml tohoto naředěného roztoku se přefiltruje přes membránový filtr (4.2). Dále se pokračuje se stanovením HPLC (5.3).
5.2.2
Podle obsahu amprolia se do 500 ml kónické baňky naváží 1–4 g premixu s přesností na 0,001 g a přidá se 200 ml extrakční směsi (3.8). Baňka se vloží na 15 minut do ultrazvukové lázně (4.4). Poté se vyjme a 1 hodinu třepe nebo míchá na magnetickém míchacím zařízení (4.5). Alikvotní část extraktu se zředí mobilní fází (3.6) na obsah amprolia mezi 0,5–2 μg/ml a promíchá se. 5–10 ml tohoto zředěného roztoku se přefiltruje přes membránový filtr (4.2). Dále se pokračuje se stanovením HPLC (5.3).
5.3 Stanovení HPLC
5.3.1
Následující podmínky jsou doporučené, mohou být použity jiné podmínky za předpokladu, že poskytují rovnocenné výsledky.
|
Kolona pro kapalinovou chromatografii (4.1.1): |
125 mm × 4 mm, katex Nucleosil 10 SA, náplň 5 nebo 10 μm nebo obdobná |
|
Mobilní fáze (3.6): |
směs acetonitrilu (3.2), roztoku dihydrogenfosforečnanu sodného (3.4) a roztoku chloristanu sodného (3.5), 450 + 450 + 100 (v + v + v). |
|
Průtok: |
0,7–1 ml/min |
|
Detekční vlnová délka: |
264 nm |
|
Objem nástřiku: |
100 μl |
Stabilita chromatografického systému se kontroluje opakovaným nástřikem kalibračního roztoku (3.7.3), který obsahuje 1,0 μg/ml, dokud se nedosáhne konstantních výšek píků a konstantních retenčních časů.
5.3.2
Provede se několikrát nástřik každého kalibračního roztoku (3.7.3) a změří se průměrná hodnota výšek (ploch) píků pro každou koncentraci. Kalibrační křivka se vytvoří vynesením průměrných hodnot výšek nebo ploch píků kalibračních roztoků proti příslušným koncentracím v μg/ml.
5.3.3
Provede se několik nástřiků extraktu vzorku (5.2) při použití stejného objemu jako u kalibračních roztoků a zjistí se průměrná hodnota výšek (ploch) píků amprolia.
6. Výpočet a vyjádření výsledků
Z průměrné hodnoty výšky (plochy) píků amprolia u roztoku vzorku se podle kalibrační křivky (5.3.2) stanoví koncentrace roztoku vzorku v μg/ml.
Obsah amprolia (w) v mg/kg ve vzorku se vypočte podle následujícího vzorce:
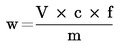 (mg/kg)
(mg/kg)
kde:
|
V |
= |
objem extrakční směsi (3.8) v ml podle 5.2 (tj. 200 ml) |
|
c |
= |
koncentrace amprolia v extraktu vzorku (5.2) v μg/ml |
|
f |
= |
faktor ředění podle 5.2 |
|
m |
= |
hmotnost navážky vzorku v g. |
7. Ověření výsledků
7.1 Identita
Identitu analytů lze potvrdit opakovaným stanovením s přídavkem (co-chromatography) nebo pomocí detektoru diodového pole, přičemž se porovnávají spektra roztoku vzorku (5.2) a kalibračního roztoku (3.7.3) obsahujícího 2,0 μg/ml.
7.1.1
K roztoku vzorku (5.2) se přidá vhodné množství kalibračního roztoku (3.7.3). Přidané množství amprolia musí odpovídat obsahu amprolia v extraktu vzorku.
Přídavek se projeví pouze zvýšením píku amprolia, a to úměrně k přidanému množství a ředění extraktu. Šířka píku v polovině maximální výšky se může odchylovat od šířky původního píku amprolia v neobohaceném extraktu vzorku nejvýše o ±10 %.
7.1.2
Výsledky jsou posuzovány podle následujících kritérií:
|
a) |
při vlnové délce maximální absorpce vzorku i standardu musí být záznam spektra zaznamenaný ve vrcholu píku na chromatogramu stejný s přesností danou rozlišovací schopností detekčního systému. Pro detektor diodového pole se udává obvykle ±2 nm; |
|
b) |
při vlnové délce mezi 210 a 320 nm se nesmí záznam spektra ve zkoušeném vzorku a standardu zaznamenaný ve vrcholu píku chromatogramu lišit od ostatních částí spektra v rozsahu 10–100 % relativní absorbance. Tohoto kritéria je dosaženo, jestliže jsou vykázána stejná maxima a jestliže odchylka mezi dvěma spektry nikde nepřesahuje 15 % absorbance standardního analytu; |
|
c) |
při vlnové délce mezi 210 a 320 nm se nesmí spektrum vzestupné části, vrcholu a sestupné části píku zkoušeného vzorku lišit od ostatních částí spektra v rozsahu 10–100 % relativní absorbance. Tohoto kritéria je dosaženo, jestliže jsou vykázána stejná maxima a jestliže ve všech sledovaných bodech odchylka mezi spektry nepřesahuje nikde 15 % absorbance spektra ve vrcholu píku. |
Není-li některé z těchto kritérií splněno, přítomnost analytu není potvrzena.
7.2 Opakovatelnost
Rozdíl mezi výsledky dvou paralelních stanovení provedených u téhož vzorku nesmí překročit
|
— |
15 % relat. z hodnoty vyššího výsledku u obsahu amprolia mezi 25 mg/kg a 500 mg/kg, |
|
— |
75 mg/kg u obsahu amprolia mezi 500 mg/kg a 1 000 mg/kg, |
|
— |
7,5 % relat. z hodnoty vyššího výsledku u obsahu amprolia vyššího než 1 000 mg/kg. |
7.3 Výtěžnost
Pro obohacený (slepý) vzorek musí být výtěžnost nejméně 90 %.
8. Výsledky kruhových testů
Při kruhových testech byla zkoušena tři krmiva pro drůbež (vzorky 1–3), minerální krmivo (vzorek 4) a premix (vzorek 5). Výsledky jsou shrnuty v níže uvedené tabulce.
|
|
vzorek 1 (slepé krmivo) |
vzorek 2 |
vzorek 3 |
vzorek 4 |
vzorek 5 |
|
L |
14 |
14 |
14 |
14 |
15 |
|
n |
56 |
56 |
56 |
56 |
60 |
|
průměr (mg/kg) |
— |
45,5 |
188 |
5 129 |
25 140 |
|
sr (mg/kg) |
— |
2,26 |
3,57 |
178 |
550 |
|
CVr (%) |
— |
4,95 |
1,90 |
3,46 |
2,20 |
|
sR (mg/kg) |
— |
2,95 |
11,8 |
266 |
760 |
|
CVR (%) |
— |
6,47 |
6,27 |
5,19 |
3,00 |
|
Deklarovaný obsah (mg/kg) |
— |
50 |
200 |
5 000 |
25 000 |
|
L |
= |
počet laboratoří |
|
n |
= |
počet jednotlivých hodnot |
|
sr |
= |
standardní odchylka opakovatelnosti |
|
CVr |
= |
variační koeficient opakovatelnosti |
|
sR |
= |
standardní odchylka reprodukovatelnosti |
|
CVR |
= |
variační koeficient reprodukovatelnosti. |
9. Poznámky
|
9.1 |
Obsahuje-li vzorek thiamin, objeví se pík thiaminu v chromatogramu krátce před píkem amprolia. Při použití této metody by se amprolium a thiamin měly oddělit. Nejsou-li amprolium a thiamin separovány kolonou (4.1.1) použitou při této metodě, upraví se mobilní fáze (3.6) nahrazením až 50 % acetonitrilu methanolem. |
|
9.2 |
Podle britského lékopisu vykazuje spektrum roztoku amprolia (c = 0,02 mol/l) v kyselině chlorovodíkové (c = 0,1 mol/l) maxima při 246 nm a 262 nm. Absorbance by měla činit 0,84 při 246 nm a 0,80 při 262 nm. |
|
9.3 |
Extrakt by se vždy měl ředit mobilní fází, jelikož jinak se retenční čas píku amprolia značně posouvá změnami iontové síly. |
D. STANOVENÍ OBSAHU CARBADOXU
Methyl-3-(2-chinoxalinylmethylen)-carbazat- N 1 ,N 4 -dioxid
1. Účel a rozsah
Tato metoda umožňuje stanovení obsahu carbadoxu v krmivech, premixech a přípravcích. Mez detekce je 1 mg/kg, mez kvantifikace je 5 mg/kg.
2. Princip
Vzorek se zvlhčí vodou a poté se extrahuje směsí methanolu a acetonitrilu. U krmiv se alikvotní část extraktu po filtraci přečistí na koloně s oxidem hlinitým. Extrakt z premixů a přípravků se přímo ředí na vhodnou koncentraci směsí vody, methanolu a acetonitrilu. Obsah carbadoxu se stanoví vysokoúčinnou kapalinovou chromatografií (HPLC) na reverzní fázi s UV detekcí.
3. Chemikálie
|
3.1 |
Methanol. |
|
3.2 |
Acetonitril, pro HPLC. |
|
3.3 |
Kyselina octová, w = 100 %. |
|
3.4 |
Oxid hlinitý: neutrální, stupeň aktivity I. |
|
3.5 |
Směs methanolu a acetonitrilu 1 + 1 (v + v). 500 ml methanolu (3.1) se smíchá s 500 ml acetonitrilu (3.2). |
|
3.6 |
Kyselina octová, σ = 10 %. 10 ml kyseliny octové (3.3) se smíchá se 100 ml vody. |
|
3.7 |
Octan sodný. |
|
3.8 |
Voda, pro HPLC. |
|
3.9 |
Octanový tlumivý roztok, c = 0,01 mol/l, pH = 6,0. 0,82 g octanu sodného (3.7) se rozpustí v 700 ml vody (3.8) a kyselinou octovou (3.6) se upraví pH na 6,0. Roztok se převede do odměrné baňky o objemu 1 000 ml, doplní se vodou (3.8) po značku a promíchá. |
|
3.10 |
Mobilní fáze pro HPLC. 825 ml octanového tlumivého roztoku (3.9) se smíchá se 175 ml acetonitrilu (3.2). Roztok se přefiltruje přes membránový filtr 0,22 μm (4.5) a odplyní se (např. v ultrazvukové lázni po dobu 10 minut). |
|
3.11 |
Standardní látka. Čistý carbadox: Methyl-3-(2-chinoxalinylmethylen)-carbazat-N1,N4-dioxid, E 850. |
|
3.11.1 |
Do 250 ml odměrné baňky se naváží 25 mg carbadoxu (3.11) s přesností na 0,1 mg. Rozpustí se ve směsi methanolu a acetonitrilu (3.5) v ultrazvukové lázni (4.7). Po působení ultrazvuku se roztok ochladí na laboratorní teplotu, doplní se směsí methanolu a acetonitrilu (3.5) po značku a promíchá. Baňka se obalí hliníkovou fólií (nebo se použije odměrná baňka z tmavého skla) a uchovává se v chladničce. Roztok je při teplotě ≤ 4 oC stálý jeden měsíc. |
|
3.11.2 |
Do sady 100 ml odměrných baněk se přenese 2,0, 5,0, 10,0 a 20,0 ml základního standardního roztoku (3.11.1). Přidá se po 30 ml vody, doplní se po značku směsí methanolu a acetonitrilu (3.5) a promíchá. Baňky se obalí hliníkovou fólií. Tyto roztoky obsahují 2,0, 5,0, 10,0 a 20,0 μg carbadoxu v 1 ml. Kalibrační roztoky se připravují čerstvé před použitím.
|
|
3.12 |
Směs vody a směsi methanolu a acetonitrilu (3.5), 300 + 700 (v + v). 300 ml vody se smíchá se 700 ml směsi methanolu a acetonitrilu (3.5). |
4. Přístroje a pomůcky
|
4.1 |
Laboratorní mechanické nebo magnetické míchací zařízení. |
|
4.2 |
Filtrační papír ze skleněných vláken (Whatman GF/A nebo obdobný). |
|
4.3 |
Skleněná kolona (délka 300–400 mm, vnitřní průměr přibližně 10 mm) s fritou a vypouštěcím kohoutem.
|
|
4.4 |
Zařízení HPLC s dávkovacím systémem pro injektované objemy 20 μl. |
|
4.4.1 |
Kolona pro kapalinovou chromatografii: 300 mm × 4 mm, C18, náplň 10 μm nebo obdobná. |
|
4.4.2 |
UV detektor s nastavitelnou vlnovou délkou nebo detektor diodového pole s měřicím rozsahem 225–400 nm. |
|
4.5 |
Membránový filtr, 0,22 μm. |
|
4.6 |
Membránový filtr, 0,45 μm. |
|
4.7 |
Ultrazvuková lázeň. |
5. Postup
|
Poznámka |
: |
Carbadox je citlivý na světlo. Celý postup se musí provádět při tlumeném světle nebo je třeba použít nádobí z tmavého skla nebo sklo obalit hliníkovou fólií. |
5.1 Obecné pokyny
5.1.1
Pro provedení zkoušky na výtěžnost (5.1.2) se provede zkouška slepého vzorku krmiva za účelem kontroly, že není přítomen ani carbadox, ani rušivé látky. Slepý vzorek krmiva má podobné složení jako zkoumaný vzorek a nesmí být zjištěn carbadox ani rušivé látky.
5.1.2
Zkouška na výtěžnost se provede na slepém vzorku krmiva (5.1.1), ke kterému bylo přidáno takové množství carbadoxu, které odpovídá množství carbadoxu ve zkoušeném vzorku. Pro obohacení na obsah 50 mg/kg se 5,0 ml základního standardního roztoku (3.11.1) převede do 200 ml kónické baňky. Roztok se v proudu dusíku odpaří na přibližně 0,5 ml. Přidá se 10 g slepého vzorku krmiva, promíchá se a po deseti minutách se může začít s extrakcí (5.2).
Není-li k dispozici slepý vzorek krmiva podobného složení jako zkoušený vzorek (viz 5.1.1), lze provést zkoušku na výtěžnost metodou standardního přídavku. V tom případě se ke zkoušenému vzorku přidá přibližně stejné množství carbadoxu, jaké je ve vzorku již obsaženo. Obohacený vzorek se potom zkouší společně se vzorkem neobohaceným a výtěžnost se vypočte odečtením.
5.2 Extrakce
5.2.1
Do 200 ml kónické baňky se naváží 10 g vzorku s přesností na 0,01 g. Přidá se 15,0 ml vody, promíchá se a nechá se 5 minut stát. Poté se přidá 35,0 ml směsi methanolu a acetonitrilu (3.5), baňka se uzavře a 30 minut se protřepává nebo míchá na magnetickém míchacím zařízení (4.1). Roztok se přefiltruje přes filtr ze skleněných vláken (4.2). Tento roztok se použije k čištění (5.3).
5.2.2
Do 200 ml kónické baňky se naváží 1 g nemletého vzorku s přesností na 0,001 g. Přidá se 15,0 ml vody, promíchá se a nechá se 5 minut stát. Poté se přidá 35,0 ml směsi methanolu a acetonitrilu (3.5), baňka se uzavře a 30 minut se protřepává nebo míchá na magnetickém míchacím zařízení (4.1). Roztok se přefiltruje přes filtr ze skleněných vláken (4.2).
Alikvotní část filtrátu se odpipetuje do 50 ml odměrné baňky. Přidá se 15,0 ml vody, doplní se po značku směsí methanolu a acetonitrilu (3.5) a promíchá. Koncentrace carbadoxu v konečném roztoku je přibližně 10 μg/ml. Alikvotní část se přefiltruje přes filtr 0,45 μm (4.6).
Dále se pokračuje se stanovením HPLC (5.4).
5.2.3
Do 250 ml kónické baňky se naváží 0,2 g nemletého vzorku s přesností na 0,001 g. Přidá se 45,0 ml vody, promíchá se a nechá se 5 minut stát. Poté se přidá 105,0 ml směsi methanolu a acetonitrilu (3.5), baňka se uzavře a homogenizuje. Baňka se vloží na 15 minut do ultrazvukové lázně (4.7) a poté se 15 minut třepe nebo míchá na magnetickém míchacím zařízení (4.1). Roztok se přefiltruje přes filtr ze skleněných vláken (4.2).
Alikvotní část filtrátu se zředí směsí vody, methanolu a acetonitrilu (3.12) tak, aby se získala koncentrace carbadoxu 10–15 μg/ml (pro 10 % přípravek je faktor ředění 10). Alikvotní část se přefiltruje přes filtr 0,45 μm (4.6).
Dále se pokračuje se stanovením HPLC (5.4).
5.3 Čištění
5.3.1
Do skleněně kolony (4.3) se naváží 4 g oxidu hlinitého (3.4).
5.3.2
Na kolonu s oxidem hlinitým se nanese 15 ml přefiltrovaného extraktu (5.2.1) a první 2 ml eluátu se odstraní. Následujících 5 ml se zachycuje, alikvotní část se přefiltruje přes filtr 0,45 μm (4.6).
Dále se pokračuje se stanovením HPLC (5.4).
5.4 Stanovení HPLC
5.4.1
Následující podmínky jsou doporučené, mohou být použity jiné podmínky za předpokladu, že poskytují rovnocenné výsledky:
|
Kolona pro kapalinovou chromatografii (4.4.1): |
300 mm × 4 mm, C18, náplň 10 μm nebo obdobná |
|
|
|
|
Mobilní fáze (3.10): |
Směs octanového tlumivého roztoku (3.9) a acetonitrilu (3.2), 825 + 175 (v+v) |
|
Průtok: |
1,5–2 ml/min |
|
Detekční vlnová délka: |
365 nm |
|
Objem nástřiku: |
20 μl |
Stabilita chromatografického systému se kontroluje opakovaným nástřikem kalibračního roztoku (3.11.2), který obsahuje 5,0 μg/ml, dokud se nedosáhne konstantních výšek (ploch) píků a konstantních retenčních časů.
5.4.2
Provede se několikrát nástřik každého kalibračního roztoku (3.11.2) a změří se výšky (plochy) píků pro každou koncentraci. Kalibrační křivka se vytvoří vynesením průměrných hodnot výšek (ploch) píků proti příslušným koncentracím v μg/ml.
5.4.3
Extrakt ze vzorku ((5.3.2) pro krmiva, (5.2.2) pro premixy a (5.2.3) pro přípravky) se opakovaně nastřikuje a stanoví se průměrná hodnota výšky (plochy) píku carbadoxu.
6. Výpočet a vyjádření výsledků
Z průměrné hodnoty výšky (plochy) píků carbadoxu v roztoku vzorku se porovnáním s kalibrační křivkou (5.4.2) stanoví koncentrace carbadoxu v roztoku vzorku v μg/ml.
6.1 Krmiva
Obsah carbadoxu (w) v mg/kg ve vzorku se vypočítá podle následujícího vzorce:
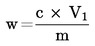 [mg/kg]
[mg/kg]
kde:
|
c |
= |
koncentrace carbadoxu v extraktu vzorku (5.3.2) v μg/ml |
|
V1 |
= |
objem extrakční směsi (tj. 50 ml) |
|
m |
= |
hmotnost navážky vzorku v g. |
6.2 Premixy a přípravky
Obsah carbadoxu (w) v mg/kg ve vzorku se vypočítá podle následujícího vzorce:
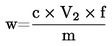 [mg/kg]
[mg/kg]
kde:
|
c |
= |
koncentrace carbadoxu v extraktu vzorku (5.2.2 nebo 5.2.3) v μg/ml |
|
V2 |
= |
objem extrakční směsi (tj. 50 ml pro premixy; 150 ml pro přípravky) |
|
f |
= |
faktor ředění podle 5.2.2 (premixy) nebo 5.2.3 (přípravky) |
|
m |
= |
hmotnost navážky vzorku v g. |
7. Ověření výsledků
7.1 Identita
Identitu analytu lze potvrdit opakovaným stanovením s přídavkem (co-chromatography) nebo pomocí detektoru diodového pole, přičemž se srovnávají spektra extraktu vzorku a kalibračního roztoku (3.11.2) obsahujícího 10,0 μg/ml carbadoxu.
7.1.1
K extraktu vzorku se přidá vhodné množství kalibračního roztoku (3.11.2). Přidané množství carbadoxu musí odpovídat obsahu carbadoxu v extraktu vzorku.
Přídavek se projeví pouze zvýšením píku carbadoxu, a to úměrně k přidanému množství a ředění extraktu. Šířka píku v polovině maximální výšky se může odchylovat od šířky původního píku carbadoxu nejvýše o ±10 %.
7.1.2
Výsledky jsou posuzovány podle následujících kritérií:
|
a) |
při vlnové délce maximální absorpce vzorku i standardu musí být záznam spektra zaznamenaný ve vrcholu píku na chromatogramu stejný s přesností danou rozlišovací schopností detekčního systému. Pro detektor diodového pole se udává obvykle ±2 nm; |
|
b) |
při vlnové délce mezi 225 a 400 nm se nesmí záznam spektra ve zkoušeném vzorku a standardu zaznamenaný ve vrcholu píku chromatogramu lišit od ostatních částí spektra v rozsahu 10–100 % relativní absorbance. Tohoto kritéria je dosaženo, jestliže jsou vykázána stejná maxima a jestliže odchylka mezi dvěma spektry nikde nepřesahuje 15 % absorbance standardního analytu; |
|
c) |
při vlnové délce mezi 225 a 400 nm se nesmí spektrum vzestupné části, vrcholu a sestupné části píku zkoušeného vzorku lišit od ostatních částí spektra v rozsahu 10–100 % relativní absorbance. Tohoto kritéria je dosaženo, jestliže jsou vykázána stejná maxima a jestliže ve všech sledovaných bodech odchylka mezi spektry nepřesahuje 15 % absorbance spektra ve vrcholu píku. |
Jestliže některé z těchto kritérií není splněno, přítomnost analytu není potvrzena.
7.2 Opakovatelnost
Rozdíl mezi výsledky dvou paralelních stanovení provedených u téhož vzorku nesmí při obsahu carbadoxu 10 mg/kg překračovat 15 % relat. z hodnoty vyššího výsledku.
7.3 Výtěžnost
Pro obohacený (slepý) vzorek musí být výtěžnost nejméně 90 %.
8. Výsledky kruhových testů
Při kruhových testech bylo 8 laboratořemi zkoušeno 6 krmiv, 4 premixy a 3 přípravky. U každého vzorku byly provedeny dvě zkoušky. (Přesnější údaje o těchto kruhových testech viz Journal of the AOAC, Volume 71, 1998, s. 484–490.) Výsledky (s výjimkou odlehlých hodnot) jsou shrnuty v níže uvedené tabulce:
Tabulka 1
Výsledky kruhových testů u krmiv
|
|
Vzorek 1 |
Vzorek 2 |
Vzorek 3 |
Vzorek 4 |
Vzorek 5 |
Vzorek 6 |
|
L |
8 |
8 |
8 |
8 |
8 |
8 |
|
n |
15 |
14 |
15 |
15 |
15 |
15 |
|
průměr (mg/kg) |
50,0 |
47,6 |
48,2 |
49,7 |
46,9 |
49,7 |
|
Sr (mg/kg) |
2,90 |
2,69 |
1,38 |
1,55 |
1,52 |
2,12 |
|
CVr (%) |
5,8 |
5,6 |
2,9 |
3,1 |
3,2 |
4,3 |
|
SR (mg/kg) |
3,92 |
4,13 |
2,23 |
2,58 |
2,26 |
2,44 |
|
CVR (%) |
7,8 |
8,7 |
4,6 |
5,2 |
4,8 |
4,9 |
|
Deklarovaný obsah (mg/kg) |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
Tabulka 2
Výsledky kruhových testů u premixů a přípravků
|
|
Premixy |
Přípravky |
|||||
|
A |
B |
C |
D |
A |
B |
C |
|
|
L |
7 |
7 |
7 |
7 |
8 |
8 |
8 |
|
n |
14 |
14 |
14 |
14 |
16 |
16 |
16 |
|
průměr (g/kg) |
8,89 |
9,29 |
9,21 |
8,76 |
94,6 |
98,1 |
104 |
|
Sr (g/kg) |
0,37 |
0,28 |
0,28 |
0,44 |
4,1 |
5,1 |
7,7 |
|
CVr (%) |
4,2 |
3,0 |
3,0 |
5,0 |
4,3 |
5,2 |
7,4 |
|
SR (g/kg) |
0,37 |
0,28 |
0,40 |
0,55 |
5,4 |
6,4 |
7,7 |
|
CVR (%) |
4,2 |
3,0 |
4,3 |
6,3 |
5,7 |
6,5 |
7,4 |
|
Deklarovaný obsah (g/kg) |
10,0 |
10,0 |
10,0 |
10,0 |
100 |
100 |
100 |
|
L |
= |
počet laboratoří |
|
n |
= |
počet jednotlivých hodnot |
|
Sr |
= |
standardní odchylka opakovatelnosti |
|
CVr |
= |
variační koeficient opakovatelnosti |
|
SR |
= |
standardní odchylka reprodukovatelnosti |
|
CVR |
= |
variační koeficient reprodukovatelnosti. |
PŘÍLOHA IX
SROVNÁVACÍ TABULKY PODLE ČLÁNKU 6
1. Směrnice 71/250/EHS
|
Směrnice 71/250/EHS |
Toto nařízení |
|
Čl. 1 první pododstavec |
Článek 3 |
|
Čl. 1 druhý pododstavec |
Článek 2 |
|
Článek 2 |
— |
|
Článek 3 |
— |
|
Příloha, část 1 |
Příloha II |
|
Příloha, část 2 |
— |
|
Příloha, část 3 |
— |
|
Příloha, část 4 |
Příloha III, část O |
|
Příloha, část 5 |
Příloha III, část M |
|
Příloha, část 6 |
Příloha III, část N |
|
Příloha, část 7 |
Příloha III, část Q |
|
Příloha, část 9 |
Příloha III, část K |
|
Příloha, část 10 |
— |
|
Příloha, část 11 |
— |
|
Příloha, část 12 |
Příloha III, část J |
|
Příloha, část 14 |
Příloha III, část D |
|
Příloha, část 16 |
— |
2. Směrnice 71/393/EHS
|
Směrnice 71/393/EHS |
Toto nařízení |
|
Článek 1 |
Článek 3 |
|
Článek 2 |
— |
|
Článek 3 |
— |
|
Příloha, část I |
Příloha III, část A |
|
Příloha, část II |
Příloha III, část E |
|
Příloha, část III |
Příloha III, část P |
|
Příloha, část IV |
Příloha III, část H |
3. Směrnice 72/199/EHS
|
Směrnice 72/199/EHS |
Toto nařízení |
|
Článek 1 |
Článek 3 |
|
Článek 2 |
— |
|
Článek 3 |
— |
|
Článek 4 |
— |
|
Příloha I, část 1 |
Příloha III, část L |
|
Příloha I, část 2 |
Příloha III, část C |
|
Příloha I, část 3 |
— |
|
Příloha I, část 4 |
— |
|
Příloha I, část 5 |
Příloha V, část A |
|
Příloha II |
— |
4. Směrnice 73/46/EHS
|
Směrnice 73/46/EHS |
Toto nařízení |
|
Článek 1 |
Článek 3 |
|
Článek 3 |
— |
|
Článek 4 |
— |
|
Příloha I, část 1 |
Příloha III, část B |
|
Příloha I, část 2 |
— |
|
Příloha I, část 3 |
Příloha III, část I |
5. Směrnice 76/371/EHS
|
Směrnice 76/371/EHS |
Toto nařízení |
|
Článek 1 |
Článek 1 |
|
Článek 2 |
— |
|
Článek 3 |
— |
|
Příloha |
Příloha I |
6. Směrnice 76/372/EHS
|
Směrnice 76/372/EHS |
Toto nařízení |
|
Článek 1 |
— |
|
Článek 2 |
— |
|
Článek 3 |
— |
|
Příloha |
— |
7. Směrnice 78/633/EHS
|
Směrnice 78/633/EHS |
Toto nařízení |
|
Článek 1 |
Článek 3 |
|
Článek 2 |
— |
|
Článek 3 |
— |
|
Příloha, část 1 |
— |
|
Příloha, část 2 |
— |
|
Příloha, část 3 |
Příloha IV, část C |
8. Směrnice 81/715/EHS
|
Směrnice 81/715/EHS |
Toto nařízení |
|
Článek 1 |
— |
|
Článek 2 |
— |
|
Článek 3 |
— |
|
Příloha |
— |
9. Směrnice 84/425/EHS
|
Směrnice 84/425/EHS |
Toto nařízení |
|
Článek 1 |
— |
|
Článek 2 |
— |
|
Článek 3 |
— |
|
Příloha |
— |
10. Směrnice 86/174/EHS
|
Směrnice 86/174/EHS |
Toto nařízení |
|
Článek 1 |
Článek 4 |
|
Článek 2 |
— |
|
Článek 3 |
— |
|
Příloha |
Příloha VII |
11. Směrnice 93/70/EHS
|
Směrnice 93/70/EHS |
Toto nařízení |
|
Článek 1 |
Článek 3 |
|
Článek 2 |
— |
|
Článek 3 |
— |
|
Příloha |
Příloha IV, část D |
12. Směrnice 93/117/ES
|
Směrnice 93/117/ES |
Toto nařízení |
|
Článek 1 |
Články 3 a 5 |
|
Článek 2 |
— |
|
Článek 3 |
— |
|
Příloha, část 1 |
Příloha IV, část E |
|
Příloha, část 2 |
Příloha VIII, část A |
13. Směrnice 98/64/ES
|
Směrnice 98/64/ES |
Toto nařízení |
|
Článek 1 |
Články 3 a 5 |
|
Článek 2 |
— |
|
Článek 3 |
— |
|
Článek 4 |
— |
|
Příloha, část A |
Příloha III, část F |
|
Příloha, část C |
Příloha VIII, část B |
14. Směrnice 1999/27/ES
|
Směrnice 1999/27/ES |
Toto nařízení |
|
Článek 1 |
Články 3 a 5 |
|
Článek 2 |
— |
|
Článek 3 |
— |
|
Článek 4 |
— |
|
Článek 5 |
— |
|
Článek 6 |
— |
|
Článek 7 |
— |
|
Příloha, část A |
Příloha VIII, část C |
|
Příloha, část B |
Příloha IV, část F |
|
Příloha, část C |
Příloha VIII, část D |
15. Směrnice 1999/76/ES
|
Směrnice 1999/76/ES |
Toto nařízení |
|
Článek 1 |
Článek 3 |
|
Článek 2 |
— |
|
Článek 3 |
— |
|
Článek 4 |
— |
|
Příloha |
Příloha IV, část G |
16. Směrnice 2000/45/ES
|
Směrnice 2000/45/ES |
Toto nařízení |
|
Článek 1 |
Článek 3 |
|
Článek 2 |
— |
|
Článek 3 |
— |
|
Článek 4 |
— |
|
Příloha, část A |
Příloha IV, část A |
|
Příloha, část B |
Příloha IV, část B |
|
Příloha, část C |
Příloha III, část G |
17. Směrnice 2002/70/ES
|
Směrnice 2002/70/ES |
Toto nařízení |
|
Článek 1 |
Článek 1 |
|
Článek 2 |
Články 2 a 3 |
|
Článek 3 |
— |
|
Článek 4 |
— |
|
Článek 5 |
— |
|
Příloha I |
Příloha I a příloha V část B bod I |
|
Příloha II |
Příloha II a příloha V část B bod II |
18. Směrnice 2003/126/ES
|
Směrnice 2003/126/ES |
Toto nařízení |
|
Článek 1 |
Článek 3 |
|
Článek 2 |
— |
|
Článek 3 |
— |
|
Článek 4 |
— |
|
Článek 5 |
— |
|
Článek 6 |
— |
|
Příloha |
Příloha VI |