(ES) č. 429/2008Nařízení Komise (ES) č. 429/2008 ze dne 25. dubna 2008 o prováděcích pravidlech k nařízení Evropského parlamentu a Rady (ES) č. 1831/2003, pokud jde o vypracování a podávání žádostí a vyhodnocování a povolování doplňkových látek (Text s významem pro EHP)
| Publikováno: | Úř. věst. L 133, 22.5.2008, s. 1-65 | Druh předpisu: | Nařízení |
| Přijato: | 25. dubna 2008 | Autor předpisu: | Evropská komise |
| Platnost od: | 11. června 2008 | Nabývá účinnosti: | 21. června 2008 |
| Platnost předpisu: | Ano | Pozbývá platnosti: | |
Text předpisu s celou hlavičkou je dostupný pouze pro registrované uživatele.
NAŘÍZENÍ KOMISE (ES) č. 429/2008
ze dne 25. dubna 2008
o prováděcích pravidlech k nařízení Evropského parlamentu a Rady (ES) č. 1831/2003, pokud jde o vypracování a podávání žádostí a vyhodnocování a povolování doplňkových látek
(Text s významem pro EHP)
KOMISE EVROPSKÝCH SPOLEČENSTVÍ,
s ohledem na Smlouvu o založení Evropského společenství,
s ohledem na nařízení Evropského parlamentu a Rady (ES) č. 1831/2003 ze dne 22. září 2003 o doplňkových látkách používaných ve výživě zvířat (1), a zejména na čl. 7 odst. 4 a 5 uvedeného nařízení,
po konzultaci s Evropským úřadem pro bezpečnost potravin v souladu s čl. 7 odst. 4 a 5 nařízení (ES) č. 1831/2003,
vzhledem k těmto důvodům:
|
(1) |
Je nezbytné stanovit prováděcí pravidla, pokud jde o postup povolování doplňkových látek podle nařízení (ES) č. 1831/2003, včetně pravidel pro vypracování a podávání žádostí a vyhodnocování a povolování těchto doplňkových látek. Tato pravidla mají nahradit ustanovení v příloze směrnice Rady 87/153/EHS (2), kterou se stanoví hlavní zásady pro vyhodnocování doplňkových látek ve výživě zvířat. |
|
(2) |
Tato pravidla by měla stanovit požadavky, které musí splňovat dokumentace připojená k žádosti. Měla by zejména určit vědecké údaje, které mají být poskytnuty k identifikaci a charakteristice dotčené doplňkové látky, a studie, jež mají být předloženy s cílem prokázat její účinnost a bezpečnost pro člověka, zvířata a životní prostředí za účelem ověření a vyhodnocení žádostí o povolení Evropským úřadem pro bezpečnost potravin (dále jen „úřad“). |
|
(3) |
Podle povahy doplňkové látky nebo požadovaných podmínek pro užití se může rozsah studií potřebných k vyhodnocení jejích vlastností a účinků lišit. Subjekty by proto měly mít možnost postupovat s určitou pružností, pokud jde o druh studií a materiály, jež mají být předloženy k prokázání bezpečnosti a účinnosti dotyčné doplňkové látky. Subjekty, které využijí této pružnosti, by měly v dokumentaci svůj výběr zdůvodnit. |
|
(4) |
Úřad by měl mít možnost popřípadě požadovat doplňkové informace, aby mohl určit, zda doplňková látka splňuje podmínky pro povolení podle článku 5 nařízení (ES) č. 1831/2003. |
|
(5) |
Je nezbytné používat příslušné normy kvality při vypracovávání dokumentace pro doplňkové látky určené k použití v krmivu (krmivech) nebo vodě, aby bylo zajištěno, že výsledky laboratorních zkoušek nejsou zpochybněny. |
|
(6) |
V případě nutnosti by měly být stanoveny zvláštní požadavky pro jednotlivé kategorie doplňkových látek uvedené v čl. 6 odst. 1 nařízení (ES) č. 1831/2003. |
|
(7) |
S cílem podnítit snahy o získání povolení pro menšinové druhy při současném zachování potřebné úrovně bezpečnosti by měly být stanoveny zvláštní podmínky pro přihlédnutí k možnosti extrapolace výsledků studií prováděných na většinových druzích na druhy menšinové. |
|
(8) |
Prováděcí pravidla týkající se žádostí o povolení by měla vzít v úvahu různé požadavky na zvířata určená k produkci potravin a ostatní zvířata, pro něž aspekty týkající se vyhodnocení bezpečnosti pro člověka jako spotřebitele nejsou důležité. |
|
(9) |
Využití postupů zahrnujících používání laboratorních zvířat k pokusným či jiným vědeckým účelům a testování na zvířatech podle směrnice Rady 86/609/EHS ze dne 24. listopadu 1986 o sbližování právních a správních předpisů členských států týkajících se ochrany zvířat používaných pro pokusné a jiné vědecké účely (3) by mělo být minimální. |
|
(10) |
Aby se zamezilo zbytečnému opakování studií, měly by být stanoveny zjednodušené postupy pro povolování doplňkových látek, které již byly povoleny pro použití v potravinách. |
|
(11) |
Co se týká doplňkových látek, které byly podle směrnice Rady 70/524/EHS (4) povoleny bez časového omezení, měla by být popřípadě stanovena možnost, aby v případě, že nejsou k dispozici žádné studie, žadatel prokázal účinnost pomocí jiných materiálů, které jsou k dispozici k prokázání účinnosti, zejména materiálů týkajících se dlouhé historie používání dotyčné doplňkové látky. |
|
(12) |
Je nutno stanovit pravidla pro žádosti o změny povolení v souladu s čl. 13 odst. 3 nařízení (ES) č. 1831/2003. |
|
(13) |
Je nutno stanovit rovněž pravidla pro žádosti o obnovení povolení podle článku 14 nařízení (ES) č. 1831/2003. |
|
(14) |
Co se týká ustanovení o studiích bezpečnosti a účinnosti, jež je nutno provést na podporu žádosti, je nezbytné stanovit přechodné období, během něhož se budou nadále používat stávající pravidla. S žádostmi podanými před vstupem tohoto nařízení v platnost by se mělo i nadále nakládat v souladu s přílohou směrnice 87/153/EHS. Pokud jde o žádosti podané během určitého období po vstupu v platnost, žadatelé by vzhledem k dlouhé době potřebné pro některé studie měli mít možnost výběru mezi pravidly stanovenými v tomto nařízení a přílohou směrnice 87/153/EHS. Prováděcí pravidla byla vypracována na základě současných vědeckých a technických poznatků a v případě nutnosti by měla být přizpůsobena případnému novému vývoji v této oblasti. |
|
(15) |
Opatření tohoto nařízení jsou v souladu se stanoviskem Stálého výboru pro potravinový řetězec a zdraví zvířat, |
PŘIJALA TOTO NAŘÍZENÍ:
Článek 1
Definice
Pro účely tohoto nařízení se použijí tyto definice:
|
1. |
„zvířaty v zájmovém chovu a ostatními zvířaty, která nejsou určena k produkci potravin“ se rozumí zvířata druhů, jež jsou obyčejně krmeny, chovány nebo drženy člověkem, nejsou však určeny pro lidskou spotřebu, vyjma koní; |
|
2. |
„menšinovými druhy“ se rozumí zvířata určená k produkci potravin jiná než skot (zvířata chovaná pro mléko a maso, včetně telat), ovce (zvířata chovaná pro maso), prasata, kuřata (včetně nosnic), krůty a ryby druhu Salmonidae. |
Článek 2
Žádost
1. Žádost o povolení doplňkové látky podle článku 7 nařízení (ES) č. 1831/2003 se předloží pomocí formuláře stanoveného v příloze I.
K žádosti se připojí dokumentace podle článku 3 (dále jen „dokumentace“), která obsahuje údaje a dokumenty uvedené v čl. 7 odst. 3 nařízení (ES) č. 1831/2003.
2. Pokud žadatel v souladu s článkem 18 nařízení (ES) č. 1831/2003 požaduje, aby se s některými částmi dokumentace podle odstavce 1 zacházelo jako s důvěrnými údaji, předloží pro každý dokument nebo část dokumentu ověřitelné odůvodnění, že zveřejnění těchto informací by mohlo významně poškodit jeho postavení vůči konkurentům. Důvěrné části dokumentace musí být předloženy odděleně od zbytku dokumentace a nesmí být zahrnuty v souhrnu dokumentace podle čl. 7 odst. 3 písm. h) nařízení (ES) č. 1831/2003. Žadatel zašle Komisi kopii částí dokumentace, s nimiž má být zacházeno jako s důvěrnými údaji, a připojeného odůvodnění.
Článek 3
Dokumentace
1. Dokumentace přiměřeně a dostatečně prokazuje, že doplňková látka splňuje podmínky pro povolení stanovené v článku 5 nařízení (ES) č. 1831/2003.
2. Obecné požadavky na vypracování a předání dokumentace jsou stanoveny v příloze II.
Zvláštní požadavky, jež dokumentace musí splňovat v dotyčném případě, jsou stanoveny v příloze III.
Minimální doba trvání dlouhodobých studií je stanovena v příloze IV.
3. Odchylně od odstavce 2 může žadatel předložit dokumentaci, která nesplňuje požadavky stanovené v odstavci 2, pokud každý prvek, jenž tyto požadavky nesplňuje, odůvodní.
Článek 4
Přechodná opatření
1. Na žádosti o povolení podané před datem vstupu tohoto nařízení v platnost se nadále vztahuje příloha směrnice 87/153/EHS.
2. U žádostí o povolení podaných před 11. červnem 2009 se mohou žadatelé rozhodnout, že budou nadále používat oddíly III a IV částí I a II přílohy směrnice 87/153/EHS místo bodů 1.3, 1.4, 2.1.3, 2.1.4, 2.2.3, 2.2.4, 3.3, 3.4, 4.1.3, 4.1.4, 4.2.3, 4.2.4, 5.3, 5.4, 6.3, 6.4, 7.3, 7.4, 8.3 a 8.4 přílohy III a místo ustanovení ve sloupci „Minimální doba trvání dlouhodobých studií účinnosti“ v tabulkách přílohy IV.
Článek 5
Vstup v platnost
Toto nařízení vstupuje v platnost dvacátým dnem po vyhlášení v Úředním věstníku Evropské unie.
Toto nařízení je závazné v celém rozsahu a přímo použitelné ve všech členských státech.
V Bruselu dne 25. dubna 2008.
Za Komisi
Androulla VASSILIOU
členka Komise
(1) Úř. věst. L 268, 18.10.2003, s. 29. Nařízení ve znění nařízení Komise (ES) č. 378/2005 (Úř. věst. L 59, 5.3.2005, s. 68).
(2) Úř. věst. L 64, 7.3.1987, s. 19. Zrušeno nařízením (ES) č. 1831/2003.
(3) Úř. věst. L 358, 18.12.1986, s. 1; opravené znění v Úř. věst. L 117, 5.5.1987, s. 31. Směrnice ve znění směrnice Evropského parlamentu a Rady 2003/65/ES (Úř. věst. 230, 16.9.2003, s. 32).
(4) Úř. věst. L 270, 14.12.1970, s. 1. Směrnice naposledy pozměněná nařízením Komise (ES) č. 1800/2004 (Úř. věst. L 317, 16.10.2004, s. 37).
PŘÍLOHA I
FORMULÁŘ ŽÁDOSTI UVEDENÝ V ČL. 2 ODST. 1 A SPRÁVNÍ ÚDAJE
1. FORMULÁŘ ŽÁDOSTI
EVROPSKÁ KOMISE
GENERÁLNÍ ŘEDITELSTVÍ
PRO ZDRAVÍ A OCHRANU SPOTŘEBITELE
(Adresa)
Datum: …
|
Věc |
: |
Žádost o povolení doplňkové látky podle nařízení (ES) č. 1831/2003. |
|
|
Povolení doplňkové látky nebo nové užití doplňkové látky (čl. 4 odst. 1 nařízení (ES) č. 1831/2003) |
|
|
Povolení existujícího produktu (čl. 10 odst. 2 nebo 7 nařízení (ES) č. 1831/2003) |
|
|
Změna stávajícího povolení (čl. 13 odst. 3 nařízení (ES) č. 1831/2003) |
|
|
Obnovení povolení doplňkové látky (článek 14 nařízení (ES) č. 1831/2003) |
|
|
Povolení v naléhavých případech (článek 15 nařízení (ES) č. 1831/2003) |
(Označte jednoznačně zaškrtnutím jednoho z rámečků)
Žadatel(é) a/nebo jeho/jejich zástupce(i) ve Společenství (čl. 4 odst. 3 nařízení (ES) č. 1831/2003) za podmínek uvedených v čl. 7 odst. 3 písm. a) nařízení (ES) č. 1831/2003 (jméno, adresa ….)
…
…
podává (podávají) tuto žádost za účelem získání povolení pro následující produkt jakožto doplňkovou látku:
1.1 Identifikace a charakteristika doplňkové látky
Název doplňkové látky (charakteristika účinné látky (látek) nebo činidla (činidel), jak je vymezeno v bodech 2.2.1.1 a 2.2.1.2 přílohy II):
…
…
Obchodní název (je-li to vhodné u povolení vázaných na držitele):
…
…
V kategorii/kategoriích a funkční skupině/skupinách doplňkových látek (1) (seznam):
…
…
Cílové druhy zvířat:
…
…
…
Jméno držitele povolení (čl. 9 odst. 6 nařízení (ES) č. 1831/2003):
…
…
Tato doplňková látka již byla povolena v právních předpisech o krmivech směrnicí.……/…/E(H)S nebo nařízením (ES) č. …/… pod číslem … jako (kategorie doplňkových látek)
…
Tato doplňková látka již byla povolena v právních předpisech o potravinách směrnicí .…/…/E(H)S nebo nařízením (ES) č. …/… pod číslem … jako
…
pro použití v
…
Pokud se produkt skládá z geneticky modifikovaných organismů nebo tyto organismy obsahuje či je z nich vyroben, uveďte tyto informace:
|
|
jednoznačný identifikační kód (nařízení Komise (ES) č. 65/2004 (2)) (je-li to vhodné): … |
|
|
údaje o případném povolení uděleném v souladu s nařízením Evropského parlamentu a Rady (ES) č. 1829/2003 (3): … |
|
|
nebo údaje o dosud nevyřízené žádosti o povolení podle nařízení (ES) č. 1829/2003: … |
1.2 Podmínky pro užití
1.2.1 Použití v kompletních krmivech
Druh nebo kategorie zvířat:
…
…
Maximální stáří nebo hmotnost:
…
…
Minimální dávka (je-li to vhodné): mg nebo jednotky aktivity (4) nebo jednotky tvořící kolonie (CFU) nebo ml/kg kompletního krmiva o obsahu vody 12 %
…
…
Maximální dávka (je-li to vhodné): mg nebo jednotky aktivity nebo CFU nebo ml/kg kompletního krmiva o obsahu vody 12 %
…
…
U tekutých krmiv je možno uvést minimální a maximální dávku na litr.
1.2.2 Použití ve vodě
Minimální dávka (je-li to vhodné): mg nebo jednotky aktivity nebo CFU nebo ml/l vody
…
…
Maximální dávka (je-li to vhodné): mg nebo jednotky aktivity nebo CFU nebo ml/l vody
…
…
1.2.3 Zvláštní podmínky pro užití (je-li to vhodné)
Druh nebo kategorie zvířat:
…
…
Maximální stáří:
…
…
Minimální dávka (je-li to vhodné): mg nebo jednotky aktivity nebo CFU/kg kompletního krmiva o obsahu vody 12 %
…
…
Maximální dávka (je-li to vhodné): mg nebo jednotky aktivity nebo CFU/kg kompletního krmiva o obsahu vody 12 %
…
…
U tekutých krmiv je možno uvést minimální a maximální dávku na litr.
Podmínky nebo omezení použití (je-li to vhodné):
…
…
…
Zvláštní podmínky nebo omezení pro manipulaci (je-li to vhodné):
…
…
…
…
Maximální limit reziduí (je-li to vhodné):
Druh nebo kategorie zvířat:
…
…
Indikátorové reziduum:
…
…
Cílové tkáně nebo produkty:
…
…
…
Maximální reziduum v tkáních nebo produktech (μg/kg):
…
…
…
Ochranná lhůta:
…
1.3 Referenční vzorky
Číslo vzorku referenční laboratoře Společenství (je-li to vhodné):
…
Číslo partie / číslo šarže:
…
Datum výroby:
…
Datum minimální trvanlivosti:
…
Koncentrace:
…
Hmotnost:
…
Fyzický popis:
…
Popis obalu:
…
Požadavky na skladování:
…
1.4 Požadovaná změna (je-li to vhodné)
…
…
…
…
Kopie této žádosti byla zaslána přímo úřadu s dokumentací a referenční laboratoři Společenství s referenčními vzorky.
Podpis …
1.5 Přílohy:
|
|
úplná dokumentace (pouze pro úřad), |
|
|
veřejný souhrn dokumentace, |
|
|
podrobné shrnutí dokumentace, |
|
|
seznam částí dokumentace, s nimiž má být zacházeno jako s důvěrnými údaji, a kopie příslušných dotyčných částí dokumentace (pouze pro Komisi a úřad), |
|
|
kopie správních údajů žadatele(ů), |
|
|
tři vzorky doplňkové látky pro referenční laboratoř Společenství podle čl. 7 odst. 3 písm. f) nařízení (ES) č. 1831 / 2003 (pouze pro referenční laboratoř Společenství), |
|
|
list s údaji o bezpečnosti materiálu (pouze pro referenční laboratoř Společenství), |
|
|
osvědčení o identifikaci a analýze (pouze pro referenční laboratoř Společenství) a |
|
|
potvrzení o uhrazení poplatku pro referenční laboratoř Společenství (článek 4 nařízení (ES) č. 378/2005 (5). |
Vyplňte příslušné části formuláře a nehodící se části škrtněte. Originál formuláře žádosti (s ostatními požadovanými přílohami) zašlete přímo Evropské komisi.
2. SPRÁVNÍ ÚDAJE ŽADATELE(Ů)
Kontaktní údaje pro předložení žádosti o povolení doplňkové látky podle nařízení (ES) č. 1831/2003
|
1. |
Společnost nebo osoba, která podává žádost
|
|
2. |
Kontaktní osoba (pro veškerou korespondenci s Komisí, úřadem a referenční laboratoří Společenství)
|
(1) U funkční skupiny „jiné zootechnické doplňkové látky“ v kategorii zootechnické doplňkové látky je nutné jednoznačně vymezit, jakou funkci má doplňková látka mít.
(2) Úř. věst. L 10, 16.1.2004, s. 5.
(3) Úř. věst. L 268, 18.10.2003, s. 1. Nařízení naposledy pozměněné nařízením (ES) č. 298/2008 (Úř. věst. L 97, 9.4.2008, s. 64).
(4) Definici „jednotky“ poskytne žadatel.
(5) Nařízení Komise (ES) č. 378/2005 ze dne 4. března 2005 o podrobných prováděcích pravidlech k nařízení Evropského parlamentu a Rady (ES) č. 1831/2003, pokud jde o povinnosti a úkoly referenční laboratoře Společenství v souvislosti s žádostmi o povolení doplňkových látek v krmivech (Úř. věst. L 59, 5.3.2005, s. 8). Nařízení ve znění nařízení (ES) č. 850/2007 (Úř. věst. L 188, 20.7.2007, s. 3).
PŘÍLOHA II
OBECNÉ POŽADAVKY, JEŽ MUSÍ SPLŇOVAT DOKUMENTACE PODLE ČLÁNKU 3
OBECNÁ USTANOVENÍ
Tato příloha stanoví požadavky na vypracování seznamu a charakteristiky studií a informace o látkách, mikroorganismech a přípravcích, jež mají být poskytnuty s dokumentací podle článku 7 nařízení (ES) č. 1831/2003 pro:
|
— |
povolení nové doplňkové látky, |
|
— |
povolení nového užití doplňkové látky, |
|
— |
změnu stávajícího povolení doplňkové látky nebo |
|
— |
obnovení povolení doplňkové látky. |
Dokumentace musí umožnit vyhodnocení doplňkových látek na základě současného stavu poznatků a ověření, že tyto doplňkové látky odpovídají základním zásadám pro jejich povolení, které jsou stanoveny v článku 5 nařízení (ES) č. 1831/2003.
Studie, jež mají být předloženy, a jejich rozsah bude záviset na povaze doplňkové látky, kategorii nebo funkční skupině, druhu povolení (povolení, které není vázané na držitele; povolení vázané na držitele), samotné látce, cílových zvířatech a podmínkách pro užití. Žadatel odkáže na tuto přílohu a přílohu III, aby bylo možno posoudit, které studie a informace mají být společně se žádostí předloženy.
Žadatel jednoznačně uvede důvody pro vynechání některých údajů nebo odchýlení se od dokumentace předepsané v této příloze, příloze III a příloze IV.
Dokumentace musí obsahovat podrobné zprávy o všech provedených studiích, předložené v souladu se systémem číslování navrženým v této příloze. Dokumentace musí obsahovat odkazy a kopie všech zveřejněných zmíněných vědeckých údajů a kopie ostatních příslušných stanovisek, která již byla vypracována uznávanou vědeckou institucí. Pokud byly tyto studie již vyhodnoceny evropskou vědeckou institucí podle právních předpisů platných ve Společenství, postačuje odkaz na výsledek hodnocení. Údaje ze studií, které byly provedeny a zveřejněny již dříve, nebo údaje vycházející z rovnocenných zkoušek se musí jednoznačně vztahovat na stejnou doplňkovou látku jako látka, která je předmětem žádosti o povolení.
Studie, včetně studií, které byly provedeny a zveřejněny již dříve nebo které vycházejí z rovnocenných zkoušek, musí být provedeny a zdokumentovány podle příslušných norem kvality (např. správná laboratorní praxe v souladu se směrnicí Evropského parlamentu a Rady 2004/10/ES ze dne 11. února 2004 o harmonizaci právních a správních předpisů týkajících se používání zásad správné laboratorní praxe a ověřování jejich používání při zkouškách chemických látek (1) nebo Mezinárodní organizace pro normalizaci (ISO).
Pokud se studie in vivo nebo in vitro provádějí mimo Společenství, žadatel musí prokázat, že dotyčná zařízení dodržují zásady správné laboratorní praxe Organizace pro hospodářskou spolupráci a rozvoj (OECD) nebo normy ISO.
Fyzikálně-chemické, toxikologické a ekotoxikologické vlastnosti se určí podle metod stanovených směrnicí Rady 67/548/EHS ze dne 27. června 1967 o sbližování právních a správních předpisů týkajících se klasifikace, balení a označování nebezpečných látek (2), naposledy pozměněnou směrnicí Komise 2004/73/ES (3), nebo pomocí aktualizovaných metod uznaných mezinárodními vědeckými institucemi. Použití jiných metod musí být zdůvodněno.
Je nutno podporovat používání metod in vitro nebo metod zpřesňujících nebo nahrazujících obvyklé testy prováděné s pomocí laboratorních zvířat nebo snižujících počet zvířat použitých při těchto testech. Tyto metody mají stejnou kvalitu a poskytují stejnou úroveň jistoty jako metoda, kterou mají nahradit.
Popis metod analýzy v krmivech nebo ve vodě musí být v souladu s pravidly správné laboratorní praxe stanovenými ve směrnici 2004/10/ES a/nebo normě EN ISO/IEC 17025. Tyto metody vyhovují požadavkům stanoveným v článku 11 nařízení Evropského parlamentu a Rady (ES) č. 882/2004 ze dne 29. dubna 2004 o úředních kontrolách za účelem ověření dodržování právních předpisů týkajících se krmiv a potravin a pravidel o zdraví zvířat a dobrých životních podmínkách zvířat (4).
Každá dokumentace musí obsahovat veřejný souhrn a podrobné vědecké shrnutí, aby bylo možno dotyčnou doplňkovou látku identifikovat a charakterizovat.
Každá dokumentace obsahuje návrh plánu monitorování v období po uvedení látky na trh, pokud se požaduje podle čl. 7 odst. 3 písm. g) nařízení (ES) č. 1831/2003, a návrh na označování podle čl. 7 odst. 3 písm. e) nařízení (ES) č. 1831/2003.
Posouzení bezpečnosti
Toto posouzení je založeno na studiích, které mají prokázat bezpečné používání doplňkové látky ve vztahu k:
|
a) |
cílovým druhům při nejvyšších navrhovaných obsazích pro zapracování do krmiv nebo vody a při násobku tohoto obsahu pro stanovení rozpětí pro bezpečnost; |
|
b) |
spotřebitelům, kteří požívají potravinářské výrobky získané ze zvířat přijímajících doplňkovou látku, její rezidua nebo metabolity. V tomto případě bude bezpečnost zajištěna stanovením maximálních limitů reziduí (MLR) a ochranných lhůt na základě přijatelné denní dávky (ADI) nebo nejvyšší povolené dávky (UL); |
|
c) |
osobám, které budou pravděpodobně vystaveny doplňkové látce vdechnutím nebo zasažením sliznice, očí či kůže při manipulaci s doplňkovou látkou jako takovou nebo při jejím zapracovávání do premixů nebo kompletních krmiv nebo vody či při používání krmiva nebo vody, které obsahují dotyčnou doplňkovou látku; |
|
d) |
zvířatům a lidským bytostem vzhledem k selekci a rozšíření genů rezistentních vůči antimikrobiálním látkám a |
|
e) |
životnímu prostředí, s ohledem na nebezpečí vyplývající z doplňkové látky samotné nebo produktů z ní odvozených, ať už přímo a/nebo vyloučených zvířaty. |
Pokud má doplňková látka více složek, každá složka může být posouzena z hlediska bezpečnosti pro spotřebitele samostatně a posléze vyhodnocena s ohledem na kumulativní účinek (lze-li prokázat, že mezi složkami nedochází k vzájemnému působení). Alternativně je možno posoudit kompletní směsi.
Posouzení účinnosti
Toto posouzení je založeno na studiích, které mají prokázat účinnost doplňkové látky, pokud jde o určené užití, jak je stanoveno v čl. 6 odst. 1 a příloze I nařízení (ES) č. 1831/2003.
1. ODDÍL I: SOUHRN DOKUMENTACE
1.1 Veřejný souhrn dokumentace podle čl. 7 odst. 3 písm. h) nařízení (ES) č. 1831/2003
Žadatel předloží souhrn s uvedením hlavních vlastností dotyčné doplňkové látky. Souhrn nesmí obsahovat žádné důvěrné údaje a musí mít tuto strukturu:
1.1.1 Obsah
|
a) |
jméno žadatele(ů); |
|
b) |
identifikace doplňkové látky; |
|
c) |
způsob výroby a metoda analýzy; |
|
d) |
studie bezpečnosti a účinnosti doplňkové látky; |
|
e) |
navrhované podmínky pro užití a |
|
f) |
návrh plánu monitorování v období po uvedení látky na trh. |
1.1.2. Popis
|
a) |
Jméno a adresa žadatele(ů) Tyto údaje musí být poskytnuty ve všech případech nezávisle na druhu povolení doplňkové látky (povolení vázané na držitele nebo povolení, které není vázané na držitele). Předkládá-li dokumentaci skupina žadatelů, musí být uvedeno jméno každého žadatele. |
|
b) |
Identifikace doplňkové látky Identifikace doplňkové látky obsahuje shrnutí požadovaných informací podle přílohy II nebo III podle druhu povolení doplňkové látky. Konkrétně: název doplňkové látky, navrhované zařazení do kategorie a funkční skupiny, cílové druhy/kategorie zvířat a dávky. |
|
c) |
Způsob výroby a metoda analýzy Je nutno popsat výrobní postup. Je nutno popsat obecné postupy analytických metod, jež mají být použity pro analýzu pro úřední kontroly doplňkové látky jako takové, v premixech a krmivech podle požadavků této přílohy a přílohy III. Popřípadě se na základě informací předložených v souladu s touto přílohou a přílohou III uvede postup metody (metod) použitých k analýze pro úřední kontroly doplňkových látek nebo jejich metabolitů v potravinách živočišného původu. |
|
d) |
Studie bezpečnosti a účinnosti doplňkové látky Je nutno uvést závěr o bezpečnosti a účinnosti doplňkové látky na základě různých provedených studií. Výsledky studií mohou být uvedeny v podobě tabulek na podporu závěru žadatele(ů). Ve shrnutí by měly být uvedeny pouze studie požadované podle přílohy III. |
|
e) |
Navrhované podmínky pro užití Žadatel(é) předloží návrh podmínek pro užití. Žadatel zejména popíše obsah použití ve vodě nebo krmivu spolu s podrobnými podmínkami pro užití v doplňkových krmivech. Požadují se rovněž informace, zda se používají jiné způsoby podávání nebo zapracování do krmiva nebo vody. Je nutno popsat případné zvláštní podmínky pro užití (např. nesnášenlivosti), zvláštní požadavky na označování a druhy zvířat, pro něž je doplňková látka určena. |
|
f) |
Návrh plánu monitorování v období po uvedení látky na trh Tato část se vztahuje pouze na doplňkové látky, které podle čl. 7 odst. 3 písm. g) nařízení (ES) č. 1831/2003 nepatří do kategorií a) nebo b) v čl. 6 odst. 1 téhož nařízení, a na doplňkové látky, které spadají do oblasti působnosti práva Společenství o uvádění na trh produktů, které se skládají z geneticky modifikovaných organismů nebo geneticky modifikované organismy obsahují nebo jsou z nich vyrobeny. |
1.2 Vědecké shrnutí dokumentace
Je nutno poskytnout vědecké shrnutí obsahující údaje o každé části dokumentů poskytnutých na podporu žádosti podle této přílohy a přílohy III. Toto shrnutí obsahuje závěry vyvozené žadatelem (žadateli).
Shrnutí musí dodržet pořadí této přílohy a zabývat se všemi jednotlivými částmi s odkazem na příslušné strany dokumentace.
1.3 Seznam dokumentů a ostatních údajů
Žadatel musí uvést počet a názvy svazků dokumentace předložené na podporu žádosti. Je nutno připojit podrobný index s odkazem na svazky a strany.
1.4 Seznam částí dokumentace, s nimiž má být zacházeno jako s důvěrnými údaji
Seznam obsahuje odkaz na příslušné svazky a strany dokumentace.
2. ODDÍL II: IDENTITA, CHARAKTERISTIKA A PODMÍNKY PRO UŽITÍ DOPLŇKOVÉ LÁTKY; METODY ANALÝZY
Doplňková látka musí být plně identifikována a charakterizována.
2.1 Identita doplňkové látky
2.1.1 Název doplňkové látky
Případně se uvede navrhovaný obchodní název doplňkových látek vázaných na držitele povolení.
2.1.2 Návrh na zařazení
Je nutno předložit návrh na zařazení doplňkové látky do jedné či několika kategorií a funkčních skupin podle jejích hlavních funkcí v souladu s článkem 6 a přílohou I nařízení (ES) č. 1831/2003.
Musí být poskytnuty údaje z jiných známých použití identických účinných látek nebo činidel (např. užití v potravinách, humánním nebo veterinárním lékařství, zemědělství a průmyslu). Je nutno uvést případná jiná povolení jako doplňková látka nebo potravinářská přídatná látka, veterinární léčivé přípravky nebo jiný druh povolení účinné látky.
2.1.3 Kvalitativní a kvantitativní složení (účinná látka/činidlo, jiné složky, nečistoty, variabilita jednotlivých šarží)
Je nutno uvést účinnou látku(y)/činidlo(a) a všechny ostatní složky doplňkové látky s uvedením hmotnostního podílu v hotovém produktu. Je nutno stanovit kvalitativní a kvantitativní variabilitu jednotlivých šarží účinné látky (látek) / činidla (činidel).
Pro mikroorganismy: je nutno stanovit počet životaschopných buněk nebo spor vyjádřený jako CFU na gram.
Pro enzymy: je nutno popsat všechny udávané (hlavní) aktivity a počet jednotek aktivity v daném hotovém produktu. Je nutno zmínit rovněž důležité vedlejší aktivity. Vymezí se jednotky aktivity, pokud možno jako μmoly produktu uvolněné za minutu ze substrátu, rovněž s uvedením pH a teploty.
Je-li účinná složka doplňkové látky směsí účinných látek nebo činidel, přičemž všechny lze jednoznačně definovat (kvalitativně a kvantitativně), musí být složky účinné látky (látek) / činidla (činidel) popsány samostatně s uvedením podílů v dané směsi.
Ostatní směsi, jejichž složky nelze popsat jedním chemickým vzorcem a/nebo není možno všechny složky identifikovat, musí být charakterizovány podle složky (složek) přispívajících k její aktivitě a/nebo podle typické hlavní složky (složek).
Aniž je dotčena případná žádost o doplňkové informace předložená úřadem podle čl. 8 odst. 2 nařízení (ES) č. 1831/2003, žadatel může vynechat popis ostatních složek, které nepředstavují bezpečnostní rizika, jiných než účinné látky nebo činidla v případě doplňkových látek, které nejsou zařazeny v kategoriích zootechnických doplňkových látek, kokcidiostatik a histomonostatik a na něž se nevztahuje oblast působnosti nařízení (ES) č. 1829/2003. Všechny studie uvedené v dokumentaci musí být každopádně založeny na konkrétní doplňkové látce, pro niž se požaduje povolení, a mohou poskytovat informace o jiných možných odlišných přípravcích. Je možno povolit interní identifikátor obsažený v dokumentech třetích stran a je nutný přehled za účelem vyjmenování identifikátorů a potvrzení, že se identifikátor(y) vztahuje(í) na složení, pro něž se podává žádost.
2.1.4 Čistota
Žadatel identifikuje a vyčíslí chemické a mikrobiální nečistoty, látky s toxickými nebo jinými nežádoucími vlastnostmi, které nejsou přidány záměrně a nepřispívají k aktivitě doplňkové látky. U produktů fermentačního procesu žadatel potvrdí nepřítomnost produkčních organismů v doplňkové látce. Je nutno popsat protokol používaný k rutinní kontrole výrobních šarží s ohledem na znečišťující látky a nečistoty.
Veškeré poskytnuté údaje musí podpořit návrh na specifikaci doplňkové látky.
Níže jsou uvedeny zvláštní požadavky v závislosti na výrobním postupu, které odpovídají stávajícím právním předpisům Společenství.
2.1.4.1 Doplňkové látky, jejichž povolení je vázané na držitele povolení
U doplňkových látek, jejichž povolení je vázané na držitele povolení, je nutno uvést příslušné informace týkající se zvláštního procesu použitého výrobcem na základě stávajících norem používaných k jiným souvisejícím účelům. Je možno použít specifikace Společného výboru odborníků FAO/WHO pro potravinářské přídatné látky (JECFA) nebo specifikace z povolení Evropského společenství pro potravinářské přídatné látky.
2.1.4.2 Doplňkové látky, jejichž povolení není vázané na držitele povolení
U doplňkových látek, jejichž povolení není vázané na držitele povolení, je možno použít stávající normy používané k jiným souvisejícím účelům nebo normy se specifikacemi pro potravinářské přídatné látky povolené v Evropském společenství nebo JECFA. Nejsou-li takové normy dostupné nebo je-li to důležité pro výrobní postup, je nutno popsat přinejmenším níže uvedené údaje a stanovit koncentrace:
|
— |
pro mikroorganismy: mikrobiologická kontaminace, mykotoxiny, těžké kovy; |
|
— |
pro produkty fermentačního procesu (které neobsahují mikroorganismy jako účinná činidla): je nutno dodržet stejné požadavky jako pro produkty mikroorganismů (viz výše). Je nutno uvést rovněž rozsah, v němž je do hotového produktu zapracováno použité kultivační medium; |
|
— |
pro látky rostlinného původu: mikrobiologická a rostlinná kontaminace (zejména např. skočec obecný, semena plevele, žitný námel), mykotoxiny, kontaminace pesticidy, maximální hodnoty pro rozpouštědla a popřípadě toxikologicky významné látky, o nichž je známo, že se vyskytují v původní rostlině; |
|
— |
pro látky živočišného původu: mikrobiologická kontaminace, těžké kovy a popřípadě maximální hodnoty pro rozpouštědla; |
|
— |
pro minerální látky: těžké kovy, dioxiny a PCB; |
|
— |
pro produkty vyrobené chemickou syntézou a chemickými procesy: určí se všechny chemické látky použité v syntetických procesech a případné meziprodukty zůstávající v hotovém produktu a uvede se jejich koncentrace. |
Popřípadě se provede selekce mykotoxinů pro analýzu podle různých matric.
2.1.5 Fyzikální vlastnosti jednotlivých forem produktu
U přípravků v tuhém stavu se poskytnou údaje o rozdělení podle velikosti částic, formě částic, hustotě, objemové hustotě, protiprašných vlastnostech a použití procesů, které mají vliv na fyzikální vlastnosti. U přípravků v kapalném stavu se poskytnou údaje o viskozitě a povrchovém napětí. Pokud je doplňková látka určena k použití ve vodě, je nutno prokázat rozpustnost nebo stupeň disperze.
2.2 Charakteristika účinné látky (látek) / činidla (činidel)
2.2.1 Popis
Je nutno uvést kvalitativní popis účinné látky nebo činidla. To zahrnuje čistotu a původ látky nebo činidla a jiné důležité charakteristiky.
2.2.1.1 Chemické látky
Chemicky přesně definované látky je nutno popsat druhovým názvem, chemickým označením podle názvosloví IUPAC (International Union of Pure and Applied Chemistry), dalšími mezinárodními druhovými názvy a zkratkami a/nebo číslem CAS (Chemical Abstract Service). Je nutno uvést strukturální a empirický vzorec a molekulovou hmotnost.
U chemicky definované sloučeniny použité jako zchutňující látka je nutno uvést číslo FLAVIS spolu s příslušnou chemickou skupinou. U rostlinných výtažků je nutno uvést fytochemické markery.
Směsi, v nichž nelze složky popsat jedním chemickým vzorcem a/nebo v nichž nelze všechny složky identifikovat, je nutno charakterizovat podle složky (složek) přispívajících k její aktivitě a/nebo podle typických hlavních složek. Je nutno identifikovat markerovou sloučeninu, aby bylo možno posoudit stabilitu a zajistit sledovatelnost.
Pro enzymy a přípravky enzymů je nutno pro každou udávanou aktivitu uvést číslo a systematický název navrhovaný Mezinárodní unií biochemie (IUB) v posledním vydání „Názvosloví enzymů“. U aktivit dosud nezahrnutých se použije systematický název odpovídající pravidlům názvosloví IUB. Obecné názvy jsou přípustné za předpokladu, že jsou jednoznačné a jsou používány konzistentně v celé dokumentaci a při jejich první zmínce je lze jednoznačně vztáhnout na systematický název a číslo IUB. Je nutno uvést biologický původ každé enzymové aktivity.
Je nutno popsat rovněž mikrobiální původ chemických látek vzniklých fermentací (viz bod 2.2.1.2 Mikroorganismy).
2.2.1.2 Mikroorganismy
Je nutno uvést původ všech mikroorganismů používaných jako produkt nebo výrobní kmen.
U mikroorganismů používaných jako produkt nebo jako výrobní kmen je nutno uvést historii změn. Uvede se název a taxonomická klasifikace každého mikroorganismu podle nejnovějších zveřejněných údajů v mezinárodních nomenklaturních kódech (ICN). Mikrobiální kmeny musí být uloženy v mezinárodně uznávané sbírce kultur (pokud možno v Evropské unii) a uchovávány sbírkou kultur po povolenou dobu používání doplňkové látky. Je nutno předložit osvědčení o uložení od sbírky, které upřesňuje depozitní číslo, pod kterým je kmen uložen. Dále je nutno popsat všechny relevantní morfologické, fyziologické a molekulární charakteristiky nezbytné pro jedinečnou identifikaci kmene a prostředky potvrzení jeho genetické stability. U geneticky modifikovaných organismů se uvede popis genetických změn. Pro každý geneticky modifikovaný organismus se uvede jednoznačný identifikační kód podle nařízení Komise (ES) č. 65/2004 ze dne 14. ledna 2004, kterým se zřizuje systém tvorby a přiřazování jednoznačných identifikačních kódů pro geneticky modifikované organismy.
2.2.2 Důležité vlastnosti
2.2.2.1 Chemické látky
Je nutno uvést popis fyzikálních a chemických vlastností. Konstanta rozkladu, pKa, elektrostatické vlastnosti, bod tání, bod varu, hustota, tlak par, rozpustnost ve vodě a v organických rozpouštědlech, Kow a Kd/Koc, hmotnostní spektrometrie a absorpční spektra, údaje NMR, případné izomery a jakékoliv jiné důležité fyzikální vlastnosti musí být uvedeny, kde je to vhodné.
Látka získaná prostřednictvím fermentačního procesu nesmí vykazovat antimikrobiální aktivity významné pro používání antibiotik u člověka nebo zvířat.
2.2.2.2 Mikroorganismy
|
— |
Toxiny a faktory virulence Je nutno prokázat neexistenci toxinů nebo faktorů virulence nebo jejich nevýznamnost. Kmeny bakterií náležející do taxonomické skupiny, která zahrnuje příslušníky, o nichž je známo, že mohou produkovat toxiny nebo jiné faktory virulence, je nutno podrobit odpovídajícím testům s cílem prokázat na molekulové a popřípadě buněčné úrovni, že neexistuje důvod k obavám. U kmenů mikroorganismů, pro něž nejsou k dispozici údaje o zřejmém bezpečném používání a jejichž biologie není dosud dostatečně známa, je nezbytný celý balíček toxikologických studií. |
|
— |
Produkce antibiotik a rezistence vůči antibiotikům Mikroorganismy používané jako doplňkové látky nebo jako výrobní kmen nesmí vykazovat antibiotickou aktivitu nebo nesmí být schopny produkovat antibiotické látky, které jsou důležité jako antibiotika u člověka a zvířat. Kmeny mikroorganismů určené k použití jako doplňkové látky nesmí dále přispívat k zdroji genů rezistence vůči antibiotikům, které se již vyskytují v střevní flóře zvířat a v životním prostředí. Všechny kmeny bakterií je proto nutno otestovat na rezistenci vůči antibiotikům používaným v humánním a veterinárním lékařství. Je-li zjištěna rezistence, je nutno zjistit genetický základ rezistence a pravděpodobnost přenosu rezistence na ostatní organismy ve střevech. Kmeny mikroorganismů nesoucích získanou antimikrobiální rezistenci se nesmí používat jako doplňkové látky, nelze-li prokázat, že rezistence je výsledkem chromozómové mutace(í) a není přenosná. |
2.3 Výrobní postup, včetně zvláštních výrobních postupů
Ke stanovení kritických míst procesu, která mohou mít vliv na čistotu účinné látky / činidla (činidel) nebo doplňkové látky, se uvede popis výrobního postupu. Je nutno předložit listy s údaji o bezpečnosti materiálu u chemických látek používaných ve výrobním postupu.
2.3.1 Účinná látky(y) / činidlo(a)
Je nutno poskytnout popis výrobního postupu (např. chemická syntéza, fermentace, kultivace, extrakce z organických materiálů nebo destilace) použitého při přípravě účinné látky (látek) / činidla (činidel) doplňkové látky, případně prostřednictvím postupového diagramu. Uvede se složení fermentačního/kultivačního média. Je nutno popsat důkladně metody čištění.
U geneticky modifikovaných mikroorganismů (GMM) používaných jako zdroj doplňkových látek a kultivovaných za podmínek uzavřeného nakládání se použije směrnice Rady 90/219/ES (5). Je nutno uvést popis fermentačních procesů (médium pro kultury, fermentační podmínky a následné zpracování produktů fermentačního procesu).
2.3.2 Doplňková látka
Předloží se podrobný popis výrobního postupu doplňkové látky. Je nutno uvést hlavní stupně výroby doplňkové látky, včetně míst (místa) zapracování účinné látky (látek) / činidla (činidel) a jiných složek a všechny následné stupně zpracování, které mají vliv na přípravu doplňkové látky, případně prostřednictvím postupového diagramu.
2.4 Fyzikálně-chemické a technologické vlastnosti doplňkové látky
2.4.1 Stabilita
Stabilita je obvykle měřena analytickým sledováním účinné látky (látek) / činidla (činidel) nebo její (jeho) aktivity/životaschopnosti. U enzymů lze stabilitu definovat s ohledem na ztrátu katalytické aktivity; u mikroorganismů s ohledem na ztrátu životaschopnosti; u zchutňujících látek s ohledem na ztrátu chuti. U ostatních chemických směsí/extraktů lze stabilitu posuzovat sledováním koncentrace jedné nebo několika vhodných indikátorových látek.
Stabilita doplňkové látky
Je nutno přezkoumat stabilitu každého složení doplňkové látky při působení podmínek vnějšího prostředí (světlo, teplota, pH, vlhkost, kyslík a obalové materiály). Předpokládaná trvanlivost doplňkové látky v obchodovatelné formě by měla být založena na nejméně dvou modelových situacích zahrnujících pravděpodobné spektrum podmínek pro užití (např. 25 oC, 60 % relativní vlhkost vzduchu a 40 oC, 75 % relativní vlhkost vzduchu).
Stabilita doplňkové látky užité v premixech a krmivech
U doplňkových látek užitých v premixech a krmivech s výjimkou zchutňujících složek se stabilita každého složení doplňkové látky ověří za běžných výrobních a skladovacích podmínek premixů a krmiv. Studie stability premixů musí trvat nejméně šest měsíců. Stabilita se testuje pokud možno u premixů obsahujících stopové prvky; v opačném případě by doplňková látka měla být označena nápisem „nemíchat se stopovými prvky“.
Studie stability u krmiv trvají normálně nejméně tři měsíce. Obvykle se stabilita kontroluje u krmiv ve formě kaše a peletovaných krmiv (včetně vlivu peletování nebo jiných forem úpravy) pro hlavní udávané druhy zvířat.
U doplňkových látek určených k použití ve vodě je nutno stabilitu každého složení doplňkové látky ověřit ve vodě za podmínek simulujících použití v praxi.
Dojde-li ke ztrátě stability a ve vhodných případech je nutno charakterizovat případné produkty degradace nebo rozkladu.
Je nutno poskytnout údaje z analýz, které zahrnují nejméně jedno pozorování na začátku a jedno na konci doby skladování.
V případě potřeby studie obsahují podrobné kvantitativní a kvalitativní složení premixů nebo krmiv použitých pro zkoušky.
2.4.2 Homogenita
Je nutno prokázat schopnost homogenního rozložení doplňkové látky (vyjma zchutňujících složek) v premixech, krmivech nebo ve vodě.
2.4.3 Ostatní vlastnosti
Je nutno popsat ostatní vlastnosti, například protiprašné a elektrostatické vlastnosti nebo schopnost disperze v kapalinách.
2.4.4 Fyzikálně-chemické nesnášenlivosti nebo interakce
Je nutno uvést fyzikálně-chemické nesnášenlivosti nebo interakce, k nimž by mohlo dojít ve styku s krmivy, nosiči, jinými povolenými doplňkovými látkami nebo léčivými přípravky.
2.5 Podmínky pro užití doplňkové látky
2.5.1 Navrhovaný způsob použití ve výživě zvířat
Je nutno uvést druh nebo kategorie zvířat, věkovou skupinu nebo produkční stadium zvířat v souladu s kategoriemi v příloze IV tohoto nařízení. Je třeba zmínit možné kontraindikace. Uvede se navrhované použití v krmivu nebo ve vodě.
Je nutno uvést navrhovaný způsob podávání a zapracování pro premixy, krmiva nebo pitnou vodu. Uvede se navrhovaná dávka v kompletním krmivu a navrhovaná doba podávání a popřípadě navrhovaná ochranná lhůta. Pokud se navrhuje konkrétní použití doplňkové látky v doplňkovém krmivu, je nutné odůvodnění.
2.5.2 Informace související s bezpečností pro uživatele/pracovníky
2.5.2.1 Chemické látky
Je nutno poskytnout listy s údaji o bezpečnosti materiálu ve formátu odpovídajícím požadavkům směrnice Komise 91/155/EHS ze dne 5. března 1991, kterou se k provedení článku 10 směrnice 88/379/EHS vymezují a stanoví podrobná opatření k systému specifických informací pro nebezpečné přípravky (6). V případě potřeby je nutno navrhnout opatření pro předcházení pracovním rizikům a ochranné prostředky při výrobě, manipulaci, užití a likvidaci.
2.5.2.2 Mikroorganismy
Je nutno uvést klasifikaci podle směrnice Evropského parlamentu a Rady 2000/54/ES ze dne 18. září 2000 o ochraně zaměstnanců před riziky spojenými s expozicí biologickým činitelům při práci (sedmá samostatná směrnice ve smyslu čl. 16 odst. 1 směrnice 89/391/EHS) (7). V případě mikroorganismů nezařazených do skupiny 1 této směrnice je nutno poskytnout spotřebitelům informace, které jim umožní přijmout příslušná opatření na ochranu pracovníků, jak je stanoveno v čl. 3 odst. 2 uvedené směrnice.
2.5.2.3 Požadavky na označování
Aniž jsou dotčena ustanovení o označování a balení obsažená v článku 16 nařízení (ES) č. 1831/2003, je nutno uvést zvláštní požadavky na označování a popřípadě zvláštní podmínky pro užití a manipulaci (včetně známých nesnášenlivostí a kontraindikací) a pokyny pro správné použití.
2.6 Metody analýzy a referenční vzorky
Metody analýzy se uvedou ve standardní úpravě podle doporučení normy ISO (tj. ISO 78-2).
Podle nařízení (ES) č. 1831/2003 a nařízení (ES) č. 378/2005 metody analýzy uvedené v tomto oddíle vyhodnotí referenční laboratoř Společenství. Referenční laboratoř Společenství předloží úřadu hodnotící zprávu, v níž se uvádí, zda jsou tyto metody vhodné pro použití za účelem úředních kontrol doplňkové látky, na niž se žádost vztahuje. Hodnocení referenční laboratoře Společenství se zaměří na metody stanovené v bodech 2.6.1 a 2.6.2.
Pokud byl stanoven MLR pro látku, na niž se vztahuje nařízení Rady (EHS) č. 2377/90 ze dne 26. června 1990, kterým se stanoví postup Společenství pro stanovení maximálních limitů reziduí veterinárních léčivých přípravků v potravinách živočišného původu (8), nebude bod 2.6.2 předmětem hodnocení referenční laboratoře Společenství. Žadatel sestaví bod 2.6.2 obsahující stejnou metodu, informace a údaje (včetně příslušných aktualizací), jež mají být předloženy Evropské agentuře pro léčivé přípravky (EMEA) v souladu s přílohou V nařízení (EHS) č. 2377/90 a podle dokumentu „Notice to Applicants and Guidelines“, svazek 8 „Rules governing medicinal products in the European Union“.
V hodnocení mohou být zahrnuty rovněž analytické metody popsané v bodě 2.6.3, pokud to referenční laboratoř Společenství, úřad nebo Komise považují za nezbytné.
V souladu s nařízením (ES) č. 378/2005 žadatel poskytne referenční vzorky přímo referenční laboratoři Společenství před vyhodnocením technické dokumentace a náhradní vzorky před uplynutím doby použitelnosti.
Žadatelé odkáží na podrobné pokyny stanovené referenční laboratoří Společenství v souladu s článkem 12 nařízení (ES) č. 378/2005.
2.6.1 Metody analýzy pro účinnou látku
Je nutno poskytnout podrobnou charakteristiku kvalitativní a popřípadě kvantitativní analytické metody (metod) pro zjištění, zda byly dodrženy maximální nebo minimální navrhované obsahy účinné látky (látek) / činidla (činidel) v doplňkové látce, premixech, krmivech a popřípadě vodě.
|
2.6.1.1 |
Tyto metody splňují stejné požadavky jako v případě metod analýzy používaných pro úřední kontroly podle článku 11 nařízení (ES) č. 882/2004. Zejména splňují nejméně jeden z těchto požadavků:
|
|
2.6.1.2 |
Podrobná charakteristika metody (metod) zahrnuje odpovídající charakteristiky stanovené v příloze III nařízení (ES) č. 882/2004. |
|
2.6.1.3 |
Pracovní charakteristiky metod validovaných interně se ověří otestováním metody v druhé akreditované a nezávislé laboratoři. Poskytnou se výsledky těchto testů spolu s případnými dalšími informacemi dokládajícími přenositelnost metody na úřední kontrolní laboratoř. Z důvodu nezávislosti a účasti na hodnocení dokumentace poskytnuté žadatelem v případě, že druhá laboratoř je účastníkem konsorcia národních referenčních laboratoří napomáhajících referenční laboratoři Společenství, jak je stanoveno v nařízení (ES) č. 378/2005, dotyčná laboratoř zašle referenční laboratoři Společenství prohlášení o zájmech (jakmile referenční laboratoř Společenství obdrží žádost) popisující práci laboratoře v souvislosti s danou žádostí a nesmí se účastnit vyhodnocení žádosti. |
|
2.6.1.4 |
Ve své hodnotící zprávě pro úřad může referenční laboratoř Společenství vybrat vhodné charakteristiky podle přílohy III nařízení (ES) č. 882/2004. |
|
2.6.1.5 |
Pracovní charakteristiky pro metody zvláštních skupin látek (například enzymy) mohou být stanoveny v podrobných pokynech poskytnutých referenční laboratoří Společenství podle článku 12 nařízení (ES) č. 378/2005. |
2.6.2 Metody analýzy pro stanovení reziduí doplňkové látky nebo jejích metabolitů v potravinách
Je nutno poskytnout podrobnou charakteristiku kvalitativních a kvantitativních analytických metod pro stanovení indikátorových reziduí a/nebo metabolitů doplňkové látky v cílových tkáních a živočišných produktech.
|
2.6.2.1 |
Tyto metody splňují stejné požadavky jako v případě metod analýzy používaných pro úřední kontroly podle článku 11 nařízení (ES) č. 882/2004. Tyto metody splňují zejména nejméně jeden z požadavků uvedených v bodě 2.6.1.1. |
|
2.6.2.2 |
Podrobná charakteristika metody (metod) zahrnuje odpovídající charakteristiky stanovené v příloze III nařízení (ES) č. 882/2004 a bere v úvahu požadavky stanovené v rozhodnutí Komise 2002/657/ES (10). Popřípadě se uváží stejné pracovní charakteristiky stanovené v rozhodnutích Komise, která stanoví analytické metody používané k zjišťování určitých látek a jejich reziduí v živých zvířatech podle směrnice Rady 96/23/ES. Limit kvantifikace u každé metody nesmí překročit polovinu odpovídajícího MLR a musí být validován v rozmezí přinejmenším od jedné poloviny po dvojnásobek MLR. |
|
2.6.2.3 |
Pracovní charakteristiky interně validovaných metod se ověří otestováním metody v druhé akreditované a nezávislé laboratoři. Je nutno uvést výsledky těchto testů. Z důvodu nezávislosti a účasti na hodnocení dokumentace poskytnuté žadatelem v případě, že druhá laboratoř je účastníkem konsorcia národních referenčních laboratoří napomáhajících referenční laboratoři Společenství, jak je stanoveno v nařízení (ES) č. 378/2005, dotyčná laboratoř zašle referenční laboratoři Společenství prohlášení o zájmech (jakmile referenční laboratoř Společenství obdrží žádost) popisující práci laboratoře v souvislosti s danou žádostí a nesmí se účastnit vyhodnocení žádosti. |
|
2.6.2.4 |
Ve své hodnotící zprávě pro úřad může referenční laboratoř Společenství vybrat vhodné charakteristiky z charakteristik uvedených v bodě 2.6.2.2. |
|
2.6.2.5 |
Pracovní charakteristiky pro metody pro zvláštní skupiny látek (například enzymy) mohou být stanoveny v podrobných pokynech poskytnutých referenční laboratoří Společenství podle článku 12 nařízení (ES) č. 378/2005. |
2.6.3 Metody analýzy týkající se identity a charakteristika doplňkové látky
Žadatel poskytne popis metod použitých ke stanovení charakteristik podle bodů 2.1.3, 2.1.4, 2.1.5, 2.2.2, 2.4.1, 2.4.2, 2.4.3 a 2.4.4.
V souladu s přílohou II nařízení (ES) č. 1831/2003 ve znění nařízení (ES) č. 378/2005 mohou být vyhodnoceny rovněž metody poskytnuté podle tohoto bodu, pokud to úřad nebo Komise považují za důležité pro posouzení žádosti.
Doporučuje se, aby metody popsané v tomto bodě byly mezinárodně uznané. Metody, které nejsou mezinárodně uznané, je nutno plně popsat. V těchto případech musí být studie provedeny akreditovanými a nezávislými laboratořemi a musí být zdokumentovány podle příslušných norem kvality (např. správná laboratorní praxe podle směrnice 2004/10/ES nebo normy ISO).
Metody pro identifikaci a charakteristiku doplňkové látky splňují stejné požadavky jako metody analýzy používané pro úřední kontroly podle článku 11 nařízení (ES) č. 882/2004, zejména pokud jsou stanoveny právní požadavky (např. nečistoty, nežádoucí látky).
3. ODDÍL III: STUDIE TÝKAJÍCÍ SE BEZPEČNOSTI DOPLŇKOVÉ LÁTKY
Studie uvedené v tomto oddíle a ve zvláštních přílohách mají umožnit vyhodnocení:
|
— |
bezpečnosti použití doplňkové látky pro cílové druhy zvířat, |
|
— |
případného rizika souvisejícího se selekcí a/nebo přenosem rezistence vůči antimikrobiálním látkám a vyšší perzistencí a uvolňováním enteropatogenů, |
|
— |
rizik pro spotřebitele u potravin získaných ze zvířat, jimž byla podávána krmiva obsahující doplňkovou látku nebo krmiva ošetřená doplňkovou látkou, nebo rizik, která mohou vyplývat z konzumace potravin obsahujících rezidua doplňkové látky nebo jejích metabolitů, |
|
— |
rizik pro osoby manipulující s doplňkovou látkou jako takovou nebo zapracovanou do premixů či krmiv, vznikajících při jejím vdechování či zasažení sliznice, očí nebo kůže a |
|
— |
rizik nežádoucích účinků vlastní doplňkové látky nebo produktů z ní odvozených, působících přímo a/nebo vyměšovaných zvířaty, na životní prostředí. |
3.1 Studie týkající se bezpečnosti používání doplňkové látky pro cílové druhy zvířat
Studie uvedené v tomto oddíle jsou určeny k vyhodnocení:
|
— |
bezpečnosti používání doplňkové látky pro cílové druhy zvířat a |
|
— |
případného rizika souvisejícího se selekcí a/nebo přenosem rezistence vůči antimikrobiálním látkám a vyšší perzistencí a uvolňováním enteropatogenů. |
3.1.1 Studie tolerance u cílových druhů zvířat
Účelem testu tolerance je poskytnout omezené vyhodnocení krátkodobé toxicity doplňkové látky u cílových zvířat. Používá se rovněž ke stanovení rozpětí pro bezpečnost, pokud se doplňková látka požije v dávkách vyšších, než je doporučeno. Tyto testy tolerance je nutno provést k doložení bezpečnosti pro všechny cílové druhy/kategorie zvířat, na něž se žádost vztahuje. V některých případech je přípustné zahrnout některé prvky testu tolerance do jedné ze zkoušek účinnosti, pokud jsou splněny níže uvedené požadavky na tyto testy. Všechny studie uvedené v tomto oddíle musí být založeny na doplňkové látce popsané v oddíle II.
3.1.1.1 Návrh testu tolerance zahrnuje nejméně tři skupiny:
|
— |
skupinu, jíž se doplňková látka nepodává, |
|
— |
skupinu s nejvyšší doporučenou dávkou a |
|
— |
pokusnou skupinu s mnohonásobným obsahem nejvyšší doporučené dávky. |
V pokusné skupině se doplňková látka obvykle podává ve výši desetinásobku nejvyšší doporučené dávky. Pokusná zvířata jsou rutinně sledována s ohledem na vizuální důkazy klinických účinků, pracovní charakteristiky, popřípadě jakost produktu, hematologii a rutinní vyšetření chemického složení krve a jiné parametry, které pravděpodobně souvisejí s biologickými vlastnostmi doplňkové látky. Vezmou se do úvahy kritické parametry známé z toxikologických studií u laboratorních zvířat. V tomto oddíle se uvedou rovněž případné nežádoucí účinky zjištěné během zkoušek účinnosti. V případě nevysvětlených úmrtí při testu tolerance se provede nekropsie a popřípadě histologie.
Pokud lze prokázat, že je snášen 100násobek maximální doporučené dávky, nepožaduje se hematologie ani rutinní vyšetření chemického složení krve. Je-li produkt snášen pouze při obsahu nižším, než je 10násobek nejvyšší doporučené dávky, je studie navržena tak, aby bylo možno vypočítat rozpětí pro bezpečnost doplňkové látky, a uvedou se doplňkové parametry (pomocí nekropsie, popřípadě histologie a jiných vhodných kritérií).
U některých doplňkových látek nemusí být v závislosti na jejich toxikologii a metabolismu nebo užití nutné provést testy tolerance.
Použité experimentální schéma musí zahrnovat úvahu o odpovídající statistické průkaznosti.
3.1.1.2 Doba trvání zkoušek tolerance
Tabulka 1
Doba trvání zkoušek tolerance: prasata
|
Cílová zvířata |
Doba trvání studií |
Charakteristika cílových zvířat |
|
Sající selata |
14 dní |
Pokud možno od 14 dnů do odstavu |
|
Odstavená selata |
42 dní |
42 dní po odstavu |
|
Výkrm prasat |
42 dní |
Tělesná hmotnost na začátku studie ≤ 35 kg |
|
Prasnice určené k reprodukci |
1 cyklus |
Od inseminace do konce odstavu |
V případě žádosti týkající se sajících a odstavených selat se považuje za dostatečnou kombinovaná studie (14denní pro sající selata a 28denní pro odstavená selata). Pokud byla prokázána tolerance u odstavených selat, nepožaduje se zvláštní studie pro výkrm prasat.
Tabulka 2
Doba trvání zkoušek tolerance: drůbež
|
Cílová zvířata |
Doba trvání studií |
Charakteristika cílových zvířat |
|
Výkrm kuřat/kuřice |
35 dní |
Od vylíhnutí |
|
Nosnice |
56 dní |
Pokud možno během první třetiny doby snášky |
|
Výkrm krůt |
42 dní |
Od vylíhnutí |
Údaje o toleranci u výkrmu kuřat nebo krůt lze použít k prokázání tolerance u odchovu kuřat a kuřic nebo u odchovu krůt.
Tabulka 3
Doba trvání zkoušek tolerance: skot
|
Cílová zvířata |
Doba trvání studií |
Charakteristika cílových zvířat |
|
Výkrm telat |
28 dní |
Výchozí tělesná hmotnost ≤ 70 kg |
|
Odchov telat; výkrm skotu nebo skot k reprodukci |
42 dní |
|
|
Dojnice |
56 dní |
|
V případě žádosti vztahující se na odchov telat a výkrm skotu se považuje za dostatečnou kombinovaná studie (28 dnů pro každé období).
Tabulka 4
Doba trvání zkoušek tolerance: ovce
|
Cílová zvířata |
Doba trvání studií |
Charakteristika cílových zvířat |
|
Odchov jehňat a výkrm jehňat |
28 dní |
|
Tabulka 5
Doba trvání zkoušek tolerance: lososovití a jiné ryby
|
Cílová zvířata |
Doba trvání studií |
Charakteristika cílových zvířat |
|
Losos a pstruh |
90 dní |
|
Jako alternativu 90denní studie by bylo možno provést studii, kdy ryby zvýší svou tělesnou hmotnost oproti výchozí tělesné hmotnosti na začátku zkoušky nejméně dvojnásobně.
Má-li být doplňková látka používána pouze pro hejno matečných ryb, testy tolerance se provedou pokud možno co nejblíže době tření. Testy tolerance trvají 90 dní a je nutno věnovat pozornost kvalitě a přežití jiker.
Tabulka 6
Doba trvání zkoušek tolerance: zvířata v zájmovém chovu a jiná zvířata, která nejsou určena k produkci potravin
|
Cílová zvířata |
Doba trvání studií |
Charakteristika cílových zvířat |
|
Psi a kočky |
28 dní |
|
Tabulka 7
Doba trvání zkoušek tolerance: králíci
|
Cílová zvířata |
Doba trvání studií |
Charakteristika cílových zvířat |
|
Výkrm králíků |
28 dní |
|
|
Chovné králice |
1 cyklus |
Od inseminace do konce doby odstavu |
Vztahuje-li se žádost na sající a odstavené králíky, považuje se za dostatečné období 49 dní (počínaje týden po narození) a musí zahrnovat králice až do odstavu.
Pokud se doplňková látka používá po specifickou a kratší dobu, než je uvedeno v definici kategorie zvířat, podává se podle navrhovaných podmínek pro užití. Období pozorování však nesmí být kratší než 28 dní a musí zahrnovat příslušný parametr (např. u prasnic určených k reprodukci počet živě narozených selat se zřetelem na období březosti nebo počet a hmotnost odstavených selat se zřetelem na období laktace).
3.1.1.3 Pokusné podmínky
Studie musí být uvedeny jednotlivě s údaji o všech pokusných skupinách. Zkušební protokol musí být pečlivě vypracován s ohledem na obecné popisné údaje. Zaznamenávají se zejména tyto položky:
|
1. |
stádo nebo hejno: místo a počet; podmínky krmení a chovu, způsob krmení; u vodních druhů velikost a počet nádrží nebo posad v hospodářství, světelné poměry a kvalita vody, včetně teploty vody a obsahu soli ve vodě; |
|
2. |
zvířata: druh (u vodních druhů určených pro lidskou spotřebu je uvedena identifikace podle jejich obecného jména a po něm v závorkách následuje latinské binomické jméno), plemeno, stáří (velikost u vodních druhů), pohlaví, způsob označení, fyziologické stadium a celkový zdravotní stav; |
|
3. |
datum a přesná doba trvání testů: datum a druh provedených zkoušek; |
|
4. |
krmné dávky: popis výroby a kvantitativního složení krmné dávky (dávek) s uvedením použitých složek, příslušných živin (analyzované hodnoty) a energetické hodnoty. Údaje o příjmu krmiva; |
|
5. |
koncentrace účinné látky (látek) nebo činidla (činidel) (a případných látek použitých pro srovnávací účely) v krmivech musí být stanovena kontrolní analýzou s použitím příslušných uznávaných metod. Referenční čísla použitých šarží; |
|
6. |
počet pokusných a kontrolních skupin, počet zvířat v každé skupině: počet zvířat použitých pro pokusy musí umožnit provedení statistické analýzy. Měly by být uvedeny použité metody statistického hodnocení. Ve zprávě musí být zahrnuta všechna zvířata a/nebo experimentální celky, s nimiž se pokusy prováděly. Případy, které není možné vyhodnotit pro nedostatek nebo ztrátu údajů, musí být ve zprávě uvedeny a jejich rozložení v jednotlivých skupinách zvířat musí být klasifikováno; |
|
7. |
okamžik a výskyt jakéhokoliv nežádoucího účinku zákroku u jedinců nebo skupin musí být zaznamenán (poskytnutí údajů o programu pozorování použitého ve studii) a |
|
8. |
léčebné/preventivní ošetření, je-li nutné, nesmí ovlivňovat navrhovaný mechanismus působení doplňkové látky a musí být zaznamenáno individuálně. |
3.1.2 Mikrobiální studie
Je nutno poskytnout studie ke stanovení schopnosti doplňkové látky způsobit křížovou rezistenci vůči antibiotikům používaným v humánním nebo veterinárním lékařství, selektovat rezistentní bakteriální kmeny za provozních podmínek u cílových druhů, vyvolat účinky u oportunních patogenů přítomných v zažívacím traktu, vyvolat uvolňování nebo vylučování zoonotických mikroorganismů.
V případě, že účinná látka (látky) vykazuje(í) antimikrobiální aktivitu při koncentraci, ve které je obsažena v krmivu, musí být v souladu se standardizovanými postupy určena minimální inhibiční koncentrace (MIK) pro příslušné druhy bakterií. Je-li prokázána příslušná antimikrobiální aktivita, je nutno stanovit schopnost doplňkové látky selektovat rezistentní bakteriální kmeny in vitro a u cílových druhů a způsobit křížovou rezistenci vůči odpovídajícím antibiotikům (11).
Zkoušky s doporučeným obsahem užití se provedou pro všechny mikrobiální doplňkové látky a pro jiné doplňkové látky, u nichž lze očekávat účinek na střevní mikroflóru. Tyto studie musí prokázat, že užití doplňkové látky nevytváří podmínky, které napomáhají nadměrnému růstu a uvolňování potenciálně patogenních mikroorganismů.
Výběr sledovaných mikroorganismů bude záviset na cílových druzích, zahrnuje však příslušné zoonotické druhy bez ohledu na to, zda u cílových zvířat vyvolávají či nevyvolávají příznaky.
3.2 Studie týkající se bezpečnosti používání doplňkové látky pro spotřebitele
Cílem je vyhodnotit bezpečnost doplňkové látky pro spotřebitele a stanovit možná rezidua doplňkové látky nebo jejích metabolitů v potravinách získaných ze zvířat, jimž bylo podáváno krmivo nebo voda obsahující doplňkovou látku nebo ošetřené touto doplňkovou látkou.
3.2.1 Studie metabolismu a reziduí
Stanovení metabolismu doplňkové látky u cílových druhů je rozhodujícím krokem při identifikaci a kvantifikaci reziduí v poživatelných tkáních nebo produktech získaných ze zvířat, jimž bylo podáváno krmivo nebo voda obsahující doplňkovou látku. Je nutno předložit studie týkající se absorpce, distribuce, metabolismu a vylučování látky (a jejích metabolitů).
Studie musí být provedeny pomocí mezinárodně validovaných zkušebních metod a v souladu s platnými evropskými právními předpisy nebo hlavními zásadami pro metodologické ukazatele OECD a podle zásad správné laboratorní praxe. Studie musí respektovat pravidla týkající se dobrých životních podmínek zvířat stanovená právními předpisy Evropského společenství a neopakují se, není-li to nezbytné.
Studie metabolismu a reziduí u cílového zvířete (zvířat) se provedou s účinnou látkou zapracovanou do krmiva (nepodávanou pomocí výživy žaludeční sondou, není-li to řádně odůvodněno).
Stanoví se strukturní identifikace metabolitů představujících více než 10 % všech reziduí v poživatelných tkáních a produktech a více než 20 % všech reziduí v exkrementech. Pokud je metabolická cesta účinné látky toxikologicky významná, je nutno identifikovat metabolity pod výše uvedenými limity.
Kinetické studie reziduí tvoří základ pro výpočet expozice spotřebitele a případně stanovení ochranné lhůty a MLR. Předloží se návrh na indikátorové reziduum.
U některých doplňkových látek nemusí být v závislosti na jejich povaze nebo užití vždy nutné provést studie metabolismu a reziduí.
3.2.1.1 Studie metabolismu
Účelem studií metabolismu je vyhodnotit absorpci, distribuci, biotransformaci a vylučování doplňkové látky u cílových druhů zvířat.
Požadované studie:
|
1. |
metabolická bilance po podání jediné dávky účinné látky v navrhovaných dávkách pro užití (celkové množství odpovídající dennímu příjmu) a případně vícenásobné dávky (je-li to odůvodněno), aby mohla být posouzena rychlost a rozsah absorpce, distribuce (plazma/krev) a vylučování (moč, žluč, výkaly, mléko nebo vejce, vydechovaný vzduch, vylučování přes žábry) u samců a samic, je-li to vhodné, a |
|
2. |
profilace do metabolismu, identifikace metabolitu(ů) v exkrementech a tkáních a distribuce v tkáních a produktech se stanoví po opakovaném podávání dávky označené látky zvířatům ve vyrovnaném stavu (metabolická rovnováha) podle obsahu v plazmě. Aplikovaná dávka odpovídá nejvyšší navrhované dávce pro užití a musí být zapracována do krmiva. |
3.2.1.2 Studie reziduí
Uváží se množství a povaha neextrahovatelných reziduí v poživatelných tkáních nebo produktech.
Studie reziduí se požadují pro všechny látky, u nichž jsou nutné studie metabolismu.
Je-li látka přirozenou složkou tělních tekutin nebo tkání nebo vyskytuje-li se v potravinách nebo krmivech přirozeně ve značném množství, je požadavek na studie reziduí omezen na porovnání obsahu v tkáních/produktu u neošetřené skupiny a u skupiny, jíž byla podávána nejvyšší udávaná dávka.
U většinových druhů studie současně vyhodnotí všechna rezidua, která jsou toxikologicky významná, a určí indikátorové reziduum účinné látky v poživatelné tkáni (játra, ledviny, svaly, kůže, kůže + tuk) a produktech (mléko, vejce a med). Indikátorové reziduum je reziduum vybrané pro zkoušku, jehož koncentrace je ve známém vztahu k veškerým toxikologicky významným reziduím v tkáních. Studie musí prokázat rovněž stálost reziduí v tkáních nebo produktech, aby bylo možno stanovit odpovídající ochrannou lhůtu.
Ke stanovení ochranné lhůty je navrhovaný minimální počet zvířat ve vzorku a/nebo produktů v každém bodu měření tento:
|
— |
poživatelné tkáně:
|
|
— |
produkty:
|
Uváží se vhodné rozložení podle pohlaví.
Rezidua se měří při nulové ochranné lhůtě (vyrovnaný stav) a nejméně ve třech jiných bodech měření.
Předloží se návrh na indikátorové reziduum.
Je nutno provést studie absorpce, distribuce a vylučování, včetně identifikace hlavních metabolitů, u laboratorních druhů zvířat, u nichž byla získána nejnižší hodnota NOAEL nebo standardně u krys (obou pohlaví). Doplňkové studie týkající se určitých metabolitů mohou být nezbytné v případě, jsou-li tyto metabolity produkovány cílovými druhy a u laboratorních druhů se ve významném rozsahu netvoří.
3.2.1.3 Studie metabolismu a ukládání
Musí být provedena studie metabolismu zahrnující metabolickou bilanci, metabolický profil a identifikaci hlavních metabolitů v moči a výkalech. Jestliže jiné laboratorní druhy vykazují oproti kryse výrazný rozdíl v citlivosti, budou požadovány doplňkové informace.
3.2.1.4 Biologická dostupnost reziduí
Při posuzování rizika pro spotřebitele spojeného s určitými vázanými rezidui obsaženými v živočišných produktech je možno vzít v úvahu doplňkový bezpečnostní faktor založený na určení jejich biologické dostupnosti za použití vhodných laboratorních zvířat a uznaných metod.
3.2.2 Toxikologické studie
Bezpečnost doplňkové látky se posuzuje na základě toxikologických studií provedených in vitro a in vivo na laboratorních zvířatech. Tyto zkoušky obvykle zahrnují měření:
|
1. |
akutní toxicity; |
|
2. |
genotoxicity (mutagenity, klastogenicity); |
|
3. |
subchronické orální toxicity; |
|
4. |
chronické orální toxicity/karcinogenity; |
|
5. |
toxicity pro reprodukci včetně teratogenity a |
|
6. |
ostatní studie. |
Existuje-li důvod k obavám, musí být provedeny další studie poskytující doplňkové informace, které jsou nezbytné pro posouzení bezpečnosti účinné látky a jejích reziduí.
Na základě výsledků těchto studií se stanoví toxikologická NOAEL.
Doplňkové studie týkající se určitých metabolitů mohou být nezbytné, pokud jsou tyto metabolity produkovány cílovými druhy a u testovaných laboratorních druhů se ve významném rozsahu netvoří. Jsou-li k dispozici studie metabolismu u člověka, vezmou se tyto údaje v úvahu při rozhodování o povaze případných doplňkových studií.
Toxikologické studie musí být provedeny s účinnou látkou. Pokud je účinná látka přítomna v produktu fermentačního procesu, testuje se produkt fermentačního procesu. Testovaný produkt fermentačního procesu musí být totožný s produktem, který má být použit v komerčním produktu.
Studie musí být provedeny pomocí mezinárodně validovaných zkušebních metod v souladu s platnými evropskými právními předpisy nebo hlavními zásadami pro metodologické ukazatele OECD a podle zásad správné laboratorní praxe. Studie zahrnující laboratorní zvířata musí respektovat pravidla týkající se dobrých životních podmínek zvířat stanovená evropskými právními předpisy a neopakují se, není-li to nezbytné.
3.2.2.1 Akutní toxicita
Studie akutní toxicity se požadují ke klasifikaci a omezené charakteristice toxicity látky.
Studie akutní toxicity musí být provedeny alespoň na dvou druzích savců. Jeden laboratorní druh může být případně nahrazen cílovým druhem.
Nebude nutné zjistit přesnou hodnotu LD50; postačuje přibližné určení minimální letální dávky. Maximální dávkování nesmí překročit 2 000 mg/kg tělesné hmotnosti.
Ke snížení počtu testovaných zvířat a zmírnění jejich utrpení se neustále vyvíjejí nové protokoly pro testování dávky vyvolávající akutní toxicitu. Studie provedené pomocí těchto nových postupů budou uznány, jsou-li řádně validovány.
Je nutno dodržet pokyny OECD 402 (akutní dermální toxicita), 420 (metoda fixní dávky), 423 (metoda stanovení třídy akutní toxicity) a 425 (postup nahoru a dolů).
3.2.2.2 Studie genotoxicity, včetně mutagenity
Za účelem identifikace účinných látek a případně jejich metabolitů a produktů degradace, které mají mutagenní a genotoxické vlastnosti, musí být provedena výběrová kombinace různých testů na genotoxicitu. V případě potřeby se testy musí provádět bez i s metabolickou aktivací u savců a vezme se v úvahu slučitelnost testovaného materiálu se zkušebním systémem.
Základní soubor zahrnuje tyto zkoušky:
|
1. |
indukce genových mutací v bakteriích a/nebo v buňkách savců (pokud možno test na tymidinkinázu u lymfomu myší); |
|
2. |
indukce chromozómových aberací v buňkách savců a |
|
3. |
zkouška in vivo u savců. |
V závislosti na výsledku výše uvedených zkoušek a s přihlédnutím k celkovému toxikologickému profilu látky a jejímu určenému užití mohou být nezbytné doplňkové zkoušky.
Protokoly by měly být v souladu s pokynem OECD 471 (test reverzních mutací u Salmonella typhimurium), 472 (test reverzních mutací u Escherichia coli), 473 (test chromozómových aberací u savců in vitro), 474 (test na mikrojádra v savčích erytrocytech), 475 (test chromozómových aberací v kostní dřeni savců), 476 (test genových mutací v savčích buňkách in vitro) nebo 482 (neplánovaná syntéza DNA v buňkách savců in vitro), jakož i jinými příslušnými pokyny OECD pro zkoušky in vitro a in vivo.
3.2.2.3 Studie subchronické orální toxicity při opakované dávce
K ověření potenciálu subchronické toxicity účinné látky se předloží nejméně jedna studie na hlodavcích s dobou trvání nejméně 90 dnů. Považuje-li se to za nutné, musí být provedena druhá studie na jiném druhu než hlodavcích. Testovaná položka musí být podávána orálně nejméně o třech různých obsazích navíc ke kontrolní skupině, aby se získala reakce na dávku. Maximální dávka musí za normálních okolností odhalit přítomnost nežádoucích účinků. Nejnižší úroveň dávkování nesmí vykazovat žádnou známku toxicity.
Protokoly pro tyto zkoušky musí být v souladu s pokyny OECD 408 (hlodavci) nebo 409 (jiné druhy než hlodavci).
3.2.2.4 Studie chronické orální toxicity (včetně karcinogenity)
K ověření potenciálu chronické toxicity a karcinogenity je nutno provést studii chronické orální toxicity nejméně na jednom druhu s dobou trvání nejméně 12 měsíců. Vybraný druh musí být nejvhodnějším druhem podle všech dostupných vědeckých údajů, včetně výsledků 90denních studií. Standardním druhem je krysa. Požaduje-li se druhá studie, použije se druh hlodavců nebo jiný druh savců než hlodavci. Testovaná položka musí být podávána orálně nejméně o třech různých obsazích navíc ke kontrolní skupině, aby se získala reakce na dávku.
Je-li studie chronické toxicity kombinována se zkouškou karcinogenity, doba trvání je prodloužena na 18 měsíců u myší a křečků a na 24 měsíců u krys.
Studie karcinogenity nemusí být nutné, pokud účinná látka a její metabolity:
|
1. |
vykazují stále negativní výsledky v testech na genotoxicitu; |
|
2. |
nejsou strukturálně propojeny se známými karcinogeny a |
|
3. |
nevyvolávají při zkouškách chronické toxicity žádné účinky (pre)neoplasie. |
Protokoly musí být v souladu s pokynem OECD 452 (studie chronické toxicity) nebo 453 (kombinovaná studie chronické toxicity/karcinogenity).
3.2.2.5 Studie toxicity pro reprodukci (včetně toxicity pro prenatální vývoj)
K zjištění možného zhoršení reprodukční funkce u samců nebo samic nebo škodlivých účinků na potomstvo vyplývajících z podávání účinné látky je nutno provést studie reprodukční funkce pomocí:
|
1. |
dvougenerační studie toxicity pro reprodukci a |
|
2. |
studie toxicity pro prenatální vývoj (zkouška teratogenity). |
U nových zkoušek mohou být použity validované alternativní metody omezující používání zvířat.
3.2.2.5.1. Dvougenerační studie toxicity pro reprodukci
Studie reprodukční funkce musí být prováděny v rozsahu nejméně dvou filiálních generací (F1, F2) u nejméně jednoho druhu, obvykle hlodavce, a mohou být kombinovány se studií teratogenity. Zkoušená látka je podávána orálně samcům a samicím ve vhodnou dobu před pářením. V podávání se musí pokračovat až do odstavu zvířat generace F2.
Všechny důležité parametry týkající se oplození, březosti, porodu, mateřského chování, kojení, růstu a vývoje zvířat generace F1 od početí až do dospělosti a vývoj generace F2 až do odstavu musí být pečlivě sledovány a zaznamenávány. Protokoly pro studii toxicity pro reprodukci by měly být v souladu s pokynem OECD 416.
3.2.2.5.2. Zkouška toxicity pro prenatální vývoj (zkouška teratogenity)
Cílem je odhalit případné nepříznivé účinky na březí samici a vývoj embrya a plodu v důsledku expozice od uhnízdění oplozeného vajíčka po celou dobu březosti. K těmto účinkům patří zvýšená toxicita u březích samic, úmrtí embrya – plodu, změněný růst plodu a strukturální abnormality a anomálie plodu.
Druhem, který se volí pro první studii, je obvykle krysa. Je-li zaznamenán negativní nebo neprůkazný výsledek pro teratogenitu, provede se další studie vývojové toxicity u druhého druhu, pokud možno u králíka. Je-li zkouška na teratogenitu na krysách pozitivní, není studie u druhého druhu nutná vyjma případu, kdy přezkum všech hlavních studií naznačuje, že ADI bude založena na teratogenitě u krys. V tomto případě by byla nutná studie u druhého druhu, aby se určil necitlivější druh pro tento parametr. Protokoly by měly být v souladu s pokynem OECD 414.
3.2.2.6 Jiné specifické toxikologické a farmakologické studie
Existují-li důvody k obavám, je nutno provést další studie poskytující doplňkové informace pro posouzení bezpečnosti účinné látky a jejích reziduí. Tyto studie mohou zahrnovat zkoušku farmakologických účinků, účinků na mladá (předpubertální) zvířata, imunotoxicity nebo neurotoxicity.
3.2.2.7 Určení množství dávky bez pozorovaného nepříznivého účinku (NOAEL)
NOAEL je obvykle založena na toxikologických účincích, příležitostně však mohou být vhodnější farmakologické účinky.
Volí se nejnižší NOAEL. Při určování nejnižší NOAEL, vyjádřené v mg/kg tělesné hmotnosti na den, se vezmou v úvahu všechna zjištění z předchozích oddílů spolu s veškerými důležitými zveřejněnými údaji (včetně jakékoliv vhodné informace o účincích účinné látky na člověka), popřípadě i informace o chemických látkách, které mají úzce související chemické struktury.
3.2.3 Hodnocení bezpečnosti pro spotřebitele
Bezpečnost pro spotřebitele se vyhodnotí srovnáním zjištěné ADI (přijatelné denní dávky) a vypočteného teoretického příjmu doplňkové látky nebo jejích metabolitů z potravy. V případě vitamínů a stopových prvků se místo ADI může použít UL (nejvyšší povolená dávka).
3.2.3.1 Návrh přijatelné denní dávky (ADI) pro účinnou látku(y)
Přijatelná denní dávka (ADI) (vyjádřená v mg doplňkové látky nebo materiálu vztahujícímu se k doplňkové látce na osobu a den) se stanoví tak, že se nejnižší hodnota NOAEL (mg/kg tělesné hmotnosti) vydělí vhodným bezpečnostním faktorem a vynásobí průměrnou hmotností lidského těla, tj. 60 kg.
Popřípadě se ADI navrhne. ADI může být rovněž „nespecifikovaná“ vzhledem k nízké toxicitě při testech na zvířatech. ADI se nenavrhne, pokud látka vykazuje genotoxické nebo karcinogenní vlastnosti důležité pro člověka.
Stanovení ADI obvykle vyžaduje obdobný metabolismus účinné látky u cílových a laboratorních zvířat (viz bod 3.2.1.4 Biologická dostupnost reziduí), což zajišťuje, že spotřebitelé jsou vystaveni stejným reziduím jako laboratorní zvířata použitá při toxikologických zkouškách. V opačném případě mohou stanovení ADI umožnit doplňkové studie u druhého laboratorního druhu zvířat nebo s metabolity, které jsou specifické pro cílový druh.
Bezpečnostní faktor použitý pro stanovení ADI u konkrétní doplňkové látky vezme v úvahu povahu biologických účinků a kvalitu údajů použitých ke stanovení NOAEL, důležitost těchto účinků pro člověka a reverzibilitu účinků a jakékoliv poznatky o přímém účinku (účincích) reziduí na člověka.
Pro výpočet ADI se použije bezpečnostní faktor nejméně 100 (pokud byl poskytnut úplný balíček toxikologických studií). Jsou-li k dispozici údaje o účinné látce vztahující se k člověku, může být přijat nižší bezpečnostní faktor. Vyšší bezpečnostní faktory se mohou použít k zohlednění dalších zdrojů nejistoty u údajů nebo v případě, je-li NOAEL stanovena na základě konkrétního kritického parametru, například teratogenity.
3.2.3.2 Nejvyšší povolená dávka (UL)
U některých doplňkových látek může být vhodnější založit hodnocení bezpečnosti na UL, což je nejvyšší množství celkového chronického denního příjmu živiny (ze všech zdrojů) považovaná (vnitrostátními nebo mezinárodními vědeckými institucemi) za příjem, který pravděpodobně nepředstavuje riziko nepříznivých účinků na zdraví u spotřebitelů nebo zvláštních skupin spotřebitelů.
Dokumentace obsahuje údaje, které prokazují, že užití doplňkové látky nepovede k situaci, kdy by mohlo dojít k překročení UL s přihlédnutím ke všem možným zdrojům živiny.
Jsou-li výsledné obsahy reziduí nutriční doplňkové látky nebo jejího metabolitu(ů) v živočišných produktech vyšší, než se u těchto produktů považuje za běžné nebo než se očekává, je nutno to jednoznačně uvést.
3.2.3.3 Expozice spotřebitelů
Celkový příjem doplňkové látky a/nebo jejích metabolitů spotřebitelem ze všech zdrojů musí být nižší než ADI nebo UL.
Výpočet teoretického příjmu z potravin živočišného původu se provede s přihlédnutím ke koncentraci (všechna rezidua jako aritmetický průměr a nejvyšší jediná hodnota) naměřené v tkáních a produktech při ukončení používání doplňkové látky. Kromě toho se v případě potřeby stanoví pro různé ochranné lhůty hodnoty denní lidské spotřeby potravin na základě nejhoršího scénáře.
U doplňkových látek určených pro více druhů se expozice z tkání vypočte nezávisle pro savce, ptáky a ryby a vezme se nejvyšší hodnota. K tomuto údaji se případně připočte expozice z mléka a vajec. Pokud se například doplňková látka používá u savců produkujících mléko a nosnic, připočítají se příslušné nejvyšší hodnoty pro poživatelné tkáně k hodnotám pro spotřebu mléka a vajec. Je-li doplňková látka používána pro ryby a nosnice a savce produkující mléko, připočítají se příslušné nejvyšší hodnoty pro poživatelné tkáně k hodnotám pro spotřebu vajec a mléka. Ostatní kombinace se pojímají stejně.
V určitých případech (např. některé nutriční a senzorické doplňkové látky nebo doplňkové látky určené pro menšinové druhy) může být vhodné upřesnit následně hodnocení expozice u člověka pomocí reálnějších údajů o spotřebě, avšak se zachováním nejkonzervativnějšího přístupu. Je-li to možné, toto upřesnění by mělo být založeno na údajích pro Společenství.
Tabulka 1
Údaje o teoretické denní lidské spotřebě (g tkání nebo produktů)
|
|
Savci |
Ptáci |
Ryby |
Ostatní |
|
Svaly |
300 |
300 |
300 (12) |
|
|
Játra |
100 |
100 |
— |
|
|
Ledviny |
50 |
10 |
— |
|
|
Tuk |
50 (13) |
90 (14) |
— |
|
|
+ Mléko |
1 500 |
— |
— |
|
|
+ Vejce |
— |
100 |
— |
|
|
+ Med |
|
|
|
20 |
3.2.3.4 Návrh maximálních limitů reziduí (MLR)
Maximálním limitem reziduí se rozumí maximální koncentrace reziduí (vyjádřena jako μg indikátorového rezidua na kg poživatelné vlhké tkáně nebo produktu), kterou může Společenství přijmout jako zákonem povolenou nebo uznanou jako přijatelnou v potravinách. Je založena na druhu a množství rezidua, o němž se usuzuje, že nepředstavuje toxikologické nebezpečí pro zdraví lidí, vyjádřeno pomocí ADI. MLR nelze bez ADI stanovit.
Při stanovení MLR pro doplňkové látky je nutno vzít v úvahu rovněž rezidua, která pocházejí z jiných zdrojů (např. potraviny rostlinného původu). MLR lze dále snížit, aby byl v souladu s podmínkami pro užití doplňkových látek, a v rozsahu, v jakém jsou dostupné praktické analytické metody.
Jednotlivé MLR (vyjádřené jako mg indikátorového rezidua na kg poživatelné přírodní tkáně nebo produktu) se popřípadě stanoví pro různé tkáně nebo produkty cílových druhů zvířat. Jednotlivé MLR v různých tkáních nebo produktech berou v úvahu kinetiku vylučování reziduí a variabilitu obsahů reziduí v těchto tkáních/produktech u živočišného druhu zamýšleného pro užití. Variabilita se obvykle zohlední použitím 95 % intervalu spolehlivosti střední hodnoty. Nelze-li interval spolehlivosti vypočítat kvůli nízkému počtu vzorků, variabilita se místo toho vyjádří použitím nejvyšší jednotlivé hodnoty.
Studie týkající se maximálních limitů reziduí u kokcidiostatik a histomonostatik musí být provedeny podle příslušných pravidel platných pro veterinární léčivé přípravky (svazek 8 „The rules governing medicinal products in European Union – Notice to applicants and guidelines. Veterinary medicinal products. Establishment of maximum residue limits (MLRs) for residues of veterinary medicinal products in foodstuffs of animal origin“, říjen 2005).
V případě potřeby se podle této přílohy předloží studie ke stanovení maximálních limitů reziduí pro kategorie doplňkových látek jiných než kokcidiostatika a histomonostatika.
Ke stanovení expozice všem reziduím u spotřebitele (podle výpočtu v bodě 3.2.3.3) berou navrhované MLR pro různé tkáně nebo produkty v úvahu poměr indikátorového rezidua ke všem reziduím (tabulka 2).
Tabulka 2
Definice použité při stanovení MLR
|
i-j |
jednotlivé tkáně/produkty (játra, ledviny, svaly, kůže + tuk, mléko, vejce, med) v různých bodech měření |
|
MLRi-j |
maximální limit reziduí v tkáních/produktech (mg indikátorové látky/kg) |
|
Qti-j |
denní lidská spotřeba jednotlivých tkání/produktů (kg) stanovená podle tabulky 1 nebo jejího zpřesnění |
|
TRCi-j |
koncentrace všech reziduí v jednotlivých tkáních/produktech (mg/kg) |
|
MRCi-j |
koncentrace indikátorového rezidua v jednotlivých tkáních/produktech (mg/kg) |
|
RMTRi-j |
poměr MRCi-j k TRCi-j pro jednotlivé tkáně/produkty |
|
DITRi-j |
dietární příjem pro jednotlivé tkáně/produkty vypočtený ze všech reziduí (mg) DITRi-j = Qti-j × TRCi-j |
|
DITRMLRi-j |
dietární příjem vypočtený z MLR (mg) jednotlivých tkání/produktů DITRMLRi-j = Qti-j × MLRi-j × RMTRi-j -1 |
Naměřené hodnoty pro TRC a MRC se vloží do vzoru uvedeného v tabulce 3 a ostatní hodnoty se vypočítají. Není-li k dispozici úplný soubor údajů, jelikož hodnoty jsou pod mezí detekce, je přípustná extrapolace RMTR.
Odvození MLR lze provést pouze v případě, je-li součet jednotlivých DITR nižší než ADI. Je-li ADI překročena, alternativou je použití údajů z delší ochranné lhůty nebo nižšího dávkování. První návrh MLR je možno získat pomocí hodnoty MRC jako vodítka a s přihlédnutím k limitu kvantifikace analytické metody. Součet DITRMLR získaný z navrhovaných MLR musí být nižší než ADI a blížit se součtu jednotlivých DITR. Je-li ADI překročena, navrhne se nižší MLR a opakuje se srovnání.
U některých doplňkových látek mohou být rezidua přítomna v mléce, vejcích nebo mase pod hodnotami MLR, mohou však narušit kvalitu potraviny při určitém jejím zpracování. U těchto doplňkových látek může být vhodné vedle stanovení hodnot MLR uvážit „maximální reziduum slučitelné se zpracováním (potravin)“ (MPCR).
Tabulka 3
Vzor pro odvození návrhu MLR
|
|
Játra |
Ledviny |
Svaly |
Kůže + tuk |
Mléko |
Vejce |
Med |
Součet |
|
TRC (15) (mg/kg) |
|
|
|
|
|
|
|
— |
|
MRC (16) (mg/kg) |
|
|
|
|
|
|
|
— |
|
RMTR (16) |
|
|
|
|
|
|
|
— |
|
DITR (17) (mg) |
|
|
|
|
|
|
|
|
|
Navrhovaný MLR (mg/kg) |
|
|
|
|
|
|
|
— |
|
DITRMLR(mg) |
|
|
|
|
|
|
|
|
3.2.3.5 Návrh ochranné lhůty
Ochranná lhůta zahrnuje dobu, jež následuje po ukončení podávání doplňkové látky, která je nezbytná k tomu, aby hladiny reziduí klesly pod MLR.
3.3 Studie týkající se bezpečnosti používání doplňkové látky pro uživatele/pracovníky
Pracovníci mohou být vystaveni působení doplňkové látky zejména při vdechnutí nebo dotyku při výrobě, manipulaci nebo užití doplňkové látky. Například zemědělští dělníci jsou potenciálně vystaveni účinkům při manipulaci nebo míchání doplňkové látky. Musí být předloženy doplňkové informace o způsobu zacházení s látkami.
Musí být zahrnuto posouzení rizika pro pracovníky. Jsou-li k dispozici, představují zkušenosti získané ve výrobním závodě často důležitý zdroj informací pro posouzení rizik pro pracovníky, která vyplývají z toho, že pracovníci jsou vystaveni účinkům doplňkové látky ze vzduchu a dotykem. Látky, jimž je zapotřebí věnovat mimořádnou pozornost, jsou doplňkové látky, krmiva zpracovaná s doplňkovými látkami a/nebo výkaly zvířat ve formě suchého prášku nebo ze kterých může suchý prášek vzniknout, a doplňkové látky, které mohou mít alergický potenciál.
3.3.1 Hodnocení toxikologického rizika pro bezpečnost uživatelů/pracovníků
Rizika pro pracovníky se posoudí v sérii studií s užitím doplňkové látky ve formě, pro niž byla podána žádost. Provedou se studie akutní inhalační toxicity, ledaže není pravděpodobné, že produkt bude tvořit prášek nebo mlhu, které by bylo možno vdechnout. Je nutno provést studii na podráždění kůže a jsou-li výsledky těchto studií negativní, posoudí se dráždivost pro sliznice (např. oči). Vyhodnotí se rovněž alergický potenciál/potenciál senzibilizace kůže. Údaje o toxicitě získané pro účely posouzení bezpečnosti pro spotřebitele (viz bod 3.2.2) se použijí k posouzení možné systémové toxicity doplňkové látky. Toto vše se v případě potřeby vyhodnotí pomocí přímého měření nebo zvláštních studií.
3.3.1.1 Účinky na dýchací ústrojí
Musí být doloženo, že hladiny prachu nebo mlhy doplňkové látky ve vzduchu nepředstavují riziko pro zdraví uživatelů/pracovníků. V případě potřeby je nutné doložit:
|
— |
inhalační testy na laboratorních zvířatech, |
|
— |
uveřejněné epidemiologické údaje a/nebo vlastní údaje žadatele o jeho výrobním zařízení a/nebo dráždivosti a |
|
— |
testy na senzibilizaci dýchacího ústrojí. |
Studie akutní inhalační toxicity se provedou, pokud částice nebo kapičky o průměru méně než 50 μm představují více než 1 % hmotnosti produktu.
Protokoly pro studie akutní inhalační toxicity by měly být v souladu s pokynem OECD 403. Pokud se považují za nezbytné studie subchronické toxicity, měly by splňovat pokyny OECD 412 (inhalační toxicita při opakované dávce: 28denní nebo 14denní studie) nebo 413 (subchronická inhalační toxicita: 90denní studie).
3.3.1.2 Účinky na oči a pokožku
Pokud jsou k dispozici, musí být předloženy přímé důkazy o tom, že nejsou známy případy dráždivosti a/nebo senzibilizace u lidí. Tyto důkazy musí být doplněny výsledky validovaných testů na zvířatech týkajících se dráždivosti pokožky a očí a možnosti senzibilizace způsobené užitím příslušné doplňkové látky. Posoudí se rovněž alergický potenciál – potenciál senzibilizace kůže. Protokoly pro tyto studie by měly být v souladu s pokyny OECD 404 (podráždění/poleptání kůže), 405 (podráždění/poleptání očí), 406 (senzibilizace kůže), 429 (senzibilizace kůže – test místních mízních uzlin).
Jsou-li známy leptavé vlastnosti buď ze zveřejněných údajů, nebo ze zvláštních zkoušek in vitro, neprovádějí se další zkoušky in vivo.
Uváží se dermální toxicita, je-li doplňková látka toxická při vdechnutí. Zkoušky musí být v souladu s pokynem OECD 402 (akutní dermální toxicita).
3.3.1.3 Systémová toxicita
Pro posouzení systémové toxicity se použijí údaje o toxicitě získané za účelem splnění požadavků na bezpečnost spotřebitelů a ostatních požadavků (včetně toxicity při opakované dávce, mutagenity, karcinogenity a testů toxicity pro reprodukci a metabolismu).
3.3.1.4 Posouzení expozice
Musí být předloženy informace o tom, zda používání doplňkové látky je schopno vyvolat vznik expozice všemi cestami (vdechnutím, kůží nebo požitím). Tyto informace musí zahrnovat kvantitativní vyhodnocení (pokud existuje), které by se mělo týkat charakteristické koncentrace ve vzduchu, kontaminace kůže nebo požití. Pokud kvantitativní údaje nejsou k dispozici, musí být předložen dostatek informací pro přiměřené posouzení expozice.
3.3.2 Opatření ke kontrole expozice
Za použití informací z hodnocení výsledků toxikologie a expozice musí být vypracován závěr o rizicích pro zdraví uživatelů/pracovníků (vdechnutí, dráždivost, senzibilizace a systémová toxicita). Je možno navrhnout bezpečnostní opatření ke snížení nebo vyloučení expozice. Použití osobních ochranných prostředků se však považuje až za poslední krok k ochraně před jakýmkoliv přetrvávajícím rizikem poté, kdy už byla zavedena kontrolní opatření. Za přednostní řešení se považuje například přepracování produktu.
3.4 Studie týkající se bezpečnosti používání doplňkové látky pro životní prostředí
Zohlednění dopadu doplňkových látek na životní prostředí je důležité z toho důvodu, že se tyto doplňkové látky podávají obvykle po dlouhou dobu, často jsou dotčeny velké skupiny zvířat a účinné látky mohou být vylučovány ve značném rozsahu jako původní látka nebo její metabolity.
Ke stanovení dopadu doplňkových látek na životní prostředí je nutno dodržet metodu postupných kroků. Všechny doplňkové látky je nutno posoudit v první fázi s cílem identifikovat doplňkové látky, které nevyžadují další testy. U jiných doplňkových látek se požaduje druhá fáze hodnocení (fáze II), aby se získaly doplňkové informace, na jejichž základě se může ukázat, že jsou nezbytné další studie. Tyto studie musí být provedeny v souladu se směrnicí 67/548/EHS.
3.4.1 Posuzování – fáze I
Účelem posuzování ve fázi I je určit, zda je pravděpodobný významný dopad doplňkové látky nebo jejích metabolitů na životní prostředí a zda je nutné posouzení ve fázi II (viz rozhodovací strom).
K fázi II se nemusí přistupovat, pokud je splněno jedno ze dvou kritérií, neexistují-li vědecky podložené důvody k obavám:
|
a) |
chemická povaha a biologický vliv doplňkové látky i podmínky jejího užití ukazují, že dopad bude zanedbatelný, tj. když doplňková látka je:
|
|
b) |
nejhorší předpokládaná koncentrace v životním prostředí (PEC) je příliš nízká, než aby musela být brána v úvahu. PEC se vyhodnotí pro každou sledovanou složku životního prostředí (viz níže) za předpokladu, že 100 % přijaté dávky je vyloučeno jako původní látka. |
Pokud žadatel nemůže prokázat, že doplňková látka spadá do jedné z vyňatých kategorií, je nutné posouzení ve fázi II.
3.4.1.1 Doplňkové látky pro suchozemská zvířata
Používají-li se exkrementy hospodářských zvířat k hnojení půdy, může užití doplňkových látek vést ke kontaminaci půdy, spodní vody a povrchové vody (prostřednictvím drenáže a odtoku).
Nejhorší PEC pro půdu (PECpůda) vyplývá ze všech vyloučených látek rozmetaných po půdě. Je-li PECpůda (standardně: hloubka 5 cm) nižší než 10 μg/kg, není nutné další posouzení.
Je-li PEC pro kontaminaci spodní vody (PECspodní voda) nižší než 0,1 μg/l, není nutné posouzení dopadu doplňkové látky na spodní vodu ve fázi II.
3.4.1.2 Doplňkové látky pro vodní živočichy
Doplňkové látky používané v akvakultuře mohou vést ke kontaminaci sedimentu a vody. Předpokládá se, že v případě ryb chovaných v klecích je významnou složkou z hlediska posouzení rizika pro životní prostředí sediment. V případě ryb chovaných v systémech založených na terénu se považuje za největší riziko pro životní prostředí voda odtékající do povrchových vod.
Nejhorší PEC pro sediment (PECsediment) vyplývá ze všech vyloučených látek uložených v sedimentu. Je-li PECsediment (standardně: hloubka 20 cm) nižší než 10 μg/kg hmotnosti za mokra, další posouzení není nutné.
Je-li PEC v povrchové vodě (PECpovrchová voda) nižší než 0,1 μg/l, další posouzení není nutné.
Fáze I – Rozhodovací strom
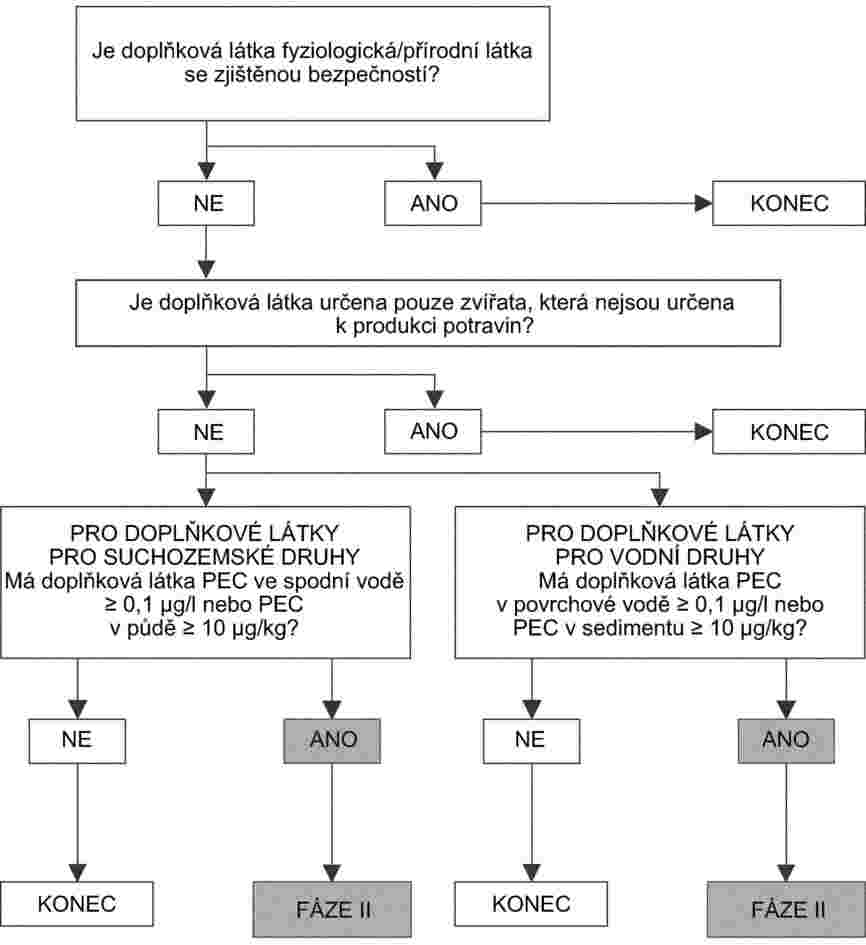
3.4.2 Posuzování – fáze II
Účelem posuzování ve fázi II je vyhodnotit možnost, že doplňkové látky budou mít vliv na necílové druhy v životním prostředí, a to vodní i suchozemské druhy, nebo se dostanou do spodní vody v nepřípustném množství. Není praktické vyhodnotit účinky doplňkových látek pro každý druh v životním prostředí, který může být doplňkové látce vystaven po jejím podání cílovému druhu. Testované taxonomické obsahy mají sloužit jako náhrady nebo ukazatele pro řadu druhů vyskytujících se v životním prostředí.
Posouzení ve fázi II je založeno na metodě rizikového koeficientu, kdy se pro každou složku životního prostředí porovnávají vypočtené hodnoty PEC a předpokládané koncentrace bez účinku (PNEC). PNEC se určí z pokusně stanovených parametrů vydělených příslušným hodnotícím faktorem. Hodnota PNEC se vypočte pro každou složku životního prostředí.
Posuzování ve fázi II začíná pokud možno zpřesněním PEC a využívá dvoustupňového přístupu k posouzení rizika pro životní prostředí.
První stupeň, fáze IIA, využívá omezeného počtu studií osudu látky v prostředí a účinku k vypracování konzervativního posouzení rizika na základě expozice a účinků ve sledované složce životního prostředí. Je-li poměr PEC k PNEC nižší než jedna (1), není nutné další posouzení, pokud se neočekává bioakumulace.
Pokud poměr PEC/PNEC předpovídá nepřijatelné riziko (poměr > 1), žadatel přikročí k fázi IIB, aby zpřesnil posouzení rizika pro životní prostředí.
3.4.2.1 Fáze IIA
Kromě složek životního prostředí posuzovaných ve fázi I je nutno vypočítat PEC pro povrchovou vodu s přihlédnutím k odtoku a drenáži.
Na základě údajů neposuzovaných ve fázi I lze vypočítat přesnější PEC pro každou sledovanou složku životního prostředí. Při určování upřesněné PEC je nutno brát v úvahu:
|
a) |
koncentrace sledované účinné látky (látek) / metabolitů v hnoji/výkalech ryb následně po podání doplňkové dávky zvířatům v navrhované dávce. Tento výpočet musí brát v úvahu míru dávkování a množství exkrementů; |
|
b) |
potenciální rozložitelnost vyloučené sledované účinné látky (látek) / metabolitů během obvyklého ošetření a uložení hnoje před jeho rozmetáním do půdy; |
|
c) |
adsorpce sledované účinné látky (látek) / metabolitů na půdu nebo sediment v případě akvakultury a desorpce, pokud možno na základě studií půdy/sedimentu (OECD 106); |
|
d) |
rozložitelnost v půdě a vodních systémech/systémech sedimentu (OECD 307 resp. 308) a |
|
e) |
jiné faktory, jako například hydrolýza, fotolýza, vypařování, rozředění při orbě. |
Pro účely posuzování rizika ve fázi II se použije nejvyšší hodnota PEC získaná z těchto výpočtů pro každou sledovanou složku životního prostředí.
Pokud se předpokládá zvýšené přetrvávání v půdě/sedimentu (doba do rozložení 90 % původní koncentrace látky: DT90 > 1 rok), posoudí se potenciál k akumulaci.
Stanoví se koncentrace doplňkových látek (nebo metabolitů) vyvolávajících závažné nežádoucí účinky pro různé trofické hladiny v dané složce životního prostředí. Tyto testy jsou většinou akutní testy a měly by dodržovat pokyny OECD nebo obdobné dobře zavedené pokyny. Studie pro suchozemské prostředí zahrnují: toxicitu pro dešťovky; tři suchozemské rostliny a půdní mikroorganismy (např. účinky na vazbu dusíku). Studie pro sladkovodní prostředí zahrnují: toxicitu pro ryby; Daphnia magna; řasy a sedimentační organismy. V případě klecí v moři se testují tři druhy různých taxonů sedimentačních organismů.
Provede se výpočet hodnoty PNEC pro každou sledovanou složku životního prostředí. PNEC je obvykle odvozena z nejnižší hodnoty toxicity zaznamenané ve výše uvedených testech a vydělí se bezpečnostním faktorem minimálně 100, v závislosti na parametru a počtu užitých testovaných druhů.
Potenciál k bioakumulaci lze odhadnout z hodnoty rozdělovacího koeficientu n-oktanol/voda, Log Kow. Hodnoty ≥ 3 udávají, že u látky může docházet k bioakumulaci. K posouzení rizika sekundární otravy se posuzuje, zda se ve fázi IIB provede studie biokoncentračního faktoru (BCF).
3.4.2.2 Fáze IIB (podrobnější ekotoxikologické studie)
U doplňkových látek, u nichž po posouzení ve fázi IIA nelze vyloučit riziko pro životní prostředí, jsou nutné podrobnější informace o účincích na biologické druhy v dané složce (složkách) životního prostředí, u kterých studie ve fázi IIA naznačily možné problémy. V tomto případě se požadují další testy, kterými se na vhodných mikrobiálních, rostlinných a živočišných druzích určí chronické a specifičtější účinky. Tyto doplňkové informace umožní použití nižšího bezpečnostního faktoru.
Vhodné doplňkové testy na ekotoxicitu jsou popsány v řadě publikací, např. v pokynech OECD. Požaduje se pečlivý výběr těchto testů, aby bylo zajištěno, že jsou vhodné pro situaci, v níž může být doplňková látka a/nebo její metabolity uvolněna a rozptýlena do prostředí. Zpřesnění posouzení účinků na půdu (PNECpůda) by mohlo být založeno na studiích chronických účinků na dešťovky, doplňkových studiích vlivu na půdní mikroflóru a řadu důležitých rostlinných druhů, studiích zaměřených na bezobratlé živočichy na pastvinách (včetně hmyzu) a volně žijící ptáky.
Zpřesnění posouzení účinků pro vodu/sediment může být založeno na testech chronické toxicity u nejcitlivějších vodních/bentických organismů určených při posuzování ve fázi IIA.
V případě potřeby se provedou studie bioakumulace podle pokynu OECD 305.
4. ODDÍL IV: STUDIE TÝKAJÍCÍ SE ÚČINNOSTI DOPLŇKOVÉ LÁTKY
Studie musí prokázat účinnost u každého navrhovaného užití a splňovat přinejmenším jednu z vlastností stanovených v čl. 5 odst. 3 nařízení (ES) č. 1831/2003, podle kategorií a funkčních skupin doplňkových látek stanovených v článku 6 a příloze I uvedeného nařízení. Tyto studie musí mimoto umožnit hodnocení účinnosti doplňkové látky podle běžné zemědělské praxe v EU.
Použité experimentální schéma musí být odůvodněné podle užití doplňkové látky, druhu a kategorie zvířat. Pokud se používají zvířata, studie musí být provedeny tak, aby jejich zdraví a podmínky chovu nepříznivě neovlivnily výklad výsledků. U každého pokusu je třeba popsat příznivé i nepříznivé účinky, a to jak v rovině technologické, tak i biologické. Je nutno prokázat rovněž neexistenci účinků, které snižují odlišující vlastnosti živočišných produktů. V ideálním případě zkoušky vyhovují kritériím stanoveným uznaným, externě auditovaným systémem zabezpečování jakosti. Pokud takovýto systém neexistuje, je nutno předložit důkazy, které prokazují, že práce byla provedena kvalifikovaným personálem, který je odpovědný jmenovanému vedoucímu studie, za použití vhodného zařízení a vybavení.
Zkušební protokol musí pečlivě vypracovat vedoucí studie s ohledem na obecné popisné údaje, např. metody, použité přístroje a materiál, údaje o druhu, plemeni nebo linii zvířat, jejich počtu a podmínkách, za nichž byla ustájena a krmena. U všech studií zahrnujících zvířata je nutno popsat zkušební podmínky podle bodu 3.1.1.3. Závěrečné zprávy, prvotní data, plány studie a přesně charakterizované a identifikované testované látky jsou archivovány pro budoucí využití.
Studie musí být navrženy tak, aby prokazovaly účinnost nejnižší doporučené dávky doplňkové látky zaměřením se na citlivé parametry v porovnání s negativní a volitelně s pozitivní kontrolní skupinou. Tyto studie zahrnují rovněž nejvyšší doporučenou dávku, pakliže se navrhuje. Nedoporučuje se žádné zvláštní schéma, určitá míra uvážení je ponechána pro vědecký názor na pojetí a provádění studií.
Pozornost je nutno věnovat rovněž známým nebo potenciálním biologickým nebo chemickým interakcím mezi doplňkovou látkou, jinými doplňkovými látkami a/nebo veterinárními léčivy a/nebo složkami krmné dávky, pokud je do důležité pro účinnost dotyčné doplňkové látky (například snášenlivost mikrobiální doplňkové látky s kokcidiostatiky a histomonostatiky nebo organickou kyselinou).
4.1 Studie in vitro
U všech technologických a některých senzorických doplňkových látek, které ovlivňují vlastnosti krmiva, je nutno účinnost prokázat pomocí laboratorní studie. Studie musí být navržena tak, aby zahrnovala reprezentativní rozsah materiálů, s nimiž se doplňková látka bude aplikovat. Výsledky se vyhodnotí pokud možno testy bez parametrů a musí prokázat očekávané změny s pravděpodobností P ≤ 0,05.
U ostatních druhů doplňkových látek je možno použít k doložení účinnosti studie in vitro, zejména studie, které simulují aspekty gastrointestinálního traktu. Tyto studie by měly umožnit statistické vyhodnocení.
4.2 Krátkodobé studie účinnosti se zvířaty
Je možno použít studie biologické dostupnosti k prokázání, nakolik nová forma nebo zdroj živiny nebo barvivo může být náhradou za rovnocennou doplňkovou látku, které již byla schválena nebo zavedena.
Studie bilanční stravitelnosti lze použít na podporu studií účinnosti u zvířat k doložení mechanismu působení. V některých případech, zejména ve vztahu k přínosům pro životní prostředí, je možno účinnost prokázat lépe bilančními studiemi a ty mohou být použity přednostně před dlouhodobými studiemi účinnosti. Při těchto pokusech se použijí počty a druhy/kategorie zvířat odpovídající navrhovaným podmínkám pro užití.
Popřípadě je možno navrhnout jiné krátkodobé studie účinnosti u zvířat a ty mohou nahradit dlouhodobé studie účinnosti u zvířat, je-li to plně odůvodněno.
4.3 Dlouhodobé studie účinnosti u zvířat
Zkoušky by měly být prováděny nejméně na dvou různých místech.
Použité experimentální schéma musí zahrnovat úvahu o odpovídající statistické průkaznosti a rizika typu 1 a 2. Protokol musí být dostatečně citlivý, aby zjistil účinky doplňkové látky při nejnižší doporučené dávce (riziko typu 1-α, P ≤ 0,05 obecně a P ≤ 0,1 pro přežvýkavce, menšinové druhy, zvířata v zájmovém chovu a zvířata, která nejsou určena k produkci potravin) s dostatečnou statistickou průkazností, aby bylo zaručeno, že experimentální protokol splňuje cíl studie. Riziko typu 2-β musí být nižší nebo rovno 20 % obecně a 25 % u pokusů s přežvýkavci, menšinovými druhy, zvířaty v zájmovém chovu a zvířaty, která nejsou určena k produkci potravin; průkaznost (1-β) je tedy větší nebo rovna 80 % (75 % u přežvýkavců, menšinových druhů, zvířat v zájmovém chovu a zvířat, která nejsou určena k produkci potravin).
Uznává se, že vzhledem k povaze některých doplňkových látek je obtížné stanovit zkušební podmínky, za nichž lze dosáhnout optimálních výsledků. Je proto nutno zvážit možnost použití metaanalýzy, je-li počet dostupných zkoušek vyšší než tři. Z tohoto důvodu by pro všechny zkoušky měly být použity podobné vzory protokolů, aby pak údaje mohly být ověřeny z hlediska jejich stejnorodosti a uspořádány (pokud to testy vyžadují) za účelem statistického hodnocení s hodnotou P ≤ 0,05.
4.4 Doba trvání dlouhodobých studií účinnosti s cílovými zvířaty
Doba trvání zkoušek účinnosti obvykle odpovídá udávané době používání.
Studie účinnosti se provádějí podle zemědělské praxe v Evropské unii a minimální doba trvání je uvedena v příloze IV.
Pokud se doplňková látka používá po specifickou a kratší dobu, než je dáno definicí kategorie zvířat, musí být podávána podle navrhovaných podmínek pro užití. Doba pozorování však nesmí být kratší než 28 dní a musí zahrnovat všechny důležité parametry (např. u prasnic k reprodukci počet selat narozených živě se zřetelem na dobu březosti, nebo počet a hmotnost odstavených selat se zřetelem na dobu laktace).
U ostatních druhů nebo kategorií zvířat, pro něž nebyla v příloze IV stanovena minimální doba trvání studií, se vezme v úvahu období podávání podle navrhovaných podmínek pro užití.
4.5 Požadavky na účinnost pro kategorie a funkční skupiny doplňkových látek
Pro všechny doplňkové látky, které mají mít účinek na zvířata, se požadují studie in vivo.
Pro kategorie zootechnických doplňkových látek a kokcidiostatik a histomonostatik musí být účinnost prokázána nejméně třemi dlouhodobými studiemi účinnosti. U některých zootechnických doplňkových látek a ostatních kategorií doplňkových látek, které mají účinek na zvířata, je možno uznat krátkodobé studie účinnosti, pokud lze účinnost jednoznačně prokázat.
Pro ostatní kategorie doplňkových látek bez přímého účinku na zvířata se předloží nejméně jedna studie účinnosti in vitro.
4.6 Studie týkající se jakosti živočišných produktů, jestliže to není udávaný účinek
S cílem prokázat, že doplňková látka nemá nepříznivý či jiný nežádoucí účinek na organoleptické a nutriční (popřípadě hygienické a technologické) vlastnosti potravin získaných ze zvířat, jež byla krmena doplňkovou látkou (pokud to není žádoucí účinek), se během jedné ze zkoušek účinnosti odeberou odpovídající vzorky. Jsou pozorovány dvě skupiny: skupina, jíž látka není podávána, a skupina s nejvyšším navrhovaným dávkováním doplňkové látky. Údaje musí umožnit statistické vyhodnocení. Vynechání těchto studií musí být přiměřeně zdůvodněno.
5. ODDÍL V: PLÁN MONITOROVÁNÍ V OBDOBÍ PO UVEDENÍ LÁTKY NA TRH
Podle čl. 7 odst. 3 písm. g) nařízení (ES) č. 1831/2003 je nutno pro některé kategorie doplňkových látek předložit návrh plánu monitorování v období po uvedení látky na trh s cílem sledovat a identifikovat případné přímé nebo nepřímé, okamžité, opožděné nebo nepředvídané účinky vyplývající z používání doplňkové látky na zdraví lidí nebo zvířat nebo na životní prostředí v souladu s vlastnostmi dotyčných produktů.
Návrh plánu monitorování musí být podrobně rozepsán podle jednotlivých případů a musí určovat, kdo (např. žadatel, uživatelé) vykonává jednotlivé úkoly v plánu stanovené a kdo odpovídá za zavedení a řádné provádění plánu, a musí stanovit způsob informování příslušných orgánů o veškerých nových skutečnostech, které souvisejí s bezpečností užívání dané doplňkové látky. Komisi a úřad je nutno informovat o případných zaznamenaných nepříznivých účincích, aniž jsou dotčena ustanovení o monitorování obsažená v článku 12 nařízení (ES) č. 1831/2003.
Je-li účinná látka rovněž uznaným antibiotikem a bylo-li prokázáno, že při jejím používání v množství, ve kterém je užita v krmivu, dochází k selekci rezistentních bakteriálních kmenů, provedou se jako součást monitorování v období po uvedení látky na trh terénní studie ke sledování rezistence bakterií vůči doplňkové látce.
U kokcidiostatik a histomonostatik se provede terénní monitorování rezistence Eimeria spp. a Histomonas meleagridis, pokud možno během druhé části doby povolení.
(1) Úř. věst. L 50, 20.2.2004, s. 44.
(2) Úř. věst. L 196, 16.8.1967, s. 1. Směrnice naposledy pozměněná směrnicí Evropského parlamentu a Rady 2006/121/ES (Úř. věst. L 396, 30.12.2006, s. 852; opraveno v Úř. věst. L 136, 29.5.2007, s. 281).
(3) Úř. věst. L 152, 30.4.2004, s. 1.
(4) Úř. věst. L 165, 30.4.2004, s. 1.
(5) Úř. věst. L 117, 8.5.1990, s. 1. Směrnice naposledy pozměněná rozhodnutím Komise 2005/174/ES (Úř. věst. L 59, 5.3.2005, s. 20)
(6) Úř. věst. L 76, 22.3.1991, s. 35. Směrnice naposledy pozměněná směrnicí 2001/58/ES (Úř. věst. L 212, 7.8.2001, s. 24).
(7) Úř. věst. L 262, 17.10.2000, s. 21.
(8) Úř. věst. L 224, 18.8.1990, s. 1. Nařízení naposledy pozměněné nařízením Komise (ES) č. 203/2008 (Úř. věst. L 60, 5.3.2008, s. 18).
(9) M. Thompson et al.: Harmonized Guidelines For Single Laboratory Validation Of Methods Of Analysis (IUPAC Technical Report) Pure Appl. Chem., sv. 74, č. 5, s. 835–855, 2002.
(10) Úř. věst. L 221, 17.8.2002, s. 8. Rozhodnutí naposledy pozměněné rozhodnutím 2004/25/ES (Úř. věst. L 6, 10.1.2004, s. 38).
(11) Nevyčerpávající seznam je k dispozici adrese: www.efsa.europa.eu/en/science/feedap/feedap_opinion/993.html.
(12) Svaly a kůže v přirozených poměrech.
(13) U prasat 50 g tuku a kůže v přirozených poměrech.
(14) Tuk a kůže v přirozených poměrech.
(15) S přihlédnutím k navrhované ochranné lhůtě.
(16) V ideálním případě stanoveno ve stejném okamžiku jako TRC.
(17) Vypočteno z hodnot TRC.
PŘÍLOHA III
ZVLÁŠTNÍ POŽADAVKY, JEŽ MUSÍ SPLŇOVAT DOKUMENTACE STANOVENÁ V ČLÁNKU 3, POKUD JDE O NĚKTERÉ KATEGORIE DOPLŇKOVÝCH LÁTEK NEBO NĚKTERÉ ZVLÁŠTNÍ SITUACE, JAK JE STANOVENO V ČL. 7 ODST. 5 NAŘÍZENÍ (ES) Č. 1831/2003
Nařízení (ES) č. 1831/2003 předpokládá doplňkovou pomoc při vypracování dokumentace (je-li to nutné) pro každou kategorii doplňkových látek nebo za jiným zvláštním účelem podle čl. 7 odst. 5 nařízení (ES) č. 1831/2003.
Seznam zvláštních požadavků na vypracování dokumentace pro:
|
1. |
technologické doplňkové látky |
|
2. |
senzorické doplňkové látky |
|
3. |
nutriční doplňkové látky |
|
4. |
zootechnické doplňkové látky |
|
5. |
kokcidiostatika a histomonostatika |
|
6. |
extrapolaci z většinových druhů na druhy menšinové |
|
7. |
zvířata v zájmovém chovu a jiná zvířata, která nejsou určena k produkci potravin |
|
8. |
doplňkové látky, které již byly povoleny pro použití v potravinách |
|
9. |
změnu povolení |
|
10. |
obnovení povolení |
|
11. |
přehodnocení některých doplňkových látek, které již byly povoleny podle směrnice 70/524/EHS. |
Žádosti je možno předložit na základě více než jednoho z výše uvedených zvláštních požadavků.
Obecné podmínky
Chybí-li v dokumentaci některý z údajů předepsaných v těchto oddílech, je třeba uvést důvody.
1. TECHNOLOGICKÉ DOPLŇKOVÉ LÁTKY
1.1 Oddíl I: Shrnutí dokumentace
Použije se celý oddíl I přílohy II.
1.2 Oddíl II: Identita, charakteristika a podmínky pro užití doplňkové látky; metody analýzy
Oddíl II přílohy II se použije takto:
|
— |
pro doplňkové látky, které nejsou vázané na držitele povolení, se použijí body 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5, 2.6, |
|
— |
pro jiné doplňkové látky, které jsou vázané na držitele povolení, se použije celý oddíl II. |
1.3 Oddíl III: Studie týkající se bezpečnosti doplňkových látek
Body 3.1, 3.2 a 3.4 přílohy II se nevztahují na doplňkové látky k silážování, pokud lze prokázat, že:
|
— |
v hotovém krmivu nepřetrvávají zjistitelná množství účinné látky (látek) nebo důležitých metabolitů nebo účinného činidla (činidel) nebo |
|
— |
účinná látka(y) a činidlo(a) se vyskytují jako běžné složky siláže a užití doplňkových látek nezvyšuje významně jejich koncentraci v porovnání se siláží připravenou bez užití doplňkové látky (např. pokud neexistuje žádná podstatná změna expozice). |
V ostatních případech se použije celý oddíl 3 přílohy II.
1.3.1 Studie týkající se bezpečnosti používání doplňkové látky pro cílová zvířata
U cizorodých (1) látek: použije se celý bod 3.1 přílohy II.
1.3.1.1 Studie tolerance u cílových druhů
Pro doplňkové látky k silážování:
|
— |
produkt se přidá do základní krmné dávky a výsledky se porovnají s negativní kontrolní skupinou se stejnou krmnou dávkou. Základní krmná dávka může obsahovat jediný zdroj siláže připravené bez použití doplňkové látky; |
|
— |
dávka zvolená pro studie tolerance je násobkem koncentrace vyskytující se v siláži v době normálního používání, pokud to lze přesvědčivě zjistit. Zvláštní pozornost se věnuje produktu, který obsahuje životaschopné mikroorganismy, a schopnosti těchto organismů přežít a množit se během silážování. |
Studie tolerance lze obvykle omezit na druhy přežvýkavců, zpravidla dojnice. Studie zahrnující jiné druhy jsou nutné pouze v případě, je-li siláž vzhledem ke své povaze vhodnější k použití pro jiné druhy než přežvýkavce.
Ostatní látky:
U ostatních látek, pro něž se požaduje povolení jako technologické doplňkové látky a které nebyly povoleny pro použití jako krmivo, je nutno prokázat, že pro zvířata nejsou škodlivé při nejvyšším navrhovaném obsahu. Toto prokázání může být omezeno na jeden pokus u jednoho z nejcitlivějších cílových druhů nebo u jednoho druhu laboratorních zvířat.
1.3.1.2 Mikrobiální testy
Použije se celý bod 3.1.2 přílohy II.
1.3.2 Studie týkající se bezpečnosti používání doplňkové látky pro spotřebitele
|
1.3.2.1 |
Studie metabolismu a reziduí Studie metabolismu a reziduí se nepožadují, pokud:
Studie metabolismu se nepožadují rovněž v případě, pokud se látka vyskytuje v potravinách nebo krmivech přirozeně ve značném množství nebo je běžnou složkou tělních tekutin nebo tkání. V těchto případech se však požadují studie reziduí, které mohou být omezeny na srovnání obsahů v tkáních/produktech u neošetřené skupiny a ve skupině, jíž byla podávána nejvyšší doporučená dávka. |
|
1.3.2.2 |
Toxikologické studie Toxikologické studie se nepožadují, pokud:
Pro mikroorganismy a enzymy, které nejsou vyňaty výše, se požadují studie genotoxicity (včetně mutagenity) a studie subchronické orální toxicity. Studie genotoxicity se nesmí provádět za přítomnosti živých buněk. Pro cizorodé látky, jež nejsou vyňaty výše, se použije celý bod 3.2.2 přílohy II. Pro ostatní látky se postupuje individuálně podle jednotlivých případů s přihlédnutím k úrovni a způsobu expozice. |
|
1.3.2.3 |
Posouzení bezpečnosti pro spotřebitele Pro doplňkové látky požadované pro zvířata, která jsou určena k produkci potravin, se použije celý bod 3.2.3 přílohy II. |
1.3.3 Studie týkající se bezpečnosti používání doplňkové látky pro uživatele/pracovníky
Použije se celý bod 3.3 přílohy II. Doplňkové látky obsahující enzymy a mikroorganismy se považují za senzibilizátory dýchacího ústrojí, pokud nejsou předloženy přesvědčivé důkazy prokazující opak.
1.3.4 Studie týkající se bezpečnosti používání doplňkové látky pro životní prostředí
Použije se celý bod 3.4 přílohy II. U doplňkových látek k silážování se posoudí účinky doplňkové látky na tvorbu silážních šťáv ze silážních jam nebo sil během silážování.
1.4 Oddíl IV: Studie týkající se účinnosti doplňkové látky
Technologické doplňkové látky mají zlepšit nebo stabilizovat vlastnosti krmiv, obvykle však nemají žádný přímý biologický účinek na živočišnou výrobu. Důkazy o účinnosti doplňkové látky musí být poskytnuty pomocí vhodných kritérií zohledněných v uznaných přípustných metodách za určených praktických podmínek pro užití v porovnání s odpovídajícím kontrolním krmivem.
Účinnost se posoudí studiemi in vitro s výjimkou látek pro kontrolu kontaminace radionuklidy. V následující tabulce jsou uvedeny vhodné parametry pro různé funkční skupiny.
Parametry pro jednotlivé technologické doplňkové látky
|
Funkční skupina |
Parametry pro prokázání účinnosti |
||||||||||
|
Inhibice mikrobiálního růstu, zejména biotických organismů a organismů přispívajících ke znehodnocení. Je nutno prokázat udávanou dobu trvání konzervačního účinku. |
||||||||||
|
Ochrana před znehodnocením způsobeným oxidací hlavních živin/složek během zpracování a/nebo skladování krmiva. Je nutno prokázat udávanou dobu trvání ochranného účinku. |
||||||||||
|
Vznik/uchování stabilních emulzí jinak nesmísitelných nebo špatně smísitelných přísad krmiva. |
||||||||||
|
Zachování fyzikálně-chemického stavu krmiv. |
||||||||||
|
Viskozita krmných materiálů nebo krmiv. |
||||||||||
|
Tvoření gelu vedoucí ke změně struktury krmiva. |
||||||||||
|
Trvanlivost pelet nebo výkonnost při vytváření pelet. |
||||||||||
|
Důkaz o omezené kontaminaci potravin živočišného původu. |
||||||||||
|
Schopnost zamezení spékání. Je nutno prokázat udávanou dobu trvání protispékavého účinku. |
||||||||||
|
pH a/nebo pufrovací kapacita krmiv. |
||||||||||
|
|
||||||||||
|
Neodstranitelná identifikace krmných materiálů. |
Doplňkové látky k silážování
Provedou se zvláštní zkoušky s cílem prokázat požadovaný účinek na silážní proces (2). Zkoušky se provedou s jedním příkladem z každé z níže uvedených kategorií (pokud jsou zahrnuta všechna nebo nespecifikovaná krmiva):
|
— |
krmiva, která lze snadno silážovat: >3 % rozpustných uhlohydrátů v čerstvém materiálu (např. celá rostlina kukuřice, jílek, sveřep nebo řepné řízky), |
|
— |
krmiva, která lze silážovat s mírnými obtížemi: 1,5–3,0 % rozpustných uhlohydrátů v čerstvém materiálu (např. lipnice, kostřava nebo zavadlá vojtěška), |
|
— |
krmiva, která lze silážovat obtížně: < 1,5 % rozpustných uhlohydrátů v čerstvém materiálu (např. srha nebo luskoviny). |
Jsou-li žádosti omezeny na dílčí kategorie krmiv popsané na základě sušiny, uvede se výslovně rozmezí sušiny. Poté se provedou tři zkoušky s materiálem reprezentativním pro dané rozmezí, pokud možno za použití příkladů různého botanického původu.
Pro zvláštní krmiva se požadují specifické zkoušky.
Doba trvání studie je obvykle 90 dnů nebo déle při stálé teplotě (doporučený rozsah 15 – 25 oC). Použití kratší doby trvání musí být zdůvodněno.
Obvykle se uvedou výsledky měření níže uvedených parametrů v porovnání s negativní kontrolní skupinou:
|
— |
sušina a vypočtené úbytky sušiny (upraveno s ohledem na těkavé látky), |
|
— |
pokles pH, |
|
— |
koncentrace těkavých mastných kyselin (např. kyselina octová, kyselina máselná a kyselina propionová) a kyseliny mléčné, |
|
— |
koncentrace alkoholů (ethanol), |
|
— |
koncentrace čpavku (g/kg celkového dusíku) a |
|
— |
obsah uhlohydrátů rozpustných ve vodě. |
Popřípadě se uvedou jiné mikrobiologické a chemické parametry k doložení zvláštního udávaného údaje (např. počet kvasinek schopných asimilovat laktát, množství klostridií, množství listérií a biogenních aminů).
Udávaný účinek v souvislosti se snížením množství odtékajících silážních šťáv se posoudí na základě celkového objemu silážních šťáv vytvořených za celé pokusné období s přihlédnutím k pravděpodobnému účinku na životní prostředí (např. ekotoxicita silážních šťáv nebo biologická potřeba kyslíku). Omezení tvorby silážních šťáv je nutno prokázat přímo. Kapacita silážní jámy musí být dostatečná, aby mohly silážní šťávy odtékat za pomoci tlaku. Doba trvání studie je obvykle 50 dní. Pokud se použije jiná doba, je nutno to odůvodnit.
Lepší aerobní stabilita musí být prokázána v porovnání s negativní kontrolní skupinou. Studie stability musí trvat nejméně sedm dnů po expozici vzduchu a doplňková látka dokládá stabilitu po dobu nejméně o dva dny delší, než je prokázáno u neošetřeného kontrolního vzorku. Doporučuje se, aby se pokus prováděl při teplotě okolí 20 oC a aby se zvýšení teploty o 3 oC nebo více oproti teplotě okolí považovalo za známku nestability. Měření teploty je možno nahradit měřením produkce CO2.
1.5 Oddíl V: Plán monitorování v období po uvedení látky na trh
Tento oddíl se použije podle ustanovení čl. 7 odst. 3 písm. g) nařízení (ES) č. 1831/2003. To znamená, že se plán monitorování v období po uvedení látky na trh požaduje pouze pro doplňkové látky, které jsou geneticky modifikované organismy nebo jsou z geneticky modifikovaných organismů vyrobeny.
2. SENZORICKÉ DOPLŇKOVÉ LÁTKY
2.1 Barviva
2.1.1 Oddíl I: Shrnutí dokumentace
Použije se celý oddíl I přílohy II.
2.1.2 Oddíl II: Identita, charakteristika a podmínky pro užití doplňkové látky; metody analýzy
Oddíl II přílohy II se použije takto:
|
— |
pro doplňkové látky, které nejsou vázané na držitele povolení, se použijí body 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5, 2.6, |
|
— |
pro jiné doplňkové látky, které jsou vázané na držitele povolení, se použije celý oddíl II. |
2.1.3 Oddíl III: Studie týkající se bezpečnosti používání doplňkové látky
Bod 3.3 přílohy II se vztahuje plně na všechny doplňkové látky.
|
1. |
Na látky, které při použití v krmivu, dávají barvu potravinám živočišného původu, se vztahuje plně oddíl III body 3.1, 3.2 a 3.4 přílohy II. |
|
2. |
U látek, které dávají nebo navracejí krmivům barvu, se studie týkající se oddílu III bodu 3.1 provedou na zvířatech, kterým byla doplňková látka podávána v doporučované dávce. Je možno poskytnout důkaz rovněž odkazem na existující vědeckou literaturu. Použije se oddíl III body 3.2 a 3.4 přílohy II. |
|
3. |
U látek, které mají pozitivní vliv na zbarvení okrasných ryb nebo ptáků, se požadují studie týkající se oddílu III bodu 3.1 přílohy II a tyto studie musí být provedeny na zvířatech, jimž byla doplňková látka podávána v doporučené dávce. Je možno poskytnout důkaz rovněž odkazem na existující vědeckou literaturu. Nepožadují se však body 3.2 a 3.4. |
2.1.4 Oddíl IV: Studie týkající se účinnosti doplňkové látky
Použije se celý oddíl IV přílohy II.
|
a) |
Pro látky, které při použití v krmivu dávají barvu potravinám živočišného původu: změny barvy produktů získaných ze zvířat, kterým byla doplňková látka podávána za doporučených podmínek pro užití, se měří pomocí vhodné metodiky. Je nutno prokázat, že používání doplňkové látky nemá nepříznivý vliv na stabilitu produktu nebo organoleptické a nutriční vlastnosti potravin. Jsou-li účinky konkrétní látky na složení/vlastnosti živočišných produktů dobře zdokumentovány, pak mohou odpovídající důkaz o účinnosti poskytnout ostatní studie (např. studie biologické dostupnosti). |
|
b) |
Pro látky, které dávají nebo navracejí krmivům barvu: důkaz o účinnosti je nutno poskytnout prostřednictvím odpovídajících laboratorních studií přihlížejících k určeným podmínkám pro užití v porovnání s kontrolními krmivy. |
|
c) |
Pro látky, které mají pozitivní vliv na zbarvení okrasných ryb nebo ptáků: studie prokazující účinek (účinky) se provedou na zvířatech, kterým byla doplňková látka podávána v doporučené výši. Změny barvy se měří pomocí vhodné metodiky. Důkaz o účinnosti je možno poskytnout rovněž jinými experimentálními studiemi (např. biologická dostupnost) nebo odkazem na vědeckou literaturu. |
2.1.5 Oddíl V: Plán monitorování v období po uvedení látky na trh
Tento oddíl se použije podle ustanovení čl. 7 odst. 3 písm. g) nařízení (ES) č. 1831/2003. To znamená, že se plán monitorování v období po uvedení látky na trh požaduje pouze pro doplňkové látky, které jsou geneticky modifikované organismy nebo jsou z geneticky modifikovaných organismů vyrobeny.
2.2 Zchutňující látky
2.2.1 Oddíl I: Shrnutí dokumentace
Použije se celý oddíl I přílohy II.
2.2.2 Oddíl II: Identita, charakteristika a podmínky pro užití doplňkové látky; metody analýzy
V případě skupiny „přírodní produkty“ se obvykle celé rostliny, zvířata a jiné organismy a jejich části nebo jejich produkty, které jsou velmi málo opracovány, jako např. drcením, mletím nebo sušením (např. mnoho bylin a koření), nepovažují za produkty spadající do funkční skupiny zchutňujících látek v kategorii senzorických doplňkových látek.
Pro účely vyhodnocení žádostí týkajících se těchto produktů jsou zchutňující látky klasifikovány takto:
|
1. |
Přírodní produkty:
|
|
2. |
Přírodní nebo odpovídající syntetické chemicky definované zchutňující látky |
|
3. |
Umělé látky Je nutno uvést příslušnou skupinu, do níž patří produkt, jehož se žádost týká. V případě produktu, který nespadá do žádné z výše uvedených skupin, je toto nutno uvést a zdůvodnit. |
|
2.2.2.1 |
Charakteristika účinné látky (látek) / činidla (činidel) Použije se celý bod 2.2 přílohy II. Mimoto: Pro všechny skupiny zchutňujících látek je nutno uvést vždy příslušné identifikační číslo (čísla) (např. FLAVIS (3), Rada Evropy (4), JECFA, CAS (5) nebo jiný mezinárodně schválený systém číslování) používané specificky k identifikaci zchutňujících produktů v krmivech a potravinách, pokud je k dispozici.
|
|
2.2.2.2 |
Způsob výroby Použije se celý bod 2.3 přílohy II. V případě přírodních produktů, které nejsou chemicky přesně definovány, obvykle komplexní směsi mnoha látek získané v procesu extrakce, je nutno uvést podrobný popis procesu extrakce. Doporučuje se v popisu použít odpovídající terminologii, např. silice, čistý, výluh, extrakt a související pojmy (7) běžně používané pro botanicky definované zchutňující produkty k popisu procesu extrakce. Upřesní se použitá extrakční rozpouštědla, přijatá bezpečnostní opatření, aby se zamezilo reziduím z rozpouštědel, a obsahy reziduí, pokud jsou toxikologicky významné, jestliže jejich výskytu nelze zabránit. Pojmy použité k charakteristice extraktu mohou zahrnovat odkaz na způsob extrakce. |
|
2.2.2.3 |
Metody analýzy
Celý bod 2.6 přílohy II se vztahuje na všechny ostatní zchutňující látky, například přírodní extrakty, které obsahují toxikologicky významné látky, přírodní nebo odpovídající syntetické chemicky definované zchutňující látky, jež jsou toxikologicky významnými látkami, a umělé zchutňující látky. |
2.2.3 Oddíl III: Studie týkající se bezpečnosti doplňkové látky
Pro všechny zchutňující látky je nutno uvést expozici zvířat a výpočty příjmu jak z přirozené expozice, tak po přidání zchutňující látky do krmiv.
Na zchutňující látky patřící do skupiny umělých látek se vztahuje celý oddíl III přílohy II.
|
2.2.3.1 |
Studie týkající se bezpečnosti používání doplňkové látky pro cílová zvířata
|
|
2.2.3.2 |
Studie týkající se bezpečnosti používání doplňkové látky pro spotřebitele Je nutno doložit, že metabolity zchutňující látky nevedou k akumulaci toxikologicky významných produktů pro člověka ve zvířeti. V případě, že užití požadovaného zchutňujícího produktu v důsledku jeho přidání do krmiv vede k reziduím v potravinách živočišného původu, je nutno poskytnout podrobný výpočet expozice spotřebitele.
|
|
2.2.3.3 |
Studie týkající se bezpečnosti používání doplňkové látky pro uživatele/pracovníky Použije se celý bod 3.3 přílohy II. |
|
2.2.3.4 |
Studie týkající se bezpečnosti používání doplňkové látky pro životní prostředí Použije se celý bod 3.4 přílohy II. |
2.2.4 Oddíl IV: Studie týkající se účinnosti doplňkové látky
Je nutno předložit důkazy o vlastnostech zchutňujících látek, obvykle na základě zveřejněné literatury. To může být prokázáno rovněž zkušenostmi z praktického používání, jsou-li k dispozici, v opačném případě mohou být požadovány studie na zvířatech.
Je nutno plně prověřit a uvést, zda produkt, kterého se žádost týká, plní v krmivu, zvířeti nebo potravinách živočišného původu jiné funkce kromě funkce v definici zchutňujících látek v příloze I nařízení (ES) č. 1831/2003.
2.2.5 Oddíl V: Plán monitorování v období po uvedení látky na trh
Tento oddíl se použije podle ustanovení čl. 7 odst. 3 písm. g) nařízení (ES) č. 1831/2003. To znamená, že se plán monitorování v období po uvedení látky na trh požaduje pouze pro doplňkové látky, které jsou geneticky modifikované organismy nebo jsou z geneticky modifikovaných organismů vyrobeny.
3. NUTRIČNÍ DOPLŇKOVÉ LÁTKY
3.1 Oddíl I: Shrnutí dokumentace
Použije se celý oddíl I přílohy II.
3.2 Oddíl II: Identita, charakteristika a podmínky pro užití doplňkové látky; metody analýzy
Oddíl II přílohy II se použije takto:
|
— |
pro doplňkové látky, které nejsou vázané na držitele povolení, se použijí body 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5, 2.6, |
|
— |
pro jiné doplňkové látky, které jsou vázané na držitele povolení, se použije celý oddíl II. |
3.3 Oddíl III: Studie týkající se bezpečnosti doplňkových látek
3.3.1 Studie týkající se bezpečnosti používání doplňkové látky pro cílové druhy
|
3.3.1.1 |
Tolerance u cílových druhů
|
|
3.3.1.2 |
Mikrobiální studie Použije se celý bod 3.1.2 přílohy II. |
3.3.2 Studie týkající se bezpečnosti použití doplňkové látky pro spotřebitele
|
3.3.2.1 |
Studie metabolismu a reziduí Studie metabolismu se obvykle nepožadují. Pro deriváty močoviny se přezkoumá metabolismus v bachoru. Studie reziduí nebo ukládání se požadují pouze u těch doplňkových látek, které spadají do funkční skupiny „vitamíny, provitamíny a chemicky přesně definované látky se srovnatelným účinkem“, které mají potenciál k akumulaci v těle, a pro funkční skupinu sloučenin stopových prvků, pokud se zvýšila biologická dostupnost. V tomto případě se nepoužije postup popsaný v oddíle 3.2.1 přílohy II. Požadavek je omezen na srovnání obsahu v tkáních nebo produktech mezi skupinou, jíž byla podávána nejvyšší dávka dotyčné látky, a pozitivní kontrolní skupinou (referenční látka). |
|
3.3.2.2 |
Toxikologické studie Ty se požadují pro produkty fermentačního procesu a pro doplňkové látky, které nebyly dosud povoleny. Pro produkty fermentačního procesu je nutno předložit studie genotoxicity a subchronické toxicity, ledaže:
Pokud produkční organismus patří do skupiny, o níž je známo, že některé kmeny produkují toxiny, je nutno výslovně vyloučit jejich přítomnost. |
|
3.3.2.3 |
Posouzení bezpečnosti pro spotřebitele Použije se celý bod 3.2.3 přílohy II. |
3.3.3 Studie týkající se bezpečnosti používání doplňkové látky pro uživatele/pracovníky
Použije se celý bod 3.3 přílohy II.
3.3.4 Studie týkající se bezpečnosti používání doplňkové látky pro životní prostředí
Celý bod 3.4 přílohy II se použije pro nové účinné látky, které patří ke skupině sloučenin stopových prvků.
3.4 Oddíl IV: Studie týkající se účinnosti doplňkové látky
Studie účinnosti se nepožadují pro močovinu, aminokyseliny, soli a analogy aminokyselin, které již byly povoleny jako doplňkové látky, sloučeniny stopových prvků, které již byly povoleny jako doplňkové látky, a vitamíny, provitamíny a chemicky přesně definované látky se srovnatelným účinkem, které již byly povoleny jako doplňkové látky.
Krátkodobá studie se požaduje k doložení účinnosti pro deriváty močoviny, soli a analogy aminokyselin, které ještě nebyly povoleny jako doplňkové látky, sloučeniny stopových prvků, které ještě nebyly povoleny jako doplňkové látky, a pro vitamíny, provitamíny a chemicky přesně definované látky se srovnatelným účinkem, které ještě nebyly povoleny jako doplňkové látky.
Pro ostatní látky, u nichž se udává nutriční účinek, se požaduje nejméně jedna dlouhodobá studie účinnosti podle ustanovení oddílu 4 přílohy II.
Studie musí případně prokázat, že doplňková látka může vyhovět nutričním požadavkům zvířat. Testy zahrnují zkušební skupinu s krmnou dávkou, která obsahuje živinu v koncentracích nižších než nutriční požadavky zvířat. Je však nutno zamezit zkouškám s použitím kontrolní skupiny trpící těžkým deficitem. Obvykle postačuje prokázat účinnost u jednoho druhu nebo kategorie zvířat, včetně laboratorních zvířat.
3.5 Oddíl V: Plán monitorování v období po uvedení látky na trh
Tento oddíl se použije podle ustanovení čl. 7 odst. 3 písm. g) nařízení (ES) č. 1831/2003.
4. ZOOTECHNICKÉ DOPLŇKOVÉ LÁTKY
4.1 Zootechnické doplňkové látky jiné než enzymy a mikroorganismy
4.1.1 Oddíl I: Shrnutí dokumentace
Použije se celý oddíl I přílohy II.
4.1.2 Oddíl II: Identita, charakteristika a podmínky pro užití doplňkové látky; metody analýzy
Použije se celý oddíl II přílohy II.
4.1.3 Oddíl III: Studie týkající se bezpečnosti doplňkových látek
|
4.1.3.1 |
Studie týkající se bezpečnosti používání doplňkové látky pro cílová zvířata Použije se celý bod 3.1 přílohy II. |
|
4.1.3.2 |
Studie týkající se bezpečnosti používání doplňkové látky pro spotřebitele
|
|
4.1.3.3 |
Studie týkající se bezpečnosti doplňkové látky pro uživatele/pracovníky Použije se celý bod 3.3 přílohy II. |
|
4.1.3.4 |
Studie týkající se bezpečnosti doplňkové látky pro životní prostředí Použije se celý bod 3.4 přílohy II. |
4.1.4 Oddíl IV: Studie týkající se účinnosti doplňkové látky
Použije se celý oddíl IV přílohy II.
|
1. |
Doplňkové látky, které mají pozitivní vliv na živočišnou výrobu, užitkovost nebo dobré životní podmínky, a funkční skupina „jiné zootechnické doplňkové látky“ Účinky lze prokázat pouze ve vztahu ke každému cílovému druhu nebo kategorii zvířat. V závislosti na vlastnostech doplňkové látky mohou být měření založena na pracovních charakteristikách (např. produkční účinnost, průměrný denní přírůstek, nárůst živočišných produktů), skladbě jatečně upraveného těla, užitkovosti stáda, reprodukčních parametrech nebo dobrých životních podmínkách zvířat. Důkaz o mechanismu působení lze poskytnout krátkodobými studiemi účinnosti nebo laboratorními studiemi měřícími příslušný parametr. |
|
2. |
Doplňkové látky, které pozitivně ovlivňují důsledky živočišné výroby pro životní prostředí U doplňkových látek, které příznivě ovlivňují životní prostředí (např. omezená exkrece dusíku a fosforu nebo omezená produkce metanu, eliminace příchutí), je možno účinnost u cílového druhu doložit třemi krátkodobými studiemi účinnosti se zvířaty prokazujícími významné prospěšné účinky. Studie vezmou v úvahu možnost přizpůsobení se doplňkové látce. |
4.1.5 Oddíl V: Plán monitorování v období po uvedení látky na trh
Tento oddíl se použije podle ustanovení čl. 7 odst. 3 písm. g) nařízení (ES) č. 1831/2003.
4.2 Zootechnické doplňkové látky: enzymy a mikroorganismy
4.2.1 Oddíl I: Shrnutí dokumentace
Použije se celý oddíl I přílohy II.
4.2.2 Oddíl II: Identita, charakteristika a podmínky pro užití doplňkové látky; metody analýzy
Použije se celý oddíl II přílohy II.
4.2.3 Oddíl III: Studie týkající se bezpečnosti doplňkových látek
|
4.2.3.1 |
Studie týkající se bezpečnosti používání doplňkové látky pro cílová zvířata Použije se celý bod 3.1.1 přílohy II. Žadatelé se vybízejí, aby pokud možno používali u pokusné skupiny přinejmenším 100násobek nadměrné dávky a snížili počet požadovaných parametrů. K tomuto účelu lze použít koncentrovanou formu doplňkové látky. Koncentrace se upraví snížením množství přítomného nosiče, poměr účinného činidla (činidel) / látky (látek) k ostatním produktům fermentačního procesu však musí být stejný jako v hotovém produktu. U enzymů krmivo poskytuje vhodný substrát(y). Celý bod 3.1.2 přílohy II se vztahuje na všechny mikroorganismy a enzymy, které mají přímý katalytický účinek na složky mikrobiální flóry nebo s ohledem na něž se udává, že ovlivňují střevní mikrobiální flóru. V případě nové expozice nebo značného zvýšení expozice mikroorganismům mohou být nezbytné doplňkové studie, aby byla prokázána neexistence nepříznivých účinků na komenzální mikrobiální flóru trávícího traktu. U přežvýkavců bude nutné přímé počítání mikrobiální flóry pouze v případě, že důkazy naznačují nepříznivé změny funkce bachoru (měřeno in vitro jako změna koncentrací těkavých mastných kyselin, snížení koncentrace propionátu nebo snížená celulolýza). |
|
4.2.3.2 |
Studie týkající se bezpečnosti používání doplňkové látky pro spotřebitele
Enzymy a mikroorganismy tvoří pouze část celé doplňkové látky, která ve většině případů může zahrnovat jiné složky pocházející z fermentačního procesu. Proto je nezbytné doplňkovou látku otestovat, aby bylo zajištěno, že neobsahuje mutagenní nebo jiné materiály, které mohou poškodit člověka požívajícího potraviny získané ze zvířat, jež byla krmena krmivy nebo vodou ošetřenými těmito doplňkovými látkami. Většina životaschopných bakterií určených pro přímé nebo nepřímé přijímání savci (včetně člověka) je však vybírána ze skupin organismů, pro něž jsou k dispozici údaje o zřejmém bezpečném používání, nebo ze skupin, pro něž jsou toxická rizika přesně vymezena. Obdobně nebezpečí spojené s mikroorganismy, které se v současnosti používají k produkci enzymů, je obvykle přesně rozpoznané a významně omezené moderními výrobními postupy. Proto se pro enzymy z mikrobiálních zdrojů a pro mikroorganismy, pro něž jsou k dispozici údaje o zřejmém bezpečném používání, a jsou-li složky fermentačního procesu přesně definované a známé, nepovažují zkoušky toxicity (např. testování orální toxicity nebo genotoxicity) za nutné. Avšak u živých organismů a organismů používaných k produkci enzymů je vždy nutno se zabývat konkrétními záležitostmi uvedenými v bodě 2.2.2.2 přílohy II. Je-li organismus nebo jeho užití nové a neexistují dostatečné poznatky o biologii (produkčního) organismu, aby byl vyloučen potenciál produkce toxických metabolitů, je nutno předložit studie genotoxicity a orální toxicity provedené s doplňkovými látkami, které obsahují životaschopné mikroorganismy nebo enzymy. V tomto případě mají podobu studií genotoxicity včetně mutagenity a studie subchronické orální toxicity. Doporučuje se, aby se tyto studie prováděly s bezbuněčnou fermentační půdou nebo v případě fermentace tuhých materiálů s vhodným extraktem. |
|
4.2.3.3 |
Studie týkající se bezpečnosti doplňkové látky pro uživatele/pracovníky Použije se celý bod 3.3 přílohy II s výjimkou těchto případů:
|
|
4.2.3.4 |
Studie týkající se bezpečnosti doplňkové látky pro životní prostředí Celý bod 3.4 přílohy II se vztahuje plně na mikroorganismy, které nejsou střevního původu nebo nejsou všudypřítomné v životním prostředí. |
4.2.4 Oddíl IV: Studie týkající se účinnosti doplňkových látek
Použije se celý oddíl IV přílohy II.
|
1. |
Doplňkové látky, které mají pozitivní vliv na živočišnou výrobu, užitkovost nebo dobré životní podmínky a pro funkční skupinu „jiné zootechnické doplňkové látky“. Účinky lze prokázat pouze ve vztahu ke každému cílovému druhu nebo kategorii zvířat. V závislosti na vlastnostech doplňkové látky mohou být měření založena na pracovních charakteristikách (např. produkční účinnost, průměrný denní přírůstek, nárůst živočišných produktů), skladbě jatečně upraveného těla, užitkovosti stáda, reprodukčních parametrech nebo dobrých životních podmínkách zvířat. Důkaz o mechanismu působení lze poskytnout krátkodobými studiemi účinnosti nebo laboratorními studiemi měřícími příslušný parametr. |
|
2. |
Doplňkové látky, které pozitivně ovlivňují důsledky živočišné výroby pro životní prostředí. U doplňkových látek, které příznivě ovlivňují životní prostředí (např. omezená exkrece dusíku nebo fosforu nebo omezená produkce metanu, eliminace příchutí), je možno účinnost u cílového druhu doložit třemi krátkodobými studiemi účinnosti se zvířaty prokazujícími významné prospěšné účinky. Studie vezmou v úvahu možnost přizpůsobení se doplňkové látce. |
4.2.5 Oddíl V: Plán monitorování v období po uvedení látky na trh
Tento oddíl se použije podle ustanovení čl. 7 odst. 3 písm. g) nařízení (ES) č. 1831/2003.
5. KOKCIDIOSTATIKA A HISTOMONOSTATIKA
5.1 Oddíl I: Shrnutí dokumentace
Použije se celý oddíl I přílohy II.
5.2 Oddíl II: Identita, charakteristika a podmínky pro užití doplňkové látky; metody analýzy
Použije se celý oddíl II přílohy II.
5.3 Oddíl III: Studie týkající se bezpečnosti doplňkových látek
5.3.1 Studie týkající se bezpečnosti používání doplňkové látky pro cílová zvířata
Použije se celý bod 3.1 přílohy II.
5.3.2 Studie týkající se bezpečnosti používání doplňkové látky pro spotřebitele
Použije se celý bod 3.2 přílohy II.
5.3.3 Studie týkající se bezpečnosti používání doplňkové látky pro uživatele/pracovníky
Použije se celý bod 3.3 přílohy II.
5.3.4 Studie týkající se bezpečnosti používání doplňkové látky pro životní prostředí
Použije se celý bod 3.4 přílohy II.
5.4 Oddíl IV: Studie týkající se účinnosti doplňkové látky
Tyto doplňkové látky chrání zvířata před důsledky invaze Eimeria spp. nebo Histomonas meleagridis. Význam se přikládá důkazům o zvláštních účincích doplňkové látky (např. kontrolovaný druh) a jejích profylaktických vlastnostech (např. snížení morbidity, mortality, počtu oocyst a výskytu chorobných změn). Popřípadě se předloží informace o účinku na růst a konverzi krmiva (výkrm drůbeže, náhrada nosnic a králíků), účincích na líhnutí vajec (plemenná drůbež).
Požadované údaje o účinnosti jsou odvozeny ze tří typů pokusů s cílovými zvířaty:
|
— |
umělé jednotlivé a smíšené infekce, |
|
— |
přirozená/umělá infekce s cílem simulovat podmínky pro užití, |
|
— |
skutečné podmínky pro užití při zkouškách v terénu. |
Pokusy s umělými jednotlivými a smíšenými infekcemi (např. klecový chov drůbeže) mají prokázat relativní účinnost proti parazitům a nevyžadují replikaci. Tři průkazné výsledky se požadují pro studie simulující podmínky pro užití (např. studie při volném ustájení s drůbeží, studie při klecovém chovu s králíky). Požadují se rovněž tři studie v terénu s určitou mírou přirozené infekce.
5.5 Oddíl V: Plán monitorování v období po uvedení látky na trh
Oddíl přílohy II se použije podle ustanovení čl. 7 odst. 3 písm. g) nařízení (ES) č. 1831/2003.
6. EXTRAPOLACE Z VĚTŠINOVÝCH DRUHŮ NA DRUHY MENŠINOVÉ
Menšinové druhy jsou vymezeny v čl. 1 odst. 2 tohoto nařízení.
U návrhu na rozšíření povoleného užití pro druh, který je z fyziologického hlediska srovnatelný s druhem, pro nějž již bylo užití doplňkové látky povoleno, se obvykle nevyžaduje předložení údajů v plném rozsahu.
Níže uvedené požadavky se vztahují pouze na požadované povolení pro menšinové druhy s ohledem na doplňkové látky, které již byly povoleny pro druhy většinové. Pro povolení nových doplňkových látek požadovaná pouze pro menšinové druhy se použijí plně všechny oddíly v závislosti na kategorii/funkční skupině doplňkové látky (viz odpovídající zvláštní požadavky v příloze III).
6.1 Oddíl I: Shrnutí dokumentace
Použije se celý oddíl I přílohy II.
6.2 Oddíl II: Identita, charakteristika a podmínky pro užití doplňkové látky; metody analýzy
Oddíl II přílohy II se použije takto:
|
— |
pro doplňkové látky, které jsou vázané na držitele povolení, se použije celý oddíl II, |
|
— |
pro jiné doplňkové látky se použijí body 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5, 2.6. |
6.3 Oddíl III: Studie týkající se bezpečnosti používání doplňkové látky
6.3.1 Studie týkající se bezpečnosti používání doplňkové látky pro cílová zvířata
|
6.3.1.1 |
Tolerance u cílových druhů Platí požadavky pro jednotlivé kategorie / funkční skupiny doplňkových látek. V zásadě se studie tolerance u menšinových druhů nepožadují, pokud doplňková látka vykazuje široké rozpětí pro bezpečnost (nejméně koeficient 10) u příslušného většinového druhu, který je z fyziologického hlediska podobný. Pokud tři většinové cílové druhy (včetně monogastrických a přežvýkavých savců a drůbeže) vykazují obdobné a široké rozpětí pro bezpečnost, nepožadují se další studie tolerance pro menšinové druhy, které nejsou z fyziologického hlediska podobné (např. koně nebo králíci). Pokud se požaduje studie tolerance, doba trvání studií pro menšinové druhy (vyjma králíků) činí nejméně 28 dní pro zvířata v růstu a 42 dní pro dospělá zvířata. U králíků platí tyto doby trvání: výkrm králíků: 28 dní; chovné králice: jeden cyklus (od inseminace do konce doby odstavu). Pokud se žádost vztahuje na sající a odstavené králíky, považuje se za dostatečné období 49 dní (počínaje jeden týden po narození) a musí zahrnovat králice až do odstavu. Pro ryby (jiné než lososovité) se požaduje 90denní období. |
6.3.2 Studie týkající se bezpečnosti používání doplňkové látky pro člověka jako spotřebitele
|
6.3.2.1 |
Studie metabolismu Platí požadavky pro jednotlivé kategorie a funkční skupiny doplňkových látek. Studie metabolismu se nepožadují, je-li doplňková látka již povolena pro užití u druhu, který je z fyziologického hlediska srovnatelný s menšinovými druhy, na něž se vztahuje žádost o povolení. Jestliže podobnost z fyziologického hlediska neexistuje, považuje se pro posouzení podobnosti metabolismu za dostatečné porovnání metabolického profilu na základě studií in vitro (např. s hepatocyty pomocí značené látky). Není-li menšinový druh z fyziologického hlediska podobný většinovému druhu, je nutno získat údaje o metabolismu doplňkové látky u menšinového druhu. |
|
6.3.2.2 |
Studie reziduí Je-li udávána nebo prokázána podobnost metabolismu, požaduje se pouze kvantifikace indikátorového rezidua v poživatelných tkáních a produktech. Ve všech ostatních případech se použije plně bod 3.2.1.2 přílohy II. |
|
6.3.2.3 |
Posouzení bezpečnosti pro spotřebitele Návrh maximálních limitů reziduí (MLR) Stanovení MLR lze provést za předpokladu, že se v poživatelných tkáních menšinového druhu v porovnání s podobným většinovým druhem nevyskytují žádné významné rozdíly v obsahu reziduí. MLR lze extrapolovat v rámci tříd zvířat takto:
MLR pro koně lze extrapolovat, pokud existují MLR pro většinového přežvýkavce a většinového monogastrického savce. Pokud byly odvozeny identické MLR u skotu (nebo ovcí), prasat a kuřat (nebo drůbeže), které představují většinové druhy s různými metabolickými schopnostmi a složením tkání, je možno stanovit stejné MLR rovněž pro ovce, koňovité a králíky, což znamená, že se považuje za možnou extrapolace na všechna zvířata, která jsou určena k produkci potravin, s výjimkou ryb. S ohledem na pokyn Výboru pro veterinární léčivé přípravky (CVMP) (10) pro stanovení MLR pro Salmonidae a ostatní ryby, který již povoluje extrapolaci z MLR ve svalu většinového druhu na Salmonidae a ostatní ryby za předpokladu, že výchozí látky jsou přijatelné jako indikátorové reziduum pro MLR ve svalu a kůži, lze MLR extrapolovat na všechna zvířata určená k produkci potravin. Jsou k dispozici analytické metody pro monitorování reziduí v poživatelných tkáních a produktech všech zvířat určených k produkci potravin. |
6.3.3 Studie týkající se bezpečnosti používání doplňkové látky pro uživatele/pracovníky
Použije se celý bod 3.3 přílohy II.
6.3.4 Studie týkající se bezpečnosti používání doplňkové látky pro životní prostředí
Posouzení rizik pro životní prostředí lze extrapolovat z hodnocení provedeného pro většinový druh, který je z fyziologického hlediska srovnatelný. Pro doplňkové látky, které mají být používány u králíků, se použije celý oddíl s přihlédnutím k požadavkům pro každou kategorii / funkční skupinu doplňkových látek.
6.4 Oddíl IV: Studie týkající se účinnosti doplňkové látky
Pokud doplňková látka byla pro stejnou funkci povolena pro většinový druh, který je z fyziologického hlediska srovnatelný, a pokud je znám nebo prokázán mechanismus působení doplňkové látky, je možno považovat za doložení účinnosti důkazy o stejném mechanismu působení u menšinových druhů. Pokud takováto spojitost neexistuje, je nutno prokázat účinnost podle obecných pravidel pro oddíl IV v příloze II. V některých případech může být vhodné kombinovat druhy zvířat ve stejné produktivní fázi (např. kozy a ovce používané k produkci mléka). V každé studii (P ≤ 0,1) nebo pomocí metaanalýzy (P ≤ 0,05), je-li to možné, by měla být prokázána významnost.
Požaduje-li se prokázání účinnosti, doba trvání studií účinnosti odpovídá srovnatelným produkčním fázím většinového druhu, který je z fyziologického hlediska srovnatelný. V ostatních případech minimální doba trvání studie splňuje příslušná ustanovení v bodě 4.4 přílohy II a přílohy IV.
6.5 Oddíl V: Plán monitorování v období po uvedení látky na trh
Tento oddíl přílohy II se použije podle ustanovení čl. 7 odst. 3 písm. g) nařízení (ES) č. 1831/2003.
7. ZVÍŘATA V ZÁJMOVÉM CHOVU A OSTATNÍ ZVÍŘATA, KTERÁ NEJSOU URČENA K PRODUKCI POTRAVIN
Zvířata v zájmovém chovu a ostatní zvířata, která nejsou určena k produkci potravin, jsou vymezena v čl. 1 odst. 1 tohoto nařízení.
7.1 Oddíl I: Shrnutí dokumentace
Použije se celý oddíl I přílohy II.
7.2 Oddíl II: Identita, charakteristika a podmínky pro užití doplňkové látky; metody analýzy
Oddíl II přílohy II se použije takto:
|
— |
pro doplňkové látky, které jsou vázané na držitele povolení, se použije celý oddíl II, |
|
— |
pro jiné doplňkové látky se použijí body 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2., 2.4.1, 2.4.2, 2.4.4, 2.5, 2.6. |
7.3 Oddíl III: Studie týkající se bezpečnosti doplňkové látky
7.3.1 Studie týkající se bezpečnosti používání doplňkové látky pro cílová zvířata
Platí požadavky pro jednotlivé kategorie / funkční skupiny doplňkových látek. Požaduje-li se studie tolerance, její doba trvání činí nejméně 28 dní.
Studie tolerance se nepožaduje, pokud doplňková látka vykazuje srovnatelné a široké rozpětí pro bezpečnost u třech většinových druhů (včetně monogastrických a přežvýkavých savců a drůbeže).
7.3.2 Studie týkající se bezpečnosti používání doplňkové látky pro spotřebitele
Tento bod se obvykle nepožaduje. Pozornost se věnuje bezpečnosti majitele.
7.3.3 Studie týkající se bezpečnosti používání doplňkové látky pro uživatele/pracovníky
Použije se celý bod 3.3 přílohy II.
7.3.4 Studie týkající se bezpečnosti používání doplňkové látky pro životní prostředí
Bod 3.4 přílohy II se nepožaduje.
7.4 Oddíl IV: Studie týkající se účinnosti doplňkové látky
Platí požadavky pro jednotlivé kategorie / funkční skupiny doplňkových látek.
Jestliže doplňková látka, pro niž se požadují studie na zvířatech, již byla předem povolena pro druh, který je z fyziologického hlediska podobný, nepožaduje se další prokázání účinnosti, jsou-li požadovaný účinek a mechanismus působení stejné. Pokud doplňková látka nebyla dosud povolena nebo požadovaný účinek či mechanismus působení jsou jiné než v případě předchozího povolení, je nutno účinnost prokázat podle obecných pravidel pro oddíl IV v příloze II.
Doba trvání dlouhodobých zkoušek účinnosti činí nejméně 28 dní.
7.5 Oddíl V: Plán monitorování v období po uvedení látky na trh
Tento oddíl přílohy II se použije podle ustanovení čl. 7 odst. 3 písm. g) nařízení (ES) č. 1831/2003.
8. DOPLŇKOVÉ LÁTKY, KTERÉ JIŽ BYLY POVOLENY PRO UŽITÍ V POTRAVINÁCH
8.1 Oddíl I: Shrnutí dokumentace
Použije se celý oddíl I přílohy II.
8.2 Oddíl II: Identita, charakteristika a podmínky pro užití doplňkové látky; metody analýzy
Oddíl II přílohy II se použije takto:
|
— |
pro doplňkové látky, které jsou vázané na držitele povolení, se použije celý oddíl II, |
|
— |
pro jiné doplňkové látky se použijí body 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2., 2.4.1, 2.4.2, 2.4.4, 2.5, 2.6. |
8.3 Oddíl III: Studie týkající se bezpečnosti doplňkových látek
Musí být uvedeno nejnovější formální posouzení bezpečnosti potravinářských přídatných látek a doplněno případnými následně získanými údaji.
Pro doplňkové látky, které jsou povoleny jako potravinářské přídatné látky nebo schváleny v Evropské unii jako složky potravin bez jakéhokoliv omezení, nejsou studie týkající se bezpečnosti pro spotřebitele a pracovníky obvykle nutné.
Uvedou se body 3.1, 3.2 a 3.3 přílohy II se zřetelem na stávající poznatky o bezpečnosti těchto látek, pokud jsou používány v potravinách. Takovéto látky, které jsou používány rovněž v potravinách, lze klasifikovat jako:
|
— |
s nespecifikovanou ADI (bez výslovného uvedení horního limitu příjmu pro látky s velmi nízkou toxicitou), |
|
— |
se stanovenou ADI nebo UL nebo |
|
— |
s nestanovenou ADI (pro látky, u nichž dostupné údaje nepostačují ke zjištění jejich bezpečnosti). |
8.3.1 Studie týkající se bezpečnosti používání doplňkové látky pro cílová zvířata
Je-li obsah užité doplňkové látky podobný jako v případě potravin, lze bezpečnost pro cílové druhy posoudit na základě dostupných toxikologických údajů ze zkoušek in vivo s přihlédnutím k chemické struktuře a metabolické schopnosti cílových druhů. Je-li obsah užití v krmivech významně vyšší než odpovídající užití v potravinách, může být podle povahy látky nutná studie tolerance u cílového zvířete.
8.3.2 Studie týkající se bezpečnosti používání doplňkové látky pro spotřebitele
Pokud užití jako doplňková látka vede k vyšší expozici spotřebitele nebo k jinému druhu metabolitů než v případě užití v potravinách, budou požadovány další toxikologické údaje a údaje o reziduích.
|
8.3.2.1 |
Pro doplňkové látky, pro něž není specifikována ADI Posouzení bezpečnosti pro spotřebitele se nepožaduje vyjma případu, kdy užití doplňkové látky v krmivech vede k odlišnému druhu metabolitů než při užití v potravinách. |
|
8.3.2.2 |
Doplňkové látky se stanovenou ADI nebo UL Bezpečnost pro spotřebitele je nutno posoudit s přihlédnutím k doplňkové expozici vyplývající z užití v krmivech nebo zvláštní expozici v souvislosti s metabolity vznikajícími u cílového druhu. To lze provést extrapolací údajů o reziduích z literatury. Jsou-li nezbytné studie reziduí, požadavek je omezen na srovnání obsahů v tkáních nebo produktech u neošetřené skupiny a skupiny, jíž byla podávána nejvyšší udávaná dávka. |
|
8.3.2.3 |
Potravinářské přídatné látky, pro něž nebyla ADI stanovena Je nutno jednoznačně uvést důvody, proč nebyla ADI stanovena. Pokud to vyvolává obavy a užití doplňkové látky v krmivech by přispělo k významnému zvýšení expozice u spotřebitele, požaduje se úplné toxikologické hodnocení. Doplňkovou expozici vyplývající z užití v krmivech lze extrapolovat z údajů o reziduích z literatury. Jsou-li nezbytné studie reziduí, požadavek je omezen na srovnání obsahů v tkáních nebo produktech u neošetřené skupiny a skupiny, jíž byla podávána nejvyšší udávaná dávka. |
8.3.3 Studie týkající se bezpečnosti používání doplňkové látky pro uživatele/pracovníky
Použije se celý bod 3.3 přílohy II.
Při posuzování bezpečnosti doplňkové látky pro uživatele se vezmou v úvahu bezpečnostní opatření stanovená pro manipulaci s těmito látkami používanými v potravinách.
8.3.4 Studie týkající se bezpečnosti používání doplňkové látky pro životní prostředí
Požaduje se bod 3.4 přílohy II.
8.4 Oddíl IV: Studie týkající se účinnosti doplňkové látky
Jestliže je funkce požadovaná pro krmivo stejná jako při užití v potravinách, nemusí být nutné další prokázání účinnosti. V opačném případě platí požadavky na účinnost uvedené v oddíle IV přílohy II.
8.5 Oddíl V: Plán monitorování v období po uvedení látky na trh
Tento oddíl přílohy II se použije podle ustanovení čl. 7 odst. 3 písm. g) nařízení (ES) č. 1831/2003.
9. ZMĚNA POVOLENÍ
Jelikož je možno využít hodnocení údajů předložených pro předchozí povolení, musí dokumentace vyhotovená pro žádost podle čl. 13 odst. 3 nařízení (ES) č. 1831/2003 splňovat pouze níže uvedené požadavky.
Žádost o změnu podmínek zahrnutých ve stávajícím povolení, např. identifikace, charakteristika nebo podmínky pro užití doplňkové látky, musí prokázat, že změna nemá žádný škodlivý účinek pro cílový druh, spotřebitele, uživatele nebo životní prostředí. Za tímto účelem je možno doplňkovou látku považovat za totožnou, pokud účinná látka(y) nebo činidlo(a) a podmínky pro užití jsou stejné, její čistota je v zásadě podobná a nebyly přidány žádné nové složky, které by vyvolávaly obavy. Pro takovéto produkty je možno podat zkrácenou žádost, jelikož obvykle nebude nutné opakovat studie k prokázání bezpečnosti pro cílový druh, spotřebitele a životní prostředí a účinnosti.
Žádost musí splňovat tyto požadavky:
|
1. |
použije se celá příloha I – to zahrnuje údaje o požadované změně; |
|
2. |
použije se celý oddíl II přílohy II; |
|
3. |
je nutno poskytnout údaje, které prokazují, že chemické nebo biologické vlastnosti doplňkové látky jsou v zásadě stejné jako u známého produktu; |
|
4. |
popřípadě je nutno předložit důkazy o bioekvivalenci, a to na základě specifikace, zveřejněné literatury nebo zvláštních studií. Není-li bioekvivalence plně prokázána, je nutno prokázat shodu ochranné lhůty s MLR; |
|
5. |
je nutno předložit důkaz, že za současných vědeckých poznatků zůstává doplňková látka za schválených podmínek bezpečná pro cílové druhy, spotřebitele, pracovníky a životní prostředí; |
|
6. |
je nutno předložit zprávu o výsledcích monitorování v období po uvedení látky na trh, jsou-li takovéto požadavky na monitorování zahrnuty v povolení; a |
|
7. |
je nutno předložit zvláštní údaje k doložení žádosti o změnu v souladu s příslušnými částmi oddílů III, IV a V přílohy II. |
10. OBNOVENÍ POVOLENÍ
Žádosti o obnovení povolení podle článku 14 nařízení (ES) č. 1831/2003 splňují tyto požadavky:
10.1 Oddíl I: Shrnutí dokumentace
Použije se celý oddíl I přílohy II. Předloží se kopie původního povolení Společenství k uvedení doplňkové látky na trh nebo poslední obnovení žádosti. Vyhotoví se aktualizovaná dokumentace podle nejnovějších požadavků a předloží se seznam s uvedením všech změn oproti původnímu povolení nebo poslednímu obnovení povolení. Žadatel musí předložit shrnutí dokumentace, které přiblíží rozsah žádosti, a případné nové informace, které se staly dostupnými od doby udělení předchozího povolení/obnovení povolení s ohledem na identitu a bezpečnost.
10.2 Oddíl II: Identita, charakteristika a podmínky pro užití doplňkové látky; metody analýzy
Oddíl II přílohy II se použije takto:
|
— |
pro doplňkové látky, které jsou vázané na držitele povolení, se použije celý oddíl II, |
|
— |
pro jiné doplňkové látky se použijí body 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5, 2.6. |
Je nutno předložit důkazy, které prokazují, že se doplňková látka významně nezměnila, pokud jde o složení, čistotu nebo aktivitu, oproti povolené doplňkové látce. Je nutno oznámit jakoukoliv změnu výrobního postupu.
10.3 Oddíl III: Studie týkající se bezpečnosti doplňkových látek
Musí být předložen důkaz, že za současného stavu poznatků zůstává doplňková látka za schválených podmínek bezpečná pro cílový druh, spotřebitele, pracovníky a životní prostředí. Musí být předložena aktualizovaná dokumentace o bezpečnosti za období od udělení původního povolení nebo od posledního obnovení povolení, která obsahuje následující informace:
|
— |
zprávy o nežádoucích účincích, včetně nahodilých jevů (dosud neznámé účinky, závažné účinky všeho druhu, zvýšený výskyt známých účinků) u cílových zvířat, spotřebitelů, uživatelů a v životním prostředí. Zpráva o nežádoucích účincích musí uvádět povahu účinku, počet postižených osob/zvířat, výsledek, podmínky pro užití a posouzení příčinné souvislosti, |
|
— |
zprávy o dosud neznámých interakcích a křížových kontaminacích, |
|
— |
údaje z monitorování reziduí, je-li to nutné, |
|
— |
údaje z epidemiologických/epizootologických a/nebo toxikologických studií, |
|
— |
jakékoliv další informace týkající se bezpečnosti doplňkové látky a rizik doplňkové látky pro zvířata, člověka a životní prostředí. |
Pokud nejsou předloženy žádné další informace o některém z těchto faktorů, musí to být přesně odůvodněno.
Zpráva o výsledcích programu monitorování v období po uvedení látky na trh se předloží, je-li takovýto požadavek na monitorování zahrnut v předchozím povolení.
Pokud podle ustanovení čl. 14 odst. 2 písm. d) nařízení (ES) č. 1831/2003 žádost o obnovení povolení zahrnuje návrh na změnu nebo doplnění podmínek původního povolení, mimo jiné podmínek týkajících se budoucího monitorování, je nutno předložit zvláštní údaje na podporu návrhu na změnu v souladu s příslušnými částmi oddílů III, IV a V přílohy II.
11. PŘEHODNOCENÍ NĚKTERÝCH DOPLŇKOVÝCH LÁTEK, KTERÉ JIŽ BYLY POVOLENY PODLE SMĚRNICE 70/524/EHS
Doplňkovými látkami, jichž se týká tento bod 11, jsou doplňkové látky, které již byly povoleny podle směrnice 70/524/EHS a mají být přehodnoceny v souladu s čl. 10 odst. 2 nařízení (ES) č. 1831/2003 a které patří do těchto skupin:
|
— |
antioxidační látky, |
|
— |
zchutňující a dochucující látky, |
|
— |
emulgátory a stabilizátory, zahušťující látky a želírující látky, |
|
— |
barviva, včetně pigmentů, |
|
— |
konzervanty, |
|
— |
vitamíny, provitamíny a chemicky přesně definované látky se srovnatelným účinkem, |
|
— |
stopové prvky, |
|
— |
pojiva, protispékavé látky a koagulanty; |
|
— |
regulátory kyselosti a |
|
— |
radionuklidní pojiva. |
Úroveň a kvalita vyhodnocení rizik u těchto doplňkových látek musí být podobná jako u jiných doplňkových látek. Avšak vzhledem k dlouhé historii jejich bezpečného používání je možno použít údaje z již zveřejněných studií podle ustanovení tohoto nařízení, aby bylo prokázáno, že za schválených podmínek doplňková látka zůstává bezpečná pro cílové druhy, spotřebitele, uživatele a životní prostředí.
11.1 Oddíl I: Shrnutí dokumentace
Použije se celý oddíl I přílohy II.
11.2 Oddíl II: Identita, charakteristika a podmínky pro užití doplňkové látky; metody analýzy
Oddíl II přílohy II se použije takto:
|
— |
pro doplňkové látky, které nejsou vázané na držitele povolení, se použijí body 2.1.2, 2.1.3, 2.1.4, 2.1.4.2, 2.2, 2.3.1, 2.3.2, 2.4.1, 2.4.2, 2.4.4, 2.5, 2.6, |
|
— |
pro jiné doplňkové látky, které jsou vázané na držitele povolení, se použije celý oddíl II. |
11.3 Oddíl III: Studie týkající se bezpečnosti doplňkových látek
Pokud byla doplňková látka posouzena s ohledem na bezpečnost pro cílové druhy, spotřebitele, uživatele/pracovníky a životní prostředí, předloží se shrnutí studií bezpečnosti předložených pro předchozí povolení a případné nové informace, které byly získány od doby udělení předchozího povolení. Pokud nebylo formální posouzení bezpečnosti provedeno pro užití látky jako doplňkové látky, lze použít studie a údaje z vědecké literatury, jestliže jsou rovnocenné údajům, které by se požadovaly v nové žádosti. V opačném případě se předloží úplný soubor studií bezpečnosti.
11.4 Oddíl IV: Studie týkající se účinnosti doplňkové látky
Shodu s požadavky na účinnost podle čl. 5 odst. 3 nařízení (ES) č. 1831/2003 je možno popřípadě prokázat předložením jiných materiálů než studie, zejména týkajících se dlouhé historie používání.
11.5 Oddíl V: Plán monitorování v období po uvedení látky na trh
Tento oddíl přílohy II se použije podle ustanovení čl. 7 odst. 3 písm. g) nařízení (ES) č. 1831/2003.
(1) Cizorodá látka je chemická látka, která není přirozenou součástí organismu, jenž je této látce vystaven. Může zahrnovat rovněž látky, které jsou přítomny v mnohem vyšších koncentracích, než je obvyklé.
(2) Pro účely tohoto nařízení se „silážním procesem“ rozumí proces, při němž je přirozené znehodnocování organické hmoty řízeno acidifikací v anaerobních podmínkách na základě přirozené fermentace a/nebo přidání doplňkových látek k silážování.
(3) Identifikační číslo pro chemicky definované zchutňující látky používané ve FLAVIS, informačním systému EU pro zchutňující látky, databáze v nařízení Komise (ES) č. 1565/2000 ze dne 18. července 2000 (Úř. věst. L 180, 19.7.2000 s. 8), kterým se stanoví opatření nezbytná pro přijetí programu hodnocení podle nařízení Evropského parlamentu a Rady (ES) č. 2232/96 (Úř. věst. L 299, 23.11.1996, s. 1).
(4) Č. Rady Evropy: číslo Rady Evropy používané pro botanicky definované produkty určené k aromatizaci ve zprávě Rady Evropy č. 1 „Natural sources of flavourings“, sv. I, Štrasburk 2000 a následujících svazcích.
(5) Číslo CAS (č. CAS) Registrační číslo podle Chemical Abstracts Service, jednoznačný identifikátor chemických látek běžně používaný v seznamech chemických látek.
(6) Pro účely tohoto oddílu nařízení se „toxikologicky významnou látkou“ rozumí látka s přípustným denním nebo týdenním příjmem (TDI nebo TWI), ADI, nebo s omezením, pokud jde o její používání, nebo účinná složka, jak je stanoveno ve směrnici Rady 88/388/EHS týkající se látek určených k aromatizaci pro použití v potravinách a výchozích materiálů pro jejich výrobu, nebo nežádoucí látka.
(7) Stanoveno v dodatku 4 zprávy Rady Evropy č. 1 „Natural sources of flavourings“, sv. I, Štrasburk, 2000.
(8) JECFA (FAO/WHO, 1996, Food additive series 35, IPCS, WHO Ženeva) odpovídající prahová hodnota pro cílové zvíře by měla být upravena s přihlédnutím k hmotnosti zvířete a příjmu krmiva.
(9) Úř. věst. L 180, 19.7.2000, s. 8.
(10) Pokyn pro stanovení maximálních limitů reziduí pro Salmonidae a ostatní ryby. Evropská agentura pro léčivé přípravky. Veterinary Medicines Evaluation Unit. EMEA/CVMP/153b/97-FINAL.
PŘÍLOHA IV
Kategorie a definice cílových zvířat a údaj o minimální době trvání studií účinnosti
1. Tabulka. Kategorie zvířat: prasata
|
Kategorie |
Definice kategorie zvířat |
Přibližná doba trvání (hmotnost/stáří) |
Minimální doba trvání dlouhodobých studií účinností |
||
|
Období/stáří |
Stáří |
Hmotnost |
|||
|
Selata (sající) |
Mladá prasata sající mléko od prasnic |
Od narození |
Až do 21–42 dní |
Až do 6–11 kg |
14 dní |
|
Selata (odstavená) |
Mladá prasata po skončení období sání chovaná k reprodukci nebo pro masnou produkci |
Od 21–42 dní |
Až do 120 dní |
Až do 35 kg |
42 dní |
|
Selata (sající a odstavená selata) |
Mladá prasata od narození chovaná k reprodukci nebo pro masnou produkci |
Od narození |
Až do 120 dní |
Až do 35 kg |
58 dní |
|
Výkrm prasat |
Prasata po odstavu určená k masné produkci až do dne odvozu na jatky |
Od 60–120 dní |
Až do 120–250 dní (nebo podle místních zvyklostí) |
80–150 kg (nebo podle místních zvyklostí) |
Až do jateční hmotnosti, nejméně však 70 dní |
|
Prasnice určené k reprodukci |
Samice prasete, které byly nejméně jednou inseminovány/připuštěny |
Od první inseminace |
|
|
Od inseminace do konce odstavu druhého vrhu (dva cykly) |
|
Prasnice určené k reprodukci selat |
Samice prasete, které byly nejméně jednou inseminovány/připuštěny |
|
|
|
Nejméně dva týdny před porodem do konce doby odstavu |
2. Tabulka. Kategorie zvířat: drůbež
|
Kategorie |
Definice kategorie zvířat |
Přibližná doba trvání (hmotnost/stáří) |
Minimální doba trvání dlouhodobých studií účinností |
||
|
Období |
Stáří |
Hmotnost |
|||
|
Výkrm kuřat |
Kuřata určená pro výkrm |
Od vylíhnutí |
Až do 35 dní |
Až do ~1 600 g (až do 2 kg) |
35 dní |
|
Odchov kuřat a kuřice |
Slepičky chované pro konzumní produkci vajec nebo pro chovné účely |
Od vylíhnutí |
Až do ~16 týdnů (až do 20 týdnů) |
— |
112 dní (nejsou-li údaje o účinnosti k dispozici pro výkrm kuřat) |
|
Nosnice |
Slepice ve snášce chované pro produkci vajec |
Od 16–21 týdnů |
Až do ~13 měsíc (až do 18 měsíců) |
Od 1 200 g (bílé), 1 400 g (hnědé) |
168 dní |
|
Výkrm krůt |
Krůty a krocani chovaní na výkrm |
Od vylíhnutí |
Až do ~14 týdnů (až do 20 týdnů) Až do ~16 týdnů (až do 24 týdnů) |
Samice: až do ~7 000 g (až do 10 000 g) Samci: až do ~12 000 g (až do 20 000 g) |
84 dní |
|
Krůty pro účely plemenitby |
Krůty a krocani chovaní pro účely plemenitby |
Celé období |
Od 30 týdnů až do ~ 60 týdnů |
Samice: od ~15 000 g Samci: od ~30 000 g |
Nejméně šest měsíců |
|
Odchov krůt |
Mladé krůty a krocani určení k chovným účelům |
Od vylíhnutí |
Až do 30 týdnů |
Samice: až do ~15 000 g Samci: až do ~30 000 g |
Celé období (nejsou-li údaje o účinnosti k dispozici pro výkrm krůt) |
3. Tabulka. Kategorie zvířat: skot (domácí skot, včetně druhů buvolec a bizon)
|
Kategorie |
Definice kategorie zvířat |
Přibližná doba trvání (hmotnost/stáří) |
Minimální doba trvání dlouhodobých studií účinností |
||
|
Období |
Stáří |
Hmotnost |
|||
|
Odchov telat |
Telata chovaná pro reprodukci nebo produkci hovězího masa |
Od narození |
Až do 4 měsíců |
Až do 60–80 kg (až do 145 kg) |
56 dní |
|
Výkrm telat |
Telata pro produkci telecího masa |
Od narození |
Až do 6 měsíců |
Až do 180 kg (až do 250 kg) |
Do porážky, nejméně však 84 dní |
|
Výkrm skotu |
Skot po odstavu určený k masné produkci do dne odvozu na jatky |
Od úplného rozvoje přežvykování |
Až do 10–36 měsíců |
Až do 350–700 kg |
168 dní |
|
Dojnice pro produkci mléka |
Samice skotu, které porodily nejméně jedno tele |
|
|
|
84 dní (je nutno uvést celé období laktace) |
|
Krávy k reprodukci |
Samice skotu, které byly nejméně jednou inseminovány/připuštěny |
Od první inseminace do konce druhého odstavu |
|
|
Dva cykly (pokud se požadují reprodukční parametry) |
4. Tabulka. Kategorie zvířat: ovce
|
Kategorie |
Definice kategorie zvířat |
Přibližná doba trvání (hmotnost/stáří) |
Minimální doba trvání dlouhodobých studií účinností |
||
|
Období |
Stáří |
Hmotnost |
|||
|
Odchov jehňat |
Jehňata chovaná pro budoucí reprodukci |
Od narození |
Až do 3 měsíců |
15–20 kg |
56 dní |
|
Výkrm jehňat |
Jehňata chovaná pro produkci jehněčího masa |
Od narození |
Až do 6 měsíců (nebo starší) |
Až do 55 kg |
Do jateční hmotnosti, nejméně však 56 dní |
|
Ovce určené k produkci mléka |
Ovce, které porodily nejméně jedno jehně |
|
|
|
84 dní (je nutno uvést celé období laktace) |
|
Bahnice k reprodukci |
Samice ovce, které byly nejméně jednou inseminovány/připuštěny |
Od první inseminace do konce druhého odstavu |
|
|
Dva cykly (pokud se požadují reprodukční parametry) |
5. Tabulka. Kategorie zvířat: kozy
|
Kategorie |
Definice kategorie zvířat |
Přibližná doba trvání (hmotnost/stáří) |
Minimální doba trvání dlouhodobých studií účinností |
||
|
Období |
Stáří |
Hmotnost |
|||
|
Odchov kůzlat |
Mladé kozy chované pro budoucí reprodukci |
Od narození |
Až do 3 měsíců |
15–20 kg |
Nejméně 56 dní |
|
Výkrm kůzlat |
Mladé kozy chované pro produkci kozího masa |
Od narození |
Až do 6 měsíců |
|
Nejméně 56 dní |
|
Kozy určené k produkci mléka |
Kozy nejméně s jedním kůzletem |
|
|
|
84 dní (je nutno uvést celé období laktace) |
|
Kozy k reprodukci |
Samice kozy, které byly nejméně jednou inseminovány/připuštěny |
Od první inseminace do konce druhého odstavu |
|
|
Dva cykly (pokud se požadují reprodukční parametry) |
6. Tabulka. Kategorie zvířat: ryby
|
Kategorie |
Definice kategorie zvířat |
Přibližná doba trvání (hmotnost/stáří) |
Minimální doba trvání dlouhodobých studií účinností |
||
|
Období |
Stáří |
Hmotnost |
|||
|
Losos a pstruh |
|
|
|
200–300 g |
90 dní nebo do doby, kdy se výchozí tělesná hmotnost zdvojnásobí |
|
Losos a pstruh |
Hejno matečných ryb |
Pokud možno co nejblíže době tření |
|
|
90 dní |
7. Tabulka. Kategorie zvířat: králíci
|
Kategorie |
Definice kategorie zvířat |
Přibližná doba trvání (hmotnost/stáří) |
Minimální doba trvání dlouhodobých studií účinností |
||
|
Období |
Stáří |
Hmotnost |
|||
|
Sající a odstavení králíci |
|
Počínaje od jednoho týdne po narození |
|
|
56 dní |
|
Výkrm králíků |
Králíci chovaní pro masnou produkci |
Po odstavu |
Až do 8–11 týdnů |
|
42 dní |
|
Chovné králice (pro reprodukci) |
Králice, které byly nejméně jednou inseminovány/připuštěny |
Od inseminace do konce druhého odstavu |
|
|
Dva cykly (pokud se požadují reprodukční parametry) |
|
Chovné králice určené k produkci mladých králíků |
Králice, které byly nejméně jednou inseminovány |
Od první inseminace |
|
|
Nejméně 2 týdny před porodem do konce doby odstavu (např. pro produkt mikroorganismů) |
8. Tabulka. Kategorie zvířat: koně
|
Kategorie |
Definice kategorie zvířat |
Přibližná doba trvání (hmotnost/stáří) |
Minimální doba trvání dlouhodobých studií účinností |
||
|
Období |
Stáří |
Hmotnost |
|||
|
Koně |
Všechny kategorie |
|
|
|
56 dní |