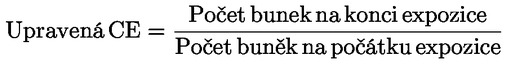|
(8)
|
V části B se doplňují tyto kapitoly:
„B.63 SCREENINGOVÁ ZKOUŠKA NA REPRODUKČNÍ/VÝVOJOVOU TOXICITU
ÚVOD
|
1.
|
Tato zkušební metoda je rovnocenná Pokynu OECD pro zkoušení (TG) č. 421 (2016). Pokyny OECD pro zkoušení chemických látek se pravidelně přezkoumávají s ohledem na vědecký pokrok. Pokyn č. 421 k původní screeningové zkoušce byl přijat v roce 1995 na základě protokolu pro „Předběžnou screeningovou zkoušku na reprodukční toxicitu“ projednaného na dvou zasedáních odborníků, a to v Londýně v roce 1990 (1) a v Tokiu v roce 1992 (2).
|
|
2.
|
Tato zkušební metoda byla aktualizována o příslušné sledované vlastnosti pro endokrinní disruptory v návaznosti na činnost s vysokou prioritou, kterou organizace OECD zahájila v roce 1998 a která byla zaměřena na revizi stávajících pokynů ke zkoušení a vypracování nových pokynů pro zjišťování a zkoušení látek, které mohou narušovat činnost endokrinních žláz (3). Například do Pokynu OECD pro zkoušení č. 407 (Studie orální toxicity u hlodavců – 28denní opakovaná aplikace, kapitola B.7 této přílohy) byly v roce 2008 doplněny parametry vhodné k detekci endokrinního působení zkoušených chemických látek. Cílem aktualizace pokynu č. 421 bylo zahrnout do pokynu ke screeningové zkoušce některé relevantní sledované vlastnosti týkající se endokrinních disruptorů, pokud doby expozice zahrnují některá citlivá období v průběhu vývoje (prenatální nebo časně postnatální období).
|
|
3.
|
Na základě studie proveditelnosti řešící vědecké a technické otázky spojené se zahrnutím relevantních sledovaných vlastností, jakož i možných úprav plánu zkoušky potřebných pro jejich zahrnutí (4) byly do pokynu č. 421 přidány vybrané další relevantní sledované vlastnosti související s endokrinními disruptory, které jsou rovněž součástí pokynu pro zkoušení č. 443 (Rozšířená jednogenerační studie toxicity pro reprodukci, kapitola B.56 této přílohy).
|
|
4.
|
Tato zkušební metoda je určena pro získání omezeného množství informací týkajících se účinků zkoušené chemické látky na reprodukční výkonnost samců a samic, jako jsou funkce pohlavních žláz, chování při páření, početí, vývoj zárodku a vrh. Není alternativou pro stávající zkušební metody B.31, B.34, B.35 nebo B.56 a ani je nahrazuje.
|
VÝCHOZÍ ÚVAHY
|
5.
|
Tuto screeningovou zkušební metodu lze použít na získání prvních informací o možných účincích na reprodukci anebo vývoj, a to v rané fázi posuzování toxikologických vlastností chemických látek, nebo informací o chemických látkách, které jsou předmětem zájmu. Rovněž může být použita jako součást sady úvodních screeningových zkoušek u stávajících chemických látek, o nichž nejsou k dispozici žádné nebo jen omezené toxikologické informace, jako studie ke stanovení dávkování pro rozsáhlejší reprodukční/vývojové studie, nebo když je to jinak považováno za relevantní. Při realizaci studie by měly být respektovány hlavní zásady a aspekty stanovené v pokynu OECD č. 19 o uznávání, posuzování a používání klinických příznaků jako humánních sledovaných vlastností pro pokusná zvířata používaná při hodnocení bezpečnosti (5).
|
|
6.
|
Tato zkušební metoda neposkytuje úplné informace o všech aspektech reprodukce a vývoje. Konkrétně nabízí jen omezené prostředky zjištění postnatálních projevů prenatální expozice nebo účinků, které mohou být vyvolány v průběhu postnatální expozice. Vzhledem (mimo jiné) k relativně malému počtu zvířat ve skupinách dávkování, selektivitě sledovaných vlastností a krátké době trvání studie neposkytuje tato metoda důkaz pro definitivní tvrzení, že nedochází k žádným účinkům. Navíc, pokud neexistují údaje z jiných zkoušek reprodukční/vývojové toxicity, jsou pozitivní výsledky užitečné pro první posouzení rizika a přispívají k rozhodnutím ohledně nezbytnosti a načasování dalších zkoušek.
|
|
7.
|
Výsledky získané pro parametry související s endokrinním působením by se měly interpretovat v kontextu „koncepčního rámce OECD pro zkoušky a hodnocení chemických látek způsobujících endokrinní poruchy“ (6). V tomto koncepčním rámci je vylepšený Pokdyn OECD pro zkoušení č. 421 zařazena na úroveň 4 jako zkouška in vivo poskytující údaje o nežádoucích účincích na relevantní endokrinní sledované vlastnosti. Endokrinní signál sám o sobě však nemusí být považován za dostatečný důkaz, že zkoušená chemická látka je endokrinním disruptorem.
|
|
8.
|
Tato zkušební metoda předpokládá orální aplikaci zkoušené chemické látky. Pokud jsou použity jiné cesty expozice, bude zřejmě nutné provést úpravy.
|
|
9.
|
Před použitím této zkušební metody ke zkoušení směsi s cílem získat údaje pro zamýšlený regulační účel by mělo být zváženo, zda tato metoda může pro tento účel poskytnout odpovídající výsledky, a pokud ano, proč tomu tak je. Takové úvahy nejsou nutné, pokud existuje právní požadavek na zkoušení dané směsi.
|
|
10.
|
Použité definice jsou uvedeny v dodatku 1.
|
PRINCIP ZKOUŠKY
|
11.
|
Zkoušená chemická látka se podává v odstupňovaných dávkách několika skupinám samců a samic. Samcům se podává dávka minimálně čtyři týdny včetně dne před plánovaným usmrcením (zahrnuje to minimálně dva týdny před pářením, v průběhu období páření a přibližně dva týdny po páření). Vzhledem k omezené době podávání před pářením u samců nemusí být fertilita zvláště citlivým ukazatelem toxicity pro varlata. Proto je velmi důležité provést podrobné histologické vyšetření varlat. Kombinace dvoutýdenní doby podávání před pářením a následné pozorování páření/fertility s celkovou dobou podávání alespoň čtyři týdny, po níž následuje podrobná histopatologie samčích gonád, se považuje za dostatečnou ke zjištění většiny účinků na fertilitu a spermatogenezi u samců.
|
|
12.
|
Samicím je třeba podávat zkoušenou chemickou látku po celou dobu studie. Zahrnuje to dva týdny před pářením (s cílem pokrýt alespoň dva celé estrální cykly), variabilní dobu do početí, dobu březosti a alespoň třináct dnů po porodu, a to včetně dne před plánovaným usmrcením.
|
|
13.
|
Délka studie následující po aklimatizaci a zhodnocení estrálního cyklu před zahájením podávání závisí na výkonnosti samice a činí přibližně 63 dnů [alespoň 14 dnů před pářením, (maximálně) 14 dnů páření, 22 dnů gestace, 13 dnů laktace].
|
|
14.
|
V průběhu období podávání se zvířata každý den pečlivě pozorují, aby se zjistily příznaky toxicity. Zvířata, která v průběhu doby zkoušky uhynula nebo byla utracena, se pitvají a na konci zkoušky se přeživší zvířata utratí a rovněž pitvají.
|
POPIS METODY
Výběr zvířecích druhů
|
15.
|
Tato zkušební metoda je určena k použití s potkany. Jestliže se parametry uvedené v této zkušební metodě zkoumají u jiných druhů hlodavců, mělo by se podat podrobné zdůvodnění. V mezinárodním validačním programu detekce endokrinních disruptorů pomocí Pokynu OECD pro zkoušení č. 407 (odpovídající kapitole B.7 této přílohy) byl jediným použitým druhem potkan. Není vhodné používat kmeny s nižší plodností nebo s typicky vysokým výskytem vývojových vad. Používají se zdravá, dosud nepřipuštěná zvířata ještě nepoužitá pro jiné experimenty. Pokusná zvířata by měla být plně charakterizována co do druhu, kmene, pohlaví, hmotnosti a věku. Na začátku studie by měly být odchylky hmotnosti zvířat minimální a neměly by překročit 20 % střední hodnoty pro každé pohlaví. Provádí-li se studie jako předběžná studie pro dlouhodobou nebo celogenerační studii, je vhodné použít v obou studiích nejlépe zvířata stejného kmene a stejného původu.
|
Umístění a krmení zvířat
|
16.
|
Všechny postupy by měly splňovat místní standardy péče o laboratorní zvířata. Teplota v experimentálních zvířecích místnostech by měla být 22o C (± 3 oC). Ačkoli by relativní vlhkost vzduchu měla být minimálně 30 % a pokud možno nepřesáhnout 70 %, kromě doby úklidu místnosti, cílem by měla být hodnota 50–60 %. Osvětlení by mělo být umělé a mělo by se střídat 12 hodin světla a 12 hodin tmy. Ke krmení lze použít konvenční laboratorní krmivo s neomezeným přísunem pitné vody. Výběr potravy může být ovlivněn potřebou zajistit dostatečné promísení s testovanou látkou, je-li látka podávána touto cestou.
|
|
17.
|
Zvířata by měla být chována v malých skupinách stejného pohlaví; je-li to z vědeckého hlediska důvodné, mohou být zvířata chována individuálně. Při chovu zvířat v klecích by v jedné kleci nemělo být umístěno více než pět zvířat. Páření by mělo probíhat v klecích, které jsou k tomuto účelu vhodné. Březí samice je třeba chovat v klecích jednotlivě a poskytnout jim materiál pro vytvoření hnízda. Samice v době laktace se chovají v klecích jednotlivě společně s potomky.
|
|
18.
|
Krmivo by se mělo pravidelně analyzovat, aby se zjistila přítomnost kontaminantů. Vzorek stravy by se měl uchovávat až do dokončení zprávy.
|
Příprava zvířat
|
19.
|
Zdravá mladá dospělá zvířata se náhodným výběrem rozdělí na kontrolní skupinu a skupinu, která se exponuje. Klece by měly být uspořádány tak, aby byl vliv umístění klecí minimalizován. Zvířata se jednoznačně identifikují a před započetím studie se nechají v laboratorních podmínkách alespoň pět dní aklimatizovat.
|
Příprava dávek
|
20.
|
Není-li jiný způsob aplikace považován za vhodnější, doporučuje se orální podávání zkoušené chemické látky. Pokud je zvolena orální cesta, zkoušená chemická látka se obvykle podává žaludeční sondou; alternativně však lze zkoušené chemické látky podávat v potravě nebo v pitné vodě.
|
|
21.
|
Je-li to nutné, je zkoušená chemická látka rozpuštěna nebo suspendována ve vhodném vehikulu. Je-li to možné, doporučuje se zvážit jako první variantu použití vodného roztoku/suspenze, poté použití roztoku/emulze v oleji (např. v kukuřičném oleji) a nakonec eventuální použití roztoku jiného vehikula. U jiného typu vehikula, než je voda, je třeba znát jeho toxické charakteristiky. Měla by být určena stabilita a homogenita zkoušené chemické látky v daném vehikulu.
|
POSTUP
Počet a pohlaví zvířat
|
22.
|
Doporučuje se, aby každá skupina měla zpočátku alespoň 10 samců a 12–13 samic. U samic se hodnotí před expozicí estrální cykly a zvířata, která nevykazují typické 4–5denní cykly, nebudou do studie zařazena; proto se doporučují náhradní samice, aby v každé skupině zůstalo 10 samic. S výjimkou případu výrazně toxických účinků se očekává, že v každé skupině bude alespoň 8 březích samic, což je obvykle minimální přijatelný počet březích samic na skupinu. Cílem je získat dostatečný počet březích samic a potomstva, a tím zajistit smysluplné hodnocení schopnosti zkoušené chemické látky ovlivnit plodnost, březost, chování samice a kojených mláďat a růst a vývoj potomstva generace F1 od doby početí do 13. dne po porodu.
|
Dávkování
|
23.
|
Obecně by měly být použity nejméně tři zkušební skupiny a kontrolní skupina. Úrovně dávek mohou vycházet z údajů ze zkoušek akutní toxicity nebo z výsledků studií po opakované dávce. S výjimkou podávání zkoušené chemické látky se se zvířaty v kontrolní skupině zachází stejně jako se zvířaty ve zkušební skupině. Používá-li se pro usnadnění aplikace zkoušené chemické látky vehikulum, podává se kontrolní skupině vehikulum v nejvyšším použitém objemu.
|
|
24.
|
Při výběru úrovní dávek by se měly vzít v úvahu veškeré existující údaje o toxických a (toxiko)kinetických vlastnostech. Rovněž je třeba vzít v úvahu, že mohou existovat rozdíly v citlivosti mezi březími a nebřezími zvířaty. Nejvyšší úroveň dávky by se měla zvolit tak, aby vyvolala toxické účinky, ne však uhynutí nebo velké utrpení. Poté by se měla zvolit sestupná řada úrovní dávek, aby se prokázala odezva související s dávkou a nepřítomnost nepříznivých účinků (NOAEL) při nejnižší úrovni dávky. Pro nastavení sestupných úrovní dávek jsou obvykle optimální intervaly lišící se faktorem 2 až 4 a často je vhodnější přidání čtvrté zkušební skupiny než používání velkých intervalů mezi jednotlivými dávkami (např. lišících se faktorem vyšším než 10).
|
|
25.
|
V přítomnosti pozorované obecné toxicity (např. snížená tělesná hmotnost, účinky na játra, srdce, plíce nebo ledviny atd.) nebo jiných změn, které nemusí být odezvou na toxicitu (např. snížený příjem krmiva, zvětšení jater), je třeba pozorované účinky na endokrinní citlivé sledované vlastnosti interpretovat obezřetně.
|
Limitní zkouška
|
26.
|
Pokud orální studie s jednou úrovní dávky v množství alespoň 1 000 mg/kg tělesné hmotnosti a den nebo rovnocenné procento látky v potravě nebo pitné vodě při aplikaci dávky v potravě nebo pitné vodě pomocí postupů popsaných pro tuto studii nevyvolá žádnou zjistitelnou toxicitu, a pokud se neočekává ani toxicita na základě existujících údajů o strukturně příbuzných látkách, pak není nutné uvažovat o kompletní studii využívající několika úrovní dávek. Limitní zkouška se použije, pokud expozice u člověka nevyžaduje použít vyšší úroveň orální dávky. U dalších způsobů podávání látky, jako je inhalační nebo kožní aplikace, může být maximální dosažitelná úroveň koncentrace často dána fyzikálně-chemickými vlastnostmi zkoušených chemických látek.
|
Podávání dávek
|
27.
|
Zvířatům se denně podává zkoušená chemická látka po dobu sedmi dnů v rámci jednoho týdne. Pokud se zkoušená chemická látka podává nitrožaludečně, podává se zvířatům v jediné dávce za použití žaludeční sondy nebo vhodné intubační kanyly. Maximální objem tekutiny, kterou lze podat najednou, závisí na velikosti pokusného zvířete. Tento objem by neměl překročit 1 ml na 100 g tělesné hmotnosti, s výjimkou vodných roztoků, kde lze použít 2 ml na 100 g tělesné hmotnosti. S výjimkou dráždivých a žíravých zkoušených chemických látek, které obvykle při vyšších koncentracích vykazují zesílené účinky, by měla být variabilita zkušebního objemu minimalizována nastavením koncentrace zajišťující konstantní objem při všech úrovních dávky.
|
|
28.
|
U zkoušených chemických látek podávaných v potravě nebo v pitné vodě je důležité zajistit, aby množství použité zkoušené chemické látky neovlivňovalo normální výživu nebo vodní rovnováhu. Podává-li se zkoušená chemická látka v krmivu, může se použít buď konstantní koncentrace (v ppm), nebo konstantní dávkování vzhledem k tělesné hmotnosti zvířete; použitá možnost by měla být specifikována. Zkoušená chemická látka podávaná žaludeční sondou se podává každý den přibližně ve stejnou dobu a alespoň jednou týdně se přizpůsobí tak, aby se udržela konstantní úroveň dávky vzhledem k tělesné hmotnosti zvířete.
|
Harmonogram zkoušky
|
29.
|
Podávání oběma pohlavím by mělo být zahájeno alespoň 2 týdny před pářením po minimálně pětidenní aklimatizaci a po screeningu samic, zda mají normální estrální cykly (v dvoutýdenním období před expozicí). Studii je třeba naplánovat tak, aby hodnocení estrálních cyklů začalo brzy poté, co zvířata dosáhnou plné pohlavní zralosti. U různých kmenů potkana v různých laboratořích se to může mírně lišit, např. potkan Sprague Dawley stáří 10 týdnů, potkan Wistar stáří přibližně 12 týdnů. Samice s potomstvem by měly být utraceny 13. den po porodu nebo krátce poté. Den porodu (tj. ten, kdy je dokončen vrh) je definován jako 0. den po porodu. Samice, které nevykazují žádné známky kopulace, se utratí 24–26 dnů po posledním dni období páření. S dávkováním se u obou pohlaví pokračuje i během období páření. U samců se dále pokračuje s podáváním zkoušené chemické látky i po období páření alespoň do doby, kdy je dokončeno minimální období podávání 28 dnů. Poté se utratí, nebo se případně uchovají a dále je jim podávána zkoušená chemická látka pro případné provedení druhého páření, je-li to považováno za vhodné.
|
|
30.
|
V denním podávání samicím rodičovské generace by se poté mělo pokračovat po celou březost a alespoň do 13. dne po porodu včetně nebo do dne před utracením. U studií, kde je zkoušená chemická látka podávána inhalací nebo dermální cestou, je třeba s dávkováním pokračovat alespoň do 19. dne gestace včetně, přičemž dávkování se obnoví co možná nejdříve, nejpozději však v PND 4.
|
|
31.
|
Diagram časového sledu zkoušky je uveden v dodatku 2.
|
Páření
|
32.
|
Obvykle se v této studii používá párování 1:1 (jeden samec a jedna samice). V případě náhodného úmrtí samců mohou nastat výjimky. Samice se umístí se stejným samcem až do doby, kdy budou pozorovány důkazy o spáření, nebo do uplynutí dvou týdnů. Každé ráno se samice vyšetří na přítomnost spermatu nebo vaginální zátky. Den 0 březosti je definován jako den, kdy je potvrzen důkaz o spáření (je nalezena vaginální zátka nebo sperma). V případě, že je páření neúspěšné, se zváží spáření se samci s osvědčenou plodností ze stejné skupiny.
|
Velikost vrhu
|
33.
|
Čtvrtý den po vrhu se velikost všech vrhů upraví náhodným vyřazením nadbytečných jedinců tak, aby bylo pokud možno dosaženo počtu čtyř až pěti samců a čtyř až pěti samic z jednoho vrhu podle obvyklé početnosti vrhu u použitého kmene potkana. Od dvou nadbytečných mláďat se odeberou krevní vzorky, smíchají se a použijí pro stanovení sérových hladin T4. Selektivní eliminace jedinců, např. na základě tělesné hmotnosti nebo anogenitální vzdálenosti (AGD), není vhodná. Pokud počet mladých samců nebo samic neumožňuje ponechat čtyři až pět od každého pohlaví a vrhu, je možná částečná úprava (např. ponechat šest samců a čtyři samice). Jestliže velikost vrhu poklesne pod cílový počet pro utracení (8 nebo 10 mláďat/vrh), nevyřazují se žádná mláďata. Jestliže zbývá pouze jedno mládě nad cílový počet pro utracení, vyřadí se pouze jedno mládě, které se použije pro odběr krve pro případné vyšetření sérové hladiny T4.
|
|
34.
|
Jestliže se velikost trhu neupravuje, 4. den po porodu se utratí dvě mláďata na vrh a odeberou se krevní vzorky pro stanovení sérové koncentrace hormonů štítné žlázy. Těmito dvěma mláďaty na vrh by měly být pokud možno samice, aby se uchovali samci pro hodnocení retenčních mléčných cyst, s výjimkou případu, že by po odstranění těchto mláďat nezbyly žádné samice pro vyšetření při ukončení zkoušky. Jestliže velikost vrhu poklesne pod 8 nebo 10 mláďat/vrh (podle normální velikosti vrhu u použitého kmene potkana), nevyřazují se žádná mláďata. Jestliže zbývá pouze jedno mládě nad normální velikost vrhu, vyřadí se pouze jedno mládě, které se použije pro odběr krve pro případné vyšetření sérové hladiny T4.
|
Pozorování živých zvířat
Poznatky z klinického pozorování
|
35.
|
Po celou dobu zkoušky by se mělo provádět všeobecné klinické pozorování nejméně jednou denně a častěji, pokud jsou zpozorovány příznaky toxicity. Pozorování se provádí denně, pokud možno ve stejnou dobu a s přihlédnutím k době očekávaného maxima účinku po podání látky. Zaznamenávají se významné změny chování, příznaky ztíženého nebo prodlouženého vrhu a všechny příznaky toxicity, včetně mortality. Tyto záznamy by měly zahrnovat čas nástupu, stupeň a délku trvání známek toxicity.
|
Tělesná hmotnost a spotřeba potravy a vody
|
36.
|
Samci a samice se váží první den podávání, poté týdně a při ukončení zkoušky. V průběhu březosti se samice zváží 0., 7., 14. a 20. den a do 24 hodin po vrhu (0. den nebo 1. den po porodu) a alespoň 4. den a 13. den po porodu. Tato pozorování se zaznamenají individuálně u každého dospělého zvířete.
|
|
37.
|
Před obdobím páření a během březosti a laktace se alespoň jednou týdně měří příjem potravy. Měření příjmu potravy v průběhu páření je volitelné. Pokud se zkoušená chemická látka podává v pitné vodě, měří se v průběhu těchto období také spotřeba vody.
|
Estrální cykly
|
38.
|
Estrální cykly je třeba sledovat před zahájením expozice, aby bylo možné vybrat pro studii samice s pravidelným cyklem (viz odstavec 22). Od začátku období expozice až do průkazu o páření je třeba také každý den sledovat vaginální stěry. V případě obav z účinků akutního stresu, které by při zahájení podávání zkoušené chemické látky mohly změnit estrální cykly, mohou laboratoře exponovat pokusná zvířata po dobu dvou týdnů, poté denně odebírat vaginální stěry za účelem sledování estrálního cyklu po dobu minimálně dvou týdnů před obdobím páření a dále je sledovat až do období páření, dokud prokazatelně nedojde k páření. Při získávání vaginálních/cervikálních buněk je třeba dávat pozor, aby se neporušila sliznice, což by mohlo vyvolat stav falešné březosti (7) (8).
|
Parametry potomstva
|
39.
|
Doba gestace se zaznamená a počítá se ode dne 0 březosti. Každý vrh se co nejdříve po porodu vyšetří a stanoví se počty a pohlaví mláďat, počet mrtvě a živě narozených mláďat, počet zakrslých mláďat (mláďat, která jsou výrazně menší než odpovídající kontrolní mláďata) a přítomnost nápadných abnormalit.
|
|
40.
|
Do 24 hodin po porodu (0. den nebo 1. den po porodu) a alespoň 4. den a 13. den po porodu se živá mláďata spočítají, stanoví se jejich pohlaví a zváží se. Kromě pozorování popsaných v odstavci 35 je třeba zaznamenat jakékoli abnormální chování potomstva.
|
|
41.
|
U každého mláděte je třeba ve stejný den po porodu v době od PND 0 do PND 4 změřit anogenitální vzdálenost (AGD). V den měření AGD by měla být zjištěna tělesná hmotnost mláděte a AGD by měla být normalizována na nějakou míru velikosti mláďat, nejlépe třetí odmocninu tělesné hmotnosti (9). Podle doporučení pokynu OECD č. 151 se v PND 12 nebo 13 spočítá počet bradavek/prsních dvorců u mladých samců (10).
|
Biochemické vyšetření
|
42.
|
Krevní vzorky z určeného místa se odebírají podle tohoto rozvrhu:
|
—
|
od alespoň dvou mláďat na vrh 4. den po porodu, pokud to umožňuje počet mláďat (viz odstavce 33-34)
|
|
—
|
od všech samic a alespoň dvou mláďat na vrh při ukončení zkoušky 13. den a
|
|
—
|
od všech dospělých samců při ukončení zkoušky.
|
|
Všechny krevní vzorky se uchovávají za vhodných podmínek. Krevní vzorky odebrané mláďatům 13. den a dospělým samcům se vyšetří na sérové hladiny hormonů štítné žlázy (T4). Ve vhodných případech se provede další vyšetření T4 v krevních vzorcích odebraných samicím a mláďatům 4. den. Je-li to vhodné, mohou být volitelně stanoveny také hladiny jiných hormonů: Krev mláďat z vrhu lze pro potřeby analýzy hormonů štítné žlázy smíchat. Hormony štítné žlázy (T4 a TSH) se měří nejlépe jako „celkové“.
|
43.
|
Následující faktory mohou ovlivnit variabilitu a absolutní koncentrace stanovovaných hormonů:
|
—
|
doba utracení vzhledem k diurnálním odchylkám v koncentracích hormonů,
|
|
—
|
způsob utracení – mělo by se zabránit zbytečnému stresu zvířat, který může ovlivnit koncentrace hormonů,
|
|
—
|
testovací soupravy pro stanovení hormonů, jejichž standardní křivky se mohou lišit.
|
|
|
44.
|
Vzorky plazmy určené speciálně k stanovení hormonů by se měly odebírat zhruba ve stejnou denní dobu. Číselné hodnoty získané při analýze koncentrací hormonů se u různých komerčních testovacích souprav liší.
|
Patologie
Celková pitva
|
45.
|
Dospělá zvířata se v okamžiku usmrcení nebo úhynu během studie makroskopicky vyšetří na abnormality nebo patologické změny. Speciální pozornost je třeba věnovat orgánům rozmnožovací soustavy. Zaznamená se počet implantací. Ráno v den pitvy se vyšetří vaginální stěry s cílem určit stadium estrálního cyklu a umožnit korelaci s histopatologickým vyšetřením vaječníků.
|
|
46.
|
Varlata, nadvarlata a prostata a semenné váčky s Cowperovými žlázami jako celek se u všech dospělých samců podle potřeby zbaví všech přirostlých tkání a co nejdříve po pitvě se ve vlhkém stavu zváží, aby nedošlo k vyschnutí. Kromě toho mohou být volitelně zváženy další orgány, jako jsou svaly m. levator ani a m. bulbocavernosus, Cowperovy žlázy a žalud penisu u samců a párové vaječníky (vlhká hmotnost) a děloha (včetně děložního hrdla) u samic; je-li toto vážení prováděno, mělo by k němu dojít co nejdříve po pitvě.
|
|
47.
|
Mrtvá mláďata a mláďata utracená 13. den po porodu nebo krátce poté je třeba alespoň pečlivě externě vyšetřit na závažné abnormality. Zvláštní pozornost je třeba věnovat vnějším rozmnožovacím orgánům, které se vyšetří na přítomnost známek změněného vývoje. 13. den se uchová štítná žláza od 1 mladého samce a 1 mladé samice z každého vrhu.
|
|
48.
|
Vaječníky a varlata, přídatné pohlavní orgány (děloha a děložní hrdlo, nadvarlata, prostata, semenné váčky s Cowperovými žlázami), štítná žláza a všechny orgány od všech dospělých zvířat vykazující makroskopické léze se uchovají. Pro rutinní vyšetření varlat a nadvarlat se nedoporučuje fixace ve formalínu. Přípustnou metodou pro tyto tkáně je použití Bouinova fixačního roztoku nebo modifikovaného Davidsonova fixačního roztoku (11). Tunica albuginea se může jemně a mělce propíchnout jehlou na obou koncích orgánu, aby se umožnilo rychlé proniknutí fixativa.
|
Histopatologie
|
49.
|
Podrobné histologické vyšetření je třeba provést u vaječníků, varlat a nadvarlat (se zvláštním důrazem na stádia spermatogeneze a histopatologii struktury intersticiálních buněk varlat) zvířat ve skupině s nejvyšší dávkou a v kontrolní skupině. V případě potřeby mohou být vyšetřeny i další uchované orgány včetně štítné žlázy od mláďat a dospělých zvířat. Hmotnost štítné žlázy lze stanovit po fixaci. Odříznutí by se mělo rovněž provádět velmi opatrně a teprve po fixaci, aby se zamezilo poškození tkáně. Poškození tkáně by mohlo narušit histopatologickou analýzu. Vyšetření by měla být provedena také u zvířat ostatních dávkových skupin, pokud jsou u skupiny s nejvyšší dávkou pozorovány změny. Pokyny k histopatologickému vyšetření (11) podrobně popisují pitvu, fixaci, tvorbu řezů a histopatologické vyšetření endokrinních tkání.
|
ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV
Údaje
|
50.
|
Měly by být uvedeny údaje pro každé jednotlivé zvíře. Dále se všechny údaje shrnou do tabulek tak, aby byl u každé experimentální skupiny uveden počet zvířat na začátku zkoušky, počet zvířat uhynulých během zkoušky nebo utracených z humánních důvodů, doba úhynu nebo humánního utracení, počet plodných samic, počet březích samic, počet zvířat vykazujících příznaky toxicity, popis pozorovaných příznaků toxicity včetně doby nástupu, trvání a závažnosti veškerých toxických účinků, druhy histopatologických změn a všechny významné údaje o vrhu. Souhrnná zpráva ve formě tabulky, která se ukázala jako velmi užitečná pro hodnocení reprodukčního/vývojového účinku, je uvedena v dodatku 3.
|
|
51.
|
Vzhledem k omezené velikosti studie mají statistické analýzy ve formě testů „významnosti“ pro mnoho sledovaných vlastností, zejména pro reprodukční sledované vlastnosti, omezenou hodnotu. Pokud se používají statistické analýzy, pak by měla být zvolená metoda vhodná pro distribuci zkoumané proměnné a měla by být zvolena před zahájením studie. Statistická analýza AGD a retenčních mléčných cyst by měla vycházet z údajů o jednotlivých mláďatech se zohledněním účinků na vrh. Ve vhodných případech je jednotkou analýzy vrh. Statistická analýza tělesné hmotnosti mláďat by měla vycházet z údajů o jednotlivých mláďatech se zohledněním velikosti vrhu. Vzhledem k malé velikosti skupin může být jako pomůcka pro interpretaci studie také užitečné použití historických kontrolních údajů (např. o velikosti vrhu), pokud jsou tyto údaje k dispozici.
|
Vyhodnocení výsledků
|
52.
|
Výsledky této studie reprodukční toxicity se vyhodnotí z hlediska pozorovaných účinků, pitvy a mikroskopických nálezů. V hodnocení musí být uveden vztah mezi dávkou zkoušené chemické látky a přítomností nebo nepřítomností, výskytem a závažností abnormalit včetně makroskopických lézí, stanovených cílových orgánů, neplodnosti, klinických abnormalit, vlivu na reprodukční výkonnost a schopnost vrhu, změny tělesné hmotnosti, vlivu na úmrtnost a další toxické účinky.
|
|
53.
|
Vzhledem ke krátké době expozice samců je třeba histopatologii varlat a nadvarlat zvažovat při posuzování reprodukčních účinků u samců společně s údaji o fertilitě. Jako pomůcka pro interpretaci studie může být také užitečné použití historických kontrolních údajů o reprodukci/vývoji (např. o velikosti vrhu, AGD, retenčních mléčných cystách, sérových hladinách T4), pokud jsou tyto údaje k dispozici.
|
|
54.
|
Za účelem kontroly jakosti se navrhuje, aby se shromáždily historické kontrolní údaje a vypočítaly se koeficienty rozptylu pro číselné údaje, zejména pro parametry spojené s detekcí endokrinních disruptorů. Tyto údaje pak lze použít pro účely srovnání při hodnocení stávajících studií.
|
ZÁVĚREČNÁ ZPRÁVA
|
55.
|
Závěrečná zpráva by měla obsahovat tyto informace:
|
|
Zkoušená chemická látka:
|
—
|
zdroj, číslo šarže a je-li k dispozici, datum použitelnosti,
|
|
—
|
stabilita zkoušené chemické látky, je-li známa.
|
|
|
|
Jednosložková látka:
|
—
|
fyzický vzhled, rozpustnost ve vodě a další relevantní fyzikálně-chemické vlastnosti,
|
|
—
|
chemická identifikace, např. název podle IUPAC nebo CAS, číslo CAS, kód SMILES nebo InChI, strukturní vzorec, čistota, případně chemická identita nečistot, je-li to prakticky možné, atd.
|
|
|
|
Vícesložkové látky, UVCB a směsi:
|
—
|
charakterizované pokud možno chemickou identitou (viz výše), kvantitativním výskytem a relevantními fyzikálně-chemickými vlastnostmi složek.
|
|
|
|
Vehikulum (je-li použito):
|
—
|
zdůvodnění výběru vehikula, pokud není použita voda.
|
|
|
|
Pokusná zvířata:
|
—
|
počet, věk a pohlaví zvířat,
|
|
—
|
původ, podmínky chovu, strava atd.,
|
|
—
|
hmotnost jednotlivých zvířat na začátku zkoušky.
|
|
—
|
zdůvodnění použití druhu, pokud je jiný než potkan.
|
|
|
|
Zkušební podmínky:
|
—
|
zdůvodnění zvolených úrovní dávek,
|
|
—
|
podrobnosti o složení zkoušené chemické látky/úpravě potravy, o použitých koncentracích, stálosti a homogenitě přípravku,
|
|
—
|
podrobné údaje o podávání zkoušené chemické látky,
|
|
—
|
popřípadě přepočet koncentrace zkoušené chemické látky v krmivu/pitné vodě (ppm) na skutečnou denní dávku (mg/kg tělesné hmotnosti/den),
|
|
—
|
podrobnosti o jakosti krmení a vody,
|
|
—
|
podrobný popis postupů randomizace při výběru mláďat pro třídění, jsou-li tříděna.
|
|
|
|
Výsledky:
|
—
|
tělesná hmotnost/změny tělesné hmotnosti,
|
|
—
|
spotřeba potravy a popřípadě spotřeba vody, je-li údaj k dispozici,
|
|
—
|
údaje o toxických reakcích podle pohlaví a dávky, včetně ukazatelů plodnosti, gestace a všech dalších známek toxicity,
|
|
—
|
toxické nebo jiné účinky na reprodukci, potomstvo, postnatální růst atd.,
|
|
—
|
povaha, závažnost a trvání klinických projevů (ať vratných, či nevratných),
|
|
—
|
počet dospělých samic s normálním nebo abnormálním estrálním cyklem a trváním cyklu,
|
|
—
|
počet živě narozených mláďat a postimplantačních ztrát,
|
|
—
|
údaje o tělesné hmotnosti mláďat,
|
|
—
|
AGD všech mláďat (a tělesná hmotnost v den měření AGD),
|
|
—
|
retenční mléčné cysty u mláďat samců,
|
|
—
|
hladiny hormonů štítné žlázy u 13denních mláďat a dospělých samců (a u samic a 4denních mláďat, jsou-li tyto hodnoty měřeny),
|
|
—
|
počet mláďat s makroskopicky viditelnými abnormalitami, orientační hodnocení vnějších genitálií, počet zakrslých mláďat,
|
|
—
|
den úhynu během studie nebo údaj, že zvířata přežila až do dne utracení,
|
|
—
|
počet implantovaných plodů, velikost vrhu a hmotnost vrhu v době pořízení záznamu,
|
|
—
|
tělesná hmotnost při utracení a údaje o hmotnosti orgánů u rodičovských zvířat,
|
|
—
|
podrobný popis histopatologických nálezů,
|
|
—
|
údaje o absorpci (jsou-li k dispozici),
|
|
—
|
statistické vyhodnocení výsledků, je-li to možné.
|
|
Rozbor výsledků.
Závěry.
|
Interpretace výsledků
|
56.
|
Studie poskytne hodnocení reprodukční/vývojové toxicity spojené s podáváním opakovaných dávek (viz odstavce 5 a 6). Mohla by naznačit nutnost provést další výzkum a poskytnout vodítko pro návrh dalších studií. Jako pomůcka při interpretaci výsledků v oblasti reprodukce a vývoje by měl posloužit pokyn OECD č. 43 (12). Pokyn OECD č. 106 týkající se histologického hodnocení endokrinních a reprodukčních zkoušek u hlodavců (11) poskytuje informace o přípravě a hodnocení (endokrinních) orgánů a vaginálních stěrů, což může být pro tento pokyn užitečné.
|
LITERATURA
|
(1)
|
OECD (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. K dispozici na vyžádání u Organizace pro hospodářskou spolupráci a rozvoj, Paříž.
|
|
(2)
|
OECD (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokyo, 27th-29th October, 1992. K dispozici na vyžádání u Organizace pro hospodářskou spolupráci a rozvoj, Paříž.
|
|
(3)
|
OECD (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10th-11th March 1998. K dispozici na vyžádání u Organizace pro hospodářskou spolupráci a rozvoj, Paříž.
|
|
(4)
|
OECD (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environment, Health and Safety Publications, Series on Testing and Assessment, (No 217) Organisation for Economic Cooperation and Development, Paris.
|
|
(5)
|
OECD (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations. Series on Testing and Assessment, (No 19), Organisation for Economic Cooperation and Development,.Paris.
|
|
(6)
|
OECD (2011). Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment(No 150), Organisation for Economic Cooperation and Development, Paris.
|
|
(7)
|
Goldman, J.M., Murr A.S., Buckalew A.R., Ferrell J.M. and Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, Part B, 80 (2), 84-97.
|
|
(8)
|
Sadleir R.M.F.S (1979). Cycles and Seasons, in Auston C.R. and Short R.V. (eds.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York.
|
|
(9)
|
Gallavan R.H. Jr, Holson J.F., Stump D.G., Knapp J.F. and Reynolds V.L. (1999). Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights, Reproductive Toxicology, 13: 383-390.
|
|
(10)
|
OECD (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environment, Health and Safety Publications, Series on Testing and Assessment (No 151), Organisation for Economic Cooperation and Development, Paris.
|
|
(11)
|
OECD (2009). Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment (No106), Organisation for Economic Cooperation and Development, Paris.
|
|
(12)
|
OECD (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 43), Organisation for Economic Cooperation and Development, Paris.
|
Dodatek 1
DEFINICE (VIZ TAKÉ POKYNY OECD Č. 150 (6))
Androgenicita je schopnost chemické látky působit v organismu savců jako přirozený androgenní hormon (např. testosteron).
Antiandrogenicita je schopnost chemické látky potlačit v organismu savců působení přirozeného androgenního hormonu (např. testosteronu).
Antiestrogenicita je schopnost chemické látky potlačit v organismu savců působení přirozeného estrogenního hormonu (např. estradiolu 17ß).
Antityreoidální působení je schopnost chemické látky potlačit v organismu savců působení přirozeného hormonu štítné žlázy (např. T3).
Chemická látka je látka nebo směs.
Vývojová toxicita: projev reprodukční toxicity představující prenatální, perinatální, postnatální, strukturální nebo funkční poruchy u potomstva.
Dávkováním se rozumí obecný termín zahrnující dávku, četnost a trvání doby, po kterou je látka podávána.
Dávkou se rozumí množství podané zkoušené chemické látky. Dávka se vyjadřuje jako hmotnost zkoušené chemické látky na jednotku tělesné hmotnosti zvířete za den (např. mg/kg tělesné hmotnosti/den) nebo jako konstantní koncentrace v krmivu.
Zřejmá toxicita je obecný termín popisující jasné příznaky toxicity po podání zkoušené chemické látky. Ty by měly být dostačující pro posouzení nebezpečnosti a měly by být takové, že při zvýšení podávané dávky lze očekávat rozvoj závažných toxických příznaků a pravděpodobnou mortalitu.
Porucha plodnosti představuje poruchy samčích nebo samičích reprodukčních funkcí nebo schopnosti.
Mateřská toxicita: nežádoucí účinky na březí samice vyskytující se buď specificky (přímý účinek), nebo nespecificky (nepřímý účinek).
NOAEL je zkratka pro angl. „no-observed-adverse-effect level“. Je to nejvyšší úroveň dávky, při které nejsou pozorovány žádné nepříznivé účinky spojené se zkouškou.
Estrogenicita je schopnost chemické látky působit v organismu savců jako přirozený estrogenní hormon (např. estradiol 17ß).
Reprodukční toxicita představuje škodlivé účinky na potomstvo a/nebo poruchy samčích nebo samičích reprodukčních funkcí nebo schopnosti.
Zkoušená chemická látka je jakákoli látka nebo směs zkoušená touto zkušební metodou.
Tyreoidální působení je schopnost chemické látky působit jako přirozený hormon štítné žlázy (např. T3) v savčím organismu.
Validace je vědecký proces, jehož cílem je charakterizovat provozní požadavky a provozní omezení zkušební metody a prokázat její spolehlivost a relevantnost pro určitý konkrétní účel.
Dodatek 2
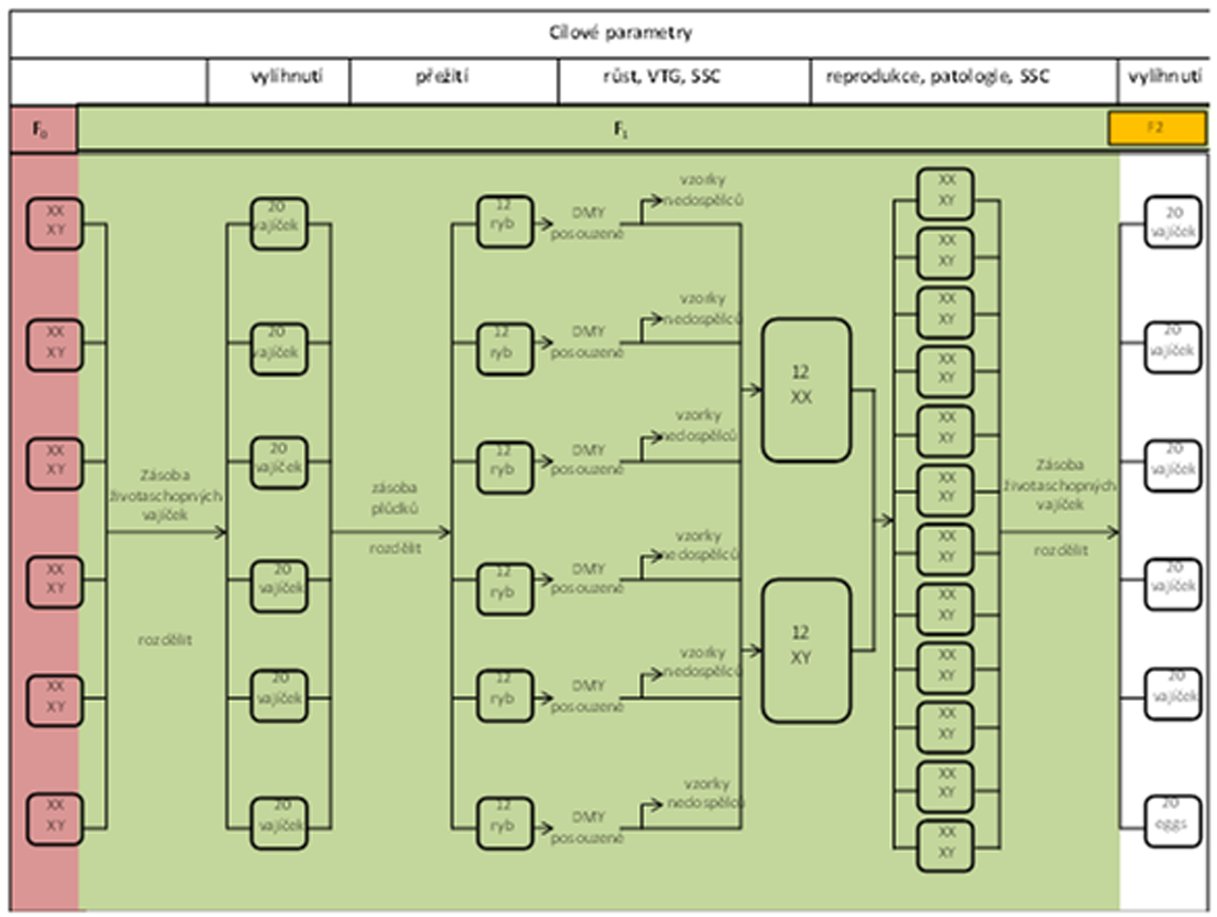
DIAGRAM ČASOVÉHO PLÁNU EXPERIMENTU SE ZNÁZORNĚNÍM MAXIMÁLNÍ DÉLKY STUDIE, NA ZÁKLADĚ CELÉHO ČTRNÁCTIDENNÍHO OBDOBÍ PÁŘENÍ
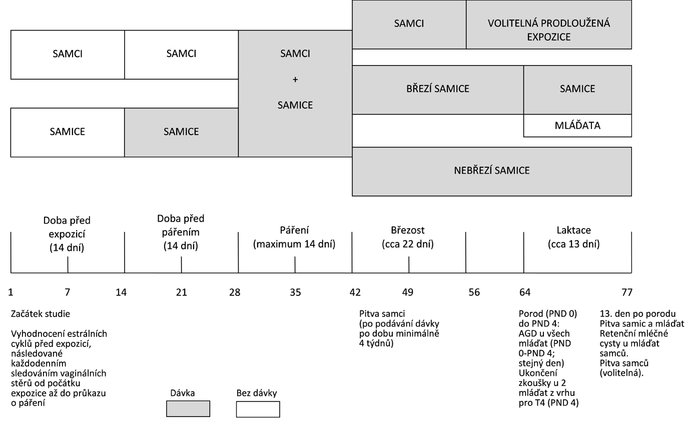
Dodatek 3
SOUHRNNÁ ZPRÁVA O ÚČINCÍCH NA REPRODUKCI/VÝVOJ VE FORMĚ TABULKY
|
POZOROVÁNÍ
|
HODNOTY
|
|
|
|
Dávka (jednotky)
|
0 (kontrola)
|
...
|
...
|
...
|
...
|
|
Páry na začátku (N)
|
|
|
|
|
|
|
Estrální cyklus (alespoň střední délka a frekvence nepravidelných cyklů)
|
|
|
|
|
|
|
Samice vykazující známky kopulace (N)
|
|
|
|
|
|
|
Zabřezlé samice (N)
|
|
|
|
|
|
|
Dny zabřeznutí 1–5 (N)
|
|
|
|
|
|
|
Dny zabřeznutí 6–... (21) (N)
|
|
|
|
|
|
|
Březost ≤ 21 dnů (N)
|
|
|
|
|
|
|
Březost = 22 dnů (N)
|
|
|
|
|
|
|
Březost ≥ 23 dnů (N)
|
|
|
|
|
|
|
Samice s živými novorozenými mláďaty (N)
|
|
|
|
|
|
|
Samice s živými novorozenými mláďaty 4. den po porodu (N)
|
|
|
|
|
|
|
Implantace/samice (průměr)
|
|
|
|
|
|
|
Živá mláďata/samice při narození (průměr)
|
|
|
|
|
|
|
Živá mláďata/samice 4. den (průměr)
|
|
|
|
|
|
|
Poměr pohlaví (samec/samice) při narození (průměr)
|
|
|
|
|
|
|
Poměr pohlaví (samec/samice) 4. den (průměr)
|
|
|
|
|
|
|
Hmotnost vrhu při narození (průměr)
|
|
|
|
|
|
|
Hmotnost vrhu 4. den (průměr)
|
|
|
|
|
|
|
Hmotnost mláďat při narození (průměr)
|
|
|
|
|
|
|
Hmotnost mláďat v době měření AGD (průměr samců, průměr samic)
|
|
|
|
|
|
|
AGD mláďat ve stejný postnatální den, při narození – 4. den (průměr samců, průměr samic, uvést PND)
|
|
|
|
|
|
|
Hmotnost mláďat 4. den (průměr)
|
|
|
|
|
|
|
Retenční mléčné cysty u mladých samců 13. den (průměr)
|
|
|
|
|
|
|
Hmotnost mláďat 13. den (průměr)
|
|
|
|
|
|
|
|
|
ABNORMÁLNÍ MLÁĎATA
|
|
Samice s 0
|
|
|
|
|
|
|
Samice s 1
|
|
|
|
|
|
|
Samice s ≥ 2
|
|
|
|
|
|
|
|
|
ZTRÁTA POTOMSTVA
|
|
|
|
Prenatální/postimplantační (implantace minus živě narozená mláďata)
|
|
Samice s 0
|
|
|
|
|
|
|
Samice s 1
|
|
|
|
|
|
|
Samice s 2
|
|
|
|
|
|
|
Samice s ≥ 3
|
|
|
|
|
|
|
|
|
Postnatální (živě narozená mláďata minus živá mláďata 13. den po porodu)
|
|
Samice s 0
|
|
|
|
|
|
|
Samice s 1
|
|
|
|
|
|
|
Samice s 2
|
|
|
|
|
|
|
Samice s ≥ 3
|
|
|
|
|
|
B.64 KOMBINOVANÁ STUDIE TOXICITY PO OPAKOVANÝCH DÁVKÁCH SE SCREENINGOVOU ZKOUŠKOU NA REPRODUKČNÍ/VÝVOJOVOU TOXICITU
ÚVOD
|
1.
|
Tato zkušební metoda je rovnocenná Pokynu OECD pro zkoušení č. 422 (2016). Pokyny OECD pro zkoušení chemických látek se pravidelně přezkoumávají s ohledem na vědecký pokrok. Pokyn č. 422 k původní screeningové zkoušce byl přijat v roce 1996 na základě protokolu pro „Kombinovanou screeningovou zkoušku na reprodukční/vývojovou toxicitu po opakovaných dávkách“ projednaného na dvou zasedáních odborníků, a to v Londýně v roce 1990 (1) a v Tokiu v roce 1992 (2).
|
|
2.
|
Tato zkušební metoda kombinuje screeningovou část na reprodukční/vývojovou toxicitu, která vychází ze zkušeností získaných v členských zemích při používání původní metody u stávajících chemických látek vyráběných ve velkém objemu a při průzkumném testování látek pozitivní kontroly (3) (4), a část zkoušení toxicity po opakovaných dávkách v souladu s Pokynem OECD pro zkoušení č. 407 (Studie orální toxicity u hlodavců s opakovaným podáváním dávky v 28denním cyklu, odpovídající kapitole B.7 této přílohy).
|
|
3.
|
Tato zkušební metoda byla aktualizována o příslušné sledované vlastnosti endokrinnch disruptorů v návaznosti na činnost s vysokou prioritou, kterou organizace OECD zahájila v roce 1998 a která byla zaměřena na revizi stávajících pokynů pro zkoušení a vypracování nových pokynů pro zjišťování a zkoušení látek, které mohou narušovat činnost endokrinních žláz (5). V této souvislosti byly do pokynu č. 407 (odpovídajícího kapitole B.7 této přílohy) v roce 2008 doplněny parametry vhodné k detekci endokrinního působení zkoušených chemických látek. Cílem aktualizace pokynu č. 422 bylo zahrnout do pokynu ke screeningové zkoušce některé relevantní sledované vlastnosti endokrinních disruptorů, pokud doby expozice zahrnují některá citlivá období v průběhu vývoje (prenatální nebo časně postnatální období).
|
|
4.
|
Na základě studie proveditelnosti řešící vědecké a technické otázky spojené se zahrnutím relevantních sledovaných vlastností, jakož i možných úprav plánu zkoušky potřebných pro jejich zahrnutí (6) byly do pokynu č. 422 přidány vybrané další relevantní sledované vlastnosti související s endokrinními disruptory, které jsou rovněž součástí pokynu pro zkoušení č. 443 (Rozšířená jednogenerační studie toxicity pro reprodukci, odpovídající kapitole B.56 této přílohy).
|
|
5.
|
Tato zkušební metoda je určena pro získání omezeného množství informací týkajících se účinků zkoušené chemické látky na reprodukční výkonnost samců a samic, jako je funkce pohlavních žláz, chování při páření, početí, vývoj zárodku a vrh. Není alternativou pro stávající zkušební metody B.31, B.34, B.35 nebo B.56 a ani je nahrazuje.
|
VÝCHOZÍ ÚVAHY
|
6.
|
Při posuzování a hodnocení toxických vlastností chemické látky lze po získání prvních informací o toxicitě z akutních zkoušek stanovit orální toxicitu pomocí opakované aplikace dávek. Tato studie poskytuje informace o možném nebezpečí pro zdraví, které by se mohlo projevit na základě opakované expozice během relativně omezeného časového období. Tato metoda se skládá ze základní studie toxicity po opakovaných dávkách, kterou lze použít u chemických látek, u nichž není důvod pro 90denní studii (např. když objem výroby nepřekračuje určité limity), nebo jako předběžnou studii k dlouhodobé studii. Při realizaci studie by měly být respektovány hlavní zásady a aspekty stanovené v pokynu OECD č. 19 o uznávání, posuzování a používání klinických příznaků jako humánních sledovaných vlastností pro pokusná zvířata používaná při hodnoceních bezpečnosti (7).
|
|
7.
|
Dále zahrnuje screeningovou zkoušku na reprodukční/vývojovou toxicitu, a proto může být použita také na získání prvních informací o možných účincích na reprodukční výkonnost samců a samic, jako je funkce pohlavních žláz, chování při páření, početí, vývoj zárodku a vrh, a to v rané fázi posuzování toxikologických vlastností zkoušených chemických látek, nebo informací o chemických látkách, které jsou předmětem zájmu. Tato zkušební metoda neposkytuje úplné informace o všech aspektech reprodukce a vývoje. Konkrétně nabízí jen omezené prostředky zjištění postnatálních projevů prenatální expozice nebo účinků, které mohou být vyvolány v průběhu postnatální expozice. Vzhledem (mimo jiné) k selektivitě sledovaných vlastností a krátké době trvání studie neposkytuje tato metoda důkaz pro definitivní tvrzení, že nedochází k žádným reprodukčním/vývojovým účinkům. Navíc, pokud neexistují údaje z jiných zkoušek reprodukční/vývojové toxicity, jsou pozitivní výsledky užitečné pro první posouzení rizika a přispívají k rozhodnutím ohledně nezbytnosti a načasování dalších zkoušek.
|
|
8.
|
Výsledky získané pro parametry související s endokrinním působením by se měly interpretovat v kontextu „koncepčního rámce OECD pro zkoušky a hodnocení chemických látek způsobujících endokrinní poruchy“ (8). V tomto koncepčním rámci je vylepšený Pokyn OECD pro zkoušení č. 422 zařazen na úroveň 4 jako zkouška in vivo poskytující údaje o nežádoucích účincích na relevantní endokrinní sledované vlastnosti. Endokrinní signál sám o sobě však nemusí být považován za dostatečný důkaz, že zkoušená chemická látka je endokrinním disruptorem.
|
|
9.
|
Tato zkušební metoda klade rovněž větší důraz na neurologické účinky jako na specifické sledované vlastnosti; důraz je kladen na potřebu pečlivého klinického pozorování zvířat, aby bylo získáno co nejvíce informací. Metoda by měla odhalit chemické látky s neurotoxickým potenciálem, u nichž může být nezbytné další hlubší zkoumání tohoto aspektu. Dále může tato metoda poskytnout základní indikaci imunologických účinků.
|
|
10.
|
Pokud neexistují údaje z jiných studií systémové toxicity, reprodukční/vývojové toxicity, neurotoxicity anebo imunotoxicity, jsou pozitivní výsledky užitečné pro první posouzení rizika a přispívají k rozhodnutím ohledně nezbytnosti a načasování dalších zkoušek. Zkouška však může být zvláště užitečná jako součást souboru údajů pro hodnocení OECD (SIDS) pro posouzení stávajících chemických látek, o nichž nejsou k dispozici žádné nebo jen omezené toxikologické informace, a může sloužit jako alternativa k provedení dvou samostatných zkoušek toxicity po opakovaných dávkách (Pokyn OECD pro zkoušení č. 407, odpovídající kapitole B.7 této přílohy), respektive reprodukční/vývojové toxicity (Pokyn OECD pro zkoušení č. 421, odpovídající kapitole B.63 této přílohy). Rovněž může být použita jako studie ke stanovení dávkování pro rozsáhlejší reprodukční/vývojové studie, nebo když je to jinak považováno za relevantní.
|
|
11.
|
Obecně se předpokládá, že existují rozdíly v citlivosti mezi březími a nebřezími zvířaty. Proto může být složitější stanovit úroveň dávek v této kombinované zkoušce, která bude adekvátní pro posouzení jak obecné systémové toxicity, tak i specifické reprodukční/vývojové toxicity, než když se jednotlivé zkoušky provádějí zvlášť. Navíc může být obtížnější interpretovat výsledky zkoušek s ohledem na obecnou systémovou toxicitu, než když se provádí samostatná studie s opakovanými dávkami, zejména když se sérové a histopatologické parametry nehodnotí ve studii současně. Vzhledem k uvedeným technickým problémům jsou pro provádění této kombinované screeningové zkoušky nutné značné zkušenosti v oblasti zkoušení toxicity. Na druhé straně může kombinovaná zkouška nabídnout kromě menšího počtu použitých zvířat lepší prostředky pro rozlišení přímých účinků na reprodukci/vývoj od účinků, které jsou sekundární k jiným (systémovým) účinkům.
|
|
12.
|
V této zkoušce je období podávání zkoušené chemické látky delší než v klasické 28denní studii s opakovanou dávkou. V porovnání se stavem, kdy se provádí klasická 28denní studie s opakovanou dávkou kromě screeningové zkoušky reprodukční/vývojové toxicity, se však používá méně zvířat každého pohlaví na skupinu.
|
|
13.
|
Tato zkušební metoda předpokládá orální aplikaci zkoušené chemické látky. Pokud jsou použity jiné cesty expozice, bude zřejmě nutné provést úpravy.
|
|
14.
|
Před použitím této zkušební metody ke zkoušení směsi s cílem získat údaje pro zamýšlený regulační účel by mělo být zváženo, zda tato metoda může pro tento účel poskytnout odpovídající výsledky, a pokud ano, proč tomu tak je. Takové úvahy nejsou nutné, pokud existuje právní požadavek na zkoušení dané směsi.
|
|
15.
|
Použité definice jsou uvedeny v dodatku 1.
|
PRINCIP ZKOUŠKY
|
16.
|
Zkoušená chemická látka se podává v odstupňovaných dávkách několika skupinám samců a samic. Samcům se podává dávka minimálně čtyři týdny včetně dne před plánovaným usmrcením (zahrnuje to minimálně dva týdny před pářením, v průběhu období páření a přibližně dva týdny po páření). Vzhledem k omezené době podávání před pářením u samců nemusí být fertilita zvláště citlivým ukazatelem toxicity pro varlata. Proto je velmi důležité provést podrobné histologické vyšetření varlat. Kombinace dvoutýdenní doby podávání před pářením a následné pozorování páření/fertility s celkovou dobou podávání alespoň čtyři týdny, po níž následuje podrobná histopatologie samčích gonád, se považuje za dostatečnou ke zjištění většiny účinků na fertilitu a spermatogenezi u samců.
|
|
17.
|
Samicím je třeba podávat zkoušenou chemickou látku po celou dobu studie. Zahrnuje to dva týdny před pářením (s cílem pokrýt alespoň dva celé estrální cykly), variabilní dobu do početí, dobu březosti a alespoň třináct dnů po porodu, a to včetně dne před plánovaným usmrcením.
|
|
18.
|
Délka studie následující po aklimatizaci a zhodnocení estrálního cyklu před zahájením podávání závisí na výkonnosti samice a činí přibližně 63 dnů [alespoň 14 dnů před pářením, (maximálně) 14 dnů páření, 22 dnů gestace, 13 dnů laktace].
|
|
19.
|
V průběhu období podávání se zvířata každý den pečlivě pozorují, aby se zjistily příznaky toxicity. Zvířata, která v průběhu zkoušky uhynula nebo byla utracena, se pitvají a na konci zkoušky se přeživší zvířata utratí a rovněž pitvají.
|
POPIS METODY
Výběr zvířecích druhů
|
20.
|
Tato zkušební metoda je určena k použití s potkany. Jestliže se parametry uvedené v tomto pokynu pro zkoušení č. 422 zkoumají u jiných druhů hlodavců, mělo by se podat podrobné zdůvodnění. V mezinárodním validačním programu detekce endokrinních disruptorů pomocí pokynu pro zkoušení č. 407 byl jediným použitým druhem potkan. Není vhodné používat kmeny s nízkou plodností nebo s typicky vysokým výskytem vývojových vad. Používají se zdravá, dosud nepřipuštěná zvířata ještě nepoužitá pro jiné experimenty. Pokusná zvířata by měla být plně charakterizována co do druhu, kmene, pohlaví, hmotnosti a věku. Na začátku studie by měly být odchylky hmotnosti zvířat minimální a neměly by překročit ± 20 % střední hodnoty pro každé pohlaví. Provádí-li se studie jako předběžná studie pro dlouhodobou nebo celogenerační studii, je vhodné použít v obou studiích nejlépe zvířata stejného kmene a stejného původu.
|
Umístění a krmení zvířat
|
21.
|
Všechny postupy by měly splňovat místní standardy péče o laboratorní zvířata. Teplota v místnosti pro pokusná zvířata by měla být 22 oC (± 3 o). Relativní vlhkost vzduchu by měla být alespoň 30 % a neměla by pokud možno přesáhnout 70 % kromě doby úklidu místnosti. Osvětlení by mělo být umělé a mělo by se střídat 12 hodin světla a 12 hodin tmy. Ke krmení lze použít konvenční laboratorní krmivo s neomezeným přísunem pitné vody. Výběr potravy může být ovlivněn potřebou zajistit dostatečné promísení s testovanou látkou, je-li látka podávána touto cestou.
|
|
22.
|
Zvířata by měla být chována v malých skupinách stejného pohlaví; je-li to z vědeckého hlediska důvodné, mohou být zvířata chována individuálně. Při chovu zvířat v klecích by v jedné kleci nemělo být umístěno více než pět zvířat. Páření by mělo probíhat v klecích, které jsou k tomuto účelu vhodné. Březí samice je třeba chovat v klecích jednotlivě a poskytnout jim materiál pro vytvoření hnízda. Samice v době laktace se chovají v klecích jednotlivě společně s potomky.
|
|
23.
|
Krmivo by se mělo pravidelně analyzovat, aby se zjistila přítomnost kontaminantů. Vzorek stravy by se měl uchovávat až do dokončení zprávy.
|
Příprava zvířat
|
24.
|
Zdravá mladá dospělá zvířata se náhodně vyberou a rozdělí do experimentálních skupin a klecí. Klece by měly být uspořádány tak, aby byl vliv umístění klecí minimalizován. Zvířata se jednoznačně identifikují a před započetím studie se nechají v laboratorních podmínkách alespoň pět dní aklimatizovat.
|
Příprava dávek
|
25.
|
Není-li jiný způsob aplikace považován za vhodnější, doporučuje se orální podávání zkoušené chemické látky. Pokud je zvolena orální cesta, zkoušená chemická látka se obvykle podává žaludeční sondou; alternativně však lze zkoušené chemické látky podávat také v potravě nebo v pitné vodě.
|
|
26.
|
Je-li to nutné, je zkoušená chemická látka rozpuštěna nebo suspendována ve vhodném vehikulu. Kdykoli je to možné, doporučuje se zvážit nejprve použití vodného roztoku/suspenze, poté použití olejového roztoku/emulze (např. kukuřičného oleje) a nakonec roztoku v jiných vehikulech. U jiného typu vehikula, než je voda, je třeba znát jeho toxické charakteristiky. Měla by být určena stabilita a homogenita zkoušené chemické látky v daném vehikulu.
|
POSTUP
Počet a pohlaví zvířat
|
27.
|
Doporučuje se, aby každá skupina měla zpočátku alespoň 10 samců a 12–13 samic. U samic se hodnotí před expozicí estrální cykly a zvířata, která nevykazují typické 4–5denní cykly, nebudou do studie zařazena; proto se doporučují náhradní samice, aby v každé skupině zůstalo 10 samic. S výjimkou případu výrazně toxických účinků se očekává, že v každé skupině bude alespoň 8 březích samic, což je obvykle minimální přijatelný počet březích samic na skupinu. Cílem je získat dostatečný počet březích samic a potomstva, a tím zajistit smysluplné hodnocení schopnosti zkoušené chemické látky ovlivnit plodnost, březost, chování samice a kojených mláďat a růst a vývoj potomstva generace F1 od doby početí do dne 13 po porodu. Pokud se budou zvířata usmrcovat v průběhu studie, je nutno zvýšit celkový počet zvířat o počet zvířat, která budou usmrcena před ukončením studie. Mělo by se zvážit přidání další satelitní skupiny pěti zvířat od každého pohlaví do kontrolní skupiny a do skupiny s nejvyšší dávkou, aby se po dobu nejméně 14 dní po ukončení aplikace mohla sledovat vratnost, přetrvávání nebo opožděný nástup systémových toxických účinků. Zvířata ze satelitních skupin se nepáří, a proto se nepoužívají pro posouzení reprodukční/vývojové toxicity.
|
Dávkování
|
28.
|
Obecně by měly být použity nejméně tři zkušební skupiny a kontrolní skupina. Nejsou-li dostupné žádné vhodné obecné údaje o toxicitě, může se provést předběžná studie (na zvířatech stejného kmene a původu) pro stanovení rozmezí dávek, které mají být použity. S výjimkou podávání zkoušené chemické látky se se zvířaty v kontrolní skupině zachází stejně jako se zvířaty ve zkušební skupině. Používá-li se pro usnadnění aplikace zkoušené chemické látky vehikulum, podává se kontrolní skupině vehikulum v nejvyšším použitém objemu.
|
|
29.
|
Při výběru úrovní dávek by se měly vzít v úvahu veškeré existující údaje o toxických a (toxiko)kinetických vlastnostech. Rovněž je třeba vzít v úvahu, že mohou existovat rozdíly v citlivosti mezi březími a nebřezími zvířaty. Nejvyšší úroveň dávky by se měla zvolit tak, aby vyvolala toxické účinky, nikoli však uhynutí ani velké utrpení. Dále by měla být zvolena sestupná řada dávek s cílem prokázat, že existuje odezva související s dávkou a že nejnižší dávka nevyvolává žádné nežádoucí účinky. Obvykle jsou optimální dvoj- až čtyřnásobné intervaly mezi dávkami a často je vhodnější přidání čtvrté zkušební skupiny než používání příliš velkých intervalů mezi jednotlivými dávkami (např. lišících se faktorem větším než 10).
|
|
30.
|
V přítomnosti pozorované obecné toxicity (např. snížená tělesná hmotnost, účinky na játra, srdce, plíce nebo ledviny atd.) nebo jiných změn, které nemusí být odezvou na toxicitu (např. snížený příjem krmiva, zvětšení jater), je třeba pozorované účinky na endokrinní citlivé sledované vlastnosti interpretovat obezřetně.
|
Limitní zkouška
|
31.
|
Pokud orální studie provedená podle postupů popsaných pro tuto studii při jedné dávce nejméně 1 000 mg na kg tělesné hmotnosti na den nebo v případě podávání v potravě nebo v pitné vodě v rovnocenné koncentraci (na základě stanovení tělesné hmotnosti) nevyvolá pozorovatelné toxické účinky a pokud se na základě údajů o látkách s podobnou strukturou nepředpokládá toxicita, není úplná studie za použití několika úrovní dávek nutná. Limitní zkouška se použije s výjimkou případu, kdy údaje o expozici člověka naznačují, že je nezbytné použití vyšší úrovně dávek. U dalších způsobů podávání látky, jako je inhalační nebo kožní aplikace, může být maximální dosažitelná expozice často dána fyzikálně-chemickými vlastnostmi zkoušených chemických látek.
|
Podávání dávek
|
32.
|
Zvířatům se denně podává zkoušená chemická látka po dobu sedmi dnů v rámci jednoho týdne. Pokud se zkoušená chemická látka podává nitrožaludečně, podává se zvířatům v jediné dávce za použití žaludeční sondy nebo vhodné intubační kanyly. Maximální objem tekutiny, kterou lze podat najednou, závisí na velikosti pokusného zvířete. Tento objem by neměl překročit 1 ml na 100 g tělesné hmotnosti, s výjimkou vodných roztoků, kde lze použít 2 ml na 100 g tělesné hmotnosti. S výjimkou dráždivých a žíravých zkoušených chemických látek, které obvykle při vyšších koncentracích vykazují zesílené účinky, by měla být variabilita zkušebního objemu minimalizována nastavením koncentrace zajišťující konstantní objem při všech úrovních dávky.
|
|
33.
|
U zkoušených chemických látek podávaných v potravě nebo v pitné vodě je důležité zajistit, aby množství použité zkoušené chemické látky neovlivňovalo normální výživu nebo vodní rovnováhu. Podává-li se zkoušená chemická látka v krmivu, může se použít buď konstantní koncentrace (v ppm), nebo konstantní dávkování vzhledem k tělesné hmotnosti zvířete; použitá možnost by měla být specifikována. Zkoušená chemická látka podávaná žaludeční sondou se podává každý den přibližně ve stejnou dobu a alespoň jednou týdně se přizpůsobí tak, aby se udržela konstantní úroveň dávky vzhledem k tělesné hmotnosti zvířete. Pokud je kombinovaná studie prováděna jako předběžná studie k dlouhodobé nebo úplné studii reprodukční toxicity, měla by být v obou studiích použita stejná potrava.
|
Harmonogram zkoušky
|
34.
|
Podávání oběma pohlavím by mělo být zahájeno 2 týdny před pářením po minimálně pětidenní aklimatizaci a po screeningu samic, zda mají normální estrální cykly (v dvoutýdenním období před expozicí). Studii je třeba naplánovat tak, aby hodnocení estrálních cyklů začalo brzy poté, co zvířata dosáhnou plné pohlavní zralosti. U různých kmenů potkana v různých laboratořích se to může mírně lišit, např. potkan Sprague Dawley stáří 10 týdnů, potkan Wistar stáří přibližně 12 týdnů. Samice s potomstvem by měly být utraceny 13. den po porodu nebo krátce poté. Aby bylo možné odebrat krev samicím nalačno (pokud je dávána přednost této možnosti), není nutné, aby byly samice a jejich potomstvo utraceny ve stejný den. Den porodu (tj. ten, kdy je dokončen vrh) je definován jako 0. den po porodu. Samice, které nevykazují žádné známky kopulace, se utratí 24–26 dnů po posledním dni období páření. S dávkováním se u obou pohlaví pokračuje i během období páření. U samců se dále pokračuje s podáváním zkoušené chemické látky i po období páření alespoň do doby, kdy je dokončeno minimální období podávání 28 dnů. Poté se utratí, nebo se případně uchovají a dále je jim podávána zkoušená chemická látka pro případné provedení druhého páření, je-li to považováno za vhodné.
|
|
35.
|
V denním podávání samicím rodičovské generace by se poté mělo pokračovat po celou březost a alespoň do 13. dne po porodu včetně nebo do dne před utracením. U studií, kde je zkoušená chemická látka podávána inhalací nebo dermální cestou, je třeba s dávkováním pokračovat alespoň do 19. dne gestace včetně, přičemž dávkování se obnoví co možná nejdříve, nejpozději však 4. postnatální den (PND).
|
|
36.
|
Pokud je zařazena satelitní skupina, pak zvířata v této skupině, která mají být podle plánu dále sledována, se nepáří. Měla by být po dalších nejméně 14 dnů po prvním naplánovaném utracení samic bez aplikace, aby se zachytil opožděný nástup či přetrvávání toxických účinků nebo zotavení se z nich.
|
|
37.
|
Diagram časového sledu zkoušky je uveden v dodatku 2.
|
Estrální cykly
|
38.
|
Estrální cykly je třeba sledovat před zahájením expozice, aby bylo možné vybrat pro studii samice s pravidelným cyklem (viz odstavec 27). Od začátku období expozice až do průkazu o páření je třeba také každý den sledovat vaginální stěry. V případě obav z účinků akutního stresu, které by při zahájení podávání zkoušené chemické látky mohly změnit estrální cykly, mohou laboratoře exponovat pokusná zvířata po dobu dvou týdnů, poté denně odebírat vaginální stěry za účelem sledování estrálního cyklu po dobu minimálně dvou týdnů před obdobím páření a dále je sledovat až do období páření, dokud prokazatelně nedojde k páření. Při získávání vaginálních/cervikálních buněk je třeba neporušit sliznice, což by mohlo vyvolat stav falešné březosti (8) (9).
|
Páření
|
39.
|
Obvykle se v této studii používá párování 1:1 (jeden samec a jedna samice). V případě náhodného úmrtí samců mohou nastat výjimky. Samice se umístí se stejným samcem až do doby, kdy budou pozorovány důkazy o spáření, nebo do uplynutí dvou týdnů. Každé ráno se samice vyšetří na přítomnost spermatu nebo vaginální zátky. Den 0 březosti je definován jako den, kdy je potvrzen důkaz o spáření (je nalezena vaginální zátka nebo sperma). V případě, že páření bylo neúspěšné, se zváží spáření se samci s osvědčenou plodností ze stejné skupiny.
|
Velikost vrhu
|
40.
|
Čtvrtý den po vrhu se velikost všech vrhů upraví náhodným vyřazením nadbytečných jedinců tak, aby bylo pokud možno dosaženo počtu čtyř až pěti samců a čtyř až pěti samic z jednoho vrhu podle obvyklé početnosti vrhu u použitého kmene potkana. Od dvou nadbytečných mláďat se odeberou krevní vzorky, smíchají se a použijí pro stanovení sérových hladin T4. Selektivní eliminace jedinců, např. na základě tělesné hmotnosti nebo anogenitální vzdálenosti (AGD), není vhodná. Pokud počet mladých samců nebo samic neumožňuje ponechat čtyři až pět od každého pohlaví a vrhu, je možná částečná úprava (např. ponechat šest samců a čtyři samice). Jestliže velikost vrhu poklesne pod cílový počet pro utracení (8 nebo 10 mláďat/vrh), nevyřazují se žádná mláďata. Jestliže zbývá pouze jedno mládě nad cílový počet pro utracení, vyřadí se pouze jedno mládě, které se použije pro odběr krve pro případné vyšetření sérové hladiny T4.
|
|
41.
|
Jestliže se velikost trhu neupravuje, 4. den po porodu se utratí dvě mláďata na vrh a odeberou se krevní vzorky pro stanovení sérové koncentrace hormonů štítné žlázy. Těmito dvěma mláďaty na vrh by měly být pokud možno samice, aby se uchovali samci pro hodnocení retenčních mléčných cyst, s výjimkou případu, že by po odstranění těchto mláďat nezbyly žádné samice pro vyšetření při ukončení zkoušky. Jestliže velikost vrhu poklesne pod 8 nebo 10 mláďat/vrh (podle normální velikosti vrhu u použitého kmene potkana), nevyřazují se žádná mláďata. Jestliže zbývá pouze jedno mládě nad normální velikost vrhu, vyřadí se pouze jedno mládě, které se použije pro odběr krve pro případné vyšetření sérové hladiny T4.
|
Pozorování
|
42.
|
Všeobecné klinické pozorování by se mělo provádět nejméně jednou denně, nejlépe ve stejnou dobu (stejné doby) a s uvážením doby očekávaného maxima účinku po podání látky. Zaznamenává se zdravotní stav zvířat. Nejméně dvakrát denně se provede prohlídka všech zvířat za účelem zjištění morbidity a mortality.
|
|
43.
|
Před expozicí látce a dále nejméně jednou týdně by se mělo provést důkladné klinické vyšetření všech rodičovských zvířat (za účelem intraindividuálního porovnání). Toto pozorování by se mělo provádět mimo chovnou klec ve standardním pozorovacím prostoru a nejlépe každý den ve stejnou dobu. Pozorování je třeba pečlivě zaznamenat, nejlépe za použití systému bodování explicitně definovaného ve zkušební laboratoři. Mělo by se usilovat o to, aby rozdíly ve zkušebních podmínkách byly co nejmenší a aby vyšetření prováděly nejlépe osoby, které nevědí, jakým testem zvířata procházejí. Vyšetření by mělo mimo jiné zahrnovat změny kůže, srsti, očí a sliznic, výskyt sekretů a exkretů a vegetativních funkcí (např. slzení, zježení srsti, velikost zornic, nezvyklé dýchání). Zaznamenávat by se měly také změny chůze, držení těla a reakce na manipulaci, dále přítomnost klonických a tonických pohybů, stereotypů v chování (např. nadměrného čištění nebo opakovaného kroužení), ztížený nebo prodlužovaný vrh nebo bizarní chování (např. sebepoškozování, chůze pozpátku) (10).
|
|
44.
|
Jednou v průběhu studie by se měly u pěti samců a pěti samic náhodně vybraných z každé skupiny vyšetřit reakce na smyslové podněty různého druhu (např. sluchové, zrakové, proprioceptivní), (8), (9), (11), síla úchopu (12) a motorická aktivita (13). Další informace o postupech, které lze použít, jsou uvedeny v příslušné literatuře. Místo postupů popsaných v této literatuře však lze použít alternativní postupy. U samců by měla tato funkční pozorování proběhnout ke konci období, kdy je jim podávána zkoušená chemická látka, krátce před naplánovaným utracením, ale před odběrem krve pro hematologické vyšetření nebo stanovení klinických biochemických ukazatelů (viz odstavce 53–56, včetně poznámky pod čarou č. 1). Samice by měly být v průběhu těchto funkčních zkoušek ve fyziologicky podobném stavu a měly by být testovány nejlépe jednou v průběhu posledního týdne laktace (např. LD 6–13), krátce před plánovaným utracením. V maximálně možné míře minimalizujte dobu oddělení samic od mláďat.
|
|
45.
|
Pozorování funkčních poruch provedené jednou ke konci studie lze případně vynechat, pokud je studie prováděna jako předběžná pro následnou subchronickou (90denní) nebo dlouhodobou studii. V takovém případě by se mělo pozorování funkčních poruch zahrnout do této dlouhodobé studie. Dostupnost údajů o pozorovaných funkčních poruchách z této studie opakovaného podávání dávky může na druhé straně usnadnit výběr úrovní dávek pro následnou subchronickou nebo dlouhodobou studii.
|
|
46.
|
Pozorování funkčních poruch lze výjimečně vynechat u skupin, které vykazují příznaky toxicity v takové míře, že by při posuzování funkčního stavu vadily.
|
|
47.
|
Doba gestace se zaznamená a počítá se ode dne 0 březosti. Každý vrh se co nejdříve po porodu vyšetří a stanoví se počty a pohlaví mláďat, počet mrtvě a živě narozených mláďat, počet zakrslých mláďat (mláďat, která jsou výrazně menší než odpovídající kontrolní mláďata) a přítomnost nápadných abnormalit.
|
|
48.
|
Do 24 hodin po porodu (0. den nebo 1. den po porodu) a alespoň 4. den a 13. den po porodu se živá mláďata spočítají, stanoví se jejich pohlaví a zváží se. Kromě pozorování zvířat rodičovské generace (viz odstavce 43 a 44) je třeba zaznamenat jakékoli abnormální chování potomstva.
|
|
49.
|
U každého mláděte je třeba ve stejný den po porodu v době od PND 0 do PND 4 změřit anogenitální vzdálenost (AGD). V den měření AGD by měla být zjištěna tělesná hmotnost mláděte a AGD by měla být normalizována na nějakou míru velikosti mláďat, nejlépe třetí odmocninu tělesné hmotnosti (14). Podle doporučení pokynu OECD č. 151 se v PND 12 nebo 13 spočítá počet bradavek/prsních dvorců u mladých samců (15).
|
Tělesná hmotnost a spotřeba potravy a vody
|
50.
|
Samci a samice se váží první den podávání, poté týdně a při ukončení zkoušky. V průběhu březosti se samice zváží 0., 7., 14. a 20. den a do 24 hodin po vrhu (0. den nebo 1. den po porodu) a alespoň 4. den a 13. den po porodu. Tato pozorování se zaznamenají individuálně u každého dospělého zvířete.
|
|
51.
|
Před obdobím páření a během březosti a laktace se alespoň jednou týdně měří příjem potravy. Měření příjmu potravy v průběhu páření je volitelné. Pokud se zkoušená chemická látka podává ve vodě, měří se v průběhu těchto období také spotřeba tohoto média.
|
Hematologická vyšetření
|
52.
|
Jednou v průběhu studie by se měla u pěti samců a pěti samic náhodně vybraných z každé skupiny tato hematologická provést vyšetření: stanovení hematokritu, koncentrace hemoglobinu, počtu erytrocytů, retikulocytů, celkového počtu a diferenciálního rozpočtu leukocytů a trombocytů a stanovení protrombinového času. Jestliže zkoušená chemická látka nebo její domnělé metabolity mají oxidační vlastnosti nebo existuje podezření, že je mají, měly by se stanovit také koncentrace methemoglobinu a Heinzových tělísek.
|
|
53.
|
Krevní vzorky by se měly odebírat z určeného místa. Samice by měly být při odběru ve fyziologicky podobném stavu. Za účelem vyloučení praktických problémů spojených s variabilitou při nástupu gestace se může samicím odebírat krev na konci období před pářením jako alternativa k odběru těsně před utracením zvířat nebo jako součást tohoto postupu. U samců se krev odebírá nejlépe těsně před utracením zvířat nebo jako součást tohoto postupu. Alternativně lze samcům odebírat krev také na konci období před pářením, pokud byla tato doba vybrána pro samice.
|
|
54.
|
Krevní vzorky je třeba skladovat za vhodných podmínek.
|
Biochemické vyšetření
|
55.
|
Klinické biochemické analýzy za účelem vyšetření hlavních toxických účinků na tkáně, a zejména na játra a ledviny, by se měly provést na krevních vzorcích od pěti vybraných samců a pěti vybraných samic z každé skupiny. Před odebráním krevních vzorků se doporučuje ponechat zvířata přes noc nalačno (22). Vyšetření plazmy a séra by mělo obsahovat stanovení sodíku, draslíku, glukózy, celkového cholesterolu, močoviny, kreatininu, celkových bílkovin a albuminu, nejméně dvou enzymů indikujících účinky na jaterní buňky (jako je alaninaminotransferáza, aspartátaminotransferáza, a sorbitoldehydrogenáza) a žlučových kyselin. Stanovení dalších enzymů (jaterního nebo jiného původu) a bilirubinu může za určitých okolností poskytnout užitečné informace.
|
|
56.
|
Krevní vzorky z určeného místa se odebírají podle tohoto rozvrhu:
|
—
|
od alespoň dvou mláďat na vrh 4. den po porodu, pokud to umožňuje počet mláďat (viz odstavce 40–41),
|
|
—
|
od všech samic a alespoň dvou mláďat na vrh při ukončení zkoušky 13. den a
|
|
—
|
od všech dospělých samců při ukončení zkoušky.
|
Všechny krevní vzorky se uchovávají za vhodných podmínek. Krevní vzorky odebrané mláďatům 13. den a dospělým samcům se vyšetří na sérové hladiny hormonů štítné žlázy (T4). Ve vhodných případech se provede další vyšetření T4 v krevních vzorcích odebraných samicím a mláďatům 4. den. Je-li to vhodné, mohou být volitelně stanoveny také hladiny jiných hormonů. Krev mláďat z vrhu lze pro potřeby analýzy hormonů štítné žlázy smíchat. Hormony štítné žlázy (T4 a TSH) se měří nejlépe jako „celek“.
|
|
57.
|
Volitelně mohou být provedena tato vyšetření moči u pěti náhodně vybraných samců z každé skupiny v průběhu posledního týdne studie za použití časově definovaného sběru moči; vyšetření se týkají vzhledu, objemu, osmolality nebo specifické hmotnosti, pH, bílkovin, glukózy a krve/krvinek.
|
|
58.
|
Dále by se mělo uvážit vyšetření indikátorů celkového poškození tkání v séru. Další vyšetření by mělo být provedeno, pokud mohou známé vlastnosti zkoušené chemické látky ovlivnit odpovídající metabolické profily, včetně vápníku, fosforu, triglyceridů na lačno a glukózy na lačno, specifických hormonů, methemoglobinu a cholinesterázy. To je třeba stanovit individuálně.
|
|
59.
|
Následující faktory mohou ovlivnit variabilitu a absolutní koncentrace stanovovaných hormonů:
|
—
|
doba utracení vzhledem k diurnálním odchylkám v koncentracích hormonů,
|
|
—
|
způsob utracení – mělo by se zabránit zbytečnému stresu zvířat, který může ovlivnit koncentrace hormonů,
|
|
—
|
testovací soupravy pro stanovení hormonů, jejichž standardní křivky se mohou lišit.
|
|
|
60.
|
Vzorky plazmy určené speciálně k stanovení hormonů by se měly odebírat zhruba ve stejnou denní dobu. Číselné hodnoty získané při analýze koncentrací hormonů se u různých komerčních testovacích souprav liší.
|
|
61.
|
Pokud nejsou údaje o výchozích hodnotách z dřívějších pozorování dostatečné, mělo by se zvážit stanovení hematologických a klinických biochemických parametrů před začátkem podávání dávky nebo přednostně u skupiny zvířat nezahrnutých do experimentálních skupin. U samic musí být údaje od zvířat v laktaci.
|
PATOLOGIE
Celková pitva
|
62.
|
U všech dospělých zvířat zařazených do studie by měla být provedena celková, podrobná pitva, zahrnující pečlivé vyšetření vnějšího povrchu těla, všech otvorů a lebeční, hrudní a břišní dutiny a jejich obsahu. Speciální pozornost je třeba věnovat orgánům rozmnožovací soustavy. Zaznamená se počet implantací. V den pitvy se vyšetří vaginální stěry s cílem určit stadium estrálního cyklu a umožnit korelaci s histopatologickým vyšetřením samičích reprodukčních orgánů.
|
|
63.
|
Varlata, nadvarlata a prostata a semenné váčky s Cowperovými žlázami jako celek se u všech dospělých samců podle potřeby zbaví všech přirostlých tkání a co nejdříve po pitvě se ve vlhkém stavu zváží, aby nedošlo k vyschnutí. Kromě toho mohou být volitelně zváženy další orgány, jako jsou svaly m. levator ani a m. bulbocavernosus, Cowperovy žlázy a žalud penisu u samců a párové vaječníky (vlhká hmotnost) a děloha (včetně děložního hrdla) u samic; je-li toto vážení prováděno, mělo by k němu dojít co nejdříve po pitvě. Vaječníky, varlata, nadvarlata, přídatné pohlavní orgány a všechny orgány od všech dospělých zvířat vykazující makroskopické léze se uchovají.
|
|
64.
|
Pro účely následného histopatologického vyšetření se uchovají v nejvhodnějším fixačním médiu štítné žlázy od všech dospělých samců a samic a od jednoho 13denního samce a jedné 13denní samice z každého vrhu. Hmotnost štítné žlázy lze stanovit po fixaci. Odříznutí by se mělo rovněž provádět velmi opatrně a teprve po fixaci, aby se zamezilo poškození tkáně. Poškození tkáně by mohlo narušit histopatologickou analýzu. Krevní vzorky by se měly odebírat z určeného místa těsně před utracením zvířat nebo v jeho průběhu a měly by se skladovat za vhodných podmínek (viz odstavec 56).
|
|
65.
|
Dále se játra, ledviny, nadledvinky, brzlík, slezina, mozek a srdce od alespoň pěti dospělých samců a samic náhodně vybraných z každé skupiny (kromě zvířat nalezených v agónii a/nebo zvířat utracených před ukončením studie) podle potřeby zbaví všech ulpělých tkání a co nejdříve po sekci se ve vlhkém stavu zváží, aby nedošlo k vyschnutí. Následující tkáně by se měly konzervovat v nejvhodnějším fixačním médiu s ohledem na typ tkáně a plánovaná následná histopatologická vyšetření: všechny makroskopicky viditelné léze, mozek (reprezentativní oblasti zahrnující hemisféry, mozeček a most), mícha, oko, žaludek, tenké a tlusté střevo (včetně Peyerových plátů), játra, ledviny, nadledvinky, slezina, srdce, brzlík, průdušnice a plíce (konzervované naplněním fixačním roztokem a následným ponořením), gonády (varlata a vaječníky), přídatné pohlavní orgány (děloha a hrdlo děložní, nadvarlata, prostata, semenné váčky s Cowperovými žlázami), vagina, močový měchýř, lymfatické uzliny (kromě nejproximálnější svodné uzliny by se měla odebrat ještě jedna uzlina dle laboratorních zkušeností (16)), periferní nerv (n. ischiadicus nebo n. tibialis) přednostně v blízkosti svalu, kosterní sval a kost, s kostní dření (řez nebo čerstvý nátěr z aspirované kostní dřeně). Doporučuje se fixovat varlata ponořením do Bouinova nebo modifikovaného Davidsonova fixačního média (16) (17) (18); pro tyto tkáně se nedoporučuje fixace ve formalínu. Tunica albuginea se může jemně a mělce propíchnout jehlou na obou koncích orgánu, aby se umožnilo rychlé proniknutí fixativa. Na základě klinických nebo jiných nálezů může být nezbytné zkoumat i další tkáně. Také by se měly uchovat všechny orgány považované na základě známých vlastností zkoušené chemické látky za možné cílové orgány.
|
|
66.
|
Následující tkáně mohou poskytnout cenné svědectví o endokrinních účincích: gonády (vaječníky a varlata), přídatné pohlavní orgány (děloha včetně děložního hrdla, nadvarlata, semenné váčky s Cowperovými žlázami, dorzolaterální a ventrální prostata), vagina, hypofýza, samčí prsní žláza a nadledvinka. Změny na samčích prsních žlázách nebyly dostatečně zdokumentovány, avšak tento parametr může být velmi citlivý na látky s estrogenním působením. Pozorování orgánů/tkání, které nejsou uvedeny v odstavci 65, jsou volitelná.
|
|
67.
|
Mrtvá mláďata a mláďata utracená 13. den po porodu nebo krátce poté je třeba alespoň pečlivě externě vyšetřit na závažné abnormality. Zvláštní pozornost je třeba věnovat vnějším rozmnožovacím orgánům, které se vyšetří na přítomnost známek změněného vývoje.
|
Histopatologie
|
68.
|
U vybraných zvířat z kontrolní skupiny a skupiny s vysokou dávkou by mělo být provedeno celkové histologické vyšetření uchovaných orgánů a tkání (se zvláštním důrazem na stádia spermatogeneze samčích gonád a histopatologii struktury intersticiálních buněk varlat). V případě potřeby může být vyšetřena štítná žláza od mláďat a zbývajících dospělých zvířat. Toto vyšetření by mělo být rozšířeno na zvířata z ostatních dávkových skupin, pokud jsou u skupiny s vysokou dávkou pozorovány změny orgánů vyvolané zkoušenou dávkou. Pokyny k histopatologickému vyšetření (10) podrobně popisují pitvu, fixaci, tvorbu řezů a histopatologické vyšetření endokrinních tkání.
|
|
69.
|
Měly by být zkoumány všechny makroskopické léze. Za účelem objasnění hodnot dávky bez pozorovaného nepříznivého účinku (NOAEL) je třeba vyšetřit cílové orgány v jiných dávkových skupinách, zejména ve skupinách, které údajně vykazují hodnotu dávky bez pozorovaného nepříznivého účinku.
|
|
70.
|
V případě použití satelitní skupiny by mělo být provedeno histopatologické vyšetření tkání a orgánů, na kterých byly pozorovány účinky u exponovaných skupin.
|
ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV
Údaje
|
71.
|
Měly by být uvedeny údaje pro každé jednotlivé zvíře. Dále se všechny údaje shrnou do tabulek tak, aby byl u každé experimentální skupiny uveden počet zvířat na začátku zkoušky, počet zvířat uhynulých během zkoušky nebo utracených z humánních důvodů, doba úhynu nebo utracení, počet plodných samic, počet březích samic, počet zvířat vykazujících příznaky toxicity, popis pozorovaných příznaků toxicity včetně doby nástupu, trvání a závažnosti veškerých toxických účinků, druhy histopatologických změn a všechny významné údaje o vrhu. Souhrnná zpráva ve formě tabulky, která se ukázala jako velmi užitečná pro hodnocení reprodukčního/vývojového účinku, je uvedena v dodatku 3.
|
|
72.
|
Je-li to možné, je třeba vyhodnotit číselné výsledky vhodnou a obecně uznávanou statistickou metodou. Srovnáním účinků v celém rozpětí dávkování by se mělo zamezit použití mnohonásobných t-testů. Statistické metody je třeba zvolit během plánování studie. Statistická analýza AGD a retenčních mléčných cyst by měla vycházet z údajů o jednotlivých mláďatech se zohledněním účinků na vrh. Ve vhodných případech je jednotkou analýzy vrh. Statistická analýza tělesné hmotnosti mláďat by měla vycházet z údajů o jednotlivých mláďatech se zohledněním velikosti vrhu. Vzhledem k omezené velikosti studie mají statistické analýzy ve formě testů „významnosti“ pro mnoho sledovaných vlastností, zejména pro reprodukční sledované vlastnosti, omezenou hodnotu. Některé nejčastěji používané metody, zejména parametrické testy na měření středního trendu, jsou nevhodné. Pokud se používají statistické analýzy, pak by měla být zvolená metoda vhodná pro distribuci zkoumané proměnné a měla by být zvolena před zahájením studie.
|
Vyhodnocení výsledků
|
73.
|
Výsledky této studie reprodukční toxicity se vyhodnotí z hlediska pozorovaných účinků, pitvy a mikroskopických nálezů. V hodnocení musí být uveden vztah mezi dávkou zkoušené chemické látky a přítomností nebo nepřítomností, výskytem a závažností abnormalit včetně makroskopických lézí, stanovených cílových orgánů, neplodnosti, klinických abnormalit, vlivu na reprodukční výkonnost a schopnost vrhu, změny tělesné hmotnosti, vlivu na úmrtnost a další toxické účinky.
|
|
74.
|
Vzhledem ke krátké době expozice samců je třeba histopatologii varlat a nadvarlat zvažovat při posuzování reprodukčních účinků u samců společně s údaji o fertilitě. Jako pomůcka pro interpretaci studie může být také užitečné použití historických kontrolních údajů o reprodukci/vývoji (např. o velikosti vrhu, AGD, retenčních mléčných cystách, sérových hladinách T4), pokud jsou tyto údaje k dispozici.
|
|
75.
|
Za účelem kontroly jakosti se navrhuje, aby se shromáždily historické kontrolní údaje a vypočítaly se koeficienty rozptylu pro číselné údaje, zejména pro parametry spojené s detekcí endokrinních disruptorů. Tyto údaje pak lze použít pro účely srovnání při hodnocení stávajících studií.
|
ZÁVĚREČNÁ ZPRÁVA
|
76.
|
Závěrečná zpráva by měla obsahovat tyto informace:
|
|
Zkoušená chemická látka:
|
—
|
zdroj, číslo šarže a, je-li k dispozici, datum použitelnosti,
|
|
—
|
stabilita zkoušené chemické látky, je-li známa.
|
|
|
|
Jednosložková látka:
|
—
|
fyzický vzhled, rozpustnost ve vodě a další relevantní fyzikálně-chemické vlastnosti,
|
|
—
|
chemická identifikace, např. název podle IUPAC nebo CAS, číslo CAS, kód SMILES nebo InChI, strukturní vzorec, čistota, případně chemická identita nečistot, je-li to prakticky možné, atd.
|
|
|
|
Vícesložkové látky, UVCB a směsi:
|
—
|
charakterizované pokud možno chemickou identitou (viz výše), kvantitativním výskytem a relevantními fyzikálně-chemickými vlastnostmi složek
|
. |
|
|
Vehikulum (je-li použito):
|
—
|
zdůvodnění výběru vehikula, pokud není použita voda.
|
|
|
|
Pokusná zvířata:
|
—
|
počet, věk a pohlaví zvířat,
|
|
—
|
původ, podmínky chovu, strava atd.,
|
|
—
|
hmotnost jednotlivých zvířat na začátku zkoušky,
|
|
—
|
zdůvodnění použití druhu, pokud je jiný než potkan.
|
|
|
|
Zkušební podmínky:
|
—
|
zdůvodnění zvolených úrovní dávek,
|
|
—
|
podrobnosti o složení zkoušené chemické látky/úpravě potravy, o použité koncentraci, stálosti a homogenitě přípravku,
|
|
—
|
podrobné údaje o podávání zkoušené chemické látky,
|
|
—
|
popřípadě přepočet koncentrace zkoušené chemické látky v krmivu/pitné vodě (ppm) na skutečnou denní dávku (mg/kg tělesné hmotnosti/den),
|
|
—
|
podrobnosti o jakosti krmení a vody,
|
|
—
|
podrobný popis postupů randomizace při výběru mláďat pro třídění, jsou-li tříděna.
|
|
|
|
Výsledky:
|
—
|
tělesná hmotnost/změny tělesné hmotnosti,
|
|
—
|
spotřeba krmiva, popřípadě spotřeba vody,
|
|
—
|
údaje o toxických reakcích podle pohlaví a dávky, včetně ukazatelů plodnosti, gestace a všech dalších známek toxicity,
|
|
—
|
toxické nebo jiné účinky na reprodukci, potomstvo, postnatální růst atd.,
|
|
—
|
povaha, závažnost a trvání klinických projevů (ať vratných, či nevratných),
|
|
—
|
posouzení reaktivity na smyslové podněty, úchopové síly a motorické aktivity,
|
|
—
|
hematologická vyšetření s příslušnými základními hodnotami (normami),
|
|
—
|
klinická biochemická vyšetření s příslušnými výchozími hodnotami (normami),
|
|
—
|
počet dospělých samic s normálním nebo abnormálním estrálním cyklem a trváním cyklu,
|
|
—
|
počet živě narozených mláďat a postimplantačních ztrát,
|
|
—
|
počet mláďat s nápadnými strukturálními abnormalitami, orientační hodnocení vnějších genitálií, počet zakrslých mláďat,
|
|
—
|
den úhynu během studie nebo údaj, že zvířata přežila až do dne utracení,
|
|
—
|
počet implantovaných plodů, velikost vrhu a hmotnost vrhu v době pořízení záznamu,
|
|
—
|
údaje o tělesné hmotnosti mláďat,
|
|
—
|
AGD všech mláďat (a tělesná hmotnost v den měření AGD),
|
|
—
|
retenční mléčné cysty u mláďat samců,
|
|
—
|
hladiny hormonů štítné žlázy u 13denních mláďat a dospělých samců (a u samic a 4denních mláďat, jsou-li tyto hodnoty měřeny),
|
|
—
|
tělesná hmotnost při utracení a údaje o hmotnosti orgánů u rodičovských zvířat,
|
|
—
|
podrobný popis histopatologických nálezů,
|
|
—
|
údaje o absorpci (jsou-li k dispozici),
|
|
—
|
statistické vyhodnocení výsledků, je-li to možné.
|
|
|
Interpretace výsledků
|
77.
|
Studie poskytne hodnocení reprodukční/vývojové toxicity spojené s podáváním opakovaných dávek. Vzhledem k tomu, že důraz je kladen na sledované vlastnosti všeobecné toxicity i na sledované vlastnosti reprodukční/vývojové toxicity, umožní výsledky této studie rozlišení mezi reprodukčními/vývojovými účinky vyskytujícími se za nepřítomnosti všeobecné toxicity a účinky projevujícími se pouze při úrovních, které jsou toxické i pro rodičovská zvířata (viz odstavce 7–11). Mohla by naznačit nutnost provést další výzkum a poskytnout vodítko pro návrh dalších studií. Jako pomůcka při interpretaci výsledků v oblasti reprodukce a vývoje by měl posloužit pokyn OECD č. 43 (19). Pokyn OECD č. 106 týkající se histologického hodnocení endokrinních a reprodukčních zkoušek u hlodavců (16) poskytuje informace o přípravě a hodnocení (endokrinních) orgánů a vaginálních stěrů, což může být pro tuto zkušební metodu užitečné.
|
LITERATURA
|
(1)
|
OECD (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. K dispozici na vyžádání u Organizace pro hospodářskou spolupráci a rozvoj, Paříž.
|
|
(2)
|
OECD (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokyo, 27th-29th October, 1992. K dispozici na vyžádání u Organizace pro hospodářskou spolupráci a rozvoj, Paříž.
|
|
(3)
|
Mitsumori K., Kodama Y., Uchida O., Takada K., Saito M. Naito K., Tanaka S., Kurokawa Y., Usami, M., Kawashima K., Yasuhara K., Toyoda K., Onodera H., Furukawa F., Takahashi M. and Hayashi Y. (1994). Confirmation Study, Using Nitro-Benzene, of the Combined Repeat Dose and Reproductive/ Developmental Toxicity Test Protocol Proposed by the Organization for Economic Cooperation and Development (OECD). J. Toxicol, Sci., 19, 141-149.
|
|
(4)
|
Tanaka S., Kawashima K., Naito K., Usami M., Nakadate M., Imaida K., Takahashi M., Hayashi Y., Kurokawa Y. and Tobe M. (1992). Combined Repeat Dose and Reproductive/Developmental Toxicity Screening Test (OECD): Familiarization Using Cyclophosphamide. Fundam. Environ. Toxicol., 18, 89-95.
|
|
(5)
|
OECD (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10th-11th March 1998, Available upon request at Organisation for Economic Cooperation and Development, Paris
|
|
(6)
|
OECD (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environment, Health and Safety Publications, Series on Testing and Assessment (No 217), Organisation for Economic Cooperation and Development, Paris.
|
|
(7)
|
OECD (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations, Environment, Health and Safety Publications, Series on Testing and Assessment, (No 19), Organisation for Economic Cooperation and Development, Paris.
|
|
(8)
|
Goldman J.M., Murr A.S., Buckalew A.R., Ferrell J.M.and Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, Part B, 80 (2), 84-97.
|
|
(9)
|
Sadleir R.M.F.S. (1979). Cycles and Seasons, in Auston C.R. and Short R.V. (Eds.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York.
|
|
(10)
|
IPCS (1986). Principles and Methods for the Assessment of Neurotoxicity Associated with Exposure to Chemicals. Environmental Health Criteria Document (No 60).
|
|
(11)
|
Moser V.C., McDaniel K.M. and Phillips P.M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of Amitraz. Toxicol. Appl. Pharmacol., 108, 267-283.
|
|
(12)
|
Meyer O.A., Tilson H.A., Byrd W.C. and Riley M.T. (1979). A Method for the Routine Assessment of Fore- and Hindlimb Grip Strength of Rats and Mice. Neurobehav. Toxicol., 1, 233-236.
|
|
(13)
|
Crofton K.M., Howard J.L., Moser V.C., Gill M.W., Reiter L.W., Tilson H.A., MacPhail R.C. (1991). Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol. 13, 599-609.
|
|
(14)
|
Gallavan R.H. Jr, J.F. Holson, D.G. Stump, J.F. Knapp and V.L. Reynolds. (1999). „Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights“, Reproductive Toxicology, 13: 383-390.
|
|
(15)
|
OECD (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environment, Health and Safety Publications, Series on Testing and Assessment (No 151). Organisation for Economic Cooperation and Development, Paris.
|
|
(16)
|
OECD (2009). Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment (No. 106) Organisation for Economic Cooperation and Development, Paris.
|
|
(17)
|
Hess RA and Moore BJ. (1993). Histological Methods for the Evaluation of the Testis. V: Methods in Reproductive Toxicology, Chapin RE and Heindel JJ (Eds.). Academic Press: San Diego, CA, pp. 52-85.
|
|
(18)
|
Latendresse JR, Warbrittion AR, Jonassen H, Creasy DM. (2002). Fixation of Testes and Eyes Using a Modified Davidson’s Fluid: Comparison with Bouin’s Fluid and Conventional Davidson’s fluid. Toxicol. Pathol. 30, 524-533.
|
|
(19)
|
OECD (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 43), Organisation for Economic Cooperation and Development, Paris.
|
|
(20)
|
OECD (2011), Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption (No 150), Organisation for Economic Cooperation and Development, Paris.
|
Dodatek 1
DEFINICE (VIZ TAKÉ (20) POKYN OECD Č. 150)
Androgenicita je schopnost chemické látky působit v organismu savců jako přirozený androgenní hormon (např. testosteron).
Antiandrogenicita je schopnost chemické látky potlačit v organismu savců působení přirozeného androgenního hormonu (např. testosteronu).
Antiestrogenicita je schopnost chemické látky potlačit v organismu savců působení přirozeného estrogenního hormonu (např. estradiolu 17ß).
Antityreoidální působení je schopnost chemické látky potlačit v organismu savců působení přirozeného hormonu štítné žlázy (např. T3).
Chemická látka je látka nebo směs.
Vývojová toxicita projev reprodukční toxicity představující prenatální, perinatální, postnatální, strukturální nebo funkční poruchy u potomstva.
Dávkou se rozumí množství podané zkoušené chemické látky. Dávka se vyjadřuje jako hmotnost zkoušené chemické látky na jednotku tělesné hmotnosti zvířete za den (např. mg/kg tělesné hmotnosti/den) nebo jako konstantní koncentrace v krmivu.
Dávkováním se rozumí obecný termín zahrnující dávku, četnost a trvání doby, po kterou je látka podávána.
Zřejmá toxicita je obecný termín popisující jasné příznaky toxicity po podání zkoušené chemické látky. Ty by měly být dostačující pro posouzení nebezpečnosti a měly by být takové, že při zvýšení podávané dávky lze očekávat rozvoj závažných toxických příznaků a pravděpodobnou mortalitu.
Porucha plodnosti představuje poruchy samčích nebo samičích reprodukčních funkcí nebo schopnosti.
Mateřská toxicita nežádoucí účinky na březí samice vyskytující se buď specificky (přímý účinek), nebo nespecificky (nepřímý účinek), které souvisí s graviditou.
NOAEL je zkratka pro angl. „no-observed-adverse-effect level“. Je to nejvyšší úroveň dávky, při které nejsou pozorovány žádné nepříznivé účinky spojené se zkouškou.
Estrogenicita je schopnost chemické látky působit v organismu savců jako přirozený estrogenní hormon (např. estradiol 17ß).
Reprodukční toxicita představuje škodlivé účinky na potomstvo a/nebo poruchy samčích nebo samičích reprodukčních funkcí nebo schopnosti.
Zkoušená chemická látka je jakákoli látka nebo směs zkoušená touto zkušební metodou.
Tyreoidální působení je schopnost chemické látky působit jako přirozený hormon štítné žlázy (např. T3) v organismu savců.
Validace je vědecký proces, jehož cílem je charakterizovat provozní požadavky a provozní omezení zkušební metody a prokázat její spolehlivost a relevantnost pro určitý konkrétní účel.
Dodatek 2
DIAGRAM ČASOVÉHO PLÁNU EXPERIMENTU SE ZNÁZORNĚNÍM MAXIMÁLNÍ DÉLKY STUDIE NA ZÁKLADĚ CELÉHO ČTRNÁCTIDENNÍHO OBDOBÍ PÁŘENÍ
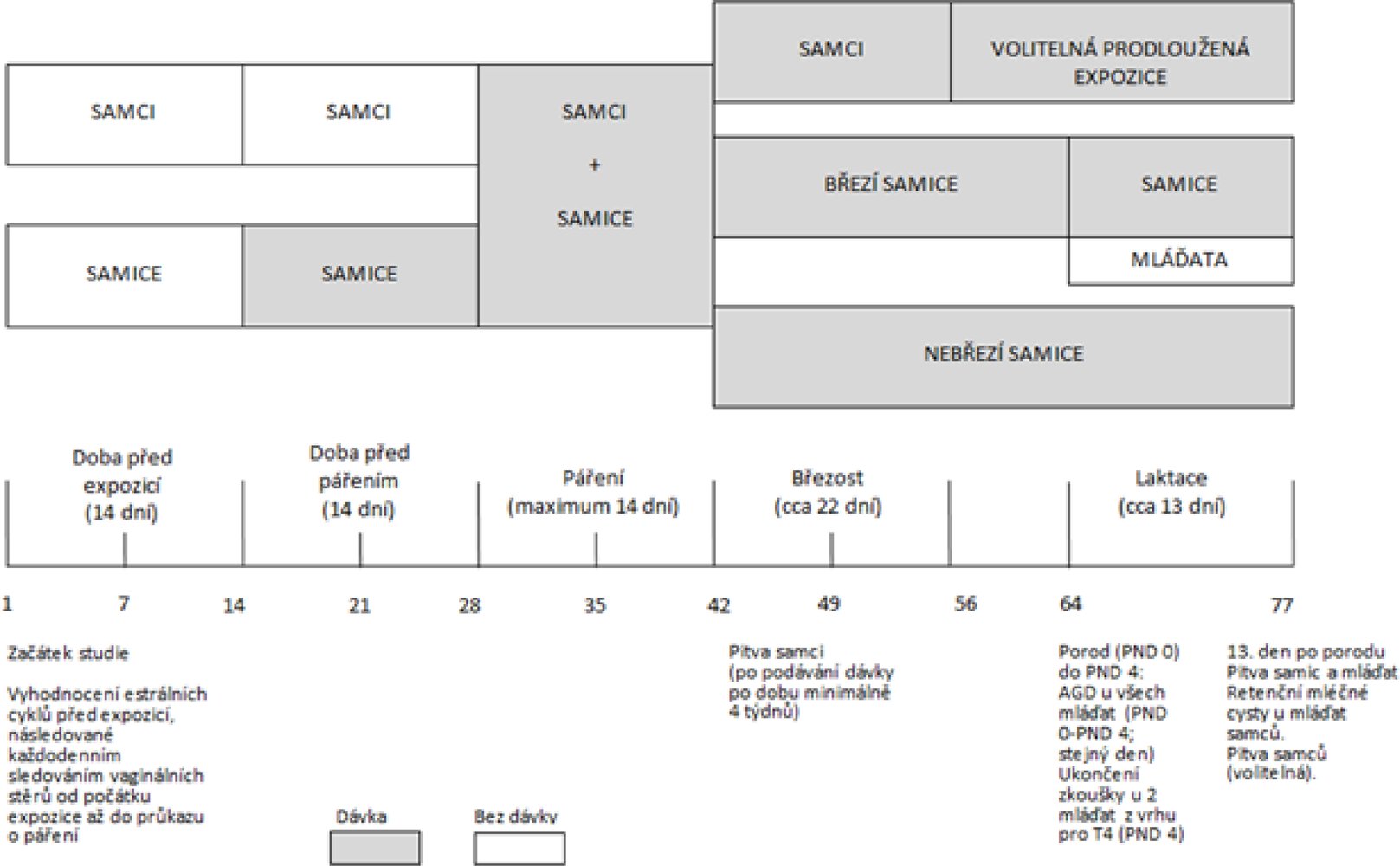
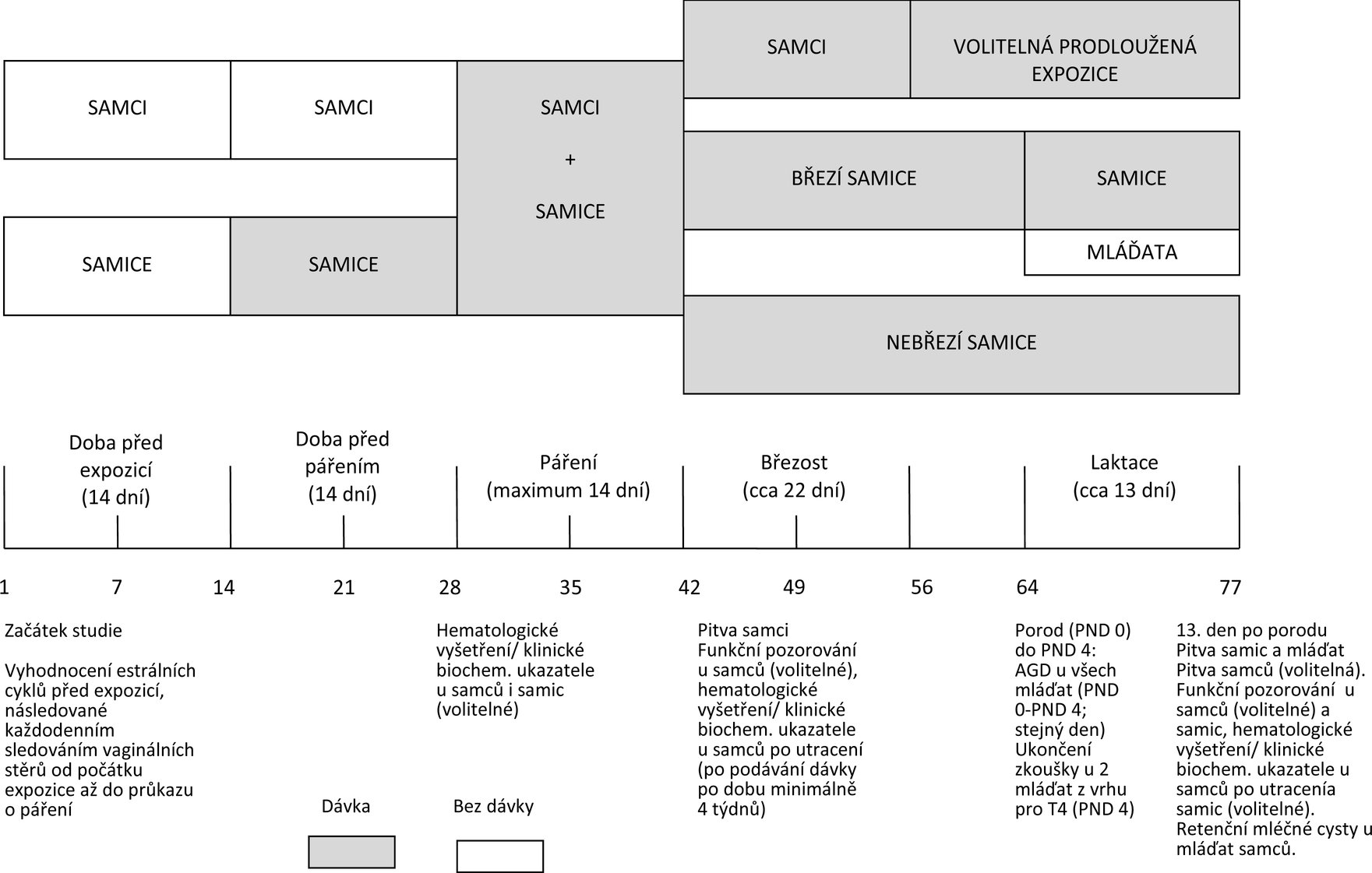
Dodatek 3
SOUHRNNÁ ZPRÁVA O ÚČINCÍCH NA REPRODUKCI/VÝVOJ VE FORMĚ TABULKY
|
POZOROVÁNÍ
|
HODNOTY
|
|
Dávka (jednotky).......
|
0 (kontrola)
|
...
|
...
|
...
|
...
|
|
Páry na začátku (N)
|
|
|
|
|
|
|
Estrální cyklus (alespoň střední délka a frekvence nepravidelných cyklů)
|
|
|
|
|
|
|
Samice vykazující známky kopulace (N)
|
|
|
|
|
|
|
Zabřezlé samice (N)
|
|
|
|
|
|
|
Dny zabřeznutí 1–5 (N)
|
|
|
|
|
|
|
Dny zabřeznutí 6–...(
(23)
) (N)
|
|
|
|
|
|
|
Březost≤21 dnů (N)
|
|
|
|
|
|
|
Březost = 22 dnů (N)
|
|
|
|
|
|
|
Březost ≥ 23 dnů (N)
|
|
|
|
|
|
|
Samice s živými novorozenými mláďaty (N)
|
|
|
|
|
|
|
Samice s živými novorozenými mláďaty 4. den po porodu (N)
|
|
|
|
|
|
|
Implantace/samice (průměr)
|
|
|
|
|
|
|
Živá mláďata/samice při narození (průměr)
|
|
|
|
|
|
|
Živá mláďata/samice 4. den (průměr)
|
|
|
|
|
|
|
Poměr pohlaví (samec/samice) při narození (průměr)
|
|
|
|
|
|
|
Poměr pohlaví (samec/samice) 4. den (průměr)
|
|
|
|
|
|
|
Hmotnost vrhu při narození (průměr)
|
|
|
|
|
|
|
Hmotnost vrhu 4. den (průměr)
|
|
|
|
|
|
|
Hmotnost mláďat při narození (průměr)
|
|
|
|
|
|
|
Hmotnost mláďat v době měření AGD (průměr samců, průměr samic)
|
|
|
|
|
|
|
AGD mláďat ve stejný postnatální den, při narození – 4. den (průměr samců, průměr samic, uvést PND)
|
|
|
|
|
|
|
Hmotnost mláďat 4. den (průměr)
|
|
|
|
|
|
|
Hmotnost mláďat 13. den (průměr)
|
|
|
|
|
|
|
Retenční mléčné cysty u mladých samců 13. den (průměr)
|
|
|
|
|
|
|
ABNORMÁLNÍ MLÁĎATA
|
|
Samice s 0
|
|
|
|
|
|
|
Samice s 1
|
|
|
|
|
|
|
Samice s ≥ 2
|
|
|
|
|
|
|
ZTRÁTA POTOMSTVA
|
|
Prenatální (implantace minus živě narozená mláďata)
|
|
Samice s 0
|
|
|
|
|
|
|
Samice s 1
|
|
|
|
|
|
|
Samice s 2
|
|
|
|
|
|
|
Samice s ≥ 3
|
|
|
|
|
|
|
Postnatální (živě narozená mláďata minus živá mláďata 13. den po porodu)
|
|
Samice s 0
|
|
|
|
|
|
|
Samice s 1
|
|
|
|
|
|
|
Samice s 2
|
|
|
|
|
|
|
Samice s ≥ 3
|
|
|
|
|
|
B.65 ZKUŠEBNÍ METODA S VYUŽITÍM MEMBRÁNOVÉ BARIÉRY IN VITRO PRO LEPTAVÉ ÚČINKY NA KŮŽI
ÚVOD
|
1.
|
Tato zkušební metoda je rovnocenná Pokynu OECD pro zkoušení č. 435 (2015). Leptavými účinky na kůži se označuje vyvolání nevratného poškození kůže, které se projevuje jako viditelná, do koria zasahující nekróza pokožky, po aplikaci zkoušené chemické látky, jak ji definuje globálně harmonizovaný systém klasifikace a označování chemických látek Organizace spojených národů (GHS OSN) (1) a nařízení Evropské unie (EU) č. 1272/2008 o klasifikaci, označování a balení látek a směsí (dále jen „nařízení CLP“) (24). Tato zkušební metoda, rovnocenná aktualizovanému Pokynu OECD pro zkoušení č. 435, představuje zkušební metodu s využitím membránové bariéry in vitro, kterou lze použít na určení žíravých chemických látek. Zkušební metoda využívá umělou membránu navrženou tak, aby reagovala na žíravé chemické látky podobně jako zvířecí kůže in situ.
|
|
2.
|
Leptavé účinky na kůži se tradičně posuzují aplikací zkoušené chemické látky na kůži živých zvířat, kdy se vyhodnotí rozsah poškození tkáně po stanoveném časovém období (2). Kromě této zkušební metody byla přijata řada jiných zkušebních metod in vitro jako alternativy (3) (4) ke standardnímu postupu zkoušení na kůži králíka in vivo (kapitola B.4 této přílohy, rovnocenná Pokynu OECD pro zkoušení č. 104), který se používá na určení žíravých chemických látek (2). Strategie odstupňovaného zkoušení a hodnocení podle systému GHS OSN pro hodnocení a klasifikaci leptavých účinků na kůži a dokument OECD k integrovanému přístupu ke zkouškám a posuzování (IATA) látek dráždivých/leptavých pro kůži doporučuje použití validovaných a uznávaných zkušebních metod in vitro podle modulů 3 a 4 (1) (5). Integrované přístupy ke zkouškám a posuzování popisují několik modulů, které seskupují informační zdroje a analytické nástroje, a i) poskytují návod na integraci a používání stávajících údajů ze zkoušek i jiných údajů pro posouzení potenciálu dráždivých a leptavých účinků chemických látek na kůži a ii) navrhují přístup, když je nutné další zkoušení, včetně případů, kdy jsou zjištěny negativní výsledky (5). V tomto modulárním přístupu mohou být pozitivní výsledky zkušebních metod in vitro použity pro klasifikaci chemické látky jako žíravé bez nutnosti zkoušek na zvířatech, čímž se snižuje a kultivuje použití zvířat a vyloučí se bolest a stres, k nimž může dojít, když jsou pro tento účel používána zvířata.
|
|
3.
|
Byly dokončeny validační studie pro model s membránovou bariérou in vitro, které jsou komerčně dostupné jako Corrositex® (6)(7)(8), vykazující celkovou přesnost predikce leptavých účinků na kůži 79 % (128/163), citlivost 85 % (76/89) a specifičnost 70 % (52/74) u databáze 163 látek a směsí (7). Na základě uznávané validity byla tato validovaná referenční metoda (VRM) doporučena pro použití jako součást strategie odstupňovaného zkoušení pro hodnocení potenciálního rizika leptavých účinků chemických látek na kůži (5) (7). Před použitím použít modelu s membránovou bariérou in vitro pro leptavé účinky na kůži pro regulační účely, je zapotřebí stanovit jeho spolehlivost, relevantnost (přesnost) a omezení týkající se jeho navrhovaného použití, aby se zajistila jeho podobnost s VRM (9) v souladu se stanovenými požadavky standardů funkčnosti (10). Vzájemné uznávání údajů bude v rámci OECD zaručeno až poté, co bude každá navrhovaná nebo aktualizovaná metoda podle standardů funkčnosti podrobena revizi a zahrnuta do rovnocenného pokynu OECD pro zkoušení. V současnosti se Pokyn OECD pro zkoušení č. 435 a tato zkušební metoda týká pouze jedné metody in vitro, a to komerčně dostupného modelu Corrositex®.
|
|
4.
|
Jiné zkušební metody pro zkoušení leptavých účinků na kůži vycházejí z použití rekonstruované lidské kůže (Pokyn OECD pro zkoušení č. 431) (3) a izolované kůže potkana (Pokyn OECD pro zkoušení č. 430) (4). Tento pokyn také stanoví podrobnější klasifikaci žíravých chemických látek do tří podkategorií žíravých látek podle systému GHS OSN a tří přepravních obalových skupin podle systému OSN pro nebezpečí žíravé látky. Tento pokyn byl původně přijat v roce 2006 a v roce 2015 byl aktualizován o odkazy na pokyny IATA a aktualizaci seznamu vhodných látek.
|
DEFINICE
|
5.
|
Použité definice jsou uvedeny v dodatku.
|
VÝCHOZÍ ÚVAHY A OMEZENÍ
|
6.
|
Zkouška popsaná v této zkušební metodě umožňuje určení žíravých zkoušených látek a dále umožňuje podrobnou klasifikaci žíravých zkoušených chemických látek podle systému GHS OSN/CLP (tabulka 1). Kromě toho může být taková zkušební metoda použita pro rozhodnutí o žíravosti a nežíravosti konkrétních tříd chemických látek, např. organických a anorganických kyselin, derivátů kyselin (25) a zásad pro některé účely zkoušení v oblasti přepravy (7)(11)(12). Tato zkušební metoda popisuje obecný postup podobný validované referenční zkušební metodě (7). I když tato zkušební metoda neposkytuje odpovídající informace o dráždivosti pro kůži, je třeba poznamenat, že zkušební metoda B.46 (rovnocenná Pokynu OECD pro zkoušení č. 439) řeší konkrétně zdravotní účinky dráždivosti pro kůži in vitro (13). Pro úplné vyhodnocení lokálních účinků na kůži po jednotlivých kožních expozicích je třeba postupovat podle pokynů OECD k integrovanému přístupu ke zkouškám a posuzování (5).
Tabulka 1
Kategorie a podkategorie leptavých účinků na kůži podle systému GHS OSN (26)
|
Kategorie žíravých látek (kategorie 1) (pro orgány nepoužívající podkategorie)
|
Podkategorie potenciálně žíravých látek (26) (pro orgány používající podkategorie, včetně nařízení CLP)
|
Žíravá u ≥ 1 ze 3 zvířat
|
|
Expozice
|
Pozorované příznaky
|
|
Žíravá
|
Podkategorie žíravosti 1A
|
≤ 3 minuty
|
≤ 1 hodina
|
|
Podkategorie žíravosti 1B
|
> 3 minuty / ≤ 1 hodina
|
≤ 14 dnů
|
|
Podkategorie žíravosti 1C
|
> 1 hodina / ≤ 4 hodiny
|
≤ 14 dnů
|
|
|
7.
|
Omezení této validované referenční metody (7) spočívá v tom, že mnoho nežíravých chemických látek a některé žíravé chemické látky nemusí být vhodné pro zkoušení na základě výsledků úvodního testu kompatibility (viz odstavec 13). Vodné chemické látky s pH v rozsahu 4,5 až 8,5 často nejsou pro zkoušení vhodné; 85 % chemických látek zkoušených v tomto rozmezí pH však nemělo ve zkouškách na zvířatech leptavé účinky (7). Metoda s membránovou bariérou in vitro může být použita na zkoušení pevných látek (rozpustných nebo nerozpustných ve vodě), kapalin (vodných a nevodných) a emulzí. Zkoušené chemické látky nevyvolávající zjistitelnou změnu při testu kompatibility (tj. barevná změna v chemickém detekčním systému (CDS) validované referenční zkušební metody) však nemohou být testovány metodou s membránovou bariérou a je třeba je testovat pomocí jiných zkušebních metod.
|
PRINCIP ZKOUŠKY
|
8.
|
Zkušební systém sestává ze dvou komponentů: syntetické makromolekulární biobariéry a chemického detekčního systému (CDS); tato zkušební metoda detekuje přes CDS poškození membránové bariéry způsobené žíravými zkoušenými chemickými látkami po aplikaci zkoušené chemické látky na povrch syntetické makromolekulární membránové bariéry (7), pravděpodobně stejným mechanismem (mechanismy) žíravosti, který působí na živou kůži.
|
|
9.
|
Penetraci membránové bariéry (neboli proniknutí) lze měřit podle počtu zákroků nebo pomocí chemického detekčního systému, včetně změny barvy pH indikačního barviva či některé jiné vlastnosti indikačního roztoku pod bariérou.
|
|
10.
|
Membránová bariéra by měla být stanovena jako validní, tj. relevantní a spolehlivá, pro zamýšlené použití. Zahrnuje to zajištění, aby byly různé přípravky shodné, pokud jde o bariérové vlastnosti, např. aby dokázaly zachovat bariéru proti nežíravým chemickým látkám a aby umožňovaly klasifikaci leptavých vlastností chemických látek do různých podkategorií žíravosti podle systému GHS OSN (1). Klasifikace vychází z doby, která je potřebná k tomu, aby chemická látka pronikla přes membránovou bariéru do indikačního roztoku.
|
PROKÁZÁNÍ ZPŮSOBILOSTI
|
11.
|
Dříve než začnou laboratoře rutinně používat metodu s membránovou bariérou in vitro, která dodržuje tuto zkušební metodu, měly by prokázat svou odbornou způsobilost správným určením klasifikace dvanácti vhodných látek doporučených v tabulce 2. V situacích, kdy některá z uvedených látek není k dispozici, lze v odůvodněných případech použít jinou látku, u níž jsou k dispozici odpovídající referenční údaje o použití in vivo a in vitro (např. ze seznamu referenčních chemických látek (10)), pokud budou použita stejná kritéria výběru jako kritéria popsaná v tabulce 1.
Tabulka 2
Vhodné látky
(27)
|
Látka (28)
|
Číslo CAS
|
Třída chemických látek
|
Podkategorie GHS OSN in vivo
(29)
|
Podkategorie GHS OSN in vitro
(29)
|
|
Dihydrát fluoridu boritého
|
13 319 -75-0 Org.
|
Anorganické kyseliny
|
1A
|
1A
|
|
Kyselina dusičná
|
7 697 -37-2
|
Anorganické kyseliny
|
1A
|
1A
|
|
Chlorid fosforečný
|
10 026 -13-8
|
Anorganické kyseliny
|
1A
|
1A
|
|
Valerylchlorid
|
638-29-9
|
Chloridy kyselin
|
1B
|
1B
|
|
Hydroxid sodný
|
1 310 -73-2
|
Anorganické zásady
|
1B
|
1B
|
|
1-(2-aminoethyl) piperazin
|
140-31-8
|
Alifatické aminy
|
1B
|
1B
|
|
Benzensulfonylchlorid
|
98-09-9
|
Chloridy kyselin
|
1C
|
1C
|
|
N,N-dimethylbenzylamin
|
103-83-3
|
Aniliny
|
1C
|
1C
|
|
Tetraethylenepentamin
|
112-57-2
|
Alifatické aminy
|
1C
|
1C
|
|
Eugenol
|
97-53-0
|
Fenoly
|
NC
|
NC
|
|
Nonylakrylát
|
2 664 -55-3
|
Akryláty/methakryláty
|
NC
|
NC
|
|
Hydrouhličitan sodný
|
144-55-8
|
Anorganické soli
|
NC
|
NC
|
|
POSTUP
|
12.
|
V dalších odstavcích jsou popsány složky a postupy zkušební metody využívající umělou membránovou bariéru na posouzení leptavosti (7) (15) a vycházející ze stávající validované referenční metody, tj. komerčně dostupného testu Corrositex®. Membránová bariéra a kompatibilní/indikační a kategorizační roztoky mohou být zhotoveny, připraveny nebo pořízeny komerčně, jako je tomu v případě validované referenční metody Corrositex®. Protokol vzorové zkušební metody pro validovanou referenční zkušební metodu je k dispozici (7). Zkoušení je třeba provádět při teplotě okolí (17–25 oC) a jednotlivé složky by měly splňovat tyto podmínky.
|
Zkouška kompatibility zkoušené chemické látky
|
13.
|
Před provedením zkoušky s membránovou bariérou se provádí zkouška kompatibility za účelem stanovení, zda chemický detekční systém dokáže zkoušenou chemickou látku detekovat. Jestliže chemický detekční systém nedetekuje zkoušenou chemickou látku, není zkušební metoda využívající membránovou bariéru vhodná pro posouzení potenciálních leptavých účinků této konkrétní zkoušené chemické látky a je třeba použít jinou zkušební metodu. Chemický detekční systém a podmínky expozice použité pro zkoušku kompatibility by měly odrážet expozici při následné zkoušce s membránovou bariérou.
|
Zkouška časové kategorie zkoušené chemické látky
|
14.
|
Pokud je to pro zkušební metodu vhodné, měla by být zkoušená chemická látka, která prošla zkouškou kompatibility, podrobena zkoušce časové kategorie, tj. screeningové zkoušce na rozlišení slabých a silných kyselin nebo zásad. Například ve validované referenční zkušební metodě se časová kategorizační zkouška používá na zjištění, který ze dvou časových rámců má být použit, a to podle toho, zda je detekována významná kyselá nebo zásaditá rezerva. Na stanovení žíravosti a podkategorie leptavých účinků na kůži podle systému GHS OSN by se měly použít dva různé limitní časové rámce, a to podle kyselé nebo zásadité rezervy zkoušené chemické látky.
|
SLOŽKY ZKUŠEBNÍ METODY S MEMBRÁNOVOU BARIÉROU
Membránová bariéra
|
15.
|
Membránová bariéra sestává ze dvou komponentů: bílkovinného makromolekulárního vodného gelu a propustné nosné membrány. Bílkovinný gel by měl být nepropustný pro kapaliny a pevné látky, ale při naleptání by měl začít propouštět. Kompletně zhotovená membránová bariéra by měla být uchovávána za předem určených podmínek, které prokazatelně vylučují znehodnocení gelu, např. vysušení, mikrobiální růst, posun, prasknutí, které by mohlo degradovat jeho funkčnost. Je třeba stanovit přijatelnou dobu uchovávání a po jejím uplynutí přípravky s membránovou bariérou nepoužívat.
|
|
16.
|
Propustná nosná membrána představuje mechanickou oporu pro bílkovinný gel v průběhu procesu vzniku gelu a expozice zkoušené chemické látce. Nosná membrána by měla zabránit průhybu nebo posunu gelu a měla by být dobře prostupná pro všechny zkoušené chemické látky.
|
|
17.
|
Bílkovinový gel složený z bílkovin, např. keratinu, kolagenu nebo směsí bílkovin, který vytváří gelovou matrici, slouží jako cíl pro zkoušenou chemickou látku. Bílkovinový materiál se umístí na povrch nosné membrány a umožní se prostup do gelu, než se membránová bariéra umístí na indikační roztok. Bílkovinový gel by měl mít všude stejnou tloušťku a hustotu a neměly by v něm být žádné vzduchové bublinky ani vady, které by mohly ovlivnit jeho funkční neporušenost.
|
Chemický detekční systém (CDS)
|
18.
|
Indikační roztok, což je stejný roztok, jaký se používá pro zkoušku kompatibility, by měl reagovat na přítomnost zkoušené chemické látky. Používá se pH indikační barvivo nebo kombinace barviv, např. kresolová červeň a methyloranž, které ukážou změnu barvy v reakci na přítomnost zkoušené chemické látky. Systém měření může být vizuální nebo elektronický.
|
|
19.
|
Je třeba posoudit relevantnost a spolehlivost detekčních systémů, které jsou vyvinuty pro detekci průchodu zkoušené chemické látky přes bariérovou membránu, aby bylo možné prokázat rozsah chemických látek, které lze detekovat, a kvantitativní limity detekce.
|
POSTUP ZKOUŠKY
Sestavení komponentů zkušební metody
|
20.
|
Membránová bariéra se vloží do lavičky (nebo zkumavky) obsahující indikační roztok tak, aby byla nosná membrána úplně v kontaktu s indikačním roztokem a aby na ní nebyly žádné vzduchové bublinky. Je třeba dbát na to, aby bariéra nebyla porušena.
|
Aplikace zkoušené chemické látky
|
21.
|
Na horní povrch membránové bariéry se pečlivě nanese a rovnoměrně rozprostře vhodné množství zkoušené chemické látky, např. 500 μl kapaliny nebo 500 mg jemně rozdrcené pevné látky (7). Pro každou zkoušenou chemickou látku a odpovídající kontroly se připraví vhodný počet replikátů, např. čtyři (7) (viz odstavce 23 až 25). Čas nanesení zkoušené chemické látky na membránovou bariéru se zaznamená. Aby bylo zajištěno přesné zaznamenání krátkých dob leptání, časy aplikace zkoušené chemické látky do lahviček s replikáty se rozloží.
|
Měření průniku přes membránovou bariéru
|
22.
|
Každá lahvička se řádně sleduje a zaznamená se čas první změny indikačního roztoku, tj. průnik přes bariéru, a určí se doba, která uplynula mezi aplikací a průnikem přes membránovou bariéru.
|
Kontroly
|
23.
|
U zkoušek, které zahrnují použití vehikula nebo rozpouštědla se zkoušenou chemickou látkou, by vehikulum nebo rozpouštědlo mělo být kompatibilní se systémem membránové bariéry, tj. nemělo by narušit neporušenost systému membránové bariéry a měnit leptavé účinky zkoušené chemické látky. V případě potřeby je třeba kontrolu s rozpouštědlem (nebo vehikulem) zkoušet souběžně se zkoušenou chemickou látkou, aby se prokázala kompatibilita rozpouštědla se systémem membránové bariéry.
|
|
24.
|
Pozitivní (leptavé účinky) kontrolu se střední žíravostí, např. 110 ± 15 mg hydroxidu sodného (podkategorie žíravých látek 1B podle systému GHS OSN) (7), je třeba provádět souběžně se zkoušenou chemickou látkou, aby bylo možné posoudit, zda zkušební systém funguje přijatelným způsobem. Pro hodnocení relativního leptavého potenciálu žíravé zkoušené chemické látky může být užitečná druhá pozitivní kontrola s látkou ze stejné chemické třídy jako zkoušená chemická látka. Pozitivní kontrolu (kontroly) je třeba volit tak, aby byly její leptavé účinky střední (např. podkategorie 1B podle GHS OSN), aby bylo možné detekovat změny doby průniku, které by mohly být nepřijatelně delší nebo kratší než stanovená referenční hodnota, což by naznačovalo, že zkušební systém nefunguje správně. Pro tento účel jsou extrémně žíravé (podkategorie 1A podle GHS OSN) nebo nežíravé chemické látky jen málo vhodné. Žíravá chemická látka podkategorie 1B podle GHS OSN by umožňovala detekci příliš rychlé nebo příliš pomalé doby průniku. Pro pozitivní kontrolu by mohla být použita slabě žíravá látka (podkategorie 1C podle GHS OSN) pro měření schopnosti zkušební metody konzistentně rozlišovat mezi slabě žíravými a nežíravými chemickými látkami. Bez ohledu na použitý přístup by měl být přijatelný rozsah odezvy pozitivní kontroly stanoven podle dosavadního rozsahu dob průniku u použité pozitivní kontroly (kontrol), například střední hodnota ± 2–3 směrodatné odchylky. V každé studii by měla být přesná doba průniku stanovena pro pozitivní kontrolu tak, aby mohly být detekovány odchylky mimo přijatelný rozsah.
|
|
25.
|
Jako další opatření pro kontrolu kvality za účelem prokázání funkčnosti membránové bariéry by měla být souběžně se zkoušenou chemickou látkou provedena také negativní (látka bez leptavých účinků) kontrola, např. 10 % kyselina citronová, 6 % kyselina propionová (7).
|
Kritéria přijatelnosti studie
|
26.
|
Na predikci leptavých účinků zkoušené chemické látky se podle stanovených časových parametrů pro jednotlivé podkategorie leptavých účinků GHS OSN použije doba (v minutách), která uplynula mezi aplikací zkoušené chemické látky na membránovou bariéru a průnikem přes bariéru. Aby mohla být studie považována za přijatelnou, měla by souběžná pozitivní kontrola ukázat očekávanou dobu průniku (např. pokud je jako pozitivní kontrola použit hydroxid sodný, je doba průniku 8–16 minut), souběžná negativní kontrola by neměla vykazovat leptavé účinky, a pokud je zařazena souběžná kontrola s rozpouštědlem, neměla by tato kontrola vykazovat leptavé účinky ani by neměla měnit potenciál žíravosti zkoušené chemické látky. Před rutinním používáním metody, která se řídí touto zkušební metodou, by laboratoře měly prokázat odbornou způsobilost při použití dvanácti látek doporučených v tabulce 2. U nových metod typu „mee-too“ vyvinutých podle této zkušební metody, které jsou strukturálně a funkčně podobné validované referenční metodě (14), by měly být použity předem definované standardy funkčnosti na prokázání spolehlivosti a přesnosti nové metody, než bude tato metoda použita pro zkoušení pro regulační účely (10).
|
Interpretace výsledků a klasifikace žíravosti zkoušených chemických látek
|
27.
|
Pro klasifikaci zkoušené chemické látky do podkategorií žíravosti podle systému GHS OSN (1), případně podle přepravních obalových skupin OSN (16) se používá doba (v minutách), která uplynula mezi aplikací zkoušené chemické látky na membránovou bariéru a průnikem přes bariéru. Pro každou navrhovanou zkušební metodu jsou stanoveny hraniční časové hodnoty pro každou ze tří podkategorií žíravosti. Konečné rozhodnutí o hraničních časových hodnotách by mělo vzít v úvahu nutnost minimalizovat podhodnocenou klasifikaci rizika leptavých účinků (tj. falešně negativní výsledky). V současném pokynu pro zkoušení by měly být použity hraniční časové hodnoty testu Corrositex® uvedené v tabulce 3, neboť tento test představuje jedinou zkušební metodu, která v současnosti odpovídá pokynu pro zkoušení (7).
Tabulka 3
Predikční model Corrositex®
|
Střední doba průniku (min)
|
Predikce podle GHS OSN (32)
|
|
Zkoušené chemické látky kategorie 1 (30) (stanovené podle kategorizační zkoušky metody)
|
Zkoušené chemické látky kategorie 2 (31) (stanovené podle kategorizační zkoušky metody)
|
|
0–3 min
|
0–3 min
|
Žíravá doplňková podkategorie 1A
|
|
> 3 až 60 min
|
> 3 až 30 min
|
Žíravá doplňková podkategorie 1B
|
|
> 60 až 240 min
|
> 30 až 60 min
|
Žíravá doplňková podkategorie 1C
|
|
> 240 min
|
> 60 min
|
Nežíravá
|
|
ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV
Údaje
|
28.
|
Doba (v minutách), která uplynula mezi aplikací a průnikem přes bariéru u zkoušené chemické látky a pozitivní kontroly (kontrol), se vykazuje ve formě tabulky jako údaje o jednotlivých replikátech a střední hodnoty ± směrodatné odchylky pro každou zkoušku.
|
ZÁVĚREČNÁ ZPRÁVA
|
29.
|
Závěrečná zpráva by měla obsahovat tyto informace:
|
|
Zkoušené chemické látky a kontrolní látky
|
—
|
jednosložková látka: chemická identifikace, např. název podle IUPAC nebo CAS, číslo CAS, kód SMILES nebo InChI, strukturní vzorec, čistota, případně chemická identita nečistot, je-li to prakticky možné, atd.
|
|
—
|
vícesložková látka, UVCB a směs: charakterizované pokud možno chemickou identitou (viz výše), kvantitativním výskytem a relevantními fyzikálně-chemickými vlastnostmi složek,
|
|
—
|
fyzický vzhled, rozpustnost ve vodě a další relevantní fyzikálně-chemické vlastnosti,
|
|
—
|
zdroj, číslo šarže, jsou-li k dispozici,
|
|
—
|
případně ošetření zkoušené chemické látky / kontrolní látky před zkoušením (např. zahřátí, mletí),
|
|
—
|
stabilita zkoušené chemické látky, datum použitelnosti nebo datum opětovné analýzy, je-li známo,
|
|
|
|
Vehikulum:
|
—
|
identifikace, koncentrace (podle potřeby), použitý objem,
|
|
—
|
zdůvodnění výběru vehikula.
|
|
|
|
Model s membránovou bariérou in vitro a použitý protokol, včetně prokázané přesnosti a spolehlivosti
|
|
|
Zkušební podmínky:
|
—
|
popis použitého zařízení a postupů přípravy,
|
|
—
|
zdroj a složení použité membránové bariéry in vitro,
|
|
—
|
složení a vlastnosti indikačního roztoku,
|
|
—
|
množství zkoušené chemické látky a kontrolní látky,
|
|
—
|
popis a odůvodnění časové kategorizační zkoušky,
|
|
—
|
popis použitých hodnotících kritérií a klasifikačních kritérií,
|
|
—
|
prokázání způsobilosti při provádění zkušební metody před rutinním použitím při zkoušení chemických látek pro prokázání způsobilosti.
|
|
|
|
Výsledky:
|
—
|
jednotlivá nezpracovaná data z jednotlivých zkoušek a kontrolních vzorků pro každý replikát ve formě tabulky,
|
|
—
|
popis ostatních pozorovaných účinků,
|
|
—
|
odvozená klasifikace s odkazem na použitý predikční model / kritéria rozhodování.
|
|
Diskuse o výsledcích
Závěry
|
LITERATURA
|
(1)
|
United Nations (UN) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS), Fifth Revised Edition, UN New York and Geneva, 2013. Dostupné na internetových stránkách: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(2)
|
Kapitola B.4 této přílohy, Akutní dráždivé/leptavé účinky na kůži.
|
|
(3)
|
Kapitola B.40a této přílohy, Leptavé účinky na kůži in vitro: zkušební metoda za použití rekonstruované lidské epidermis (RHE).
|
|
(4)
|
Kapitola B.40 této přílohy, Leptavé účinky na kůži in vitro: Transkutánní elektrický odpor (TER).
|
|
(5)
|
OECD (2015). Guidance Document on Integrated Approaches to Testing and Assessment of Skin Irritation/Corrosion. Environment, Health and Safety Publications, Series on Testing and Assessment, (No 203). Organisation for Economic Cooperation and Development, Paris.
|
|
(6)
|
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.-G. and Liebsch, M. (1998). The ECVAM International Validation Study on In Vitro Tests for Skin Corrosivity. 2. Results and Evaluation by the Management Team. Toxicology In Vitro 12, 483-524.
|
|
(7)
|
ICCVAM (1999). Corrositex®. An In Vitro Test Method for Assessing Dermal Corrosivity Potential of Chemicals. The Results of an Independent Peer Review Evaluation Coordinated by ICCVAM, NTP and NICEATM. NIEHS, NIH Publication (No 99-4495.)
|
|
(8)
|
Gordon V.C., Harvell J.D. and Maibach H.I. (1994). Dermal Corrosion, the Corrositex® System: A DOT Accepted Method to Predict Corrosivity Potential of Test Materials. In vitro Skin Toxicology-Irritation, Phototoxicity, Sensitization. Alternative Methods in Toxicology 10, 37-45.
|
|
(9)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications. Series on testing and Assessment (No 34).
|
|
(10)
|
OECD (2014). Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Membrane Barrier Test Method for Skin Corrosion in Relation to TG 435. Organisation for Economic Cooperation and Development, Paris. Dostupné na webových stránkách: http://www.oecd.org/chemicalsafety/testing/PerfStand-TG430-June14.pdf.
|
|
(11)
|
ECVAM (2001). Statement on the Application of the CORROSITEX® Assay for Skin Corrosivity Testing. 15th Meeting of ECVAM Scientific Advisory Committee (ESAC), Ispra, Italy. ATLA 29, 96-97.
|
|
(12)
|
U.S. DOT (2002). Exemption DOT-E-10904 (Fifth Revision). (September 20, 2002). Washington, D.C., U.S. DOT.
|
|
(13)
|
Kapitola B.46 této přílohy, Dráždivost pro kůži in vitro: Zkušební metoda za použití rekonstruované lidské epidermis. ICCVAM (2004). ICCVAM Recommended Performance Standards for In Vitro Test Methods for Skin Corrosion. NIEHS, NIH Publication No 04-4510. Dostupné na internetových stránkách: http://www.ntp.niehs.nih.gov/iccvam/docs/dermal_docs/ps/ps044510.pdf.
|
|
(14)
|
U.S. EPA (1996). Method 1120, Dermal Corrosion. Dostupné na internetových stránkách: http://www.epa.gov/osw/hazard/testmethods/sw846/pdfs/1120.pdf.
|
|
(15)
|
United Nations (UN) (2013). UN Recommendations on the Transport of Dangerous Goods, Model Regulations, 18th Revised Edition (Part, Chapter 2.8), UN, 2013. Dostupné na internetových stránkách: http://www.unece.org/fileadmin/DAM/trans/danger/publi/unrec/rev18/English/Rev18_Volume1_Part2.pdf.
|
Dodatek
DEFINICE
Přesnost
: přesnost shody mezi výsledky zkušební metody a přijatými referenčními hodnotami. Je to míra funkčnosti zkušební metody a jeden z aspektů její relevantnosti. Tento pojem se často používá místo pojmu „soulad“ a rozumí se jím podíl správných výsledků zkušební metody (9).
Chemická látka
: látka nebo směs.
Chemický detekční systém (CDS)
: vizuální nebo elektronický měřicí systém s indikačním roztokem, který reaguje na přítomnost zkoušené chemické látky, např. změnou indikačního barviva pH nebo kombinace barviv, jež ukáže barevnou změnu v reakci na přítomnost zkoušené chemické látky, nebo jinými druhy chemické či elektrochemické reakce.
Soulad
: je měřítkem funkčnosti u zkušebních metod poskytujících kategorické výsledky a je jedním z aspektů relevantnosti. Tento pojem se někdy používá místo pojmu „přesnost“ a je definován jako podíl všech zkoušených chemických látek, které jsou správně klasifikovány jako pozitivní nebo negativní. Soulad závisí do velké míry na prevalenci pozitivních výsledků u druhů studovaných zkoušených chemických látek (9).
GHS (Globálně harmonizovaný systém klasifikace a označování chemických látek)
: systém navrhující klasifikaci chemických látek (látek a směsí) podle standardizovaných typů a úrovní fyzikální, zdravotní a environmentální nebezpečnosti a zabývající se příslušnými komunikačními prvky, jako jsou piktogramy, signální slova, výroky o nebezpečí, výroky o bezpečnostních opatřeních a bezpečnostní datové listy, které obsahují informace o jejich nežádoucích účincích s ohledem na ochranu lidí (včetně zaměstnavatelů, pracovníků, přepravců, spotřebitelů a osob reagujících na nouzové situace) a životního prostředí (1).
IATA
: integrovaný přístup ke zkoušení a posuzování.
Směs
: směs nebo roztok složený ze dvou nebo více látek.
Jednosložková látka
: látka definovaná svým kvantitativním složením, v níž je přítomna jedna hlavní složka v míře alespoň 80 % hmot.
Vícesložková látka
: látka definovaná svým kvantitativním složením, v níž je přítomna více než jedna hlavní složka v koncentraci ≥ 10 % hmot. a < 80 % hmot. Vícesložková látka je výsledkem výrobního procesu. Rozdíl mezi směsí a vícesložkovou látkou spočívá v tom, že směs se získá smísením dvou nebo více látek bez chemické reakce. Vícesložková látka je výsledkem chemické reakce.
NC („non-corrosive“):: nežíravá.
Standardy funkčnosti
: standardy založené na validované referenční metodě, které slouží jako základ pro hodnocení srovnatelnosti navrhované zkušební metody, která je mechanicky a funkčně podobná. Zahrnují i) zásadní složky zkušební metody; ii) minimální seznam referenčních chemických látek vybraných z chemických látek používaných k prokázání přijatelné funkčnosti validované referenční metody; a iii) srovnatelné hladiny spolehlivosti a přesnosti založené na hodnotách získaných u validované referenční metody, jež by navrhovaná zkušební metoda měla prokázat při vyhodnocení za použití minimálního seznamu referenčních chemických látek (9).
Relevantnost
: popis vztahu zkoušky k účinku, který je předmětem zájmu, a také toho, zda je zkouška smysluplná a použitelná pro konkrétní účel. Vyjadřuje, do jaké míry zkušební metoda správně měří nebo predikuje biologický účinek, který je předmětem zájmu. Relevantnost zahrnuje také posouzení přesnosti (shody) zkušební metody (9).
Spolehlivost
: měření rozsahu, v jakém může být zkušební metoda v průběhu času reprodukovatelná v laboratořích a mezi laboratořemi, když se provádí s použitím stejného protokolu. Hodnotí se na základě výpočtu vnitrolaboratorní a mezilaboratorní reprodukovatelnosti (9).
Citlivost
: podíl všech pozitivních/aktivních chemických látek, které jsou na základě zkušební metody správně klasifikovány. Je měřítkem přesnosti zkušební metody, která poskytuje kategorické výsledky, a je důležitým prvkem při hodnocení relevantnosti zkušební metody (9).
Leptavé účinky na kůži in vivo
: vyvolání nevratného poškození kůže; zejména viditelné, do koria zasahující nekrózy pokožky do čtyř hodin po aplikaci zkoušené chemické látky. Reakce po poleptání se projevují vředy, krvácením, krvavými strupy a v závěru čtrnáctidenního pozorování ztrátou barvy v důsledku vyblednutí kůže, úplnými ložisky alopecie a jizvami. K vyhodnocení sporných lézí by se měla uvážit histopatologie.
Specifičnost
: podíl všech negativních/neaktivních chemických látek, které jsou na základě zkušební metody správně klasifikovány. Je měřítkem přesnosti zkušební metody, která poskytuje kategorické výsledky, a je důležitým prvkem při hodnocení relevantnosti zkušební metody (9).
Látka
: chemický prvek a jeho sloučeniny v přírodním stavu nebo získané výrobním postupem, včetně jakýchkoliv aditiv potřebných pro zachování jeho stability a všech nečistot pocházejících z použitého postupu, ale s vyloučením všech rozpouštědel, která je možno oddělit bez ovlivnění stability dané látky nebo změny jejího složení.
Zkoušená chemická látka
: jakákoli látka nebo směs zkoušená touto zkušební metodou.
UVCB
: látky s neznámým nebo proměnlivým složením, komplexní reakční produkty nebo biologický materiál.
B.66 ZKOUŠKY IN VITRO FORMOU STABILNĚ TRANSFEKOVANÉ TRANSAKTIVACE SLOUŽÍCÍ K DETEKCI AGONISTŮ A ANTAGONISTŮ ESTROGENOVÝCH RECEPTORŮ
VŠEOBECNÝ ÚVOD
Pokyn OECD vycházející z funkční způsobilosti
|
1.
|
Tato zkušební metoda odpovídá pokynu OECD pro zkoušení (TG) 455 (2016). Pokyn č. 455 je pokyn vycházející z funkční způsobilosti (PBTG), který popisuje metodiku zkoušek in vitro formou stabilně transfekované transaktivace sloužící k detekci agonistů a antagonistů estrogenových receptorů (analýzy ER TA). Skládá se z několika mechanisticky a funkčně podobných zkušebních metod zjišťování agonistů a antagonistů estrogenových receptorů (tj. ERα a/nebo ERβ) a měl by usnadnit vypracování nových podobných nebo modifikovaných zkušebních metod podle zásad pro validaci stanovených v dokumentu OECD o validaci a mezinárodním uznání nových nebo aktualizovaných zkušebních metod pro posuzování nebezpečnosti (1). Plně validovanými referenčními zkušebními metodami (dodatek 2 a dodatek 3), které představují základ pro tento pokyn vycházející z funkční způsobilosti, jsou:
|
—
|
analýza STTA (2) s využitím buněčné linie (h) ERα-HeLa-9903, a
|
|
—
|
analýza VM7Luc ER TA (3) s využitím buněčné linie (33) VM7Luc4E2, která převážně exprimuje hERα s určitým příspěvkem od hERββ(4)(5).
|
Pro vývoj a validaci podobných analýz stejného rizika jsou k dispozici standardy funkčnosti (PS) (6)(7), které by se měly používat. Umožňují včasnou změnu pokynu PBTG 455, aby mohly být do aktualizovaného pokynu přidány nové podobné analýzy; podobné analýzy však budou přidány až po přezkumu a dohodě s OECD, že splňují standardy funkčnosti. Analýzy zařazené do pokynu 455 mohou být použity libovolně pro řešení požadavků členských zemí OECD na výsledky zkoušek na transaktivaci estrogenových receptorů při využití vzájemného uznávání údajů podle postupu dohodnutého v rámci OECD.
|
Obecné informace a principy analýz zahrnutých do této zkušební metody
|
2.
|
V roce 1998 zahájila OECD činnost vysoké priority, jejíž podstatou je revize stávajících a vytvoření nových pokynů pro screening a zkoušení potenciálních endokrinních disruptorů. V roce 2012 byl revidován koncepční rámec OECD pro zkoušky a hodnocení potenciálních endokrinních disruptorů. Původní i revidovaný koncepční rámec je uveden v přílohách dokumentu OECD o standardizovaných pokynech pro zkoušení a posuzování endokrinních disruptorů (Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption) (8). Koncepční rámec je tvořen pěti úrovněmi, které odpovídají různým úrovním biologické komplexnosti. Analýzy ER transaktivace (TA) popsané v této zkušební metodě představují úroveň 2, která zahrnuje „analýzy in vivo poskytující údaje o vybraných endokrinních mechanismech/drahách“. Tato zkušební metoda je pro analýzy transaktivace (TA) in vitro určené na zjištění agonistů a antagonistů estrogenových receptorů (ER).
|
|
3.
|
Interakce estrogenů s estrogenovými receptory může ovlivnit transkripci estrogenem kontrolovaných genů, což může vést k indukci nebo inhibici buněčných procesů, včetně procesů nezbytných pro buněčnou proliferaci, normální vývoj plodu a reprodukční funkci (9)(10)(11). Narušení normálních estrogenních systémů může spustit nežádoucí účinky v oblasti normálního vývoje (ontogeneze), reprodukčního zdraví a správné funkce reprodukčního systému.
|
|
4.
|
Analýzy TA in vitro vycházejí z přímé nebo nepřímé interakce látek se specifickým receptorem, který reguluje transkripci reportérového genového produktu. Tyto analýzy se široce používají na hodnocení genové exprese regulované specifickými jadernými receptory, jako jsou estrogenové receptory (12)(13)(14)(15)(16). Byly navrženy pro detekci estrogenní transaktivace regulované estrogenovým receptorem (17)(18)(19). Existují minimálně dva hlavní podtypy jaderných estrogenových receptorů, α a β, které jsou kódovány odlišnými geny. Příslušné bílkoviny mají různé biologické funkce i různou distribuci v tkáních a vazebnou afinitu ligandů (20)(21)(22)(23)(24)(25)(26). Jaderné ERα zprostředkovávají klasickou estrogenní odpověď (27)(28)(29)(30), a proto je většina modelů, které jsou v současnosti vyvíjeny na měření aktivace nebo inhibice estrogenových receptorů, specifická pro ERα. Analýzy se používají k určení chemických látek, které aktivují (nebo inhibují) estrogenové receptory po vazbě na ligandy, kdy se poté komplex receptor–ligand naváže na specifické responzivní elementy DNA a transaktivuje reportérový gen, přičemž výsledkem je zvýšená buněčná exprese proteinového markeru. V těchto analýzách mohou být použity různé odpovědi reportérových genů. V systémech na bázi luciferázy transformuje enzym luciferáza substrát luciferin na bioluminiscenční produkt, který může být kvantitativně změřen luminometrem. Mezi další příklady běžných reportérových genů patří fluorescenční protein a gen LacZ, který kóduje β-galaktosidázu, enzym, který dokáže transformovat bezbarvý substrát X-gal (5- bromo-4-chloro-indolyl-galaktopyranosid) na modrý produkt, který lze kvantifikovat pomocí spektrofotometru. Tyto reportérové geny lze rychle a levně vyhodnotit pomocí komerčně dostupných testů.
|
|
5.
|
Validační studie analýz STTA a VM7Luc TA prokázaly svoji relevanci a spolehlivost pro zamýšlený účel (3)(4)(5)(30). Standardy funkčnosti pro analýzy ER TA na bázi luminiscence využívající buněčné linie prsu jsou uvedeny v hodnotící zprávě výboru ICCVAM týkající se zkušební metody LUMI-CELL® ER (VM7Luc ER TA): An In Vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals (3). Tyto standardy funkčnosti byly upraveny, aby je bylo možné použít na analýzu STTA i analýzu VM7Luc TA (2).
|
|
6.
|
Definice a zkratky používané v této zkušební metodě jsou popsány v dodatku 1.
|
Platnost a omezení analýz TA
|
7.
|
Tyto analýzy jsou navrženy pro účely screeningu a stanovení priorit, ale mohou také poskytovat mechanistické informace, které lze použít při analýze průkaznosti výsledků. Týkají se transaktivace indukované chemickou vazbou na estrogenové receptory v systému in vitro. Výsledky proto nelze přímo extrapolovat na komplexní signalizaci a regulaci intaktního endokrinního systému in vivo.
|
|
8.
|
Transaktivace zprostředkovaná estrogenovými receptory se považuje za jeden z klíčových mechanismů narušení endokrinního systému (ED), i když existují i jiné mechanismy, jejichž prostřednictvím může k tomuto narušení dojít, včetně i) interakcí s jinými receptory a enzymatickými systémy v endokrinním systému, ii) syntézy hormonů, iii) metabolické aktivace anebo inaktivace hormonů, iv) distribuce hormonů do cílových tkání a v) clearance hormonů z těla. Žádná z analýz podle této zkušební metody se nezabývá těmito způsoby účinku.
|
|
9.
|
Tato zkušební metoda se zabývá schopností chemických látek aktivovat (tj. účinkovat jako agonisté) a také potlačovat (tj. účinkovat jako antagonisté) transkripci závislou na estrogenových receptorech. Některé chemické látky mohou v závislosti na druhu buněk vykazovat agonistické i antagonistické účinky a jsou známy jako selektivní modulátory estrogenových receptorů (SERM). Chemické látky, které jsou v těchto analýzách negativní, by před vyvozením závěru, že daná chemická látka se na receptor neváže, mohly být posouzeny v testu vazby na estrogenové receptory. Dále je pravděpodobné, že tyto analýzy budou informovat pouze o účinku původní molekuly, přičemž nelze zapomínat na omezenou metabolizační kapacitu buněčných systémů in vitro. Vzhledem k tomu, že při validaci byly použity pouze jednotlivé látky, nebyla řešena použitelnost na zkoušené směsi. Zkušební metoda je nicméně teoreticky použitelná i na zkoušení vícesložkových látek, UVCB a směsí. Před použitím této zkušební metody ke zkoušení vícesložkové látky, UVCB nebo směsi s cílem získat údaje pro zamýšlené regulační účely by mělo být zváženo, zda tato metoda může pro tento účel poskytnout přiměřené výsledky, a pokud ano, proč je tomu tak. Takové úvahy nejsou nutné, pokud existuje právní požadavek na zkoušení dané směsi.
|
|
10.
|
V tabulce 1 jsou pro informaci uvedeny výsledky testu na agonistické účinky 34 látek, které byly zkoušeny oběma plně validovanými referenčními zkušebními metodami popsanými v této zkušební metodě. Z těchto látek je na základě publikovaných zpráv, včetně analýz na vazbu na ER a TA in vitro, anebo uterotrofní analýzy 26 klasifikováno s určitostí jako agonisté ER a 8 negativně (2)(3)(18)(31) (32)(33)(34). V tabulce 2 uvedeny výsledky testu na antagonistické účinky 15 látek, které byly zkoušeny oběma plně validovanými referenčními zkušebními metodami popsanými v této zkušební metodě. Z těchto látek jsou na základě publikovaných zpráv, včetně analýz na vazbu na ER a TA in vitro, 4 klasifikovány s určitostí/pravděpodobností jako antagonisté ER a 10 negativně (2)(3)(18)(31). Pokud jde o údaje uvedené v tabulce 1 a v tabulce 2, byla mezi oběma referenčními zkušebními metodami u klasifikace všech látek s výjimkou jedné (Mifepristone) 100 % shoda u analýz antagonistů, přičemž každá látka byla správně klasifikována jako agonista/antagonista ER nebo negativně. Doplňkové informace o této skupině chemických látek i o dalších chemických látkách zkoušených v analýzách STTA a VM7Luc ER TA v průběhu validačních studií jsou uvedeny ve standardech funkčnosti pro zkoušku ERTA (6)(7), dodatek 2 (tabulky 1, 2 a 3).
|
Tabulka 1
Přehled výsledků z analýz STTA a VM7Luc ER TA u látek zkoušených pomocí obou analýz na agonistické účinky a klasifikovaných jako agonisté ER (POS) nebo negativní (NEG)
|
|
Látka
|
Číslo CAS
|
STTA Assay (35)
|
VM7Luc ER TA Assay (36)
|
Zdroj údajů pro klasifikaci (38)
|
|
ER TA aktivita
|
Hodnota PC10 (M)
|
Hodnota PC50
(2) (M)
|
ER TA aktivita
|
Hodnota EC50
(2), (37) (M)
|
Jiné ER TA (34)
|
ER vazba
|
Uterotrofní
|
|
1
|
17ß-estradiol (1)
|
50-28-2
|
POS
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
POS
|
5,63 × 10-12
|
POS (227/227)
|
POS
|
POS
|
|
2
|
17α-estradiol (1)
|
57-91-0
|
POS
|
7,24 × 10-11
|
6,44 × 10-10
|
POS
|
1,40 × 10-9
|
POS(11/11)
|
POS
|
POS
|
|
3
|
17-alfa-ethinylestradiol (1)
|
57-63-6
|
POS
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
POS
|
7,31 × 10-12
|
POS(22/22)
|
POS
|
POS
|
|
4
|
17β-trenbolon
|
10161-33-8
|
POS
|
1,78 × 10-8
|
2,73 × 10-7
|
POS
|
4,20 × 10-8
|
POS (2/2)
|
NT
|
NT
|
|
5
|
19-nortestosteron (1)
|
434-22-0
|
POS
|
9,64 × 10-9
|
2,71 × 10-7
|
POS
|
1,80 × 10-6
|
POS(4/4)
|
POS
|
POS
|
|
6
|
4-kumylfenol (1)
|
599-64-4
|
POS
|
1,49 × 10-7
|
1,60 × 10-6
|
POS
|
3,20 × 10-7
|
POS(5/5)
|
POS
|
NT
|
|
7
|
4-terc-oktylfenol (1)
|
140-66-9
|
POS
|
1,85 × 10-9
|
7,37 × 10-8
|
POS
|
3,19 × 10-8
|
POS(21/24)
|
POS
|
POS
|
|
8
|
Apigenin (1)
|
520-36-5
|
POS
|
1,31 × 10-7
|
5,71 × 10-7
|
POS
|
1,60 × 10-6
|
POS(26/26)
|
POS
|
NT
|
|
9
|
Atrazin (1)
|
1912-24-9
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (30/30)
|
NEG
|
NT
|
|
10
|
Bisfenol A (1)
|
80-05-7
|
POS
|
2,02 × 10-8
|
2,94 × 10-7
|
POS
|
5,33 × 10-7
|
POS (65/65)
|
POS
|
POS
|
|
11
|
Bisfenol B (1)
|
77-40-7
|
POS
|
2,36 × 10-8
|
2,11 × 10-7
|
POS
|
1,95 × 10-7
|
POS(6/6)
|
POS
|
POS
|
|
12
|
Butylbenzylfthalát (1)
|
85-68-7
|
POS
|
1,14 × 10-6
|
4,11 × 10-6
|
POS
|
1,98 × 10-6
|
POS(12/14)
|
POS
|
NEG
|
|
13
|
Kortikosteron (1)
|
50-22-6
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG(6/6)
|
NEG
|
NT
|
|
14
|
Kumestrol (1)
|
479-13-0
|
POS
|
1,23 × 10-9
|
2,00 × 10-8
|
POS
|
1,32 × 10-7
|
POS(30/30)
|
POS
|
NT
|
|
15
|
Daidzein (1)
|
486-66-8
|
POS
|
1,76 × 10-8
|
1,51 × 10-7
|
POS
|
7,95 × 10-7
|
POS(39/39)
|
POS
|
POS
|
|
16
|
Diethylstilbestrol (1)
|
56-53-1
|
POS
|
< 1,00 × 10-11
|
2,04 × 10-11
|
POS
|
3,34 × 10-11
|
POS(42/42)
|
POS
|
NT
|
|
17
|
Di-n-butylftalát
|
84-74-2
|
POS
|
4,09 × 10-6
|
|
POS
|
4,09 × 10-6
|
POS(6/11)
|
POS
|
NEG
|
|
18
|
Ethylparaben
|
120-47-8
|
POS
|
5,00 × 10-6
|
(žádná hodnota PC50)
|
POS
|
2,48 × 10-5
|
POS
|
|
NT
|
|
19
|
Estron (1)
|
53-16-7
|
POS
|
3,02 × 10-11
|
5,88 × 10-10
|
POS
|
2,34 × 10-10
|
POS(26/28)
|
POS
|
POS
|
|
20
|
Genistein (1)
|
446-72-0
|
POS
|
2,24 × 10-9
|
2,45 × 10-8
|
POS
|
2,71 × 10-7
|
POS(100/102)
|
POS
|
POS
|
|
21
|
Haloperidol
|
52-86-8
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
22
|
Kaempferol (1)
|
520-18-3
|
POS
|
1,36 × 10-7
|
1,21 × 10-6
|
POS
|
3,99 × 10-6
|
POS(23/23)
|
POS
|
NT
|
|
23
|
Kepon (1)
|
143-50-0
|
POS
|
7,11 × 10-7
|
7,68 × 10-6
|
POS
|
4,91 × 10-7
|
POS(14/18)
|
POS
|
NT
|
|
24
|
Ketokonazol
|
65277-42-1
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
25
|
Linuron (1)
|
330-55-2
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (8/8)
|
NEG
|
NT
|
|
26
|
meso-Hexestrol (1)
|
84-16-2
|
POS
|
< 1,00 × 10-11
|
2,75 × 10-11
|
POS
|
1,65 × 10-11
|
POS(4/4)
|
POS
|
NT
|
|
27
|
Methyltestosteron (1)
|
58-18-4
|
POS
|
1,73 × 10-7
|
4,11 × 10-6
|
POS
|
2,68 × 10-6
|
POS(5/6)
|
POS
|
NT
|
|
28
|
Morin
|
480-16-0
|
POS
|
5,43 × 10-7
|
4,16 × 10-6
|
POS
|
2,37 × 10-6
|
POS (2/2)
|
POS
|
NT
|
|
29
|
Norethynodrel (1)
|
68-23-5
|
POS
|
1,11 × 10-11
|
1,50 × 10-9
|
POS
|
9,39 × 10-10
|
POS (5/5)
|
POS
|
NT
|
|
30
|
p,p’-Methoxychlor (1)
|
72-43-5
|
POS
|
1,23 × 10-6
|
(žádná hodnota PC50) (2)
|
POS
|
1,92 × 10-6
|
POS (24/27)
|
POS
|
POS
|
|
31
|
Fenobarbital (1)
|
57-30-7
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
32
|
Reserpin
|
50-55-5
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (4/4)
|
NEG
|
NT
|
|
33
|
Spironolakton (1)
|
52-01-7
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (4/4)
|
NEG
|
NT
|
|
34
|
Testosteron
|
58-22-0
|
POS
|
2,82 × 10-8
|
9,78 × 10-6
|
POS
|
1,75 × 10-5
|
POS (5/10)
|
POS
|
NT
|
|
Zkratky: CASRN = Chemical Abstracts Service Registry Number (registrační číslo CAS); M = molární; EC50 = poloviční maximální účinná koncentrace zkoušené látky; NEG = negativní; POS = pozitivní; NT = nezkoušeno; PC10 (a PC50) = koncentrace zkoušené látky, při níž je odpověď 10 % (nebo 50 % u PC50) odpovědi indukované pozitivní kontrolou (E2, 1nM) na každé destičce.
|
Tabulka 2
Porovnání výsledků z analýz STTA a VM7Luc ER TA u látek zkoušených pomocí obou analýz na antagonistické účinky a klasifikovaných jako antagonisté ER (POS) nebo negativní (NEG)
|
|
Látka (3)
|
CASRN
|
Analýza ER STTA (39)
|
Analýza VM7Luc ER TA (40)
|
Kandidát na účinky ER STTA (42)
|
ICCVAM (43) Dohoda o klasifikaci
|
MeSH (44) Třída chemických látek
|
Třída produktu (45)
|
|
ER TA Činnost
|
Hodnota IC50
(4) (M)
|
ER TA Činnost
|
Hodnota IC50
(4), (41) (M)
|
|
1
|
4-Hydroxytamoxifen
|
68047-06-3
|
POS
|
3,97 × 10-9
|
POS
|
2,08 × 10-7
|
středně POS
|
POS
|
Uhlovodík (cyklický)
|
Léčivý přípravek
|
|
2
|
Dibenzo[a,h]anthracen
|
53-70-3
|
POS
|
žádná hodnota IC50
|
POS
|
žádná hodnota IC50
|
POS
|
PP
|
Polycyklická sloučenina
|
Laboratorní chemická látka, přírodní produkt
|
|
3
|
Mifepriston
|
84371-65-3
|
POS
|
5,61 × 10-6
|
NEG
|
—
|
mírně POS
|
NEG
|
Steroid
|
Léčivý přípravek
|
|
4
|
Raloxifen-hydrochlorid
|
82640-04-8
|
POS
|
7,86 × 10-10
|
POS
|
1,19 × 10-9
|
středně POS
|
POS
|
Uhlovodík (cyklický)
|
Léčivý přípravek
|
|
5
|
Tamoxifen
|
10540-29-1
|
POS
|
4,91 × 10-7
|
POS
|
8,17 × 10-7
|
POS
|
POS
|
Uhlovodík (cyklický)
|
Léčivý přípravek
|
|
6
|
17β-estradiol
|
50-28-2
|
NEG
|
—
|
NEG
|
—
|
PN
|
PN
|
Steroid
|
Léčivý přípravek, veterinární látka
|
|
7
|
Apigenin
|
520-36-5
|
NEG
|
—
|
NEG
|
—
|
NEG
|
NEG
|
Heterocyklická sloučenina
|
Barvivo, přírodní produkt, farmaceutický meziprodukt
|
|
8
|
Atrazin
|
1912-24-9
|
NEG
|
—
|
NEG
|
—
|
NEG
|
PN
|
Heterocyklická sloučenina
|
Herbicid
|
|
9
|
Di-n-butylftalát
|
84-74-2
|
NEG
|
—
|
NEG
|
—
|
NEG
|
NEG
|
Ester, kyselina ftalová
|
Kosmetická složka, průmyslová chemická látka, plastifikátor
|
|
10
|
Fenarimol
|
60168-88-9
|
NEG
|
—
|
NEG
|
—
|
nezkoušeno
|
PN
|
Heterocyklická sloučenina, pyrimidin
|
Fungicid
|
|
11
|
Flavon
|
525-82-6
|
NEG
|
—
|
NEG
|
—
|
PN
|
PN
|
Flavonoid, heterocyklická sloučenina
|
Přírodní produkt, léčivý přípravek
|
|
12
|
Flutamid
|
13311-84-7
|
NEG
|
—
|
NEG
|
—
|
NEG
|
PN
|
Amid
|
Léčivý přípravek, veterinární látka
|
|
13
|
Genistein
|
446-72-0
|
NEG
|
—
|
NEG
|
—
|
PN
|
NEG
|
Flavonoid, heterocyklická sloučenina
|
Přírodní produkt, léčivý přípravek
|
|
14
|
p-n-nonylfenol
|
104-40-5
|
NEG
|
—
|
NEG
|
—
|
nezkoušeno
|
NEG
|
Fenol
|
Chemický meziprodukt
|
|
15
|
Resveratrol
|
501-36-0
|
NEG
|
—
|
NEG
|
—
|
PN
|
NEG
|
Uhlovodík (cyklický)
|
Přírodní produkt
|
|
Zkratky: CASRN = Chemical Abstracts Service Registry Number (registrační číslo CAS); M = molární; IC50 = poloviční maximální inhibiční koncentrace zkoušené chemické látky; NEG = negativní; PN = pravděpodobně negativní; POS = pozitivní; PP = pravděpodobně pozitivní.
|
SLOŽKY ANALÝZY ER TA
Zásadní složky zkoušky
|
11.
|
Tato zkušební metoda se vztahuje na analýzy využívající stabilně transfekovaný nebo endogenní receptor ERα a stabilně transfekovanou reportérovou genovou konstrukci pod kontrolou jednoho nebo několika estrogenových responzivních elementů; mohou však být přítomny i jiné receptory, jako je ERβ. Jsou to zásadní složky analýzy.
|
Kontroly
|
12.
|
Je třeba popsat východisko pro navrhované souběžné referenční standardy pro každou analýzu agonistů a antagonistů. Souběžné kontroly (negativní, rozpouštědlo a pozitivní) slouží podle potřeby jako indikace, že analýza je za zkušebních podmínek funkční a poskytuje základ pro porovnání jednotlivých experimentů; obvykle jsou součástí kritérií přijatelnosti pro daný experiment (1).
|
Standardní postupy kontroly kvality
|
13.
|
Standardní postupy kontroly kvality by měly být prováděny tak, jak je popsáno pro každou analýzu, aby bylo zajištěno, že buněčná linie zůstane stabilní i po opakovaných pasážích, nebudou v ní mykoplazmata (tj. nebude obsahovat bakteriální kontaminaci) a uchová si schopnost podávat očekávané odpovědi zprostředkované estrogenovými receptory v průběhu času. U buněčných linií je třeba dále zkontrolovat správnou identitu a další kontaminanty (např. houby, kvasinky a viry).
|
Prokázání odborné způsobilosti laboratoře
|
14.
|
Před zkoušením neznámých chemických látek pomocí některé z analýz v rámci této zkušební metody by měla každá laboratoř prokázat odbornou způsobilost v používání analýzy. Aby laboratoř prokázala svou odbornou způsobilost, měla by analyzovat 14 vhodných látek uvedených v tabulce 3 na agonistické účinky a 10 vhodných látek z tabulky 4 na antagonistické účinky. Toto vyzkoušení odborné způsobilosti rovněž potvrdí reakční schopnost zkušebního systému. Seznam vhodných látek představuje podmnožinu referenčních látek uvedených ve standardech funkčnosti pro analýzy ER TA (6). Tyto látky jsou komerčně dostupné, představují třídy chemických látek běžně souvisejících s ER agonistickými nebo antagonistickými účinky, vykazují vhodný rozsah síly očekávané od ER agonistů/antagonistů (tj. silné až slabé) a zahrnují negativní kontroly. Zkoušení vhodných látek by mělo být alespoň dvakrát zopakováno, a to v různé dny. Odborná způsobilost je prokázána správnou klasifikací (pozitivní/negativní) každé vhodné látky. Zkoušení odborné způsobilosti by měl zopakovat každý technik, který se analýzy učí. Podle typu buňky se mohou některé z těchto vhodných látek chovat jako selektivní modulátory estrogenových receptorů a vykazovat agonistické i antagonistické účinky. Vhodné látky jsou však v tabulce 3 a 4 klasifikovány podle známé převažující aktivity, která by měla být použita při hodnocení odborné způsobilosti.
|
|
15.
|
Pro prokázání funkčnosti a pro účely kontroly kvality by si měla každá laboratoř sestavovat historické databáze agonistů a antagonistů s údaji o referenčním standardu (např. 17β-estradiol a tamoxifen), chemických látkách pro pozitivní a negativní kontrolu a kontrolu rozpouštědla (např. DMSO). Na začátku by měla být databáze vytvořena z alespoň 10 nezávislých zkoušek na agonistické účinky (např. 17β-estradiol) a 10 nezávislých zkoušek na antagonistické účinky (např. tamoxifen). Dále se přidají výsledky z budoucích analýz těchto referenčních standardů a kontrol rozpouštědla, aby se databáze rozšířila a byla tak zajištěna konzistence a funkčnost bioanalýz prováděných v laboratoři v průběhu času.
|
Tabulka 3
Seznam (14) vhodných látek pro analýzu agonistických účinků
(53)
|
č. (52)
|
Látka
|
CASRN
|
Předpokládaná odpověď (46)
|
Analýza STTA
|
Analýza VM7Luc ER TA
|
Třída chemických látek MeSH (50)
|
Třída produktu (51)
|
|
Hodnota PC10 (M) (47)
|
Hodnota PC50 (M) (47)
|
Rozpětí zkušebních konc. (M)
|
Hodnota VM7Luc EC50 (M) (48)
|
Nejvyšší konc. pro zjištění rozmezí (M) (49)
|
|
14
|
Diethylstilbestrol
|
56-53-1
|
POS
|
< 1,00 × 10-11
|
2,04 × 10-11
|
10-14 × 10-8
|
3,34 × 10-11
|
3,73 × 10-4
|
Uhlovodík (cyklický)
|
Léčivý přípravek, veterinární látka
|
|
12
|
17α-estradiol
|
57-91-0
|
POS
|
4,27 × 10-11
|
6,44 × 10-10
|
10-11 × 10-5
|
1,40 × 10-9
|
3,67 × 10-3
|
Steroid
|
Léčivý přípravek, veterinární látka
|
|
15
|
meso-Hexestrol
|
84-16-2
|
POS
|
< 1,00 × 10-11
|
2,75 × 10-11
|
10-11 × 10-5
|
1,65 × 10-11
|
3,70 × 10-3
|
Uhlovodík (cyklický), fenol
|
Léčivý přípravek, veterinární látka
|
|
11
|
4-terc-oktylfenol
|
140-66-9
|
POS
|
1,85 × 10-9
|
7,37 × 10-8
|
10-11 × 10-5
|
3,19 × 10-8
|
4,85 × 10-3
|
Fenol
|
Chemický meziprodukt
|
|
9
|
Genistein
|
446-72-0
|
POS
|
2,24 × 10-9
|
2,45 × 10-8
|
10-11 × 10-5
|
2,71 × 10-7
|
3,70 × 10-4
|
Flavonoid, heterocyklická sloučenina
|
Přírodní produkt, léčivý přípravek
|
|
6
|
Bisfenol A
|
80-05-7
|
POS
|
2,02 × 10-8
|
2,94 × 10-7
|
10-11 × 10-5
|
5,33 × 10-7
|
4,38 × 10-3
|
Fenol
|
Chemický meziprodukt
|
|
2
|
Kaempferol
|
520-18-3
|
POS
|
1,36 × 10-7
|
1,21 × 10-6
|
10-11 × 10-5
|
3,99 × 10-6
|
3,49 × 10-3
|
Flavonoid, heterocyklická sloučenina
|
Přírodní produkt
|
|
3
|
Butylbenzylfthalát
|
85-68-7
|
POS
|
1,14 × 10-6
|
4,11 × 10-6
|
10-11 × 10-5
|
1,98 × 10-6
|
3,20 × 10-4
|
Kyselina karboxylová, ester, kyselina ftalová
|
Plastifikátor, průmyslová chemická látka
|
|
4
|
p,p’- Methoxychlor
|
72-43-5
|
POS
|
1,23 × 10-6
|
—
|
10-11 × 10-5
|
1,92 × 10-6
|
2,89 × 10-3
|
Uhlovodík (halogenovaný)
|
Léčivý přípravek, veterinární látka
|
|
1
|
Ethylparaben
|
120-47-8
|
POS
|
5,00 × 10-6
|
—
|
10-11 × 10-5
|
2,48 × 10-5
|
6,02 × 10-3
|
Kyselina karboxylová, fenol
|
Léčivý přípravek, konzervant
|
|
17
|
Atrazin
|
1912-24-9
|
NEG
|
—
|
—
|
10-10 - 10-4
|
—
|
4,64 × 10-4
|
Heterocyklická sloučenina
|
Herbicid
|
|
20
|
Spironolakton
|
52-01-7
|
NEG
|
—
|
—
|
10-11 - 10-5
|
—
|
2,40 × 10-3
|
Lakton, steroid
|
Léčivé přípravky
|
|
21
|
Ketokonazol
|
65277-42-1
|
NEG
|
—
|
—
|
10-11 - 10-5
|
—
|
9,41 × 10-5
|
Heterocyklická sloučenina
|
Léčivé přípravky
|
|
22
|
Reserpin
|
50-55-5
|
NEG
|
—
|
—
|
10-11 - 10-5
|
—
|
1,64 × 10-3
|
Heterocyklická sloučenina, indol
|
Léčivý přípravek, veterinární látka
|
|
Zkratky: CASRN = Chemical Abstracts Service Registry Number (registrační číslo CAS); EC50 = poloviční maximální účinná koncentrace zkoušené látky; NEG = negativní; POS = pozitivní; PC10 (a PC50) = koncentrace zkoušené látky, při níž je odpověď 10 % (nebo 50 % u PC50) odpovědi indukované pozitivní kontrolou (E2, 1nM) na každé destičce.
|
Tabulka 4
Seznam (10) vhodných látek pro analýzu antagonistů
|
|
Látka (5)
|
CASRN
|
Analýza ER STTA (54)
|
Analýza VM7Luc ER TA (55)
|
Kandidát na účinky ER STTA (54)
|
ICCVAM (58) Dohoda o klasifikaci
|
MeSH (59) Třída chemických látek
|
Třída produktu (60)
|
|
ER TA aktivita
|
IC50 (M)
|
Rozpětí zkušebních konc. (M)
|
ER TA aktivita
|
IC50
(56) (M)
|
Nejvyšší konc. pro zjištění rozmezí (M) (57)
|
|
1
|
4-Hydroxytamoxifen
|
68047-06-3
|
POS
|
3,97 × 10-9
|
10-12–10-7
|
POS
|
2,08 × 10-7
|
2,58 × 10-4
|
středně POS
|
POS
|
Uhlovodík (cyklický)
|
Léčivé přípravky
|
|
2
|
Raloxifen-hydrochlorid
|
82640-04-8
|
POS
|
7,86 × 10-10
|
10-12–10-7
|
POS
|
1,19 × 10-9
|
1,96 × 10-4
|
středně POS
|
POS
|
Uhlovodík (cyklický)
|
Léčivé přípravky
|
|
3
|
Tamoxifen
|
10540-29-1
|
POS
|
4,91 × 10-7
|
10-10–10-5
|
POS
|
8,17 × 10-7
|
2,69 × 10-4
|
POS
|
POS
|
Uhlovodík (cyklický)
|
Léčivé přípravky
|
|
4
|
17β-estradiol
|
50-28-2
|
NEG
|
—
|
10-9–10-4
|
NEG
|
—
|
3,67 × 10-3
|
negativní (*4)
|
PN
|
Steroid
|
Léčivý přípravek, veterinární látka
|
|
5
|
Apigenin
|
520-36-5
|
NEG
|
—
|
10-9–10-4
|
NEG
|
—
|
3,70 × 10-4
|
NEG
|
NEG
|
Heterocyklická sloučenina
|
Barvivo, přírodní produkt, farmaceutický meziprodukt
|
|
6
|
Di-n-butylftalát
|
84-74-2
|
NEG
|
—
|
10-8–10-3
|
NEG
|
—
|
3,59 × 10-3
|
NEG
|
NEG
|
Ester, kyselina ftalová
|
Kosmetická složka, průmyslová chemická látka, plastifikátor
|
|
7
|
Flavon
|
525-82-6
|
NEG
|
—
|
10-8–10-3
|
NEG
|
—
|
4,50 × 10-4
|
negativní (*4)
|
PN
|
Flavonoid, heterocyklická sloučenina
|
Přírodní produkt, léčivý přípravek
|
|
8
|
Genistein
|
446-72-0
|
NEG
|
—
|
10-9–10-4
|
NEG
|
—
|
3,70 × 10-4
|
negativní (*4)
|
NEG
|
Flavonoid, heterocyklická sloučenina
|
Přírodní produkt, léčivý přípravek
|
|
9
|
p-n-nonylfenol
|
104-40-5
|
NEG
|
—
|
10-9–10-4
|
NEG
|
—
|
4,54 × 10-4
|
nezkoušeno
|
NEG
|
Fenol
|
Chemický meziprodukt
|
|
10
|
Resveratrol
|
501-36-0
|
NEG
|
—
|
10-8–10-3
|
NEG
|
—
|
4,38 × 10-4
|
negativní (*4)
|
NEG
|
Uhlovodík (cyklický)
|
Přírodní produkt
|
|
Zkratky: CASRN = Chemical Abstracts Service Registry Number (registrační číslo CAS); M = molární; IC50 = poloviční maximální inhibiční koncentrace zkoušené chemické látky; NEG = negativní; PN = pravděpodobně negativní; POS = pozitivní.
|
Kritéria přijatelnosti provedení zkoušky
|
16.
|
Přijetí nebo odmítnutí provedené zkoušky vychází z hodnocení výsledků získaných pro referenční standardy a kontroly použité pro každý experiment. Hodnoty pro PC50 (EC50) nebo IC50 pro referenční standardy by měly splňovat kritéria přijatelnosti stanovená pro zvolenou analýzu (pro STTA viz dodatek 2, pro VM7Luc ER TA viz dodatek 3) a všechny pozitivní/negativní kontroly by měly být pro každý přijatý experiment správně klasifikovány. Schopnost konzistentního provedení analýzy by měla být prokázána vytvořením a údržbou historické databáze referenčních standardů a kontrol (viz odstavec 15). Jako měřítko vnitrolaboratorní reprodukovatelnosti mohou být použity směrodatné odchylky (SD) nebo variační koeficienty (CV) pro střední hodnoty parametrů stanovené křivky referenčních standardů z vícečetných experimentů. Kromě toho by měly být splněny tyto zásady, pokud jde o kritéria přijatelnosti:
|
—
|
Údaje by měly být dostačující pro kvantitativní posouzení aktivace (u analýzy agonistů) nebo suprese (u analýzy antagonistů) ER (tj. účinnost a síla).
|
|
—
|
Střední aktivita reportérových genů pro referenční koncentraci referenčního estrogenu by měla dosahovat alespoň minima stanoveného v analýzách týkajících se aktivity kontroly s vehikulem (rozpouštědlem), aby byla zajištěna adekvátní citlivost. Pro analýzy STTA a VM7Luc ER TA je to čtyřnásobek střední kontroly s vehikulem na každé destičce.
|
|
—
|
Zkoušené koncentrace by měly zůstat v rozmezí rozpustnosti zkoušených chemických látek a neměly by vykazovat cytotoxicitu.
|
|
Analýza údajů
|
17.
|
Pro klasifikaci pozitivní a negativní odpovědi by měl být použit definovaný postup interpretace údajů pro každou analýzu.
|
|
18.
|
Splnění kritérií přijatelnosti (odstavec 16) ukazuje, že analýza funguje správně, ale nezaručuje to, že konkrétní provedení zkoušky podá přesné údaje. Opakování výsledků první zkoušky je nejlepším důkazem, že údaje byly přesné. Jestliže jsou při dvou provedeních zkoušky získány reprodukovatelné výsledky (např. výsledky z obou provedení ukazují, že zkoušená chemická látka je pozitivní), není nutné provádět zkoušku potřetí.
|
|
19.
|
Jestliže dvě provedení zkoušky nepodají reprodukovatelné výsledky (např. zkoušená chemikálie je při jednom provedení pozitivní a při druhém provedení negativní) nebo jestliže je potřebný větší stupeň jistoty, pokud jde o výsledek této analýzy, je třeba provést alespoň tři nezávislé zkoušky. V takovém případě vychází klasifikace ze dvou shodných výsledků ze tří.
|
Obecná kritéria interpretace údajů
|
20.
|
V současné době neexistuje žádná univerzálně akceptovaná metoda pro interpretaci údajů ER TA. Kvalitativní (např. pozitivní/negativní) anebo kvantitativní (např. EC50, PC50, IC50) posouzení aktivity zprostředkované estrogenovými receptory by však mělo vycházet z empirických údajů a fundovaného vědeckého úsudku. Pokud je to možné, měly by být pozitivní výsledky charakterizovány jak intenzitou účinku v porovnání s kontrolou vehikulem (rozpouštědlem) nebo referenčním estrogenem, tak i koncentrací, při níž k účinku dochází (např. EC50, PC50, RPCMax, IC50 atd.).
|
ZÁVĚREČNÁ ZPRÁVA
|
21.
|
Závěrečná zpráva by měla obsahovat tyto informace:
|
|
Analýza:
|
—
|
kontrola / referenční standard / zkoušená chemická látka,
|
|
—
|
zdroj, číslo šarže a datum použitelnosti, je-li k dispozici,
|
|
—
|
stabilita zkoušené chemické látky samotné, je-li známa,
|
|
—
|
rozpustnost a stabilita zkoušené chemické látky v rozpouštědle, jsou-li známy,
|
|
—
|
měření pH, případně osmolality a sraženiny v kultivačním médiu, do něhož byla přidána zkoušená chemická látka.
|
|
|
|
Jednosložková látka:
|
—
|
fyzický vzhled, rozpustnost ve vodě a další relevantní fyzikálně-chemické vlastnosti,
|
|
—
|
chemická identifikace, např. název podle IUPAC nebo CAS, číslo CAS, kód SMILES nebo InChI, strukturní vzorec, čistota, případně chemická identita nečistot, je-li to prakticky možné, atd.
|
|
|
|
Vícesložková látka, látka s neznámým nebo proměnlivým složením, komplexní reakční produkt nebo biologický materiál a směsi:
|
—
|
charakterizované pokud možno chemickou identitou (viz výše), kvantitativním výskytem a relevantními fyzikálně-chemickými vlastnostmi složek.
|
|
|
|
Rozpouštědlo/vehikulum:
|
—
|
charakterizace (povaha, dodavatel a šarže),
|
|
—
|
zdůvodnění volby rozpouštědla/vehikula,
|
|
—
|
rozpustnost a stabilita zkoušené chemické látky v rozpouštědle/vehikulu, jsou-li známy.
|
|
|
|
Buňky:
|
—
|
typ a zdroj buněk:
|
—
|
Je ER endogenně exprimován? Pokud není, které receptory byly transfekovány?
|
|
—
|
použitá (použité) reportérová (reportérové) konstrukce (včetně zdrojových druhů),
|
|
—
|
selekční metoda pro udržení stabilní transfekce (ve vhodných případech),
|
|
—
|
je metoda transfekce relevantní pro stabilní linie?
|
|
|
—
|
počet pasáží (z rozmrazených buněk),
|
|
—
|
číslo pasáže buněk při rozmrazení,
|
|
—
|
metody udržování buněčných kultur.
|
|
|
|
Zkušební podmínky:
|
—
|
popis použitých metod posuzování životaschopnosti,
|
|
—
|
složení média, koncentrace CO2,
|
|
—
|
koncentrace zkoušené chemické látky,
|
|
—
|
objem vehikula a přidané zkoušené chemické látky,
|
|
—
|
inkubační teplota a vlhkost,
|
|
—
|
hustota buněk na začátku expozice a během expozice,
|
|
—
|
pozitivní a negativní referenční standardy,
|
|
—
|
reportérové reagencie (název přípravku, dodavatel a šarže),
|
|
—
|
kritéria klasifikace jednotlivých provedení zkoušky na pozitivní, negativní nebo neurčitou.
|
|
|
|
Kontrola přijatelnosti:
|
—
|
násobné indukce pro každou destičku, a zda splňují minimum požadované pro analýzu na základě dosavadních kontrol,
|
|
—
|
skutečné hodnoty pro kritéria přijatelnosti, např. log10EC50, log10PC50, logIC50 a hodnoty Hillova koeficientu pro souběžné pozitivní kontroly / referenční standardy.
|
|
|
|
Výsledky:
|
—
|
nezpracovaná a normalizovaná data,
|
|
—
|
maximální úroveň násobku indukce,
|
|
—
|
pokud existuje, nejnižší účinná koncentrace (LEC),
|
|
—
|
hodnoty RPCMax, PCMax, PC50, IC50 a/nebo EC50 podle potřeby,
|
|
—
|
závislost účinku na koncentraci, pokud je to možné,
|
|
—
|
případné statistické analýzy společně s měřítkem chyby a spolehlivosti (např. SEM, SD, CV nebo 95 % CI) a popis, jak byly tyto hodnoty získány.
|
|
|
LITERATURA
|
(1)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 34.), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
OECD (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environment, Health and Safety Publications, Series on Testing and Assessment (No 225), Organisation for Economic Cooperation and Development, Paris.
|
|
(3)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method, an In Vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(4)
|
Pujol P. et al. (1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer. Res., 58(23): s. 5367-73.
|
|
(5)
|
Rogers J.M. and Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In Vitro and Molecular Toxicology: Journal of Basic and Applied Research, 13(1): s. 67-82.
|
|
(6)
|
OECD (2012). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Agonists (for TG 455). Environment, Health and Safety Publications, Series on Testing and Assessment (No 173.), Organisation for Economic Cooperation and Development, Paris.
|
|
(7)
|
OECD (2015). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Antagonists. Environment, Health and Safety Publications, Series on Testing and Assessment (No 174.), Organisation for Economic Cooperation and Development, Paris.
|
|
(8)
|
OECD (2012). Guidance Document on Standardized Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment (No 150.), Organisation for Economic Cooperation and Development, Paris.
|
|
(9)
|
Cavailles V. (2002). Estrogens and Receptors: an Evolving Concept. Climacteric, 5 Suppl 2: s. 20- 6.
|
|
(10)
|
Welboren W.J. et al. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and how are they Regulated? Endocr. Relat. Cancer, 16(4): s. 1073-89.
|
|
(11)
|
Younes M. and Honma N. (2011). Estrogen Receptor Beta, Arch. Pathol. Lab. Med., 135(1): s. 63- 6.
|
|
(12)
|
Jefferson W.N., et al. (2002). Assessing Estrogenic Activity of Phytochemicals Using Transcriptional Activation and Immature Mouse Uterotrophic Responses, Journal of Chromatography B, 777(1-2): s. 179-189.
|
|
(13)
|
Sonneveld E. et al. (2006). Comparison of In Vitro and In Vivo Screening Models for Androgenic and Estrogenic Activities, Toxicol. Sci., 89(1): s. 173-187.
|
|
(14)
|
Takeyoshi M. et al. (2002). The Efficacy of Endocrine Disruptor Screening Tests in Detecting Anti- Estrogenic Effects Downstream of Receptor-Ligand Interactions, Toxicology Letters, 126(2): s. 91- 98.
|
|
(15)
|
Combes R.D. (2000). Endocrine Disruptors: a Critical Review of In Vitro and In Vivo Testing Strategies for Assessing their Toxic Hazard to Humans, ATLA Alternatives to Laboratory Animals,28(1): s. 81-118.
|
|
(16)
|
Escande A. et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol,71(10): s. 1459-69.
|
|
(17)
|
Gray L.E. Jr. (1998). Tiered Screening and Testing Strategy for Xenoestrogens and Antiandrogens, Toxicol. Lett, 102-103, 677-680.
|
|
(18)
|
EDSTAC (1998). Endocrine Disruptor Screening and Testing Advisory Committee (EDSTAC) Final Report.
|
|
(19)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(20)
|
Gustafsson J.Ö. (1999). Estrogen Receptor ß - A New Dimension in Estrogen Mechanism of Action, Journal of Endocrinology, 163(3): s. 379-383.
|
|
(21)
|
Ogawa S. et al. (1998). The Complete Primary Structure of Human Estrogen Receptor ß (hERß) and its Heterodimerization with ERαIn Vivo and In Vitro, Biochemical and Biophysical Research Communications, 243(1): s. 122-126.
|
|
(22)
|
Enmark E. et al. (1997). Human Estrogen Receptor ß-Gene Structure, Chromosomal Localization, and Expression Pattern, Journal of Clinical Endocrinology and Metabolism,82(12): s. 4258-4265.
|
|
(23)
|
Ball L.J. et al. (2009). Cell Type- and Estrogen Receptor-Subtype Specific Regulation of Selective Estrogen Receptor Modulator Regulatory Elements, Molecular and Cellular Endocrinology, 299(2): s. 204-211.
|
|
(24)
|
Barkhem T. et al. (1998). Differential Response of Estrogen Receptor Alpha and Estrogen Receptor Beta to Partial Estrogen Agonists/Antagonists, Mol. Pharmacol, 54(1): s. 105-12.
|
|
(25)
|
Deroo B.J. and Buensuceso A.V. (2010). Minireview: Estrogen Receptor-ß: Mechanistic Insights from Recent Studies, Molecular Endocrinology, 24(9): s. 1703-1714.
|
|
(26)
|
Harris D.M. et al. (2005). Phytoestrogens Induce Differential Estrogen Receptor Alpha- or Beta- Mediated Responses in Transfected Breast Cancer Cells, Experimental Biology and Medicine, 230(8): s. 558-568.
|
|
(27)
|
Anderson J.N. Clark J.H. and Peck E.J.Jr. (1972). The Relationship Between Nuclear Receptor- Estrogen Binding and Uterotrophic Responses, Biochemical and Biophysical Research Communications, 48(6): s. 1460-1468.
|
|
(28)
|
Toft D. (1972). The Interaction of Uterine Estrogen Receptors with DNA, Journal of Steroid Biochemistry, 3(3): s. 515-522.
|
|
(29)
|
Gorski J. et al. (1968), Hormone Receptors: Studies on the Interaction of Estrogen with the Uterus, Recent Progress in Hormone Research, 24: s. 45-80.
|
|
(30)
|
Jensen E.V. et al. (1967), Estrogen-Receptor Interactions in Target Tissues, Archives d’Anatomie Microscopique et de Morphologie Experimentale, 56(3): s. 547-569.
|
|
(31)
|
ICCVAM (2002). Background Review Document: Estrogen Receptor Transcriptional Activation (TA) Assay. Appendix D, Substances Tested in the ER TA Assay, NIH Publication Report (No 03-4505.).
|
|
(32)
|
Kanno J. et al. (2001). The OECD Program to Validate the Rat Uterotrophic Bioassay to Screen Compounds for In Vivo Estrogenic Responses: Phase 1, Environ. Health Persp., 109:785-94.
|
|
(33)
|
Kanno J. et al. (2003). The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two Dose -Response Studies, Environ. Health Persp., 111:1530-1549.
|
|
(34)
|
Kanno J. et al. (2003), The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two – Coded Single-Dose Studies, Environ. Health Persp., 111:1550-1558.
|
|
(35)
|
Geisinger et al. (1989) Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors, Cancer 63, 280-288.
|
|
(36)
|
Baldwin et al. (1998) BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In Vitro Cell. Dev. Biol. – Animal, 34, 649-654.
|
|
(37)
|
Li, Y. et al. (2014) Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, 2072-2081.
|
|
(38)
|
Rogers, J.M. and Denison, M.S. (2000) Recombinant cell bioassays for endocrine disruptors: development of a stably transfected human ovarian cell line for the detection of estrogenic and anti-estrogenic chemicals, In Vitro & Molec. Toxicol. Chem. 13, 67-82.
|
Dodatek 1
DEFINICE A ZKRATKY
Kritéria přijatelnosti
: Minimální pravidla pro výkonnost experimentálních kontrol a referenčních standardů. Aby se experiment mohl považovat za platný, měla by být splněna všechna kritéria přijatelnosti.
Přesnost (soulad)
: Přesnost shody mezi výsledky analýzy a přijatými referenčními hodnotami. Je to měřítko funkčnosti analýzy a jeden z aspektů relevance. Tento pojem se často používá místo pojmu „soulad“ a rozumí se jím podíl správných výsledků analýzy (1).
Agonista
: Látka, která vyvolá odpověď, např. transkripci, když se naváže na specifický receptor.
Antagonista
: Druh receptorového ligandu nebo chemická látka, která sama nevyvolává biologickou odpověď po navázání na receptor, ale blokuje nebo tlumí odpovědi zprostředkované agonisty.
Antiestrogenní aktivita
: je schopnost chemické látky potlačit působení 17β-estradiolu zprostředkované přes estrogenové receptory.
Buněčná morfologie
: Tvar a vzhled buněk rostoucích v monovrstvě v jedné jamce destičky pro tkáňové kultury. Buňky, které odumírají, často vykazují abnormální buněčnou morfologii.
CF
: Koncepční rámec OECD pro zkoušky a hodnocení endokrinních distruptorů.
Ošetření aktivním uhlím/dextranem
: Ošetření séra použitého v buněčné kultuře. Ošetření aktivním uhlím/dextranem (často označované jako „stripping“) odstraňuje endogenní hormony a bílkoviny, na které se hormony vážou.
Chemická látka
: Látka nebo směs.
Cytotoxicita
: Škodlivé účinky pro buněčnou strukturu nebo funkci, které mohou nakonec způsobit buněčnou smrt a vyvolat snížení počtu buněk přítomných v jamce na konci doby expozice nebo snížení kapacity pro měření buněčné funkce při porovnání se souběžnou kontrolou vehikulem.
CV
: Variační koeficient.
DCC-FBS
: Fetální bovinní sérum ošetřené aktivním uhlím, které je potaženo dextranem.
DMEM
: Dulbeccova modifikace Eaglova média.
DMSO
: Dimethylsulfoxid.
E2
: 17β-estradiol.
EC50
: Poloviční maximální účinná koncentrace zkoušené chemické látky.
ED
: Narušení endokrinní činnosti.
hERα
: Lidský estrogenní receptor alfa.
hERß
: Lidský estrogenní receptor beta.
EFM
: Médium bez estrogenů Dulbeccova modifikace Eaglova média (DMEM) doplněná 4,5 % FBS ošetřeného aktivním uhlím/dextranem, 1,9 % L-glutaminu a 0,9 % Pen-Strepu.
ER
: Estrogenový receptor.
ERE
: Estrogen responzivní element.
Estrogenní aktivita
: Schopnost chemické látky napodobit schopnost 17β-estradiolu vázat se na estrogenové receptury a aktivovat je. Touto zkušební metodou lze detekovat estrogenní aktivitu zprostředkovanou hERα.
ERTA
: Transaktivace estrogenových receptorů.
FBS
: Fetální bovinní sérum.
HeLa
: Imortalizovaná buněčná linie z lidského děložního hrdla.
HeLa9903
: Subklon buněk HeLa, do nichž byly stabilně transfekovány hERααa reportérový gen luciferázy.
IC
50
: Poloviční maximální účinná koncentrace inhibiční zkoušené chemické látky.
ICCVAM
: Meziagenturní koordinační výbor pro validaci alternativních metod.
Mezilaboratorní reprodukovatelnost
: Míra, v jaké mohou různé kvalifikované laboratoře za použití stejného protokolu a při testování stejných látek získat kvalitativně a kvantitativně podobné výsledky. Mezilaboratorní reprodukovatelnost se určuje během předvalidačních a validačních postupů a ukazuje, do jaké míry lze analýzu úspěšně přenášet mezi různými laboratořemi (1).
Vnitrolaboratorní reprodukovatelnost
: Určení míry, v jaké může kvalifikovaný personál ve stejné laboratoři jindy úspěšně dosáhnout stejných výsledků za použití určitého protokolu. Označuje se také jako „reprodukovatelnost v rámci laboratoře“ (1).
LEC
: Nejnižší účinná koncentrace je nejnižší koncentrace zkoušené chemické látky, která vyvolává odpověď (tj. nejnižší koncentrace zkoušené chemické látky, při níž je násobná indukce statisticky odlišná od souběžné kontroly vehikulem).
Zkouška typu „me-too“
: Hovorový výraz pro analýzu, která je strukturálně a funkčně podobná validované a uznávané referenční zkušební metodě. Používá se namísto podobné zkušební metody.
MT
: Metallothionein.
MMTV
: Virus myšího nádoru prsní žlázy.
OHT
: 4-Hydroxytamoxifen.
PBTG
: Pokyn vycházející z funkční způsobilosti.
PC
: (pozitivní kontrola): Silně aktivní látka, nejlépe 17ß-estradiol, která je zařazena do všech zkoušek, a pomáhá tak zajistit správné fungování analýzy.
PC
10
: Koncentrace zkoušené chemické látky, při níž činí naměřená aktivita v analýze agonistů 10 % maximální aktivity indukované pozitivní kontrolou (E2 při 1nM pro analýzu STTA) na každé destičce.
PC
50
: Koncentrace zkoušené chemické látky, při níž činí naměřená aktivita v analýze agonistů 50 % maximální aktivity indukované pozitivní kontrolou (E2 při referenční koncentraci stanovené ve zkušební metodě) na každé destičce.
PC
Max
: Koncentrace zkoušené chemické látky vyvolávající RPCMax.
Standardy funkčnosti
: Standardy založené na validované analýze, které slouží jako základ pro hodnocení srovnatelnosti navrhované analýzy, která je mechanicky a funkčně podobná. Zahrnují 1) zásadní složky analýzy; 2) minimální seznam srovnávacích látek vybraných z chemických látek používaných k prokázání přijatelné funkčnosti validované referenční metody; a 3) srovnatelné hladiny přesnosti a spolehlivosti založené na hodnotách získaných u validované zkušební metody, jež by navrhovaná analýza měla prokázat při vyhodnocení za použití minimálního seznamu srovnávacích látek (1).
Vhodné látky
: Podsoubor ze seznamu referenčních látek zařazených do standardů funkčnosti, které mohou laboratoře použít k prokázání způsobilosti k provádění standardizované zkušební metody. Kritéria výběru u těchto látek obvykle zahrnují to, že reprezentují celou škálu odpovědí, jsou komerčně dostupné a jsou k nim dostupné kvalitní referenční údaje.
Způsobilost
: Prokázaná schopnost řádně provést analýzu před zkoušením neznámých látek.
Referenční estrogen (pozitivní kontrola, PC)
: 17β-estradiol (E2, CAS 50-28-2).
Referenční standard
: Referenční látka použitá na prokázání vhodnosti analýzy. Pro analýzy STTA a VM7Luc ER TA je referenčním standardem 17β-estradiol.
Referenční zkušební metody
: Analýzy, z nichž vychází pokyn PBTG č. 455.
Relevantnost
: Popis vztahu analýzy k účinku, který je předmětem zájmu, a také toho, zda je analýza smysluplná a použitelná pro konkrétní účel. Vyjadřuje, do jaké míry analýza správně měří nebo predikuje biologický účinek, který je předmětem zájmu. Relevantnost zahrnuje také posouzení přesnosti (shody) analýzy (1).
Spolehlivost
: Míra rozsahu, v jakém může být analýza časem reprodukovatelná v laboratořích a mezi laboratořemi, když se provádí s použitím stejného protokolu. Hodnotí se to výpočtem laboratorní a mezilaboratorní reprodukovatelnosti.
RLU
: Relativní luminiscenční jednotky.
RNA
: Kyselina ribonukleová.
RPC
Max
: Maximální úroveň odpovědi indukované zkoušenou chemickou látkou, vyjádřená jako % odpovědi indukované 1 nM E2 na stejné destičce.
RPMI
: Médium RPMI 1 640 doplněné 0,9 % Pen-Strepu a 8,0 % fetálního bovinního séra (FBS).
Provedení zkoušky
: Individuální experiment, kterým se hodnotí chemické působení na biologický výsledek analýzy. Každé provedení představuje úplný experiment provedený v replikovaných jamkách s buňkami kultivovanými ze stejného zdroje buněk ve stejném čase.
Nezávislé provedení zkoušky
: Samostatný nezávislý experiment, při němž se hodnotí chemické působení na biologický výsledek analýzy s použitím buněk z jiného zdroje, čerstvě naředěných chemických látek, provedený v jiné dny nebo ve stejný den jiným pracovníkem.
SD
: Směrodatná odchylka.
Citlivost
: Podíl všech pozitivních/aktivních látek, které jsou na základě analýzy správně klasifikovány. Je to měřítko přesnosti analýzy, která dává kategorické výsledky, a je důležitým prvkem při hodnocení relevance analýzy (1).
Specifičnost
: Podíl všech negativních/neaktivních látek, které jsou na základě zkoušky správně klasifikovány. Je to měřítko přesnosti analýzy, která dává kategorické výsledky, a je důležitým prvkem při hodnocení relevance analýzy (1).
Stabilní transfekce
: Když je do kultivovaných buněk transfekována DNA takovým způsobem, aby byla stabilně intregrována do genomu buněk, čímž dojde ke stabilní expresi transfekovaných genů. Klony stabilně transfekovaných buněk se selektují stabilními markery (např. resistence na G418).
Analýza STTA
: Analýza stabilně transfekované transaktivace, analýza transkripční aktivace ERα s využitím buněčné linie HeLa 9 903.
Studie
: Experimentální práce v plném rozsahu provedená za účelem hodnocení jedné specifické látky s využitím specifické analýzy. Studie zahrnuje všechny kroky včetně testů ředění zkoušené látky ve zkušebním médiu, předběžné zkoušky ke stanovení rozsahu, všechny nezbytné komplexní zkoušky, analýzy údajů, zajištění kvality, posouzení cytotoxicity atd. Provedení studie umožňuje klasifikovat aktivitu zkoušené chemické látky z hlediska toxicity (tj. účinná, neúčinná nebo neprůkazná), což se hodnotí použitou analýzou a odhadem síly ve vztahu k pozitivní referenční chemické látce.
Látka
: Podle nařízení REACH (61) je látka definována jako chemický prvek a jeho sloučeniny v přírodním stavu nebo získané výrobním procesem, včetně všech přídatných látek nezbytných k uchování jeho stability a všech nečistot vznikajících v použitém procesu, avšak s vyloučením všech rozpouštědel, která lze oddělit bez ovlivnění stability látky nebo změny jejího složení. Velmi podobná definice se používá v rámci GHS OSN (1).
TA (transaktivace):: Iniciace syntézy mRNA v reakci na specifický chemický signál, jako je navázání estrogenu na estrogenový receptor.
Analýza
: V souvislosti s touto zkušební metodou je analýza jednou z metodik, která je uznávána jako validní z hlediska plnění stanovených kritérií výkonnosti. Složky analýzy zahrnují například specifickou buněčnou linii se souvisejícími růstovými podmínkami, specifické médium, v němž je zkouška prováděna, podmínky uspořádání destiček, sestavu a ředění zkoušených chemických látek společně s dalšími požadovanými opatřeními v oblasti kontroly kvality a související kroky při hodnocení údajů.
Zkoušená chemická látka
: Jakákoli látka nebo směs zkoušená touto zkušební metodou.
Transkripce
: Syntéza mRNA.
UVCB
: Chemické látky s neznámým nebo proměnlivým složením, komplexní reakční produkty nebo biologický materiál.
Validovaná zkušební metoda
: Analýza, u níž byly provedeny validační studie k určení její relevantnosti (včetně přesnosti) a spolehlivosti pro konkrétní účel. Je důležité si uvědomit, že validovaná zkušební metoda nemusí mít dostatečnou funkčnost, pokud jde o přesnost a spolehlivost, aby mohla být shledána přijatelnou pro navrhovaný účel (1).
Validace
: Proces, při němž je stanovena spolehlivost a relevance konkrétního přístupu, metody, analýzy, procesu nebo hodnocení pro definovaný účel (1).
VC (kontrola s vehikulem)
: Rozpouštědlo, které se používá na rozpuštění zkoušené chemické látky a kontrolních chemických látek a zkouší se výhradně jako vehikulum bez rozpuštěné chemické látky.
VM7
: Imortalizovaná buňka adenokarcinomu, která endogenně exprimuje estrogenový receptor.
VM7Luc4E2
: Buněčná linie VM7Luc4E2 byla odvozena z imortalizovaných buněk získaných z lidského adenokarcinomu VM7, které endogenně exprimují obě formy estrogenového receptoru (ERα a ERβ) a jsou stabilně transfekovány plasmidem pGudLuc7.ERE. Tento plasmid obsahuje čtyři kopie syntetického oligonukleotidu obsahujícího estrogen responzivní element před promotorem viru myšího nádoru prsní žlázy a genu luciferázy světlušek.
Slabá pozitivní kontrola
: Slabě aktivní látka vybraná ze seznamu referenčních chemických látek, která je zařazena do všech zkoušek, a pomáhá tak zajistit správné fungování analýzy.
Dodatek 2
ANALÝZA STABILNĚ TRANSFEKOVANÉ TRANSAKTIVACE ESTROGENOVÉHO RECEPTORU Α PRO DETEKCI ESTROGENOVÝCH AGONISTICKÝCH A ANTAGONISTICKÝCH ÚČINKŮ CHEMICKÝCH LÁTEK S POUŽITÍM BUNĚČNÉ LINIE HERΑ-HELA-9903
VÝCHOZÍ ÚVAHY A OMEZENÍ (VIZ TAKÉ VŠEOBECNÝ ÚVOD)
|
1.
|
Při této transaktivační analýze (TA) se používá buněčná linie hERα-HeLa-9 903 na detekci estrogenní agonistické aktivity zprostředkované lidským estrogenovým receptorem alfa (hERα). Validační studie analýzy stabilně transfekované transaktivace (STTA) provedená japonským Ústavem pro hodnocení a výzkum chemických látek (CERI) s použitím buněčné linie hERα-HeLa-9 903 na detekci estrogenových agonistických a antagonistických účinků zprostředkovaných lidským estrogenovým receptorem alfa (hERα) prokázala relevanci a spolehlivost analýzy pro zamýšlený účel (1).
|
|
2.
|
Tato analýza je specificky určena na detekci TA zprostředkované hERα měřením chemiluminiscence jako sledované vlastnosti. Při koncentracích fytoestrogenů vyšších než 1 μM však byly hlášeny luminiscenční signály nezprostředkované receptorem v důsledku nadměrné aktivace reportérového genu luciferázy (2) (3). Přestože křivka závislosti odpovědi na dávce naznačuje, že ke skutečné aktivaci systému ER dochází při nižších koncentracích, je nutné v systémech analýzy stabilně transfekované ER TA pečlivě zkoumat exprese luciferázy získané při vysoké koncentraci fytoestrogenů nebo podobných sloučenin, u nichž existuje podezření, že vyvolávají nadměrnou aktivaci reportérového genu luciferázy podobně jako fytoestrogeny (dodatek 1).
|
|
3.
|
Před použitím této analýzy pro regulační účely je třeba přečíst si části „VŠEOBECNÝ ÚVOD“ a „SLOŽKY ANALÝZY ER TA“. Definice a zkratky používané v této zkušební metodě jsou popsány v dodatku 2.1.
|
PRINCIP ANALÝZY (VIZ TAKÉ VŠEOBECNÝ ÚVOD)
|
4.
|
Analýza se používá na signalizaci vazby ligandu na estrogenový receptor. Po vazbě na ligandy se komplex receptor-ligand přesune do jádra, kde se naváže na specifické responzivní elementy DNA a transaktivuje reportérový gen luciferázy světlušek, přičemž výsledkem je zvýšená buněčná exprese enzymu luciferáza. Luciferin je substrát, který enzym luciferáza transformuje na bioluminiscenční produkt, jenž může být kvantitativně změřen luminometrem. Aktivitu luciferázy lze rychle a levně vyhodnotit pomocí řady komerčně dostupných testů.
|
|
5.
|
Zkušební systém používá buněčnou linii hERα-HeLa-9 903, která je odvozena z lidského tumoru děložního hrdla, se dvěma stabilně vloženými konstrukcemi: (i) konstrukcí pro expresi hERαα (kódující lidský receptor v celé délce) a (ii) reportérovou konstrukcí luciferázy světlušek nesoucí pět tandemových repeticí vitelogeninového estrogen-responzivního elementu (ERE) řízeného myším methalothioneinovým (MT) promotorem TATA elementu. Prokázalo se, že myší genová konstrukce MT TATA podává nejlepší výkon, a proto se běžně používá. Proto může tato buněčná linie hERα-HeLa-9 903 měřit schopnost testované chemické látky indukovat hERα-zprostředkovanou transaktivaci exprese luciferázového genu.
|
|
6.
|
V případě analýzy ER agonistů vychází interpretace údajů z toho, zda je maximální úroveň odpovědi indukované zkoušenou chemickou látkou rovná nebo vyšší než odpověď agonisty rovnající se 10 % odpovědi indukované maximálně indukující (1 nM) koncentrací pozitivní kontroly (PC) 17β-estradiol (E2) (tj. PC10). V případě analýzy ER antagonistů vychází interpretace údajů z toho, zda odpověď vykazuje alespoň 30 % snížení aktivity oproti odpovědi indukované obohacenou kontrolou (25 pM E2) bez cytotoxicity. Analýza a interpretace údajů je podrobně probrána v odstavcích 34–48.
|
POSTUP
Buněčné linie
|
7.
|
Pro analýzu by se měla používat stabilně transfekovaná buněčná linie hERα-HeLa-9 903. Buněčnou linii lze získat z buněčné banky Japonské sbírky výzkumných biozdrojů (Japanese Collection of Research Bioresources (JCRB) (62) po podpisu smlouvy o prodeji materiálu.
|
|
8.
|
Při zkoušení by měly být používány pouze buňky charakterizované jako buňky bez mykoplazmat. Metodou volby pro citlivou detekci mykoplazmové infekce je RT-PCR (polymerázová řetězová reakce v reálném čase) (4)(5)(6).
|
Stabilita buněčné linie
|
9.
|
Pro monitorování stability buněčné linie by se jako referenční standardy pro analýzu agonistů měly používat E2, 17α-estradiol, 17α-methyltestosteron a kortikosteron a alespoň jednou je třeba při každém provedení analýzy změřit kompletní křivku závislosti odpovědi na koncentraci v rozsahu zkoušených koncentrací uvedených v tabulce 1, přičemž výsledky by měly být v souladu s výsledky uvedenými v tabulce 1.
|
|
10.
|
V případě analýzy antagonistů je třeba změřit současně s každým provedením zkoušky úplné křivky koncentrace pro dva referenční standardy, tamoxifen a flutamid. U obou chemických látek je třeba sledovat správnou kvalitativní klasifikaci na pozitivní nebo negativní.
|
Buněčná kultura a podmínky na destičce
|
11.
|
Buňky se udržují v Eaglově minimálním esenciálním médiu (EMEM) bez fenolové červeně, doplněném 60 mg/l antibiotika kanamycinu a 10 % fetálního bovinního séra ošetřeného aktivním uhlím, které je potaženo dextranem (DCC-FBS) v inkubátoru s CO2 (5 % CO2) při 37±1°C. Po dosažení 75–90 % konfluence mohou být buňky pasážovány s 10 ml 0,4 x 105 – 1 x 105 buněk/ml pro 100 mm nádobu s buněčnou kulturou. Buňky by měly být suspendovány s 10 % FBS-EMEM (což je stejné médium jako EMEM s DCC-FBS) a poté umístěny do jamek mikrotitrační destičky v hustotě 1 x 104 jamek/(100 μl x jamka). Dále by měly být buňky předběžně inkubovány v inkubátoru s 5 % CO2 při teplotě 37°±1°C po dobu 3 hodin před expozicí chemické látce. Plastové pomůcky by neměly vykazovat estrogenní aktivitu.
|
|
12.
|
Aby se zachovala integrita odpovědi, měly by buňky růst více než jednu pasáž ze zmrazené zásoby v upraveném médiu a neměly by být kultivovány déle než 40 pasáží. Pro buněčnou linii hERα-HeLa-9 903 to bude méně než tři měsíce. Výkonnost buněk však může být snížena, pokud jsou kultivovány v nevhodných podmínkách.
|
|
13.
|
Médium DCC-FBS lze připravit podle popisu v dodatku 2.2 nebo pořídit z komerčních zdrojů.
|
Kritéria přijatelnosti
Pozitivní a negativní referenční standardy pro analýzu agonistů ER
|
14.
|
Před provedením studie a v jejím průběhu je třeba ověřit reaktivitu zkušebního systému pomocí vhodných koncentrací silného estrogenu: E2, slabého estrogenu (17α-estradiol), velmi slabého agonisty (17α-methyltestosteron) a negativní látky (kortikosteron). Hodnoty rozsahu přijatelnosti odvozené od validované studie (1) jsou uvedeny v tabulce 1. Tyto 4 souběžné referenční standardy by měly být zařazeny do každého experimentu a výsledky by měly spadat do uvedených přijatelných mezí. Pokud tomu tak není, je třeba stanovit příčinu nesplnění kritérií přijatelnosti (např. manipulace s buňkami a kvalita a koncentrace séra a antibiotik) a analýzu zopakovat. Po dosažení kritérií přijatelnosti je pro zajištění minimální variability hodnot EC50, PC50 a PC10 zásadně důležité konzistentní používání materiálů pro kultivaci buněk. Čtyři souběžné referenční standardy, které by měly být zařazeny do každého experimentu (provedeného za stejných podmínek včetně materiálů, úrovně pasážování a techniků), mohou zajistit citlivost analýzy, protože hodnoty PC10 tří pozitivních referenčních standardů by měly spadat do přijatelného rozsahu, stejně jako hodnoty PC50 a EC50, pokud je lze vypočítat (viz tabulka 1).
|
Tabulka 1
Rozsah přijatelných hodnot čtyř referenčních standardů pro analýzu agonistů ER
|
Název
|
logPC50
|
logPC10
|
logEC50
|
Hillův koeficient
|
Zkušební rozsah
|
|
17β-estradiol (E2) CAS č.: 50-28-2
|
–11,4 ~ –10,1
|
< –11
|
–11,3 ~ –10,1
|
0,7 ~ 1,5
|
10-14 ~ 10-8M
|
|
17α-estradiol CAS č.: 57-91-0
|
–9,6 ~ –8,1
|
–10,7 ~ –9,3
|
–9,6 ~ –8,4
|
0,9 ~ 2,0
|
10-12 ~ 10-6M
|
|
Kortikosteron CAS č.: 50-22-6
|
–
|
–
|
–
|
–
|
10-10 ~ 10-4M
|
|
17α-methyltestosteron CAS č.: 58-18-4
|
–6,0 ~ –5,1
|
–8,0 ~ –6,2
|
–
|
–
|
10-11 ~ 10-5M
|
Pozitivní a negativní referenční standardy pro analýzu antagonistů ER
|
15.
|
Před provedením studie a v jejím průběhu je třeba ověřit reaktivitu zkušebního systému pomocí vhodných koncentrací pozitivní látky (tamoxifen) a negativní látky (flutamid). Hodnoty rozsahu přijatelnosti odvozené od validované studie (1) jsou uvedeny v tabulce 2. Tyto dva souběžné referenční standardy by měly být zařazeny do každého experimentu a výsledky by měly být správně posouzeny, jak je uvedeno v kritériích. Pokud tomu tak není, je třeba stanovit příčinu nesplnění kritérií (např. manipulace s buňkami a kvalita a koncentrace séra a antibiotik) a analýzu zopakovat. Dále by měly být vypočítány hodnoty IC50 pro pozitivní látku (tamoxifen) a výsledky by měly spadat do uvedených přijatelných mezí. Po dosažení kritérií přijatelnosti je pro zajištění minimální variability hodnot EC50 zásadně důležité konzistentní používání materiálů pro kultivaci buněk. Dva souběžné referenční standardy, které by měly být zařazeny do každého experimentu (provedeného za stejných podmínek včetně materiálů, úrovně pasážování a techniků), mohou zajistit citlivost analýzy (viz tabulka 2).
|
Tabulka 2
Kritéria a rozsah přijatelných hodnot dvou referenčních standardů pro analýzu antagonistů ER
|
Název
|
Kritéria
|
logIC50
|
Zkušební rozsah
|
|
Tamoxifen CAS č.: 10 540 -29-1
|
Pozitivní: IC50 nemohla být vypočtena
|
-–5,942 ~ –7,596
|
10-10 ~ 10-5M
|
|
Flutamid CAS č.: 13 311 -84-7
|
Negativní: IC30 nemohla být vypočtena
|
-
|
10-10 ~ 10-5M
|
Pozitivní a negativní kontroly
|
16.
|
Pozitivní kontrola (PC) pro analýzu agonistů ER (1 nM E2) a pro analýzu antagonistů ER (10μM TAM) by měla být testována na každé destičce alespoň třikrát. Rozpouštědlo, které se používá na rozpuštění zkoušené chemické látky, by mělo být testováno jako kontrola vehikula (VC) na každé destičce alespoň třikrát. Kromě této kontroly vehikula by měla být kontrola vehikula provedena na stejné destičce alespoň třikrát se stejnou pozitivní kontrolou, pokud se u pozitivní kontroly používá jiné vehikulum než u zkoušené chemické látky.
|
Kvalitativní kritéria pro analýzu agonistů ER
|
17.
|
Střední aktivita luciferázy u pozitivní kontroly (1 nM E2) by měla být alespoň čtyřnásobná oproti střední aktivitě kontroly vehikula na každé destičce. Toto kritérium je stanoveno na základě spolehlivosti hodnot cílových ukazatelů z validační studie (historicky mezi čtyřnásobkem a třicetinásobkem).
|
|
18.
|
Pokud jde o kontrolu kvality analýzy, měla by být násobná indukce odpovídající hodnotě PC10 souběžné pozitivní kontroly (1 nM E2) větší než 1+2SD hodnoty násobné indukce (=1) souběžné kontroly vehikula. Za účelem stanovení priorit může být hodnota PC10 užitečná pro zjednodušení analýzy dat potřebné na porovnání se statistickou analýzou. Ačkoli statistická analýza poskytuje informace o významnosti, tato analýza není kvantitativním parametrem, pokud jde o potenciál vycházející z koncentrace, a proto je méně užitečná pro účely stanovení priorit.
|
Kvalitativní kritéria pro analýzu antagonistů ER
|
19.
|
Střední aktivita luciferázy u obohacené kontroly (25 pM E2) by měla být alespoň čtyřnásobná oproti střední aktivitě kontroly vehikula na každé destičce. Toto kritérium je stanoveno na základě spolehlivosti hodnot cílových ukazatelů z validační studie.
|
|
20.
|
Pokud jde o kontrolu kvality analýzy, měla by být relativní transkripční aktivace (RTA) 1 nM E2 větší než 100 %, RTA 1μM 4-hydroxytamoxifenu (OHT) by měla být menší než 40,6 % a RTA 100 μM Digitoninu (Dig) by měla být menší než 0 %.
Prokázání odborné způsobilosti laboratoře (viz odstavec 14 a tabulky 3 a 4 v části «SLOŽKY ANALÝZY ER TA» této zkušební metody).
|
Vehikulum
|
21.
|
Jako souběžná kontrola vehikula by měl být použit dimethylsulfoxid (DMSO) nebo vhodné rozpouštědlo ve stejné koncentraci použité pro různé pozitivní a negativní kontroly a zkoušené chemické látky. Zkoušené chemické látky by měly být rozpuštěny v rozpouštědle, které danou zkoušenou chemickou látku rozpouští a je mísitelné s kultivačním médiem. Vhodnými vehikuly jsou voda, etanol (čistota 95 % až 100 %) a DMSO. Jestliže se používá DMSO, neměla by úroveň překročit 0,1 % (obj.). Pro každé vehikulum je třeba prokázat, že maximální použitý objem není cytotoxický a nenarušuje provedení analýzy.
|
Příprava zkoušených chemických látek
|
22.
|
Obecně je třeba zkoušené chemické látky rozpustit v DMSO nebo jiném vhodném rozpouštědle, postupně naředit stejným rozpouštědlem ve společném poměru 1:10, a připravit tak roztoky pro ředění médiem.
|
Rozpustnost a cytotoxicita: Důvody pro zjištění rozsahu
|
23.
|
Měla by být provedena předběžná zkouška, aby bylo možné stanovit vhodný rozsah koncentrace chemické látky, jež má být testována, a zkontrolovat, zda zkoušená chemická látka nevykazuje nějaké problémy v oblasti rozpustnosti a cytotoxicity. Nejdříve se chemické látky zkoušejí do maximální koncentrace 1 μl/ml, 1 mg/ml, nebo 1 mM podle toho, která z uvedených hodnot je nejnižší. Podle rozsahu cytotoxicity nebo nerozpustnosti pozorované při předběžné zkoušce by se chemická látka měla při prvním definitivním provedení zkoušky testovat v log-sériovém ředění, počínaje maximální přijatelnou koncentrací (např. 1 mM, 100μM, 10μM atd.) a měla by se zaznamenat přítomnost zákalu nebo sraženiny či cytotoxicity. Při druhém a v případě potřeby i třetím provedení zkoušky by se měla podle potřeby upravit koncentrace, aby lépe charakterizovala křivku závislosti odpovědi na koncentraci a aby byly vyloučeny koncentrace, které byly zjištěny jako nerozpustné nebo vyvolávající nadměrnou cytotoxicitu.
|
|
24.
|
U agonistů a antagonistů ER může přítomnost zvyšujících se úrovní cytotoxicity významně změnit nebo eliminovat typickou sigmoidní odpověď, což je třeba vzít v úvahu při interpretaci dat. Měly by se používat metody zkoušení cytotoxicity, které mohou poskytnout informace o 80 % životaschopnosti buněk, s využitím vhodné analýzy na základě zkušeností laboratoře.
|
|
25.
|
Pokud výsledky zkoušky cytotoxicity ukazují, že koncentrace zkoušené chemické látky snížila počet buněk o 20 % nebo více, je třeba tuto koncentraci považovat za cytotoxickou a koncentrace na cytotoxické úrovni nebo vyšší je třeba z hodnocení vyloučit.
|
Expozice chemické látce a organizace testovacích destiček
|
26.
|
Postup ředění chemických látek (kroky 1 a 2) a expozici buněk (krok 3) lze provést takto:
Krok 1: Každou zkoušenou chemickou látku je třeba postupně naředit v DMSO nebo vhodném rozpouštědle a přidat do jamek mikrotitrační destičky, aby byla dosažena finální sériová koncentrace stanovená při předběžné zkoušce ke stanovení rozsahu (obvykle v sérii například 1 mM, 100 μM, 10 μM, 1μM, 100 nM, 10 nM, 1 nM, 100 pM a 10 pM (10-3–10-11 M)) pro trojí testování.
Krok 2: Ředění chemické látky: Nejprve rozpusťte 1,5 μl zkoušené chemické látky v rozpouštědle na objem 500 μl média.
Krok 3: Expozice buněk chemické látce: Přidejte 50 μl roztoku s médiem (připraveného v kroku 2) do testovací jamky obsahující 104 buněk/100 μl/ jamku.
Doporučený konečný objem média potřebného pro každou jamku je 150 μl. Zkušební vzorky a referenční standardy lze zařadit tak, jak je uvedeno v tabulce 3 a v tabulce 4.
|
Tabulka 3
Příklad uspořádání koncentrací referenčních standardů na testovací destičce při analýze agonistů ER
|
Řádek
|
17α-methyltestosteron
|
Kortikosteron
|
17α-estradiol
|
E2
|
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
konc. 1 (10 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
konc. 2 (1 μM)
|
→
|
→
|
10 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
konc. 3 (100 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
konc. 4 (10 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
konc. 5 (1 nM)
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
konc. 6 (100 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
konc. 7 (10 pM)
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VC
|
→
|
→
|
→
|
→
|
→
|
PC
|
→
|
→
|
→
|
→
|
→
|
|
VC: Kontrola vehikulem (0,1 % DMSO); PC: Pozitivní kontrola (1 nM E2)
|
|
27.
|
Při každém provedení zkoušky by měly být testovány referenční standardy (E2, 17α-estradiol, 17-methyltestosteron a kortikosteronmm(tabulka 3)((Na každé testovací destičce by měly být zařazeny jamky PC exponované 1 nM E2, který dokáže vyvolat maximální indukci E2, a jamky VC exponované pouze DMSO (nebo vhodným rozpouštědlem) (tabulka 4). Pokud jsou použity ve stejném experimentu buňky z různých zdrojů (například různý počet pasáží, různá šarže atd.), je třeba referenční standardy testovat pro každý zdroj buněk.
|
Tabulka 4
Příklad uspořádání koncentrací zkoušených a kontrolních chemických látek na testovací destičce při analýze agonistů ER
|
Řádek
|
Zkoušená chemická látka 1
|
Zkoušená chemická látka 2
|
Zkoušená chemická látka 3
|
Zkoušená chemická látka 4
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
konc. 1 (10 μM)
|
→
|
→
|
1 mM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
konc. 2 (1 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
konc. 3 (100 nM)
|
→
|
→
|
10 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
konc. 4 (10 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
konc. 5 (1 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
konc. 6 (100 nM)
|
→
|
→
|
10 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
konc. 7 (10 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VC
|
→
|
→
|
→
|
→
|
→
|
PC
|
→
|
→
|
→
|
→
|
→
|
|
VC: Kontrola vehikulem (0,1 % DMSO); Pozitivní kontrola: Pozitivní kontrola (1 nM E2)
|
Tabulka 5
Příklad uspořádání koncentrací referenčních standardů na testovací destičce při analýze antagonistů ER
|
28.
|
Aby mohla být posouzena antagonistická aktivita chemických látek, je třeba jamky umístěné v řadách A až G obohatit 25pM E2. Při každém provedení zkoušky by měly být testovány referenční standardy (tamoxifen a flutamid). Na každé testovací destičce by měly být zařazeny jamky s PC exponované 1 nM E2, který může být použit jako kontrola kvality buněčné linie hERα-HeLa-9 903, jamky s VC exponované DMSO (nebo vhodným rozpouštědlem), jamky s 0,1 % DMSO exponované přídavkem DMSO do obohaceného E2 odpovídající „obohacené kontrole“, jamky exponované finální koncentrací 1 μM OHT a jamky exponované 100 μM Dig (tabulka 5). Další testovací destička by měla mít stejné uspořádání bez buněk s referenčními standardy (tabulka 6). Pokud jsou použity ve stejném experimentu buňky z různých zdrojů (například různý počet pasáží, různá šarže atd.), je třeba referenční standardy testovat pro každý zdroj buněk.
|
Tabulka 6
Příklad uspořádání koncentrací zkoušených a kontrolních chemických látek na testovací destičce při analýze antagonistů ER
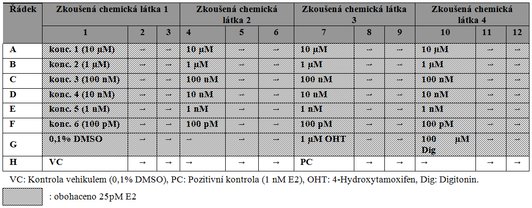
|
29.
|
Podle potřeby je třeba potvrdit nepřítomnost okrajových účinků, a pokud je podezření na takové účinky, je třeba uspořádání destičky změnit, aby k nim nedocházelo. Může být například použito uspořádání destičky s vyloučením okrajových jamek.
|
|
30.
|
Po přidání chemických látek by testovací destičky měly být inkubovány v inkubátoru s 5 % CO2 při teplotě 37°±1°C po dobu 20–24 hodin, aby se indukovaly reportérové genové produkty.
|
|
31.
|
Zvláštní pozornost je třeba věnovat sloučeninám, které jsou vysoce těkavé. V těchto případech mohou vedlejší kontrolní jamky generovat falešně pozitivní výsledky, což je třeba vzít v úvahu s ohledem na očekávané a dosavadní kontrolní hodnoty. V ojedinělých případech, kdy může být těkavost problémem, může pomoci použití „zapečetěných destiček“ účinně izolovat jednotlivé jamky v průběhu zkoušení, a proto je v těchto případech doporučováno.
|
|
32.
|
Opakované definitivní testy pro stejnou chemickou látku by měly být prováděny v různé dny, aby byla zajištěna nezávislost.
|
Luciferázová analýza
|
33.
|
Pro analýzu může být použito komerční luciferázové činidlo [např. systém Steady-Glo® Luciferase Assay (Promega, E2510 nebo ekvivalentní)] nebo standardní luciferázový systém (např. Promega, E1500 nebo ekvivalentní), pokud jsou splněna kritéria přijatelnosti. Činidla pro analýzu je třeba zvolit podle citlivosti luminometru, který bude použit. Když se používá standardní luciferázový systém, mělo by být před přidáním substrátu použito lyzační činidlo na buněčnou kulturu (např. Promega, E1531 nebo ekvivalentní). Luciferázové činidlo je třeba aplikovat podle pokynů výrobce.
|
ANALÝZA ÚDAJŮ
Analýza agonistů ER
|
34.
|
Aby byla získána relativní transkripční aktivita u PC (1 nM E2), mohou být v případě analýzy agonistů ER luminiscenční signály ze stejné destičky analyzovány podle těchto kroků (přijatelné jsou i jiné ekvivalentní matematické procesy):
Krok 1. Vypočítejte střední hodnotu pro VC.
Krok 2. Odečtěte střední hodnotu VC od hodnoty každé jamky, abyste normalizovali data.
Krok 3. Vypočítejte střední hodnotu pro normalizovanou PC.
Krok 4. Vydělte normalizovanou hodnotu každé jamky na destičce střední hodnotou normalizované PC (PC = 100 %).
Konečná hodnota každé jamky je relativní transkripční aktivita pro tuto jamku v porovnání s odpovědí PC.
Krok 5. Vypočtěte střední hodnotu relativní transkripční aktivity pro každou skupinu koncentrace zkoušené chemické látky. Odpověď má dva rozměry: zprůměrovaná transkripční aktivita (odpověď) a koncentrace, při níž dochází k odpovědi (viz následující část).
|
Poznámky k indukci EC50, PC50 a PC10
|
35.
|
Pro výpočet EC50 je nutná celá křivka závislosti koncentrace na odpovědi, ale to nemusí být vždy dosažitelné nebo praktické vzhledem k mezím rozsahu zkušebních koncentrací (například v důsledku problémů s cytotoxicitou nebo rozpustností). Protože jsou však EC50 a maximální indukční úroveň (odpovídající horní hodnotě Hillovy rovnice) informativními parametry, měly by být tyto parametry pokud možno uvedeny. Pro výpočet EC50 a maximální indukční úrovně by měl být použit vhodný statistický software (např. statistický software Graphpad Prism). Jestliže lze na údaje o koncentraci a odpovědi aplikovat Hillovu logistickou rovnici, je třeba vypočítat EC50 takto (7):
Y= dolní + (horní–dolní) / (1+10 exp ((log EC50–X) x Hillův koeficient)) Kde:
X je logaritmus koncentrace; a
Y je odpověď, přičemž Y začíná na dolní hranici a pokračuje nahoru sigmoidní křivkou. Dolní hranice je v Hillově logistické rovnici pevně stanovena na nule.
|
|
36.
|
Pro každou zkoušenou chemickou látku by měly být zajištěny tyto podmínky:
RPCMax, což je maximální úroveň odpovědi indukované zkoušenou chemickou látkou, vyjádřená jako procento odpovědi indukované 1 nM E2 na stejné destičce, stejně jako PCMax (koncentrace související s RPCMax); a
u pozitivních chemických látek koncentrace, které indukují PC10, případně PC50.
|
|
37.
|
Hodnotu PCx lze vypočítat interpolací mezi 2 body do bodu o souřadnici X–Y, přičemž jeden je bezprostředně nad a jeden bezprostředně pod hodnotou PCx. Pokud mají datové body ležící bezprostředně nad a pod hodnotou PCx souřadnice (a,b), respektive (c,d), pak lze vypočítat hodnotu PCx podle této rovnice:
log[PCx] = log[c]+(x-d)/(d-b)
|
|
38.
|
Popisy hodnot PC jsou uvedeny níže na obrázku 1.
Obrázek 1
Příklad odvození hodnot PC. Na každé testovací destičce je zařazena PC (1 nM E2)
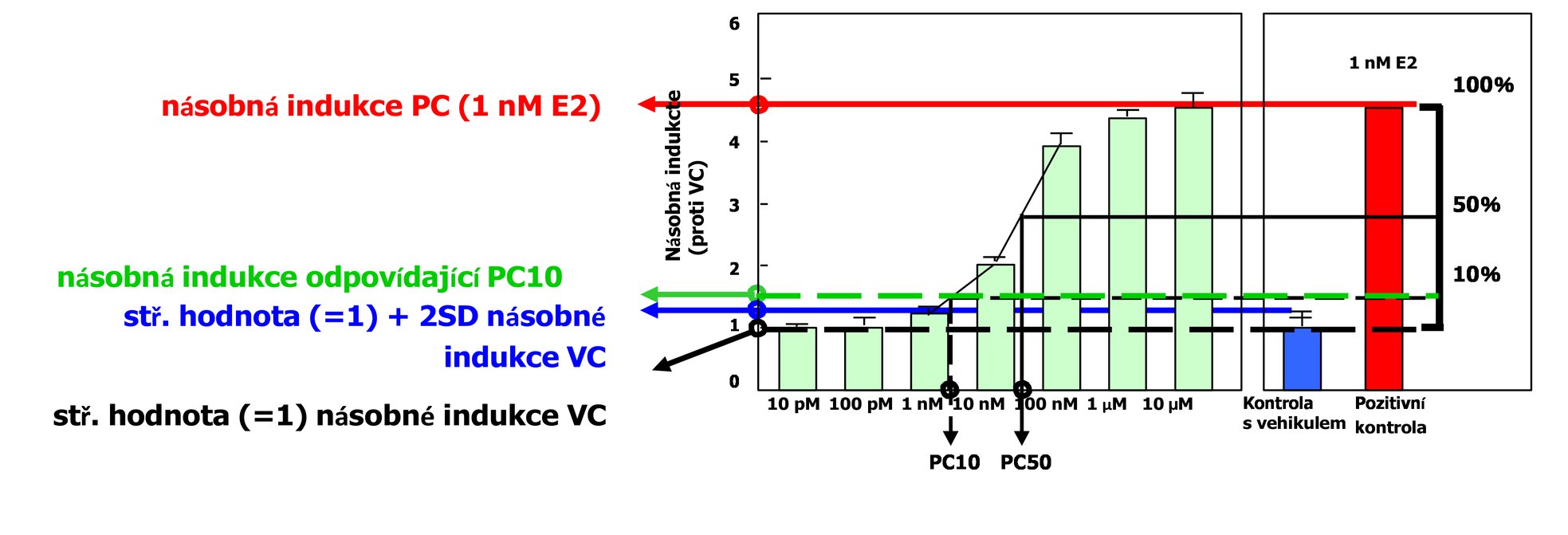
|
Analýza antagonistů ER
|
39.
|
Aby byla získána relativní transkripční aktivita (RTA) u obohacené kontroly (25 pM E2), mohou být v případě analýzy antagonistů ER luminiscenční signály ze stejné destičky analyzovány podle těchto kroků (přijatelné jsou i jiné ekvivalentní matematické procesy):
Krok 1. Vypočítejte střední hodnotu pro VC.
Krok 2. Odečtěte střední hodnotu VC od hodnoty každé jamky, abyste normalizovali data. Krok 3. Vypočítejte střední hodnotu pro normalizovanou obohacenou kontrolu.
Krok 4. Vydělte normalizovanou hodnotu každé jamky na destičce střední hodnotou normalizované obohacené kontroly (obohacená kontrola = 100 %).
Konečná hodnota každé jamky je relativní transkripční aktivita pro tuto jamku v porovnání s odpovědí obohacené kontroly.
Krok 5. Vypočtěte střední hodnotu relativní transkripční aktivity pro každou expozici.
|
Poznámky k indukci IC30 a IC50
|
40.
|
U pozitivních chemických látek je třeba uvést koncentrace, které indukují IC30, případně IC50.
|
|
41.
|
Hodnotu ICx lze vypočítat interpolací mezi 2 body do bodu o souřadnici X–Y, přičemž jeden je bezprostředně nad a jeden bezprostředně pod hodnotou ICx. Pokud mají datové body ležící bezprostředně nad a pod hodnotou ICx souřadnice (c,d), respektive (a,b), pak lze vypočítat hodnotu ICx podle této rovnice:
lin ICx = a-(b-(100-x)) (a-c) /(b-d)
Obrázek 2
Příklad odvození hodnot IC. Na každé testovací destičce je zařazena obohacená kontrola (25 nM E2)
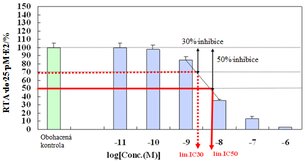
RTA: relativní transkripční aktivita
|
|
42.
|
Výsledky by měly vycházet ze dvou (nebo tří) nezávislých provedení zkoušek. Jestliže jsou při dvou provedeních zkoušky získány srovnatelné, a tedy reprodukovatelné výsledky, není nutné provádět zkoušku potřetí. Aby byly výsledky přijatelné, měly by:
|
—
|
splňovat kritéria přijatelnosti (viz kritéria přijatelnosti v odstavcích 14–20),
|
|
Kritéria interpretace údajů
Tabulka 7
Pozitivní a negativní kritéria rozhodování při analýze agonistů ER
|
Pozitivní
|
Jestliže je získána RPCMax, která se rovná nebo je vyšší než 10 % odpovědi pozitivní kontroly alespoň dvou ze dvou nebo dvou ze tří provedení zkoušek.
|
|
Negativní
|
Jestliže RPC
|
Tabulka 8
Pozitivní a negativní kritéria rozhodování při analýze antagonistů ER
|
Pozitivní
|
Jestliže je vypočtena IC30 alespoň ve dvou ze dvou nebo dvou ze tří provedení zkoušek.
|
|
Negativní
|
Jestliže IC30 není vypočtena alespoň ve dvou ze dvou nebo dvou ze tří provedení zkoušek.
|
|
43.
|
Kritéria interpretace údajů jsou uvedena v tabulkách 7 a 8. Pozitivní výsledky budou charakterizovány jak intenzitou účinku, tak i koncentrací, při níž k účinku dochází. Oba tyto cíle splňuje vyjádření výsledků jako koncentrace, při níž je u analýzy agonistů dosaženo 50 % (PC50) nebo 10 % (PC10) hodnot PC a u analýzy antagonistů je 50 % (IC50) nebo 30 % (IC30) hodnoty obohacené kontroly inhibováno. Zkoušená chemická látka je však určena jako pozitivní, jestliže se maximální odpověď indukovaná touto zkoušenou chemickou látkou (RPCMax) rovná nebo je vyšší než 10 % odpovědi PC alespoň ve dvou ze dvou nebo dvou ze tří provedení zkoušek, zatímco za negativní je zkoušená chemická látka považována, jestliže RPCMax nedosáhne alespoň 10 % odpovědi pozitivní kontroly ve dvou ze dvou nebo dvou ze tří provedení zkoušek.
|
|
44.
|
Výpočty PC10, PC50 a PCMax v analýze agonistů ER a IC30 a IC50 v analýze antagonistů ER mohou být provedeny pomocí tabulkového kalkulátoru, který je dostupný u příslušného pokynu na veřejných webových stránkách OECD (63).
|
|
45.
|
Získání hodnot PC10 nebo PC50 a IC30 nebo IC50 alespoň dvakrát by mělo stačit. Pokud však výsledná výchozí hodnota pro data ve stejném rozsahu koncentrace vykazují variabilitu s nepřijatelně vysokým variačním koeficientem (CV; %), nelze tato data považovat za spolehlivá a je třeba najít zdroj vysoké variability. Variační koeficient prvních tří opakování (tj. údaje o intenzitě luminiscence) datových bodů, které jsou použity pro výpočet PC10, by měl být menší než 20 %.
|
|
46.
|
Splnění kritérií přijatelnosti ukazuje, že analytický systém funguje správně, ale nezaručuje to, že konkrétní provedení zkoušky podá přesné údaje. Opakování výsledků první zkoušky je nejlepším důkazem, že údaje byly přesné.
|
|
47.
|
V případě analýzy agonistů ER, kdy jsou potřebné další informace kromě informací pro účely screeningu a stanovení priorit tohoto pokynu pro pozitivní zkoušené chemické látky, zvláště pro chemické látky PC10–PC49, jakož i chemické látky, u nichž existuje podezření, že nadměrně stimulují luciferázu, může být potvrzeno, že pozorovaná aktivita luciferázy je výhradně ERα-specifická odpověď pomocí antagonisty ERα (viz dodatek 2.1).
|
ZÁVĚREČNÁ ZPRÁVA
|
48.
|
Viz odstavec 20 „SLOŽKY ANALÝZY ER TA“.
|
LITERATURA
|
(1)
|
OECD (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environment, Health and Safety Publications, Series on Testing and Assessment (No 225), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol., 71, 1 459-1 469.
|
|
(3)
|
Kuiper G.G., et al. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta, Endocrinol., 139, 4 252-4 263.
|
|
(4)
|
Spaepen M., et al. (1992). Detection of Bacterial and Mycoplasma Contamination in Cell Cultures by Polymerase Chain Reaction, FEMS Microbiol. Lett., 78(1), 89-94.
|
|
(5)
|
Kobayashi H., et al. (1995). Rapid Detection of Mycoplasma Contamination in Cell Cultures by Enzymatic Detection of Polymerase Chain Reaction (PCR) Products, J. Vet. Med. Sci., 57(4), 769- 71.
|
|
(6)
|
Dussurget O. and Roulland-Dussoix D. (1994). Rapid, Sensitive PCR-Based Detection of Mycoplasmas in Simulated Samples of Animal Sera, Appl. Environ. Microbiol., 60(3), 953-9.
|
|
(7)
|
De Lean A., Munson P.J. and Rodbard D. (1978). Simultaneous Analysis of Families of Sigmoidal Curves: Application to Bioassay, Radioligand Assay, and Physiological Dose-Response Curves, Am. J. Physiol., 235, E97-El02.
|
Dodatek 2.1
FALEŠNĚ POZITIVNÍ: POSOUZENÍ LUMINISCENČNÍCH SIGNÁLŮ NEZPROSTŘEDKOVANÝCH RECEPTOREM
|
1.
|
Falešně pozitivní výsledky mohou být při analýze agonistů ER generovány aktivací luciferázového genu nezprostředkovanou ER nebo přímou aktivací genového produktu či nesouvisející fluorescencí. Tyto účinky indikuje neúplná nebo neobvyklá křivka závislosti odpovědi na dávce. V případě podezření na tyto účinky je třeba vyšetřit účinek antagonisty ER (např. 4-hydroxytamoxifen (OHT) při netoxické koncentraci) na odpověď. K tomuto účelu nemusí být vhodný čistý antagonista ICI 1827 80, neboť dostatečná koncentrace ICI 1827 80 může snížit hodnotu VC, což ovlivní analýzu dat.
|
|
2.
|
Aby byla zajištěna validita tohoto přístupu, je třeba na stejné destičce otestovat:
|
—
|
agonistickou aktivitu neznámé chemické látky s/bez 10 μM OHT
|
|
—
|
1 nM E2 (tři opakování) jako agonistická PC
|
|
—
|
1 nM E2 + OHT (tři opakování)
|
|
KRITÉRIA INTERPRETACE ÚDAJŮ
Poznámka: Do všech jamek by měla být aplikována stejná koncentrace vehikula.
|
—
|
Jestliže aplikace antagonisty ER NEOVLIVNÍ agonistickou aktivitu neznámé chemické látky, klasifikuje se jako „negativní“.
|
|
—
|
Jestliže je agonistická aktivita neznámé chemické látky zcela inhibována, použijte kritéria rozhodování.
|
|
—
|
Jestliže se agonistická aktivita při nejnižší koncentraci rovná nebo je vyšší než odpověď PC10, je neznámá chemická látka inhibována při odpovědi rovnající se nebo vyšší než PC10. Vypočte se rozdíl odpovědí mezi jamkami neošetřenými a ošetřenými antagonistou ER a jamkami, do nichž antagonista ER byl aplikován, a tento rozdíl je třeba považovat za skutečnou odpověď a použít jej pro výpočet vhodných parametrů, aby mohlo být rozhodnuto o klasifikaci.
|
ANALÝZA ÚDAJŮ
Zkontrolujte standard funkčnosti.
Zkontrolujte variační koeficient mezi jamkami ošetřenými za stejných podmínek.
|
1.
|
Vypočítejte střední hodnotu VC.
|
|
2.
|
Odečtěte střední hodnotu VC od hodnoty každé jamky neošetřené OHT.
|
|
3.
|
Vypočítejte střední hodnotu OHT.
|
|
4.
|
Odečtěte střední hodnotu VC od hodnoty každé jamky ošetřené OHT.
|
|
5.
|
Vypočítejte střední hodnotu PC.
|
|
6.
|
Vypočítejte relativní transkripční aktivitu všech ostatních jamek ve vztahu k PC.
|
Dodatek 2.2
PŘÍPRAVA SÉRA OŠETŘENÉHO AKTIVNÍM UHLÍM POTAŽENÝM DEXTRANEM (DCC)
|
1.
|
Ošetření séra aktivním uhlím potaženým dextranem (DCC) je obecná metoda na odstranění estrogenních sloučenin ze séra, které se přidává do kultivačního média, aby se vyloučila zkreslená odpověď související s reziduálními estrogeny v séru. Tímto postupem lze ošetřit 500 ml fetálního bovinního séra (FBS).
|
SLOŽKY
|
2.
|
Potřebné budou tyto materiály a vybavení:
|
Materiály
|
Hexahydrát chloridu hořečnatého (MgCl2· 6H2O)
|
|
1 M HEPES pufrační roztok (pH 7,4)
|
|
Ultračistá voda z filtračního systému
|
Vybavení
Sterilizovaná skleněná nádoba (velikost podle potřeby) běžná laboratorní odstředivka (s možností nastavení teploty na 4 °C)
POSTUP
|
3.
|
Při použití 50ml odstředivkových zkumavek se upraví tento postup:
[Den 1] Připravte suspenzi aktivního uhlí potaženého dextranem s 1 litrem ultračisté vody obsahující 1,5 mM MgCl2, 0,25 M sacharózy, 2,5 g aktivního uhlí, 0,25 g dextranu a 5 mM HEPES a nechte promíchávat přes noc při 4 °C.
[Den 2] Nadávkujte suspenzi do 50ml odstředivkových zkumavek a odstřeďujte při 10 000 ot./min. při 4 °C po dobu 10 minut. Odstraňte supernatant a uložte polovinu sedimentu aktivního uhlí při 4 °C pro použití 3. den. Druhou polovinu aktivního uhlí rozpusťte v FBS, které jste nechali pomalu roztát, aby nevznikla sraženina, a inaktivovali teplem při teplotě 56 °C po dobu 30 minut, poté přemístěte do sterilizované nádoby, například Erlenmeyerovy baňky. Tuto suspenzi nechte lehce promíchávat přes noc při teplotě 4 °C.
[Den 3] Nadávkujte suspenzi s FBS do odstředivkových zkumavek a odstřeďujte při 10 000 ot./min. při 4 °C po dobu 10 minut. Seberte FBS a uložte jej do nového sedimentu aktivního uhlí připraveného a uloženého 2. den. Rozpusťte sediment aktivního uhlí a tuto suspenzi nechte přes noc lehce promíchávat ve sterilizované nádobě při teplotě 4 °C.
[Den 4] Nadávkujte suspenzi k odstřeďování při 10 000 ot./min. při teplotě 4 °C po dobu 10 minut a sterilizujte supernatant filtrací přes sterilní filtr 0,2 μm. Tento FBS ošetřený DCC by měl být uchováván při teplotě -20 °C a může být používán po dobu jednoho roku.
|
Dodatek 3
ANALÝZA TRANSAKTIVACE ESTROGENOVÝCH RECEPTORŮ VM7LUC KE ZJIŠTĚNÍ AGONISTŮ A ANTAGONISTŮ ESTROGENOVÝCH RECEPTORŮ
VÝCHOZÍ ÚVAHY A OMEZENÍ (VIZ TAKÉ VŠEOBECNÝ ÚVOD)
|
1.
|
Při této analýze se používá buněčná linie VM7Luc4E2 (64). Byla validována Meziagenturním střediskem pro hodnocení alternativních toxikologických metod Národního toxikologického programu (National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods – NICEATM) a Meziagenturním koordinačním výborem pro validaci alternativních metod (Interagency Coordinating Committee on the Validation of Alternative Methods – ICCVAM) (1). Buněčné linie VM7Luc exprimují převážně endogenní ERα a menší množství endogenního ERβ (2) (3) (4).
|
|
2.
|
Tuto analýzu lze použít u široké škály látek, pokud mohou být rozpuštěny v dimethylsulfoxidu (DMSO; CASRN 67-68-5), nereagují s DMSO ani s kultivačním médiem a nejsou cytotoxické ve zkoušených koncentracích. Pokud není možné použít DMSO, může být použito jiné vehikulum, jako je etanol nebo voda (viz odstavec 12). Prokázaná funkční způsobilost analýzy (ant)agonistů VM7Luc ER TA naznačuje, že údaje získané při této analýze mohou poskytnout informace o mechanismech působení zprostředkovaných ER a mohou být vzaty v úvahu při určování priorit látek k dalšímu zkoušení.
|
|
3.
|
Tato analýza je specificky určena na detekci TA zprostředkované hERα a hERß měřením chemiluminiscence jako sledované vlastnosti. Použití chemiluminiscence při bioanalýzách je rozšířené, protože luminiscence má vysoký poměr odstupu signálu od pozadí. Aktivitu luciferázy světlušek v buněčných testech však mohou zkreslovat látky, které inhibující enzym luciferázu, a vyvolávají tak zdánlivou inhibici nebo vyšší luminiscenci v důsledku stabilizace proteinů. Kromě toho v některých analýzách reportérového genu ER na bázi luciferázy byly při koncentracích fytoestrogenů vyšších než 1 μM hlášeny luminiscenční signály nezprostředkované receptorem v důsledku nadměrné aktivace reportérového genu luciferázy (9) (11). Přestože křivka závislosti odpovědi na dávce naznačuje, že ke skutečné aktivaci systému ER dochází při nižších koncentracích, je nutné v systémech analýzy stabilně transfekované ER TA pečlivě zkoumat exprese luciferázy získané při vysoké koncentraci fytoestrogenů nebo podobných sloučenin, u nichž existuje podezření, že vyvolávají nadměrnou aktivaci reportérového genu luciferázy podobně jako fytoestrogeny.
|
|
4.
|
Před použitím této analýzy pro regulační účely je třeba přečíst si „VŠEOBECNÝ ÚVOD“ a „SLOŽKY ANALÝZY ER TA“. Definice a zkratky používané v této zkušební metodě jsou popsány v dodatku 1.
|
PRINCIP ANALÝZY (VIZ TAKÉ VŠEOBECNÝ ÚVOD)
|
5.
|
Analýza se používá na indikaci vazby ligandu na estrogenový receptor, po níž následuje přesun komplexu receptor-ligand do jádra. V jádru se komplex receptor-ligand naváže na specifické responzivní elementy DNA a transaktivuje reportérový gen (luc), což vyvolá tvorbu luciferázy a následnou emisi světla, které lze kvantifikovat pomocí luminometru. Aktivitu luciferázy lze rychle a levně vyhodnotit pomocí řady komerčně dostupných testů. Test VM7Luc ER TA využívá ER responzivní buněčnou linii lidského adenokarcinomu prsu, VM7, která je stabilně transfekována reportérovou konstrukcí světlušek luc pod kontrolou čtyř estrogenových responzivních elementů umístěných před promotorem viru myšího nádoru prsní žlázy (MMTV), k detekci látek s agonistickou nebo antagonistickou aktivitou ER in vitro. Tento promotor MMTV vykazuje jen malou křížovou reaktivitu s jinými steroidními a nesteroidními hormony (8). Kritéria pro interpretaci údajů jsou podrobně popsána v odstavci 41. Stručně řečeno, pozitivní odpověď se určuje podle křivky závislosti odpovědi na koncentraci obsahující alespoň tři body s nepřekrývajícími se chybovými úsečkami (střední hodnota ± SD) a podle změny amplitudy (normalizovaná relativní luminiscenční jednotka [RLU]) ve výši alespoň 20 % maximální hodnoty pro referenční standard (17β-estradiol [E2; CASRN 50-28-2] při analýze agonistů, raloxifen-hydrochlorid [Ral; CASRN 84 449-90-1]/E2 při analýze antagonistů).
|
POSTUP
Buněčná linie
|
6.
|
Pro analýzu by se měla používat stabilně transfekovaná buněčná linie VM7Luc4E2. Tato buněčná linie je v současnosti k dispozici pouze na základě technické licenční smlouvy od Kalifornské univerzity, Davis, Kalifornie, USA (65) a společnosti Xenobiotic Detection Systems Inc., Durham, North Carolina, USA (66).
|
Stabilita buněčné linie
|
7.
|
Aby se zachovala stabilita a integrita buněčné linie, měly by buňky růst více než jednu pasáž ze zmrazené zásoby v udržovacím médiu (viz odstavec 9). Buňky by neměly být kultivovány déle než 30 pasáží. Pro buněčnou linii VM7Luc4E2 bude 30 pasáží trvat zhruba tři měsíce.
|
Buněčná kultura a podmínky na destičce
|
8.
|
Měly být dodržovány postupy uvedené v pokynu ke správné praxi při kultivaci buněk (5)(6), aby byla zajištěna kvalita všech materiálů a metod a zachována integrita, validita a reprodukovatelnost veškeré provedené práce.
|
|
9.
|
Buňky VM7Luc4E2 se uchovávají v médiu RPMI 1 640 doplněném 0,9 % přípravkem Pen-Strep a 8,0 % fetálním bovinním sérem (FBS) v inkubátoru vyhrazeném pro tkáňové kultury při teplotě 37 oC ± 1 oC, vlhkosti vzduchu 90 % ± 5 % a 5,0 % ± 1 % CO2/vzduch.
|
|
10.
|
Po dosažení konfluence ~ 80 % se buňky VM7Luc4E2 subkultivují a po dobu 48 hodin upravují v prostředí bez estrogenů, než jsou umístěny na 96jamkovou mikrotitrační destičku, kde jsou exponovány zkoušenými chemickými látkami a analyzovány na indukci luciferázové aktivity závislé na estrogenu. Médium bez estrogenů (EFM) obsahuje Dulbeccovu modifikaci Eaglova média bez fenolové červeně (DMEM), doplněného 4,5 % FBS ošetřeného aktivním uhlím/dextranem, 1,9 % L-glutaminu a 0,9 % Pen-Strepu. Žádné plastové pomůcky by neměly vykazovat estrogenní aktivitu [viz podrobný protokol (7)].
|
Kritéria přijatelnosti
|
11.
|
Přijetí nebo odmítnutí zkoušky vychází z hodnocení referenčního standardu a kontrolních výsledků z každého experimentu na 96jamkové destičce. Každý referenční standard se zkouší v několika koncentracích a na několika vzorcích každé referenční i kontrolní koncentrace. Výsledky se porovnají s kvalitativními kontrolami (QC) u těch parametrů, které byly odvozeny z dosavadních databází agonistů a antagonistů, které si každá laboratoř vypracovala při prokazování způsobilosti. Dosavadní databáze se průběžně aktualizují o hodnoty referenčních standardů a kontrol. Při změnách vybavení nebo podmínek v laboratoři může být nutná aktualizace dosavadních databází.
|
Zkouška agonistů
Zkouška ke stanovení rozsahu
|
•
|
Indukce: Indukce na destičce se měří vydělením průměrné nejvyšší referenční standardní hodnoty relativní luminiscenční jednotky (RLU) E2 průměrnou kontrolní hodnotou RLU DMSO. Obvykle je dosažena pětinásobná indukce, ale pro účely přijetí by měla být indukce větší než nebo rovna čtyřnásobku.
|
|
•
|
Výsledky kontroly DMSO: Kontrolní hodnoty RLU rozpouštědla by měly být v rozsahu 2,5násobku směrodatné odchylky dosavadní kontrolní střední hodnoty RLU rozpouštědla.
|
|
•
|
Experiment, při němž není splněno ani jedno kritérium, je třeba vyřadit a zopakovat.
|
Souhrnný test
Zahrnuje kritéria přijatelnosti ze zkoušky ke stanovení rozsahu agonistů a tyto údaje:
|
•
|
Referenční standardní výsledky: Tvar křivky závislosti odpovědi na koncentraci u referenčního standardu E2 by měl být sigmoidní a křivka by měla mít alespoň tři hodnoty v lineární části křivky závislosti odpovědi na koncentraci.
|
|
•
|
Výsledky pozitivní kontroly: Kontrolní hodnoty RLU methoxychloru by měly být větší než střední hodnota DMSO plus trojnásobek směrodatné odchylky od střední hodnoty DMSO.
|
|
•
|
Experiment, při němž není splněno některé z kritérií přijatelnosti, je třeba vyřadit a zopakovat.
|
Zkouška antagonistů
Zkouška ke stanovení rozsahu
|
•
|
Redukce: Redukce na destičce se měří vydělením průměrné nejvyšší referenční standardní hodnoty RLU Ral/E2 průměrnou kontrolní hodnotou RLU DMSO. Obvykle je dosažena pětinásobná redukce, ale pro účely přijetí by měla být redukce větší než nebo rovna trojnásobku.
|
|
•
|
Výsledky kontroly E2: Kontrolní hodnoty RLU E2 by měly být v rozsahu 2,5násobku směrodatné odchylky dosavadní kontrolní střední hodnoty RLU E2.
|
|
•
|
Výsledky kontroly DMSO: Kontrolní hodnoty RLU DMSO by měly být v rozsahu 2,5násobku směrodatné odchylky dosavadní kontrolní střední hodnoty RLU rozpouštědla.
|
|
•
|
Experiment, při němž není splněno některé z kritérií přijatelnosti, se vyřadí a zopakuje.
|
Souhrnný test
Zahrnuje kritéria přijatelnosti ze zkoušky ke stanovení rozsahu antagonistů a tyto údaje:
|
•
|
Referenční standardní výsledky: Tvar křivky závislosti odpovědi na koncentraci u referenčního standardu Ral/E2 by měl být sigmoidní a křivka by měla mít alespoň tři hodnoty v lineární části křivky závislosti odpovědi na koncentraci.
|
|
•
|
Výsledky pozitivní kontroly: Kontrolní hodnoty RLU tamoxifenu/E2 by měly být menší než kontrolní střední hodnota E2 minus trojnásobek směrodatné odchylky od střední hodnoty E2.
|
|
•
|
Experiment, při němž není splněno některé z kritérií přijatelnosti, se vyřadí a zopakuje.
|
Referenční standardy, pozitivní kontrola a kontrola vehikula
Kontrola vehikula (analýzy agonistů a antagonistů)
|
12.
|
Rozpouštědlo, které se používá na rozpuštění zkoušených chemických látek, by mělo být testováno jako kontrola s vehikulem. Vehikulem použitým při validaci analýzy VM7Luc ER TA byl 1 % (obj.) dimethylsulfoxid (DMSO, CASRN 67-68-5) (viz odstavec 24). Jestliže je použito jiné vehikulum než DMSO, měly by být všechny referenční standardy, kontroly a zkoušené chemické látky testovány podle potřeby ve stejném vehikulu.
|
Referenční standardy (zjištění rozsahu agonistů)
|
13.
|
Referenčním standardem je E2 (CASRN 50-28-2). Při zkouškách ke stanovení rozsahu sestává referenční standard z postupného ředění čtyř koncentrací E2 (1,84 x 10-10, 4,59 x 10-11, 1,15 x 10-11 a 2,87 x 10-12 M), přičemž každá koncentrace se zkouší v duplikátních jamkách.
|
Referenční standardy (souhrnný test agonistů)
|
14.
|
E2 pro souhrnný test sestává z postupného ředění 1: 2 a zahrnuje 11 koncentrací (v rozsahu od 3,67 x 10-10 do 3,59 x 10-13 M) E2 v duplikátních jamkách.
|
Referenční standardy (zjištění rozsahu antagonistů)
|
15.
|
Referenčním standardem je kombinace Ral (CASRN 84 449-90-1) a E2 (CASRN 50-28-2). Ral/E2 pro zkoušky ke stanovení rozsahu sestává z postupného ředění tří koncentrací Ral (3,06 x 10-9, 7,67 x 10-10 a 1,92 x 10-10M) plus fixní koncentrace E2 (9,18 × 10-11 M) v duplikátních jamkách.
|
Referenční standard (souhrnný test antagonistů)
|
16.
|
Ral/E2 pro souhrnný test sestává z postupného ředění Ral 1: 2 (v rozsahu od 2,45x 10-8 do 9,57 x 10-11M) plus fixní koncentrace (9,18 × 10-11 M) E2 zahrnujícího devět koncentrací Ral/E2 v duplikátních jamkách.
|
Slabá pozitivní kontrola (agonista)
|
17.
|
Slabá pozitivní kontrola je 9,06 x 10-6 M p,p’-methoxychlor (methoxychlor; CASRN 72-43-5) v EFM.
|
Slabá pozitivní kontrola (antagonista)
|
18.
|
Slabá pozitivní kontrola sestává z tamoxifenu (CASRN 10 540-29-1) 3,36 x 10-6 M s 9,18 × 10-11 M E2 v EFM.
|
Kontrola E2 (pouze analýza antagonistů)
|
19.
|
Kontrola E2 je 9,18 × 10-11 M E2 v EFM a používá se jako výchozí negativní kontrola.
|
Násobná indukce (agonista)
|
20.
|
Indukce luciferázové aktivity referenčního standardu (E2) se měří vydělením průměrné nejvyšší referenční standardní hodnoty RLU E2 průměrnou kontrolní hodnotou RLU DMSO, přičemž výsledek by měl být větší než čtyřnásobek.
|
Násobná indukce (antagonista)
|
21.
|
Střední luciferázová aktivita referenčního standardu (Ral/E2) se měří vydělením průměrné nejvyšší referenční standardní hodnoty RLU Ral/E2 průměrnou kontrolní hodnotou RLU DMSO a měla by být větší než trojnásobek.
Prokázání odborné způsobilosti laboratoře (viz odstavec 14 a tabulky 3 a 4 v části «SLOŽKY ANALÝZY ER TA» této zkušební metody)
|
Vehikulum
|
22.
|
Zkoušené chemické látky by měly být rozpuštěny v rozpouštědle, které zkoušenou chemickou látku rozpouští a je mísitelné s kultivačním médiem. Vhodnými vehikuly jsou voda, etanol (čistota 95 % až 100 %) a DMSO. Jestliže se používá DMSO, neměla by úroveň překročit 1 % (obj.). Pro každé vehikulum je třeba prokázat, že maximální použitý objem není cytotoxický a nenarušuje provedení analýzy. Referenční standardy a kontroly se rozpustí ve 100 % rozpouštědle a poté naředí na vhodné koncentrace v EFM.
|
Příprava zkoušených chemických látek
|
23.
|
Zkoušené chemické látky se rozpustí ve 100 % DMSO (nebo vhodném rozpouštědle) a poté naředí na vhodné koncentrace v EFM. Před rozpuštěním a naředěním by se měly všechny zkoušené chemické látky nechat ustálit na pokojovou teplotu. Pro každý experiment by se měly připravit čerstvé roztoky zkoušených chemických látek. V roztocích by neměla být znatelná žádná sraženina ani zákal. Zásoba referenčních standardů a kontrol může být připravována hromadně; pro každý experiment by však měly být připraveny čerstvé konečné referenční standardní a kontrolní roztoky a zkoušené chemické látky a měly by být použity do 24 hodin od přípravy.
|
Rozpustnost a cytotoxicita: Důvody pro zjištění rozmezí dávek
|
24.
|
Zkoušky ke stanovení rozsahu zahrnují sedmibodové postupné ředění – 1:10 a provádějí se dvakrát. Nejdříve se chemické látky zkoušejí do maximální koncentrace 1 mg/ml (~1 mM) u zkoušek agonistů a 20 μg/ml (~10 μM) u zkoušek antagonistů. Experimenty ke stanovení rozsahu se používají k určení:
|
|
—
|
počátečních koncentrací zkoušené chemické látky, jež mají být použity při souhrnném testování,
|
|
—
|
ředění zkoušených chemických látek (1: 2 nebo 1: 5), jež mají být použity při souhrnném testování.
|
|
25.
|
Posouzení životaschopnosti buněk/cytotoxicity je zahrnuto v protokolech o zkouškách agonistů a antagonistů (7) a je součástí zkoušek ke stanovení rozsahu a souhrnného testování. Metodou posouzení cytotoxicity, která byla použita pro posouzení životaschopnosti buněk při validaci analýzy VM7Luc ER TA (1), byla škálovaná kvalitativní vizuální observační metoda; ke stanovení cytotoxicity však může být použita kvantitativní metoda (viz protokol (7). Údaje z koncentrací zkoušených chemických látek, které vyvolávají více než 20 % redukci životaschopnosti, nelze použít.
|
Expozice zkoušené chemické látce a organizace testovacích destiček
|
26.
|
Buňky se spočítají a umístí na 96jamkové mikrotitrační destičky (2 x 105 buněk na jamku) v EFM a ponechají se inkubovat po dobu 24 hodin, aby se umožnilo přichycení buněk na destičku. EFM se odstraní a nahradí zkoušenými a referenčními chemickými látkami v EFM a ponechají se inkubovat po dobu 19–24 hodin. Zvláštní pozornost je třeba věnovat sloučeninám, které jsou vysoce těkavé, neboť vedlejší kontrolní jamky mohou generovat falešně pozitivní výsledky. V těchto případech může pomoci použití „zapečetěných destiček“ účinně izolovat jednotlivé jamky v průběhu zkoušení, a proto je doporučováno.
|
Zkoušky ke stanovení rozsahu
|
27.
|
Při zkouškách ke stanovení rozsahu se používají všechny jamky 96jamkové mikrotitrační destičky na testování až šesti zkoušených chemických látek v sedmibodovém postupném ředění 1:10 duplicitně (viz obrázky 1 a 2).
|
|
—
|
Při zkouškách ke stanovení rozsahu agonistů se používají čtyři koncentrace E2 duplicitně jako referenční standard a čtyři replikované jamky pro kontrolu DMSO.
|
|
—
|
Při zkouškách ke stanovení rozsahu antagonistů se používají tři koncentrace Ral/E2 s 9,18 × 10-11 M E2 duplicitně jako referenční standard a tři replikované jamky pro kontrolu E2 a DMSO.
|
Obrázek 1
Uspořádání 96jamkové mikrotitrační destičky při zkoušce ke stanovení rozsahu agonistů
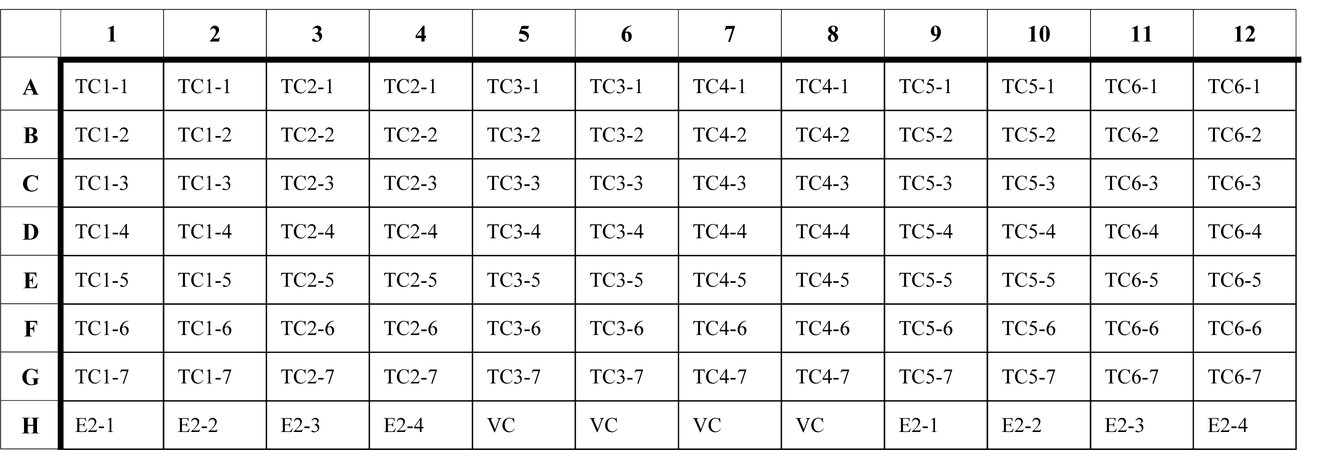
Zkratky: E2-1 až E2-4 = koncentrace referenčního standardu E2 (od vysoké po nízkou); TC1-1 až TC1-7 = koncentrace (od vysoké po nízkou) zkoušené chemické látky 1 (TC1); TC2-1 až TC2-7 = koncentrace (od vysoké po nízkou) zkoušené chemické látky 2 (TC2); TC3-1 až TC3-7 = koncentrace (od vysoké po nízkou) zkoušené chemické látky 3 (TC3); TC4-1 až TC4-7 = koncentrace (od vysoké po nízkou) zkoušené chemické látky 4 (TC4); TC5-1 až TC5-7 = koncentrace (od vysoké po nízkou) zkoušené chemické látky 5 (TC5); TC6-1 až TC6-7 = koncentrace (od vysoké po nízkou) zkoušené chemické látky 6 (TC6); VC = kontrola vehikula (DMSO [1 % obj. EFM.]).
Obrázek 2
Uspořádání 96jamkové mikrotitrační destičky při zkoušce ke stanovení rozsahu antagonistů
Zkratky: E2 = E2 kontrola; Ral-1 až Ral-3 = koncentrace raloxifenu/referenčního standardu E2 (od vysoké po nízkou); TC1-1 až TC1-7 = koncentrace (od vysoké po nízkou) zkoušené chemické látky 1 (TC1); TC2-1 až TC2-7 = koncentrace (od vysoké po nízkou) zkoušené chemické látky 2 (TC2); TC3-1 až TC3-7 = koncentrace (od vysoké po nízkou) zkoušené chemické látky 3 (TC3); TC4-1 až TC4-7 = koncentrace (od vysoké po nízkou) zkoušené chemické látky 4 (TC4); TC5-1 až TC5-7 = koncentrace (od vysoké po nízkou) zkoušené chemické látky 5 (TC5); TC6-1 až TC6-7 = koncentrace (od vysoké po nízkou) zkoušené chemické látky 6 (TC6); VC = kontrola vehikula (DMSO [1 % obj. EFM.]).
Poznámka: Všechny zkoušené chemické látky jsou testovány v přítomnosti 9,18 × 10-11 M E2.
|
28.
|
Doporučený konečný objem média potřebného pro každou jamku je 200 μl. Používejte pouze testovací destičky, v nichž buňky ve všech jamkách vykazují životaschopnost 80 % a více.
|
|
29.
|
Stanovení počátečních koncentrací pro souhrnné testování
agonistů
je podrobně popsáno v protokolu pro agonisty (7). Stručně řečeno, použijí se tato kritéria:
|
|
—
|
Jestliže na křivce koncentrace zkoušené chemické látky nejsou žádné body, které by byly větší než střední hodnota plus trojnásobek směrodatné odchylky kontroly DMSO, provede se souhrnné testování s použitím 11bodového postupného ředění 1: 2, počínaje maximální rozpustnou koncentrací.
|
|
—
|
Jestliže na křivce koncentrace zkoušené chemické látky jsou body větší než střední hodnota plus trojnásobek směrodatné odchylky kontroly DMSO, měla by být počáteční koncentrace, která se použije pro 11bodové postupné ředění při souhrnném testování o jeden logaritmus vyšší než koncentrace vykazující nejvyšší upravenou hodnotu RLU ve zkoušce ke zjištění rozsahu. 11bodové ředění vychází z ředění 1: 2 nebo 1: 5 podle těchto kritérií:
|
Postupné 11bodové ředění 1: 2 by se mělo používat, pokud bude výsledné rozmezí koncentrace zahrnovat celý rozsah odpovědí vycházejících z křivky závislosti odpovědi na koncentraci vytvořené při zkoušce ke zjištění rozsahu. Jinak používejte ředění 1: 5.
|
|
|
—
|
Jestliže zkoušená chemická látka vykazuje při zkoušce ke zjištění rozsahu dvoufázovou křivku závislosti odpovědi na koncentraci, měly by být při souhrnném testování vyřešeny také obě fáze.
|
|
30.
|
Stanovení počátečních koncentrací pro souhrnné testování
antagonistů
je podrobně popsáno v protokolu pro antagonisty (7). Stručně, použijí se tato kritéria:
|
|
—
|
Jestliže na křivce koncentrace zkoušené chemické látky nejsou žádné body, které by byly menší než střední hodnota minus trojnásobek směrodatné odchylky kontroly E2, provede se souhrnné testování s použitím 11bodového postupného ředění 1: 2, počínaje maximální rozpustnou koncentrací.
|
|
—
|
Jestliže na křivce koncentrace zkoušené chemické látky jsou body menší než střední hodnota minus trojnásobek směrodatné odchylky kontroly E2, měla by počáteční koncentrace, která se použije pro 11bodové postupné ředění při souhrnném testování, odpovídat jedné z těchto možností:
|
—
|
Koncentrace vykazující při zkoušce ke zjištění rozsahu nejnižší upravenou hodnotou RLU
|
|
—
|
Maximální rozpustná koncentrace (viz protokol pro antagonisty (7), obrázek 14-2)
|
|
—
|
Nejnižší cytotoxická koncentrace (viz protokol pro antagonisty (7), obrázek 14-3, kde je uveden podobný příklad).
|
|
|
—
|
11bodové ředění vychází z postupného ředění 1: 2 nebo 1: 5 podle těchto kritérií:
|
Postupné 11bodové ředění 1: 2 by se mělo používat, pokud bude výsledné rozmezí koncentrace zahrnovat celý rozsah odpovědí vycházejících z křivky závislosti odpovědi na koncentraci vytvořené při zkoušce ke zjištění rozsahu. Jinak se použije ředění 1: 5.
|
|
Souhrnné testování
|
31.
|
Souhrnné testování sestává z jedenáctibodového postupného ředění (postupné ředění buď 1: 2, nebo 1: 5 na základě počáteční koncentrace pro kritéria souhrnného testování), přičemž každá koncentrace se testuje ve třech jamkách 96jamkové mikrotitrační destičky (viz obrázky 3 a 4).
|
|
—
|
Při souhrnném testování agonistů se používá 11 koncentrací E2 duplicitně jako referenční standard. Na každé destičce jsou zahrnuty čtyři replikované jamky pro kontrolu s DMSO a čtyři replikované jamky pro kontrolu s methoxychlorem (9,06 x 10-6 M).
|
|
—
|
Při zkouškách ke stanovení rozsahu antagonistů se používá devět koncentrací Ral/E2 s 9,18 × 10-11 M E2 duplicitně jako referenční standard a čtyři replikované jamky pro kontrolu E2 9,18 x 10-11 M, čtyři replikované jamky pro kontrolu DMSO a čtyři replikované jamky pro tamoxifen 3,36 x 10-6 M.
|
Obrázek 3
Uspořádání 96jamkové mikrotitrační destičky při souhrnném testování agonistů
Zkratky: TC1-1 až TC1-11 = koncentrace (od vysoké po nízkou) zkoušené chemické látky 1; TC2-1 až TC2-11 = koncentrace (od vysoké po nízkou) zkoušené chemické látky 2; E2-1 až E2-11 = koncentrace referenčního standardu E2 (od vysoké po nízkou); Meth = p,p’ methoxychlor slabá pozitivní kontrola; VC = DMSO (1 % obj.) EFM kontrola vehikula
Obrázek 4
Uspořádání 96jamkové mikrotitrační destičky při souhrnném testování antagonistů
Zkratky: E2 = E2 kontrola; Ral-1 až Ral-9 = koncentrace raloxifenu/referenčního standardu E2 (od vysoké po nízkou); Tam = tamoxifen/E2 slabá pozitivní kontrola; TC1-1 až TC1-11 = koncentrace (od vysoké po nízkou) zkoušené chemické látky 1 (TC1); TC2-1 až TC2-11 = koncentrace (od vysoké po nízkou) zkoušené chemické látky 2 (TC2); VC = kontrola vehikula (DMSO [1 % obj. EFM.]).
Poznámka: Jak bylo uvedeno, všechny referenční a testovací jamky obsahují pevně stanovenou koncentraci E2 (9,18 x 10-11 M).
|
32.
|
Opakované souhrnné testy pro stejnou chemickou látku by měly být prováděny v různé dny, aby byla zajištěna nezávislost. Měly by být provedeny nejméně dva souhrnné testy. Jestliže jsou výsledky testů vzájemně rozporné (například jeden test je pozitivní, druhý negativní) nebo jestliže jeden z testů není adekvátní, měl by být proveden dodatečný třetí test.
|
Měření luminiscence
|
33.
|
Luminiscence se měří v rozsahu 300 až 650 nm, a to pomocí injekčního luminometru a softwaru, který řídí objem vstřikování a interval měření (7). Světelné emise z každé jamky se vyjadřují jako RLU na jamku.
|
ANALÝZA ÚDAJŮ
Stanovení EC50 /IC50
|
34.
|
Hodnota EC50 (poloviční maximální účinná koncentrace zkoušené chemické látky [agonisté]) a hodnota IC50 (poloviční maximální inhibiční koncentrace zkoušené chemické látky [antagonisté]) se stanoví z údajů o závislosti odpovědi na koncentraci. U zkoušených chemických látek, které jsou pozitivní při jedné nebo několika koncentracích, se koncentrace zkoušené chemické látky, která vyvolává poloviční maximální odpověď (IC50 nebo EC50) vypočítá pomocí analýzy Hillovy funkce nebo vhodným alternativním způsobem. Hillova funkce je čtyřparametrový logistický matematický model týkající se závislosti odpovědi na koncentraci zkoušené chemické látky (obvykle podle sigmoidní křivky) s využitím níže uvedené rovnice:
|
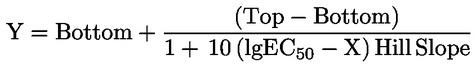
kde:
Y= odpověď (tj. RLU);
X= logaritmus koncentrace;
Bottom= minimální odpověď;
Top= maximální odpověď;
log EC50 (nebo log IC50)= logaritmus X jako odpověď uprostřed mezi maximální a minimální;
Hillův koeficient= sklon křivky.
Model počítá nejlépe odpovídající hodnoty pro parametry maximální odpovědi, minimální odpovědi, sklon křivky a IC50 a EC50. Pro výpočet hodnot EC50 a IC50 by měl být použit vhodný statistický software (např. statistický software Graphpad PrismR).
Stanovení odlehlých hodnot
|
35.
|
Správné statistické posouzení by mohlo být usnadněno zařazením (například) Q-testu (viz protokoly pro agonisty a antagonisty (7) pro stanovení „nepoužitelných“ jamek, které budou vyřazeny z analýzy dat.
|
|
36.
|
U referenčních standardních replikátů E2 (velikost vzorku dva) se každá upravená hodnota RLU pro replikáty při dané koncentraci E2 považuje za odlehlou hodnotu, jestliže je více než 20 % nad nebo pod upravenou hodnotou RLU pro tuto koncentraci v dosavadní databázi.
|
Sběr a korekce dat z luminometru pro testování ke stanovení rozsahu
|
37.
|
Nezpracovaná data z luminometru je třeba převést do vzorového tabulkového editoru určeného pro analýzu. Mělo by být stanoveno, zda jsou zde odlehlé datové body, které je třeba odstranit. (Viz kritéria přijatelnosti zkoušky, kde jsou uvedeny parametry určované v analýzách.) Provádějí se tyto výpočty:
|
Agonista
|
Krok 1
|
Vypočítejte střední hodnotu pro kontrolu s vehikulem DMSO (VC).
|
|
Krok 2
|
Odečtěte střední hodnotu VC DMSO od hodnoty každé jamky, abyste normalizovali data.
|
|
Krok 3
|
Vypočítejte střední hodnotu násobné indukce pro referenční standard (E2).
|
|
Krok 4
|
Vypočítejte střední hodnotu EC50 pro zkoušené chemické látky.
|
Antagonista
|
Krok 1
|
Vypočítejte střední hodnotu pro VC DMSO.
|
|
Krok 2
|
Odečtěte střední hodnotu VC DMSO od hodnoty každé jamky, abyste normalizovali data.
|
|
Krok 3
|
Vypočítejte střední násobnou redukci pro referenční standard (Ral/E2).
|
|
Krok 4
|
Vypočítejte střední hodnotu pro referenční standard E2.
|
|
Krok 5
|
Vypočítejte střední hodnotu IC50 pro zkoušené chemické látky.
|
Sběr a korekce dat z luminometru pro souhrnné testování
|
38.
|
Nezpracovaná data z luminometru je třeba převést do vzorového tabulkového editoru určeného pro analýzu. Mělo by být stanoveno, zda jsou zde odlehlé datové body, které je třeba odstranit. (Viz kritéria přijatelnosti zkoušky, kde jsou uvedeny parametry určované v analýzách.) Provádějí se tyto výpočty:
|
Agonista
|
Krok 1
|
Vypočítejte střední hodnotu pro VC DMSO.
|
|
Krok 2
|
Odečtěte střední hodnotu VC DMSO od hodnoty každé jamky, abyste normalizovali data.
|
|
Krok 3
|
Vypočítejte střední hodnotu násobné indukce pro referenční standard (E2).
|
|
Krok 4
|
Vypočítejte střední hodnotu EC50 pro E2 a zkoušené chemické látky.
|
|
Krok 5
|
Vypočítejte střední upravenou hodnotu RLU pro methoxychlor.
|
Antagonista
|
Krok 1
|
Vypočítejte střední hodnotu pro VC DMSO.
|
|
Krok 2
|
Odečtěte střední hodnotu VC DMSO od hodnoty každé jamky, abyste normalizovali data.
|
|
Krok 3
|
Vypočítejte střední násobnou indukci pro referenční standard (Ral/E2).
|
|
Krok 4
|
Vypočítejte střední hodnotu IC50 pro Ral/E2 a zkoušené chemické látky.
|
|
Krok 5
|
Vypočítejte střední upravenou hodnotu RLU pro tamoxifen.
|
|
Krok 6
|
Vypočítejte střední hodnotu pro referenční standard E2.
|
Kritéria interpretace údajů
|
39.
|
Analýza VM7Luc ER TA je součástí přístupu založeného na průkazných důkazech a má pomoci stanovit priority u látek pro zkoušení ED in vivo. Součásti tohoto postupu stanovení priorit bude klasifikace zkoušené chemické látky jako pozitivní nebo negativní pro ER agonistickou nebo antagonistickou aktivitu. Pozitivní a negativní rozhodovací kritéria použitá ve validační studii analýzy VM7Luc ER TA jsou popsána v tabulce 1.
|
Tabulka 1
Pozitivní a negativní kritéria rozhodování
|
|
|
AGONISTICKÁ AKTIVITA
|
|
Pozitivní
|
|
—
|
Všechny zkoušené chemické látky klasifikované jako pozitivní u ER agonistické aktivity by měly mít křivku závislosti odpovědi na koncentraci sestávající z výchozí hodnoty, po níž následuje pozitivní sklon a křivku ukončuje plató nebo vrchol. V některých případech mohou být definovány pouze dvě z těchto charakteristik (výchozí hodnota – sklon nebo sklon – vrchol).
|
|
—
|
Přímka definující pozitivní sklon by měla obsahovat alespoň tři body s nepřekrývajícími se chybovými úsečkami (střední hodnota ± SD). Body vytvářející výchozí hodnotu se vyloučí, ale lineární část křivky může zahrnovat vrchol prvního bodu plató.
|
|
—
|
Pozitivní klasifikace vyžaduje amplitudu odpovědi, rozdíl mezi výchozí hodnotou a vrcholem, alespoň 20 % maximální hodnoty pro referenční standard, E2 (tj. 2 000 RLU nebo více, když je hodnota maximální odpovědi referenčních standardů [E2] upravena na 10 000 RLU).
|
|
—
|
Pro každou pozitivní zkoušenou chemickou látku by měla být vypočítána pokud možno hodnota EC50.
|
|
|
Negativní
|
Průměrná upravená hodnota RLU pro danou koncentraci je na nebo pod střední hodnotou RLU kontroly DMSO plus trojnásobek směrodatné odchylky hodnoty RLU DMSO.
|
|
Neadekvátní
|
Data, která nelze interpretovat jako platná podle vykazované přítomnosti nebo nepřítomnosti aktivity vzhledem k velkým kvalitativním nebo kvantitativním omezením, se považují za neadekvátní a nelze je použít na stanovení, zda je zkoušená chemická látka pozitivní nebo negativní. Chemické látky je třeba otestovat znovu.
|
|
|
|
ANTAGONISTICKÁ AKTIVITA
|
|
Pozitivní
|
|
—
|
Data zkoušené chemické látky vytvářejí křivku závislosti odpovědi na koncentraci sestávající z výchozí hodnoty, po níž následuje negativní sklon.
|
|
—
|
Přímka definující negativní sklon by měla obsahovat alespoň tři body s nepřekrývajícími se chybovými úsečkami; body vytvářející výchozí hodnotu se vyloučí, ale lineární část křivky může zahrnovat první bod plató.
|
|
—
|
Mělo by zde být alespoň 20 % snížení aktivity oproti maximální hodnotě pro referenční standard, Ral/E2 (tj. 8 000 RLU nebo méně, když je hodnota maximální odpovědi referenčních standardů [Ral/E2] upravena na 10 000 RLU).
|
|
—
|
Nejvyšší necytotoxické koncentrace zkoušené chemické látky by měly být menší než nebo rovny 1 x 10-5 M.
|
|
—
|
Pro každou pozitivní zkoušenou chemickou látku by měla být vypočítána pokud možno hodnota IC50.
|
|
|
Negativní
|
Všechny datové body nad hodnotou EC80 (80 % odpovědi E2, nebo 8 000 RLU) při koncentracích menších než 1,0 x 10-5 M.
|
|
Neadekvátní
|
Data, která nelze interpretovat jako platná podle vykazované přítomnosti nebo nepřítomnosti aktivity vzhledem k velkým kvalitativním nebo kvantitativním omezením, se považují za neadekvátní a nelze je použít na stanovení, zda je zkoušená chemická látka pozitivní nebo negativní. Chemickou látku je třeba otestovat znovu.
|
|
40.
|
Pozitivní výsledky budou pokud možno charakterizovány jak intenzitou účinku, tak i koncentrací, při níž k účinku dochází. Příklady pozitivních, negativních a neadekvátních dat jsou uvedeny na obrázcích 5 a 6.
|
Obrázek 5
Příklady agonistů: Pozitivní, negativní a neadekvátní data
Tečkovaná čára označuje 20 % odpovědi E2, 2 000 upravených a normalizovaných RLU.
Obrázek 6
Příklady antagonistů: Pozitivní, negativní a neadekvátní data
Tečkovaná čára označuje 80 % odpovědi Ral/E2, 8 000 upravených a normalizovaných RLU.
Plná čára označuje 1,00 10-5 M. Aby byla odpověď považována za pozitivní, měla by být pod čarou 8 000 RLU, a to při koncentracích menších než 1,00 10-5M.
Koncentrace označené hvězdičkou na grafu meso-hexestrolu označují skóre životaschopnosti „2“ nebo větší.
Výsledky zkoušek u meso-hexestrolu se považuji za neadekvátní data, protože jediná odpověď, která je pod 8 000 RLU, se vyskytuje při 1,00 10-5M.
|
41.
|
Výpočty EC50 a IC50 lze provést pomocí čtyřparametrové Hillovy funkce (viz protokol pro agonisty a protokol pro antagonisty, kde jsou uvedeny podrobnější informace (7)). Splnění kritérií přijatelnosti ukazuje, že systém funguje správně, ale nezaručuje to, že konkrétní provedení zkoušky podá přesné údaje. Opakování výsledků první zkoušky je nejlepším důkazem, že byly získány přesné údaje (viz odstavec 19„SLOŽKY ANALÝZY ER TA“.
|
ZÁVĚREČNÁ ZPRÁVA
|
42.
|
Viz odstavec 20 „SLOŽKY ANALÝZY ER TA“.
|
LITERATURA
|
(1)
|
ICCVAM. (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.(1)
|
|
(2)
|
Monje P., Boland R. (2001). Subcellular Distribution of Native Estrogen Receptor α and β Isoforms in Rabbit Uterus and Ovary, J. Cell Biochem., 82(3): 467-479.
|
|
(3)
|
Pujol P., et al. (1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer Res., 58(23): 5 367-5 373.
|
|
(4)
|
Weihua Z., et al. (2000). Estrogen Receptor (ER) β, a Modulator of ERα in the Uterus, Proceedings of the National Academy of Sciences of the United States of America 97(11): 936-5 941.
|
|
(5)
|
Balls M., et al. (2006). The Importance of Good Cell Culture Practice (GCCP), ALTEX, 23(Suppl): 270-273.
|
|
(6)
|
Coecke S., et al. (2005). Guidance on Good Cell Culture Practice: a Report of the Second ECVAM Task Force on Good Cell Culture Practice, Alternatives to Laboratory Animals, 33: 261-287.
|
|
(7)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report, The LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals, NIH Publication No 11-7 850.
|
|
(8)
|
Rogers J.M., Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In Vitro Mol. Toxicol.,13(1):67-82.
|
|
(9)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol., 71(10):1 459-69.
|
|
(10)
|
Thorne N., Inglese J., Auld D.S. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology,17(6):646-57.
|
|
(11)
|
Kuiper G.G, et al. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta, Endocrinology,139(10):4 252-63.
|
|
(12)
|
Geisinger, et al. (1989). Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors, Cancer 63, 280-288.
|
|
(13)
|
Baldwin, et al. (1998). BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In Vitro Cell. Dev. Biol. – Animal, 34, 649-654.
|
|
(14)
|
Li, Y., et al. (2014). Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, 2072-2081.
|
|
(15)
|
Rogers, J.M. and Denison, M.S. (2000). Recombinant cell bioassays for endocrine disruptors:development of a stably transfected human ovarian cell line for the detection of estrogenicand anti-estrogenic chemicals, In Vitro & Molec. Toxicol. Chem. 13, 67-82.
|
Dodatek 4
ANALÝZA STABILNĚ TRANSFEKOVANÉ TRANSAKTIVACE ESTROGENOVÉHO RECEPTORU Α PRO DETEKCI ESTROGENOVΎCH AGONISTICKΎCH A ANTAGONISTICKΎCH ΪΘINKΩ CHEMICKΎCH LΑTEK S POUžITΝM BUNΜΘNΙ LINIE ERΑ CALUX
VÝCHOZÍ ÚVAHY A OMEZENÍ (VIZ TAKÉ VŠEOBECNÝ ÚVOD)
|
1.
|
Při transaktivační analýze ERα CALUX se používá buněčná linie U2OS na detekci estrogenní agonistické a antagonistické aktivity zprostředkované lidským estrogenovým receptorem alfa (hERα). Relevanci a spolehlivost analýzy pro zamýšlený účel prokázala validační studie biotestu stabilně transfekovaného ERα CALUX od společnosti BioDetection Systems BV (Amsterdam, Nizozemsko) (1). Buněčná linie ERα CALUX exprimuje pouze stabilně transfekovaný lidský ERα (2)(3).
|
|
2.
|
Tato analýza je specificky určena k detekci transaktivace zprostředkované hERα měřením bioluminiscence jako sledované vlastnosti. Použití bioluminiscence je v biotestech běžné vzhledem k vysokému poměru signálu k šumu (4).
|
|
3.
|
Bylo hlášeno, že koncentrace fytoestrogenů vyšší než 1 μM nadměrně aktivují reportérový gen luciferázy, přičemž výsledkem je luminiscence nezprostředkovaná receptorem (5)(6)(7). Proto je třeba vyšší koncentrace fytoestrogenů nebo podobných sloučenin, které mohou nadměrně aktivovat expresi luciferázy, pečlivě prozkoumat v analýzách stabilně transfekované transaktivace ER (viz dodatek 2).
|
|
4.
|
Před použitím této analýzy pro regulační účely je třeba přečíst si „VŠEOBECNÝ ÚVOD“ a „SLOŽKY ANALÝZY ER TA“. Definice a zkratky používané v této zkušební metodě jsou popsány v dodatku 1.
|
PRINCIP ANALÝZY (VIZ TAKÉ VŠEOBECNÝ ÚVOD)
|
5.
|
Biotest se používá k posouzení vazby ligandu na estrogenový receptor a následného přesunu komplexu receptor-ligand do jádra. V jádru se komplex receptor-ligand naváže na specifické responzivní elementy DNA a transaktivuje reportérový gen luciferázy světlušek, přičemž výsledkem je zvýšená buněčná exprese enzymu luciferáza. Po přidání substrátu luciferázy luciferinu se luciferin transformuje na bioluminiscenční produkt. Vzniklé světlo lze snadno detekovat a kvantifikovat pomocí luminometru.
|
|
6.
|
Zkušební systém využívá stabilně transfekované buňky ERα CALUX. Buňky ERα CALUX pocházejí z buněčné linie lidského osteoblastického osteosarkomu U2OS. Lidské buňky U2OS byly stabilně transfekovány 3xHRE-TATA-Luc a pSG5-neo-hERα pomocí koprecipitace s využitím fosforečnanu vápenatého. Buněčná linie U2OS byla vybrána jako nejlepší kandidát na estrogen- (a jiný steroidní hormon) responzivní reportérovou buněčnou linii na základě pozorování, že buněčná linie U2OS vykazovala jen malou nebo žádnou endogenní receptorovou aktivitu. Absence endogenních receptorů byla hodnocena pouze pomocí luciferázových reportérových plazmidů, kdy po přidání ligandů pro receptory nebyla vykázána žádná aktivita. Navíc tato buněčná linie podporovala silné, hormonem zprostředkované odpovědi, když byly přechodně vneseny příbuzné receptory (2)(3)(8).
|
|
7.
|
Testování chemických látek na estrogenní nebo antiestrogenní aktivitu pomocí buněčné linie ERα CALUX zahrnuje předběžnou screeningovou zkoušku a souhrnné zkoušky. Při předběžné screeningové zkoušce se stanoví rozpustnost a cytotoxicita a přesně se určí rozmezí koncentrace zkoušených chemických látek pro souhrnné testování. V průběhu souhrnného testování se zkouší přesně určené rozmezí koncentrace zkoušených chemických látek v biotestech ERα CALUX, po nichž následuje klasifikace zkoušených chemických látek na agonistickou nebo antagonistickou aktivitu.
|
|
8.
|
Kritéria pro interpretaci údajů jsou podrobně popsána v odstavci 59. Stručně řečeno, zkoušená chemická látka se považuje za pozitivní na agonistickou aktivitu v případě, že alespoň dvě po sobě jdoucí koncentrace zkoušené chemické látky vykazují odpověď, která se rovná nebo je vyšší než 10 % maximální odpovědi referenčního standardu 17β-estradiolu (PC10). Zkoušená chemická látka se považuje za pozitivní na antagonistickou aktivitu v případě, že alespoň dvě po sobě jdoucí koncentrace zkoušené chemické látky vykazují odpověď, která se rovná nebo je menší než 80 % maximální odpovědi referenčního standardu tamoxifenu (PC80).
|
POSTUP
Buněčné linie
|
9.
|
Pro analýzu by se měla používat stabilně transfekovaná buněčná linie U2OS ERα CALUX. Buněčnou linii lze získat od společnosti BioDetection Systems BV, Amsterdam, Nizozemsko na základě technické licenční smlouvy.
|
|
10.
|
Používat by se měly pouze buněčné kultury bez mykoplazmat. Použité šarže buněk by měly být certifikovány jako negativní na kontaminaci mykoplazmatem, nebo by měl být před použitím proveden test na mykoplazma. Pro citlivou detekci mykoplazmové infekce se používá RT-PCR (polymerázová řetězová reakce v reálném čase) (9).
|
Stabilita buněčné linie
|
11.
|
Aby se udržela stabilita a integrita buněk CALUX, měly by být buňky uchovávány v tekutém dusíku (–80 °C). Po rozmrazení je třeba buňky pro přípravu nové kultury subkultivovat alespoň dvakrát, než se použijí pro hodnocení estrogenní agonistické a antagonistické aktivity chemických látek. Buňky by neměly být subkultivovány déle než 30 pasáží.
|
|
12.
|
Pro monitorování stability buněčné linie v čase by měla být reaktivita zkušebního systému agonistické a antagonistické aktivity ověřena hodnocením EC50 nebo IC50 referenčního standardu. Dále by měla být monitorována relativní indukce pozitivního kontrolního vzorku (PC) a negativního kontrolního vzorku (NC). Výsledky by měly odpovídat kritériím přijatelnosti pro biotest ERα CALUX agonistické (tabulka 3C) nebo antagonistické (tabulka 4C) aktivity. Referenční standardy, pozitivní a negativní kontroly jsou uvedeny v tabulce 1 a v tabulce 2 pro agonistický, respektive antagonistický režim.
|
Buněčná kultura a podmínky na destičce
|
13.
|
Buňky U2OS se kultivují v růstovém médiu (médium DMEM/F12 (1:1) s fenolovou červení jako indikátorem pH, doplněném fetálním bovinním sérem (7,5 %), neesenciálními aminokyselinami (1 %), 10 jednotkami/ml penicilinu, streptomycinu a geneticinu (G-418) jako selekčním markerem). Buňky se umístí do inkubátoru CO2 (5 % CO2) při teplotě 37 °C a 100 % vlhkosti vzduchu. Když buňky dosáhnou konfluence 85–95 %, buď se subkultivují, nebo připraví pro nasazení do 96jamkových mikrotitračních destiček. Ve druhém případě je třeba buňky resuspendovat ve zkušebním médiu 1 x 105 buněk/ml (DMEM/F12 (1:1) neobsahujícím estrogeny bez fenolové červeně, doplněném fetálním bovinním sérem ošetřeným aktivním uhlím, které je potaženo dextranem (5 % obj.), neesenciálními aminokyselinami (1 % obj.), 10 jednotkami/ml penicilinu a streptomycinu) a umístit do jamek 96jamkových mikrotitračních destiček (100 μl homogenizované buněčné suspenze). 24 hodin před expozicí by buňky měly být předběžně inkubovány v inkubátoru (5 % CO2, 37 °C, 100 % vlhkost vzduchu). Plastové pomůcky by neměly obsahovat estrogen.
|
Kritéria přijatelnosti
|
14.
|
Agonistická a antagonistická aktivita zkoušených chemických látek se testuje ve zkušební sérii. Zkušební série sestává maximálně z 6 mikrotitračních destiček. Každá zkušební série obsahuje alespoň 1 celou sérii ředění referenčního standardu, pozitivní kontrolní vzorek, negativní kontrolní vzorek a kontroly s rozpouštědlem. Na obrázku 1 a 2 je znázorněno uspořádání destiček pro agonistickou a antagonistickou zkušební sérii.
|
|
15.
|
Každé ředění referenčních standardů, zkoušených chemických látek, všechny kontroly s rozpouštědlem a pozitivní a negativní kontroly by měly být analyzovány třikrát. Každá ze tří analýz by měla splňovat požadavky uvedené v tabulce 3A v tabulce 4A.
|
|
16.
|
Na první destičce v každé zkušební sérii se měří celá série ředění referenčního standardu (17β-estradiol na agonistickou aktivitu; tamoxifen na antagonistickou aktivitu). Aby bylo možné porovnat výsledky analýzy zbývajících 5 mikrotitračních destiček s první mikrotitrační destičkou obsahující celou křivku závislosti odpovědi referenčního standardu na koncentraci, měly by všechny destičky obsahovat 3 kontrolní vzorky: kontrolu s rozpouštědlem, nejvyšší koncentraci zkoušeného referenčního standardu a přibližnou koncentraci referenčního standardu EC50 (agonistická aktivita) nebo IC50 (antagonistická aktivita). Poměr průměrných kontrolních vzorků na první destičce a na zbývajících 5 destičkách by měl splňovat požadavky uvedené v tabulce 3C (agonistická aktivita) nebo v tabulce 4C (antagonistická aktivita).
|
|
17.
|
Pro každou mikrotitrační destičku ve zkušební sérii se vypočítá faktor z (10). Faktor z se počítá s použitím odpovědí při nejvyšší a nejnižší koncentraci referenčního standardu. Mikrotitrační destička se považuje za platnou v případě, že splňuje požadavky uvedené v tabulce 3C (agonistická aktivita) nebo v tabulce 4C (antagonistická aktivita).
|
|
18.
|
Referenční standard by měl vykazovat sigmoidní křivku závislosti odpovědi na dávce. Hodnoty EC50 nebo IC50 odvozené od odpovědi při sérii ředění referenčního standardu by měly splňovat požadavky uvedené v tabulce 3C (agonistická aktivita) nebo v tabulce 4C (antagonistická aktivita).
|
|
19.
|
Každá zkušební série by měla obsahovat pozitivní kontrolní a negativní kontrolní vzorek. Vypočtená relativní indukce pozitivního i negativního kontrolního vzorku by měla splňovat požadavky uvedené v tabulce 3C (agonistická aktivita) nebo v tabulce 4C (antagonistická aktivita).
|
|
20.
|
V průběhu všech měření se indukční faktor nejvyšší koncentrace referenčního standardu měří vydělením průměrné nejvyšší referenční standardní hodnoty relativní luminiscenční jednotky (RLU) 17β-estradiolu průměrnou odpovědí RLU referenční kontroly s rozpouštědlem. Tento indukční faktor by měl splňovat formální požadavky na násobnou indukci uvedené v tabulce 3C (agonistická aktivita) nebo v tabulce 4C (antagonistická aktivita).
|
|
21.
|
Za platné se považují pouze mikrotitrační destičky, které splňují všechna výše uvedená kritéria přijatelnosti, a tyto mikrotitrační destičky mohou být použity pro hodnocení odpovědi zkoušených chemických látek.
|
|
22.
|
Kritéria přijatelnosti jsou použitelná pro předběžné screeningové zkoušky i pro souhrnné zkoušky.
|
Tabulka 1
Koncentrace referenčního standardu, pozitivní kontroly (PC) a negativní kontroly (NC) pro biotest agonistické aktivity CALUX
|
|
Látka
|
Číslo CAS
|
Zkušební rozsah (M)
|
|
Referenční standard
|
17β-estradiol
|
50-28-2
|
1*10-13–1*10-10
|
|
Pozitivní kontrola (PC)
|
17α-methyltestosteron
|
58-18-4
|
3*10-6
|
|
Negativní kontrola (NC)
|
kortikosteron
|
50-22-6
|
1*10-8
|
Tabulka 2
Koncentrace referenčního standardu, pozitivní kontroly (PC) a negativní kontroly (NC) pro biotest antagonistické aktivity CALUX
|
|
Látka
|
Číslo CAS
|
Zkušební rozsah (M)
|
|
Referenční standard
|
tamoxifen
|
10540-29-1
|
3*10-9–1*10-5
|
|
Pozitivní kontrola (PC)
|
4-hydroxytamoxifen
|
68047-06-3
|
1*10-9
|
|
Negativní kontrola (NC)
|
resveratrol
|
501-36-0
|
1*10-5
|
Tabulka 3
Kritéria přijatelnosti pro biotest agonistické aktivity ERα CALUX
|
A - individuální vzorky na destičce
|
Kritérium
|
|
1
|
Maximální % SD trojích jamek (pro NC, PC, každé ředění zkoušené chemické látky a referenčního standardu s výjimkou C0)
|
< 15 %
|
|
2
|
Maximální % SD trojích jamek (pro referenční standard a kontroly zkoušené chemické látky s rozpouštědlem (C0, SC))
|
< 30 %
|
|
3
|
Maximální průnik LDH jako míra cytoxicity.
|
< 120 %
|
|
B - v rámci jedné mikrotitrační destičky
|
|
|
4
|
Poměr kontroly referenčního standardu s rozpouštědlem (C0; destička 1) a kontroly zkoušené chemické látky s rozpouštědlem; destičky 2 až x)
|
0,5 až 2,0
|
|
5
|
Poměr přibl. hodnoty EC50 a nejvyšších koncentrací referenčního standardu na destičce 1 a přibl. hodnoty EC50 a nejvyšších koncentrací referenčního standardu na destičkách 2 až x (C4, C8)
|
0,70 až 1,30
|
|
6
|
Faktor Z pro každou destičku
|
> 0,6
|
|
C - v rámci jedné série analýz (všechny destičky v rámci jedné série)
|
|
|
7
|
Sigmoidní křivka referenčního standardu
|
ano (17ß-estradiol)
|
|
8
|
Rozsah EC50 referenčního standardu 17ß-estradiol
|
4*10-12–4*10-11 M
|
|
9
|
Minimální násobná indukce nejvyšší koncentrace 17ß-estradiolu vzhledem ke kontrole referenčního standardu s rozpouštědlem.
|
5
|
|
10
|
Relativní indukce (%) PC.
|
> 30 %
|
|
11
|
Relativní indukce (%) NC
|
< 10 %
|
PC: pozitivní kontrola; NC: negativní kontrola; SC: kontrola s rozpouštědlem chemické látky; C0: kontrola referenčního standardu s rozpouštědlem; SD: směrodatná odchylka; LDH: laktát dehydrogenáza
Tabulka 4
Kritéria přijatelnosti pro biotest antagonistické aktivity ERα CALUX
|
A – individuální vzorky na destičce
|
Kritérium
|
|
1
|
Maximální % SD trojích jamek (pro NC, PC, každé ředění zkoušené chemické látky a referenčního standardu, kontrola s rozpouštědlem (C0))
|
< 15 %
|
|
2
|
Maximální % SD trojích jamek (pro kontrolu s vehikulem (VC) a nejvyšší koncentrace referenčního standardu (C8))
|
< 30 %
|
|
3
|
Maximální průnik LDH jako míra cytoxicity.
|
≥ 120%
|
|
B – v rámci jedné mikrotitrační destičky
|
|
|
4
|
Poměr kontroly referenčního standardu s rozpouštědlem (C0; destička 1) a kontroly zkoušené chemické látky s rozpouštědlem; destičky 2 až x)
|
0,70 až 1,30
|
|
5
|
Poměr přibl. hodnoty IC50 koncentrací referenčního standardu na destičce 1 a přibl. hodnoty IC50 koncentrací referenčního standardu na destičkách 2 až x (C4)
|
0,70 až 1,30
|
|
6
|
Poměr nejvyšších koncentrací referenčního standardu na destičce 1 a nejvyšších koncentrací referenčního standardu na destičkách 2 až x (C8)
|
0,50 až 2,0
|
|
7
|
Faktor z pro každou destičku
|
> 0,6
|
|
C – v rámci jedné série analýz (všechny destičky v rámci jedné série)
|
|
|
8
|
Sigmoidní křivka referenčního standardu
|
ano (tamoxifen)
|
|
9
|
Rozsah IC50 referenčního standardu (tamoxifen)
|
1*10-8–1*10-7 M
|
|
10
|
Minimální násobná indukce kontroly referenčního standardu s rozpouštědlem vzhledem k nejvyšší koncentraci tamoxifenu.
|
2,5
|
|
11
|
Relativní indukce (%) PC.
|
< 70 %
|
|
12
|
Relativní indukce (%) NC
|
> 85 %
|
PC: pozitivní kontrola; NC: negativní kontrola; VC: kontrola s vehikulem (kontrola s rozpouštědlem bez pevné koncentrace agonistického referenčního standardu); SC: kontrola s rozpouštědlem chemické látky; C0: kontrola referenčního standardu s rozpouštědlem; SD: směrodatná odchylka; LDH: laktát dehydrogenáza
Kontrola s rozpouštědlem/vehikulem, referenční standardy, pozitivní kontroly, negativní kontroly
|
23.
|
Při předběžných screeningových zkouškách i souhrnných zkouškách by se měla používat stejná kontrola s rozpouštědlem/vehikulem, referenční standardy, pozitivní kontroly a negativní kontroly. Dále by měla být stejná koncentrace referenčních standardů, pozitivních kontrol a negativních kontrol.
|
Kontrola s rozpouštědlem
|
24.
|
Rozpouštědlo, které se používá na rozpuštění zkoušených chemických látek, by mělo být testováno jako kontrola s rozpouštědlem. Při validaci biotestu ERα CALUX byl jako vehikulum použit dimethylsulfoxid (DMSO, 1 % (obj.); CASRN 67-68-5). Jestliže je použito jiné rozpouštědlo než DMSO, měly by být všechny referenční standardy, kontroly a zkoušené chemické látky testovány ve stejném vehikulu. Nezapomeňte, že kontrola s rozpouštědlem u antagonistických studií obsahuje pevnou koncentraci agonistického referenčního standardu 17β-estradiol (přibližně koncentrace EC50). Pro testování rozpouštědla použitého pro antagonistické studie je třeba připravit a otestovat kontrolu s vehikulem.
|
Kontrola s vehikulem (antagonistická aktivita)
|
25.
|
Při testování antagonistické aktivity se zkušební médium doplňuje pevnou koncentrací agonistického referenčního standardu 17β-estradiol (přibližně koncentrace EC50). Pro testování rozpouštědla použitého k rozpuštění zkoušených chemických látek při zkoušce na antagonistickou aktivitu je třeba připravit zkušební médium bez pevné koncentrace agonistického referenčního standardu 17β-estradiol. Tento kontrolní vzorek se označuje jako kontrola s vehikulem. Při validaci biotestu ERα CALUX byl jako vehikulum použit dimethylsulfoxid (DMSO, 1 % (obj.); CASRN 67-68-5). Jestliže je použito jiné rozpouštědlo než DMSO, měly by být všechny referenční standardy, kontroly a zkoušené chemické látky testovány ve stejném vehikulu.
|
Referenční standardy
|
26.
|
Agonistickým referenčním standardem je 17β-estradiol (tabulka 1). Referenční standardy sestávají ze série ředění osmi koncentrací 17β-estradiolu (1*10-13, 3*10-13, 1*10-12, 3*10-12, 6*10-12, 1*10-11, 3*10-11, 1*10-10 M).
|
|
27.
|
Antagonistickým referenčním standardem je tamoxifen (tabulka 2). Referenční standardy sestávají ze série ředění osmi koncentrací tamoxifenu (3*10-9, 1*10-8, 3*10-8, 1*10-7, 3*10-7, 1*10-6, 3*10-6, 1*10-5 M). Každá koncentrace antagonistického referenčního standardu se inkubuje společně s pevnou koncentrací agonistického referenčního standardu 17β-estradiol (3*10-12 M).
|
Pozitivní kontrola
|
28.
|
Pozitivní kontrolou pro agonistické studie je 17α-methyltestosteron (tabulka 1).
|
|
29.
|
Pozitivní kontrolou pro antagonistické studie je 4-hydroxytamoxifen (tabulka 2). Antagonistická pozitivní kontrola se inkubuje společně s pevnou koncentrací agonistického referenčního standardu 17β-estradiol (3*10-12 M).
|
Negativní kontrola
|
30.
|
Negativní kontrolou pro agonistické studie je kortikosteron (tabulka 1).
|
|
31.
|
Negativní kontrolou pro antagonistické studie je resveratrol (tabulka 2). Antagonistická negativní kontrola se inkubuje společně s pevnou koncentrací agonistického referenčního standardu 17β-estradiol (3,0*10-12 M).
|
Prokázání odborné způsobilosti laboratoře (viz odstavec 14 a tabulky 3 a 4 v části «SLOŽKY ANALÝZY ER TA» této zkušební metody)
Vehikulum
|
32.
|
Rozpouštědlo použité na rozpuštění zkoušených chemických látek by mělo zkoušenou chemickou látku zcela rozpustit a mělo by být mísitelné s kultivačním médiem. Vhodnými rozpouštědly jsou DMSO, voda a etanol (čistota 95 % až 100 %). Jestliže se jako rozpouštědlo používá DMSO, neměla by maximální koncentrace DMSO při inkubaci překročit 1 % (obj.). Před použitím by rozpouštědlo mělo být otestováno, zda není cytotoxické a zda neinterferuje s provedením analýz.
|
Příprava referenčních standardů, pozitivních kontrol, negativních kontrol a zkoušených chemických látek
|
33.
|
Referenční standardy, pozitivní kontroly, negativní kontroly a zkoušené chemické látky se rozpustí ve 100 % DMSO (nebo ve vhodném rozpouštědle). Poté se ve stejném rozpouštědle připraví příslušná (postupná) ředění. Před rozpuštěním by se měly všechny látky nechat ustálit na pokojovou teplotu. V čerstvě připravené zásobě roztoků referenčních standardů, pozitivních kontrol, negativních kontrol a zkoušených chemických látek by neměla být znatelná sraženina ani zákal. Zásoba referenčních standardů a kontrol může být připravována hromadně. Před každým experimentem by se měly připravit čerstvé roztoky zkoušených chemických látek. Konečná ředění referenčních standardů, pozitivních kontrol, negativních kontrol a zkoušených chemických látek by se měla připravit čerstvá pro každý experiment a použít do 24 hodin od přípravy.
|
Rozpustnost, cytotoxicita a zjištění rozsahu
|
34.
|
Při předběžné screeningové zkoušce se stanoví rozpustnost zkoušených chemických látek ve zvoleném rozpouštědle. Připraví se zásoba v maximální koncentraci 0,1 M. Pokud tato koncentrace vykazuje problémy s rozpustností, je třeba připravit zásobní roztoky s nižší koncentrací, dokud nebudou zkoušené chemické látky zcela rozpustné. Při předběžné screeningové zkoušce se testují postupná ředění zkoušené chemické látky 1:10. Maximální koncentrace pro testování agonistů nebo antagonistů je 1 mM. Po předběžné screeningové zkoušce se odvodí vhodný upřesněný rozsah koncentrací pro zkoušené chemické látky, jež mají být testovány v souhrnných zkouškách. Ředění použitá pro souhrnné testování by měla být 1x, 3x, 10x, 30x, 100x, 300x, 1000x a 3000x.
|
|
35.
|
V protokolu analýzy agonistů a antagonistů je zařazeno testování cytotoxicity (11). Testování cytotoxicity je začleněno do předběžné screeningové zkoušky i do souhrnných zkoušek. Při validaci biotestu ERα CALUX se pro posouzení cytotoxicity použila zkouška na průnik laktát dehydrogenázy v kombinaci s kvalitativní vizuální kontrolou buněk (viz dodatek 4.1) po expozici zkoušeným chemickým látkám. Mohou však být použity i jiné kvantitativní metody pro stanovení cytotoxicity (např. kolorimetrická analýza na bázi tetrazolia (MTT) nebo biotest cytotoxicity CALUX). Obecně, koncentrace zkoušených chemických látek, které vykazují více než 20 % redukci životaschopnosti, se považují za cytotoxické, a proto nemohou být použity pro hodnocení dat. Pokud jde o analýzu průniků LDH, považuje se koncentrace zkoušené chemické látky za cytotoxickou, když je procento průniku LDH větší než 120 %.
|
Expozice zkoušené chemické látce a organizace testovacích destiček
|
36.
|
Po přidání trypsinu do konfluentní lahve s kultivovanými buňkami se buňky resuspendují na 1 x 105 buněk/ml ve zkušebním médiu bez estrogenů. 100 μl resuspendovaných buněk se umístí do vnitřních jamek 96jamkové mikrotitrační destičky. Vnější jamky se naplní 200 μl fosfátového pufru (PBS) (viz obrázky 1 a 2). Buňky na destičkách se předběžně inkubují po dobu 24 hodin dne v inkubátoru s CO2 (5 % CO2, 37 °C, 100 % vlhkost vzduchu).
|
|
37.
|
Po předběžné inkubaci se destičky prohlédnou, zda nejeví vizuální známky cytotoxicity (viz dodatek 4.1), kontaminace a konfluence. Pro testování se použijí pouze destičky, které nejeví žádné vizuální známky cytotoxicity a kontaminace a které mají minimálně 85 % konfluenci. Médium z vnitřních jamek se opatrně odstraní a nahradí 200 μl zkušebního média bez estrogenu obsahujícího příslušnou sérii ředění referenčních standardů, zkoušených chemických látek, pozitivních kontrol, negativních kontrol a kontrol s rozpouštědlem (tabulka 5: studie agonistů; tabulka 6: studie antagonistů). Všechny referenční standardy, zkoušené chemické látky, pozitivní kontroly, negativní kontroly a kontroly s rozpouštědlem se testují třikrát. Na obrázku 1 je uvedeno uspořádání destičky pro zkoušení agonistů. Na obrázku 2 je uvedeno uspořádání destičky pro zkoušení antagonistů. Uspořádání destičky pro předběžné screeningové testování a souhrnné testování je stejné. Při testování antagonistů obsahují všechny vnitřní jamky, s výjimkou jamek pro kontrolu vehikula (VC), také pevnou koncentraci agonistického referenčního standardu 17β-estradiolu (3*10-12 M). Nezapomeňte, že na každou destičku TC je třeba přidat referenční standardy C8 a C4.
|
|
38.
|
Po expozici buněk všem chemickým látkám se 96jamkové mikrotitrační destičky inkubují po dobu dalších 24 hodin v inkubátoru s CO2 (5 % CO2, 37 °C, 100 % vlhkost vzduchu).
|
Obrázek 1
Uspořádání 96jamkových mikrotitračních destiček pro předběžné screeningové testování a posouzení agonistického účinku
C0 = referenční standardní rozpouštědlo
C(1–8) = série ředění (1–8, koncentrace od nízké po vysokou) referenčního standardu
PC = pozitivní kontrola
NC = negativní kontrola
TCx-(1–8) = ředění (1–8, koncentrace od nízké po vysokou) zkoušené chemické látky pro předběžnou screeningovou zkoušku a posouzení agonistického účinku zkoušené chemické látky x
SC = kontrola zkoušené chemické látky s rozpouštědlem (optimálně stejné rozpouštědlo jako v C0, ale může být z jiné šarže)
Šedé buňky: = vnější buňky, naplněné 200 μl PBS
Obrázek 2
Uspořádání 96jamkových mikrotitračních destiček pro předběžné screeningové testování a posouzení antagonistického účinku
C0 = referenční standardní rozpouštědlo
C(1–8) = série ředění (1–8, koncentrace od nízké po vysokou) referenčního standardu
NC = negativní kontrola
PC = pozitivní kontrola
TCx–(1–8) = ředění (1–8, koncentrace od nízké po vysokou) zkoušené chemické látky pro předběžnou screeningovou zkoušku a posouzení agonistického účinku zkoušené chemické látky x
SC = kontrola zkoušené chemické látky s rozpouštědlem (optimálně stejné rozpouštědlo jako v C0, ale může být z jiné šarže)
VC = kontrola s vehikulem (kontrola s rozpouštědlem bez pevné koncentrace agonistického referenčního standardu 17β-estradiolu)
Šedé buňky: = vnější buňky, naplněné 200 μl PBS
Poznámka: Všechny vnitřní jamky, s výjimkou jamek pro kontrolu vehikula (VC), obsahují také pevnou koncentraci agonistického referenčního standardu 17β-estradiolu (3,0*10–12 M).
Měření luminiscence
|
39.
|
Měření luminiscence je podrobně popsáno v protokolu analýzy agonistů a antagonistů (10). Médium by se mělo z jamek odstranit a buňky by se po 24 hodinách inkubace měly štěpit, aby se otevřela buněčná membrána a umožnilo měření luciferázové aktivity.
|
|
40.
|
Pro měření luminiscence vyžaduje tento postup luminometr vybavený dvěma injektory. Reakce luciferázy začíná vstříknutím substrátu luciferin. Reakce se zastaví přidáním 0,2 M NaOH. Reakce je zastavena, aby nedocházelo k přenosu luminiscence z jedné jamky do druhé.
|
|
41.
|
Světlo emitované z každé jamky se vyjadřuje v relativních luminiscenčních jednotkách (RLU) na jamku.
|
Předběžná screeningová zkouška
|
42.
|
Výsledky předběžné screeningové analýzy se použijí k určení přesného rozmezí koncentrace zkoušených chemických látek pro souhrnné testování. Hodnocení výsledků předběžné screeningové analýzy a určení přesného rozmezí koncentrace zkoušených chemických látek pro souhrnné testování je podrobně popsáno v protokolech pro analýzu agonistů a antagonistů (10). Zde je uveden stručný souhrn postupů pro určení rozmezí koncentrace zkoušených chemických látek pro analýzu agonistů a antagonistů. Viz tabulky 5 a 6, kde je uveden návod na způsob postupného ředění.
|
Volba koncentrací pro posouzení agonistických účinků
|
43.
|
Při předběžné screeningové zkoušce se zkoušené chemické látky testují pomocí série ředění, jak je uvedeno v tabulce 5 (agonistická aktivita) a v tabulce 6 (antagonistická aktivita). Všechny koncentrace je třeba testovat v trojích jamkách podle rozvržení destičky, jak je znázorněno na obrázku 1 (agonistická aktivita) nebo 2 (antagonistická aktivita).
|
|
44.
|
Za platné se považují pouze výsledky analýzy, které splňují kritéria přijatelnosti (tabulka 3), a tyto výsledky mohou být použity pro hodnocení odpovědi zkoušených chemických látek. V případě, že jedna nebo více mikrotitračních destiček v sérii analýz nesplní kritéria přijatelnosti, je třeba příslušné mikrotitrační destičky analyzovat znovu. Pokud první destička obsahující celou sérii ředění referenčního standardu nesplní kritéria přijatelnosti, je třeba znovu analyzovat celou zkušební sérii (6 destiček).
|
|
45.
|
Původní rozmezí koncentrací zkoušených chemických látek se upraví a předběžná screeningová zkouška se zopakuje v případě, že:
|
—
|
je pozorována cytotoxicita. Předběžný screeningový postup se zopakuje s nižšími necytotoxickými koncentracemi zkoušené chemické látky,
|
|
—
|
předběžný screening zkoušené chemické látky nevykazuje celou křivku závislosti odpovědi na dávce, protože testované koncentrace generují maximální indukci. Předběžná screeningová zkouška se zopakuje s nižšími koncentracemi zkoušené chemické látky.
|
|
|
46.
|
Když je pozorována platná odpověď ve vztahu k dávce, zvolí se (nejnižší) koncentrace, při níž je pozorována maximální indukce a která nevykazuje cytotoxicitu. Nejvyšší koncentrace zkoušené chemické látky, která se testuje v souhrnných zkouškách, by měla být trojnásobkem této zvolené koncentrace.
|
|
47.
|
Připraví se celá upřesněná série ředění zkoušené chemické látky, a to podle kroků ředění uvedených v tabulce 5, počínaje nejvyšší koncentrací, jak je stanoveno výše.
|
|
48.
|
Zkoušená chemická látka, která nevyvolává žádný agonistický účinek, se testuje v souhrnných zkouškách počínaje nejvyšší necytotoxickou koncentrací určenou v průběhu předběžného screeningu.
|
Volba koncentrací pro posouzení antagonistických účinků
|
49.
|
Za platné se považují pouze výsledky analýzy, které splňují kritéria přijatelnosti (tabulka 4), a tyto výsledky mohou být použity pro hodnocení odpovědi zkoušených chemických látek. V případě, že jedna nebo více mikrotitračních destiček v sérii analýz nesplní kritéria přijatelnosti, je třeba příslušné mikrotitrační destičky analyzovat znovu. Pokud první destička obsahující celou sérii ředění referenčního standardu nesplní kritéria přijatelnosti, je třeba znovu analyzovat celou zkušební sérii (6 destiček).
|
|
50.
|
Původní rozmezí koncentrací zkoušených chemických látek se upraví a předběžná screeningová zkouška se zopakuje v případě, že:
|
—
|
je pozorována cytotoxicita. Předběžný screeningový postup se zopakuje s nižšími necytotoxickými koncentracemi zkoušené chemické látky,
|
|
—
|
předběžný screening zkoušené chemické látky nevykazuje celou křivku závislosti odpovědi na dávce, protože testované koncentrace generují maximální inhibici. Předběžná screeningová zkouška se zopakuje s nižšími koncentracemi zkoušené chemické látky.
|
|
|
51.
|
Když je zjištěna platná odpověď ve vztahu k dávce, zvolí se (nejnižší) koncentrace, při níž je pozorována maximální inhibice a která nevykazuje cytotoxicitu. Nejvyšší koncentrace zkoušené chemické látky, která se testuje v souhrnných zkouškách, by měla být trojnásobkem této zvolené koncentrace.
|
|
52.
|
Připraví se celá upřesněná série ředění zkoušené chemické látky, a to podle kroků ředění uvedených v tabulce 6, počínaje nejvyšší koncentrací, jak je stanoveno výše.
|
|
53.
|
Zkoušené chemické látky, které nevyvolávají žádný antagonistický účinek, se testují v souhrnných zkouškách počínaje nejvyšší necytotoxickou koncentrací otestovanou v průběhu předběžného screeningu.
|
Souhrnné zkoušky
|
54.
|
Po výběru zpřesněných rozmezí koncentrací se zkoušené chemické látky testují souhrnně pomocí série ředění, jak je uvedeno v tabulce 5 (agonistická aktivita) a v tabulce 6 (antagonistická aktivita). Všechny koncentrace je třeba testovat v trojích jamkách podle rozvržení destičky, jak je znázorněno na obrázku 1 (agonistická aktivita) nebo 2 (antagonistická aktivita).
|
|
55.
|
Za platné se považují pouze výsledky analýzy, které splňují kritéria přijatelnosti (tabulka 3 a 4), a tyto výsledky mohou být použity pro hodnocení odpovědi zkoušených chemických látek. V případě, že jedna nebo více mikrotitračních destiček v sérii analýz nesplní kritéria přijatelnosti, je třeba příslušné mikrotitrační destičky analyzovat znovu. Pokud první destička obsahující celou sérii ředění referenčního standardu nesplní kritéria přijatelnosti, je třeba znovu analyzovat celou zkušební sérii (6 destiček).
|
Tabulka 5
Koncentrace a ředění referenčních standardů, kontrol a zkoušených chemických látek použité pro testování agonistů
|
Referenční 17β-estradiol
|
TCx – předběžná screeningová zkouška
|
TCx – souhrnná zkouška
|
Kontroly
|
|
konc. (M)
|
ředění
|
ředění
|
konc. (M)
|
|
C0
|
0
|
TCx-1
|
10000000 x
|
TCx-1
|
3000 x
|
PC
|
3*10-6
|
|
C1
|
1*10-13
|
TCx-2
|
1000000 x
|
TCx-2
|
1000 x
|
NC
|
1*10-8
|
|
C2
|
3*10-13
|
TCx-3
|
100000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
1*10-12
|
TCx-4
|
10000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
3*10-12
|
TCx-5
|
1000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
6*10-12
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
|
|
|
C6
|
1*10-11
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
|
|
|
C7
|
3*10-11
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
|
|
|
C8
|
1*10-10
|
|
|
|
|
|
|
|
TCx – zkoušená chemická látka x
PC – pozitivní kontrola (17α-methyltestosteron)
NC – negativní kontrola (kortikosteron)
C0 – kontrola referenčního standardu s rozpouštědlem
SC – kontrola zkoušené chemické látky s rozpouštědlem
|
Tabulka 6
Koncentrace a ředění referenčních standardů, kontrol a zkoušených chemických látek použité pro testování antagonistů
|
Referenční tamoxifen
|
TCx – předběžná screeningová zkouška
|
TCx – souhrnná zkouška
|
Kontroly
|
|
konc. (M)
|
zředění
|
zředění
|
konc. (M)
|
|
C0
|
0
|
TCx-1
|
10000000 x
|
TCx-1
|
3000 x
|
PC
|
1*10-9
|
|
C1
|
3*10-9
|
TCx-2
|
1000000 x
|
TCx-2
|
1000 x
|
NC
|
1*10-5
|
|
C2
|
1*10-8
|
TCx-3
|
100000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
3*10-8
|
TCx-4
|
10000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
1*10-7
|
TCx-5
|
1000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
3*10-7
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
Doplněný agonista
|
|
C6
|
1*10-6
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
konc. (M)
|
|
C7
|
3*10-6
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
17β-estradiol
|
3*10-12
|
|
C8
|
1*10-5
|
|
|
|
|
|
|
|
TCx – zkoušená chemická látka x
PC – pozitivní kontrola (4-hydroxytamoxifen)
NC – negativní kontrola (resveratrol)
C0 – kontrola referenčního standardu s rozpouštědlem
SC – kontrola zkoušené chemické látky s rozpouštědlem
VC – kontrola s vehikulem (neobsahuje pevnou koncentraci agonistického referenčního standardu 17β-estradiol (3,0*10-12 M)
|
Sběr a analýza dat
|
56.
|
Po předběžné screeningové zkoušce a souhrnných zkouškách se stanoví EC10, EC50, PC10, PC50 a maximální indukce (TCxmax) zkoušené chemické látky pro testování agonistických účinků. Pro testování antagonistických účinků se stanoví IC20, IC50, PC80, PC50 a minimální indukce (TCxmin). Na obrázku 3 (agonistická aktivita) a 4 (antagonistická aktivita) jsou tyto parametry znázorněny graficky. Potřebné parametry se vypočítají podle relativní indukce každé zkoušené chemické látky (ve vztahu k maximální indukci referenčního standardu (= 100 %)). Pro hodnocení dat se použije nelineární regrese (variabilní sklon, 4 parametry), a to podle následující rovnice:
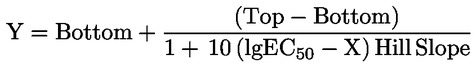
kde:
X = logaritmus dávky nebo koncentrace
Y = odpověď (relativní indukce (%))
Top = maximální indukce (%)
Bottom = minimální indukce (%)
logEC50 = logaritmus koncentrace, při níž je pozorováno 50 % maximální odpovědi
Hillův koeficient = míra sklonu nebo Hillova křivka
|
|
57.
|
Nezpracovaná data z luminometru, vyjádřená jako relativní luminiscenční jednotky (RLU), je třeba převést do tabulkového editoru pro analýzu dat určeného pro předběžné screeningové a souhrnné zkoušky. Nezpracovaná data by měla splňovat kritéria přijatelnosti uvedená v tabulce 3A a 3B (agonistická aktivita) nebo 4A a 4B (antagonistická aktivita). V případě, že nezpracovaná data splňují kritéria přijatelnosti, provede se výpočet v následujících krocích pro stanovení potřebných parametrů:
|
Agonistická aktivita
|
—
|
Odečtěte průměrné jednotky RLU kontroly referenčního standardu s rozpouštědlem od každé analýzy nezpracovaných dat referenčních standardů.
|
|
—
|
Odečtěte průměrné jednotky RLU kontroly zkoušené chemické látky s rozpouštědlem od každé analýzy nezpracovaných dat zkoušených chemických látek.
|
|
—
|
Vypočtěte relativní indukci každé koncentrace referenčního standardu. Stanovte indukci nejvyšší koncentrace referenčního standardu na 100 %.
|
|
—
|
Vypočtěte relativní indukci každé koncentrace zkoušené chemické látky v porovnání s nejvyšší koncentrací referenčního standardu jako 100 %.
|
|
—
|
Vyhodnoťte výsledky analýzy podle nelineární regrese (variabilní sklon, 4 parametry).
|
|
—
|
Určete hodnoty EC50 a EC10 referenčního standardu.
|
|
—
|
Určete hodnoty EC50 a EC10 zkoušených chemických látek.
|
|
—
|
Určete maximální relativní indukci zkoušené chemické látky (TCmax).
|
|
—
|
Určete hodnoty PC10 a PC50 zkoušených chemických látek.
|
U zkoušených chemických látek nemusí být vždy dosažena celá křivka závislosti odpovědi na dávce, např. vzhledem k cytotoxicitě nebo problémům s rozpustností. Proto nelze určit hodnoty EC50, EC10 a PC50. V takovém případě lze určit pouze hodnoty PC10 and TCmax.
Antagonistická aktivita
|
—
|
Odečtěte průměrné jednotky RLU nejvyšší koncentrace referenčního standardu od každé analýzy nezpracovaných dat referenčních standardů.
|
|
—
|
Odečtěte průměrné jednotky RLU nejvyšší koncentrace referenčního standardu od každé analýzy nezpracovaných dat zkoušených chemických látek.
|
|
—
|
Vypočtěte relativní indukci každé koncentrace referenčního standardu. Stanovte indukci nejnižší koncentrace referenčního standardu na 100 %.
|
|
—
|
Vypočtěte relativní indukci každé koncentrace zkoušené chemické látky v porovnání s nejnižší koncentrací referenčního standardu jako 100 %.
|
|
—
|
Vyhodnoťte výsledky analýzy podle nelineární regrese (variabilní sklon, 4 parametry).
|
|
—
|
Určete hodnoty IC50 a IC20 referenčního standardu.
|
|
—
|
Určete hodnoty IC50 a IC20 zkoušených chemických látek.
|
|
—
|
Určete maximální relativní indukci zkoušené chemické látky (TCmin).
|
|
—
|
Určete hodnoty PC80 a PC50 zkoušených chemických látek.
|
Obrázek 3
Přehled parametrů stanovených v analýze agonistů
EC10 = koncentrace látky, při níž je pozorováno 10 % její maximální odpovědi
EC50 = koncentrace látky, při níž je pozorováno 50 % její maximální odpovědi
PC10 = koncentrace zkoušené chemické látky, při níž se její odpověď rovná EC10 referenčního standardu
PC50 = koncentrace zkoušené chemické látky, při níž se její odpověď rovná EC50 referenčního standardu
TCxmax = maximální relativní indukce zkoušené chemické látky.
Obrázek 4
Přehled parametrů stanovených v analýze antagonistů
IC20 = koncentrace látky, při níž je pozorováno 80 % její maximální odpovědi (20 % inhibice)
IC50 = koncentrace látky, při níž je pozorováno 50 % její maximální odpovědi (50 % inhibice)
PC80 = koncentrace zkoušené chemické látky, při níž se její odpověď rovná IC20 referenčního standardu
PC50 = koncentrace zkoušené chemické látky, při níž se její odpověď rovná IC50 referenčního standardu
TCxmin = minimální relativní indukce zkoušené chemické látky.
U zkoušených chemických látek nemusí být vždy dosažena celá křivka závislosti odpovědi na dávce, např. vzhledem k cytotoxicitě nebo problémům s rozpustností. Proto nelze určit hodnoty IC50, IC20 a PC50. V takovém případě lze určit pouze hodnoty PC20 a TCmin.
|
58.
|
Výsledky by měly vycházet ze dvou (nebo tří) nezávislých provedení zkoušek. Jestliže jsou při dvou provedeních zkoušky získány srovnatelné, a tedy reprodukovatelné výsledky, není nutné provádět zkoušku potřetí. Aby byly výsledky přijatelné, měly by:
|
—
|
splňovat kritéria přijatelnosti (viz kritéria přijatelnosti, odstavce 14–22),
|
|
Kritéria interpretace údajů
|
59.
|
Pro interpretaci údajů a rozhodnutí, zda je zkoušená chemická látka považována za pozitivní, nebo negativní, se používají tato kritéria:
|
U každé souhrnné zkoušky se zkoušená chemická látka považuje za pozitivní v případě:
|
1
|
TCmax se rovná nebo je vyšší než 10 % maximální odpovědi referenčního standardu (REF10).
|
|
2
|
Alespoň 2 po sobě jdoucí koncentrace zkoušené chemické látky se rovnají nebo jsou vyšší než REF10.
|
|
|
U každé souhrnné zkoušky se zkoušená chemická látka považuje za negativní v případě:
|
1
|
TCmax není vyšší než 10 % maximální odpovědi referenčního standardu (REF10).
|
|
2
|
Méně než 2 koncentrace zkoušené chemické látky se rovnají nebo jsou vyšší než REF10.
|
|
|
U každé souhrnné zkoušky se zkoušená chemická látka považuje za pozitivní v případě:
|
1
|
TCmin se rovná nebo je nižší než 80 % maximální odpovědi referenčního standardu (REF80 = 20 % inhibice).
|
|
2
|
Alespoň 2 po sobě jdoucí koncentrace zkoušené chemické látky se rovnají nebo jsou nižší než REF80.
|
|
|
U každé souhrnné zkoušky se zkoušená chemická látka považuje za negativní v případě:
|
1
|
TCmin je vyšší než 80 % maximální odpovědi referenčního standardu (REF80 = 20 % inhibice).
|
|
2
|
Méně než 2 koncentrace zkoušené chemické látky se rovnají nebo jsou nižší než REF80.
|
|
|
|
60.
|
Aby bylo možné charakterizovat sílu pozitivní odpovědi zkoušené chemické látky, uvedou se velikost účinku (agonistická aktivita: TCmax; antagonistická aktivita: TCmin) a koncentrace, při níž k účinku dochází (agonistická aktivita: EC10, EC50, PC10, PC50; antagonistická aktivita: IC20, IC50, PC80, PC50).
|
ZÁVĚREČNÁ ZPRÁVA
|
61.
|
Viz odstavec 20 „SLOŽKY ANALÝZY ER TA“.
|
LITERATURA
|
(1)
|
OECD (2016). Draft Validation report of the (anti-) ERα CALUX bioassay - transactivation bioassay for the detection of compounds with (anti)estrogenic potential. Environmental Health and Safety Publications, Series on Testing and Assessment (No 240). Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
Sonneveld E, Jansen HJ, Riteco JA, Brouwer A, van der Burg B. (2005). Development of androgen- and estrogen-responsive bioassays, members of a panel of human cell line-based highly selective steroid-responsive bioassays. Toxicol Sci. 83(1), 136-148.
|
|
(3)
|
Quaedackers ME, van den Brink CE, Wissink S, Schreurs RHMM, Gustafsson JA, van der Saag PT, and van der Burg B. (2001). 4-Hydroxytamoxifen trans-represses nuclear factor-kB Activity in human osteoblastic U2OS cells through estrogen receptor (ER)α and not through ERβ. Endocrinology 142(3), 1156-1166.
|
|
(4)
|
Thorne N, Inglese J and Auld DS. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology17(6):646-57.
|
|
(5)
|
Escande A, Pillon A, Servant N, Cravedi JP, Larrea F, Muhn P, Nicolas JC, Cavaillès V and Balaguer P. (2006). Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta. Biochem. Pharmacol., 71, 1459-1469.
|
|
(6)
|
Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der Burg B and Gustafsson JA. (1998). Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinol., 139, 4252-4263.
|
|
(7)
|
Sotoca AM, Bovee TFH, Brand W, Velikova N, Boeren S, Murk AJ, Vervoort J, Rietjens IMCM. (2010). Superinduction of estrogen receptor mediated gene expression in luciferase based reporter gene assays is mediated by a post-transcriptional mechanism. J. Steroid. Biochem. Mol. Biol., 122, 204–211.
|
|
(8)
|
Sonneveld E, Riteco JAC, Jansen HJ, Pieterse B, Brouwer A, Schoonen WG, and van der Burg B. (2006). Comparison of in vitro and in vivo screening models for androgenic and estrogenic activities. Toxicol. Sci., 89(1), 173–187.
|
|
(9)
|
Kobayashi H, Yamamoto K, Eguchi M, Kubo M, Nakagami S, Wakisaka S, Kaizuka M and Ishii H. (1995). Rapid detection of mycoplasma contamination in cell cultures by enzymatic detection of polymerase chain reaction (PCR) products. J. Vet. Med. Sci., 57(4), 769-771.
|
|
(10)
|
Zhang J-H, Chung TDY, and Oldenburg KR. (1999). A simple statistical parameter for use in evaluation and validation of high throuphut screening assays. J. Biomol. Scr., 4, 67-73
|
|
(11)
|
Besselink H, Middelhof I, and Felzel, E. (2014). Transactivation assay for the detection of compounds with (anti)estrogenic potential using ERα CALUX cells. BioDetection Systems BV (BDS). Amsterdam, the Netherlands.
|
Dodatek 4.1:
VIZUÁLNÍ KONTROLA ŽIVOTASCHOPNOSTI BUNĚK
B.67 ZKOUŠKA NA GENOVÉ MUTACE V BUŇKÁCH SAVCŮ IN VITRO S POUŽITÍM GENU THYMIDINKINÁZY
ÚVOD
|
1.
|
Tato zkušební metoda je rovnocenná Pokynu OECD pro zkoušení (TG) č. 490 (2016). Zkušební metody se pravidelně přezkoumávají a revidují s ohledem na vědecký pokrok, potřeby regulace a dobré životní podmínky zvířat. Test myšího lymfomu (MLA) a test TK6 využívající lokus thymidinkinázy (TK) byly původně obsaženy ve zkušební metodě B.17. Následně expertní pracovní skupina pro MLA Mezinárodního semináře o zkoušení genotoxicity (IWGT) vyvinula mezinárodní harmonizovaná doporučení pro kritéria přijatelnosti tohoto testu a interpretaci dat MLA (1)(2)(3)(4)(5) a uvedená doporučení jsou začleněna do této nové zkušební metody B.67. Tato zkušební metoda je napsána pro test MLA, a protože využívá také lokus TK, pro test TK6. Zatímco test MLA se používá běžně pro regulační účely, test TK6 se používá mnohem méně často. Je třeba uvést, že i přes podobnost mezi cílovými parametry nejsou tyto dvě buněčné linie vzájemně zaměnitelné a regulační programy mohou platně vyjádřit preferenci jedné nebo druhé pro konkrétní regulační použití. Například validace testu MLA prokázala jeho vhodnost pro detekci nejen genových mutací, ale také schopnosti zkoušené chemické látky indukovat strukturální chromozomální poškození. Tato zkušební metoda je součástí série zkušebních metod genetické toxikologie. Byl vypracován dokument, který obsahuje stručné informace o zkoušení v oblasti genetické toxikologie a přehled posledních změn, které OECD provedla ve svých pokynech ke zkoušení genotoxicity (6).
|
|
2.
|
Účelem zkoušek na genové mutace v buňkách savců in vitro je detekce genových mutací indukovaných chemickými látkami. Buněčné linie používané při těchto zkouškách měří přímé mutace reportérových genů, a to endogenního genu thymidinkinázy (TK pro lidské buňky a Tk pro buňky hlodavců, společně v této zkušební metodě nazývané jen TK). Tato zkušební metoda je určena k použití se dvěma buněčnými liniemi: buněčnou linií myšího lymfomu L5178Y TK+/--3.7.2C (obecně nazývanou L5178Y) a buněčnou linií lidského lymfoblastoidu TK6 (obecně nazývanou TK6). I když se tyto dvě buněčné linie liší vzhledem ke svému původu, růstu buněk, stavu p53 atd., mohou být testy genových mutací TK prováděny podobným způsobem u obou typů buněk, jak je popsáno v této zkušební metodě.
|
|
3.
|
Autozomální a heterozygotní povaha genu thymidinkinázy umožňuje detekci životaschopných kolonií, jejichž buňky jsou deficitní na enzym thymidinkinázu po mutaci z TK+/-
na TK-/-
. Tento deficit může vyplývat z genetických jevů ovlivňujících gen TK včetně genových mutací (bodové mutace, posunové mutace, malé delece atd.) i chromozomálních změn (velké delece, přestavby chromozomu a mitotické rekombinace). Posledně jmenované změny se projevují jako ztráta heterozygotnosti, což je běžná genetická změna genů tlumících nádory v lidské tumorigenezi. Teoreticky může být testem MLA detekována ztráta celého chromozomu nesoucího gen TK v důsledku poškození vřetene a/nebo mitotické nondisjunkce. Kombinace cytogenetické a molekulární analýzy jasně ukazuje, že některé mutace MLA TK jsou výsledkem nondisjunkce. Průkazné důkazy však ukazují, že testy genové mutace TK nemohou spolehlivě detekovat aneugeny při aplikaci standardních kritérií cytotoxicity (jak je popsáno v této zkušební metodě), a proto není vhodné používat tyto testy na detekci aneugenů (7)(8)(9).
|
|
4.
|
Při testech genových mutací TK se generují dvě odlišné fenotypové třídy mutantů TK; normálně rostoucí mutanti rostoucí stejnou rychlostí jako heterozygotní buňky TK a pomalu rostoucí mutanti rostoucí s prodlouženou dobou zdvojení. Normálně rostoucí i pomalu rostoucí mutanti se v testu MLA rozpoznávají jako mutanti tvořící velké kolonie a mutanti tvořící malé kolonie a v testu TK6 jako mutanti tvořící časné kolonie a mutanti tvořící pozdní kolonie. Podrobně byla zkoumána molekulární a cytogenetická povaha mutantů MLA tvořících jak velké kolonie, tak malé kolonie (8)(10)(11)(12)(13). Rovněž byla rozsáhle studována molekulární a cytogenetická povaha mutantů TK6 tvořících časné kolonie i pozdní kolonie (14)(15)(16)(17). Pomalu rostoucí mutanti u obou buněčných druhů vykazovali genetické poškození, které zahrnuje putativní gen(y) regulující růst v blízkosti lokusu TK, což má za následek prodlouženou dobu zdvojení a vznik pozdních nebo malých kolonií (18). Indukce pomalu rostoucích mutantů se spojuje s chemickými látkami, které indukují silné strukturální změny na úrovni chromozomů (26). Buňky, jejichž poškození nezahrnuje putativní gen(y) regulující růst v blízkosti lokusu TK, rostou podobnou rychlostí jako mateřské buňky a vzniknou z nich normálně rostoucí mutanti. Indukce primárně normálně rostoucích mutantů se spojuje s chemickými látkami, které primárně působí jako bodové mutageny. Proto je velmi důležité spočítat jak pomalu rostoucí, tak i normálně rostoucímu mutanty, aby se zjistily všechny mutace a získaly informace o druhu (druzích) poškození (mutageny oproti klastogenům) indukovaného zkoušenou chemickou látkou (10)(12)(18)(19).
|
|
5.
|
Zkušební metoda je uspořádána tak, aby poskytla obecné informace, které platí pro test MLA i TK6, a specializovaný návod pro individuální testy.
|
|
6.
|
Použité definice jsou uvedeny v dodatku 1.
|
VÝCHOZÍ ÚVAHY A OMEZENÍ
|
7.
|
Zkoušky prováděné in vitro obecně vyžadují použití vnějšího zdroje metabolické aktivace. Systém exogenní metabolické aktivace zcela nenapodobuje podmínky in vivo.
|
|
8.
|
Je třeba zabránit vzniku podmínek, které by mohly vést k uměle pozitivním výsledkům (tj. možná interakce se zkušebním systémem), které nejsou způsobeny interakcí mezi zkoušenou chemickou látkou a genetickým materiálem buňky; takové podmínky zahrnují změny pH nebo osmolality, interakce se složkami média (20) (21) nebo nadměrné úrovně cytotoxicity (22)(23)(24). Pro testy MLA a TK6 se cytotoxicita přesahující doporučené horní úrovně cytotoxicity stanovené v odstavci 28 považuje za nadměrnou. Dále je třeba poznamenat, že zkoušené chemické látky, které jsou analogy thymidinu nebo které se chovají jako analogy thymidinu, mohou zvyšovat frekvenci mutací selektivním růstem spontánních mutantů na pozadí při ošetření buněk a vyžadovat tak další zkušební metody pro adekvátní hodnocení (25).
|
|
9.
|
Při zkoušení vyráběných nanomateriálů mohou být potřebné určité úpravy metody, které však v této zkušební metodě nejsou popsány.
|
|
10.
|
Před použitím této zkušební metody ke zkoušení směsi za účelem získání údajů pro zamýšlené použití v právních předpisech by mělo být zváženo, zda tato metoda může pro tento účel poskytnout přiměřené výsledky, a pokud ano, proč je tomu tak. Takové posouzení není nutné, pokud existuje právní požadavek na zkoušení dané směsi.
|
|
11.
|
Mutantní buňky, které v důsledku mutace TK
+/- na TK
-/- vykazují deficit aktivity enzymu thymidinkináza, jsou rezistentní k cytostatickým účinkům analogu pyrimidinu trifluorthymidinu (TFT). Buňky produkující TK jsou citlivé na TFT, což způsobuje inhibici buněčného metabolismu a zastavuje další buněčné dělení. Mutantní buňky jsou tedy schopny proliferace za přítomnosti TFT a tvoří viditelné kolonie, zatímco buňky, které obsahují TK, tuto schopnost nemají.
|
PRINCIP ZKOUŠKY
|
12.
|
Buňky v suspenzi jsou vhodnou dobu (viz odstavec 33) vystaveny zkoušené chemické látce jak s exogenním zdrojem metabolické aktivace (viz odstavec 19), tak bez něj a poté subkultivovány za účelem stanovení cytotoxicity a za účelem exprese fenotypu před selekcí mutantů. Cytotoxicita se stanoví podle relativního celkového růstu (RTG – viz odstavec 25) v testu MLA a podle relativního přežití (RS – viz odstavec 26) v testu TK6. Exponované kultury se udržují v růstovém médiu po vhodnou dobu charakteristickou pro každý zvolený typ buněk (viz odstavec 37), aby nastala přibližně optimální fenotypová exprese indukovaných mutací. Po fenotypové expresi se četnost mutantů stanoví tak, že se nasadí známý počet buněk do média obsahujícího selekční činidlo pro detekci mutantních kolonií a do média bez selekčního činidla, aby byla stanovena účinnost klonování (životaschopnost). Po vhodné inkubační době se spočítají kolonie. Četnost mutantů se vypočítá podle počtu mutantních kolonií korigovaného účinností klonování v době selekce mutantů.
|
POPIS METODY
Přípravky
Buňky
|
13.
|
Pro test MLA: Protože test MLA byl vyvinut a charakterizován pomocí sublinie TK
+/- -3.7.2C buněk L5178Y, musí být pro test MLA použita tato specifická sublinie. Buněčná linie L5178Y byla odvozena od lymfomu brzlíku indukovaného methylcholanthrenem z DBA-2 myši (26). Clive a spolupracovníci ošetřili buňky L5178Y (které Clive označil jako TK+/+ -3) ethyl-metansulfonátem a izolovali klon TK-/- (označený jako TK-/- -3.7) pomocí bromodeoxyuridinu jako selekčního činidla. Z klonu TK-/- byly izolovány spontánní klon TK+/- (označený jako TK+/- -3.7.2.) a subklon (označený jako TK+/--3.7.2C) a charakterizovány pro použití v testu MLA (27). Karyotyp pro buněčnou linii byl zveřejněn (28)(29)(30)(31). Modální hodnota počtu chromozomů je 40. Je zde jeden metacentrický chromozom (t12;13), který by se měl počítat jako jeden chromozom. Myší lokus TK je umístěn na distálním konci chromozomu 11. Buněčná linie L5178Y TK
+/- -3.7.2C má mutace v obou alelách p53 a vytváří mutantní protein p53 (32)(33). Za schopnost testu detekovat rozsáhlé poškození je pravděpodobně odpovědný stav p53 buněčné linie TK+/--3.7.2C (17).
|
|
14.
|
Pro TK6: TK6 je buněčná linie lidského lymfoblastoidu. Mateřskou buněčnou linií je transformovaná buněčná linie viru Epstein-Barrové, WI-L2, která byla původně získána od pětiletého chlapce s hereditární sférocytózou. První izolovaný klon, HH4, byl zmutován ICR191 a byla vytvořena heterozygotní TK buněčná linie, TK6 (34). Buňky TK6 jsou téměř diploidní a reprezentativní karyotyp je 47, XY, 13+, t(14; 20), t(3; 21 (35). Na dlouhém rameni chromozomu 17 se nachází lidský lokus TK. TK6 je p53-kompetentní buněčná linie, protože má v obou alelách sekvenci p53 divokého typu a exprimuje pouze protein p53 divokého typu (36).
|
|
15.
|
Pro MLA i TK6 se doporučuje, aby zkoušející laboratoř při prvním vytváření nebo doplňování základní zásoby zkontrolovala nepřítomnost mykoplazmové kontaminace, určila karyotyp buněk nebo obarvila chromozomy s lokusem TK a zkontrolovala dobu zdvojení populace. Pro buňky použité ve zkušební laboratoři by měla být stanovena normální délka buněčného cyklu, která by měla být v souladu s buněčnými charakteristikami uvedenými v literatuře (16)(19)(37). Tuto základní zásobu je třeba skladovat při –150 oC nebo méně a používat na přípravu veškeré pracovní zásoby buněk.
|
|
16.
|
Buď před vytvořením velkého množství zmrazené pracovní zásoby, nebo bezprostředně před použitím v experimentu může být nutné očistit kulturu od již existujících mutantních buněk [pokud již není frekvence mutací (MF) v kontrole s rozpouštědlem v přijatelném rozsahu – viz tabulka 2 pro MLA)]. Provádí se to pomocí methotrexátu (aminopterin), kterým se vyselektují TK-deficitní buňky, a přidáním thymidinu, hypoxanthinu a glycinu (L5178Y) nebo 2’-deoxycytidinu (TK6) do kultury pro zajištění optimálního růstu TK-kompetentních buněk (19)(38)(39) a (40) pro TK6). Obecné rady týkající se správného postupu udržování buněčných kultur i konkrétní rady pro buňky L5178Y a TK6 lze nalézt v (19)(31)(37)(39)(41). Laboratoře, které potřebují základní zásobu buněk, aby mohly iniciovat MLA nebo TK6, nebo si chtějí pořídit novou základní zásobu buněk, mají k dispozici buněčný archív s dobře charakterizovanými buňkami (37).
|
Média a podmínky kultivace
|
17.
|
U obou zkoušek by pro udržování kultur mělo být použito vhodné kultivační médium a inkubační podmínky (např. kultivační nádoby, zvlhčená atmosféra s 5 % CO2, inkubační teplota 37 oC). Buněčné kultury by měly být vždy uchovávány za podmínek, při nichž je zajištěn jejich logaritmický růst. Je zvláště důležité zvolit média a podmínky kultivace tak, aby byly zajištěny optimální růst buněk během období exprese a klonování jak mutovaných, tak nemutovaných buněk. Pro MLA i TK6 je také důležité, aby podmínky kultivace zajišťovaly optimální růst velkých kolonií/časných kolonií i malých kolonií/pozdních kolonií mutantů TK. Podrobnější informace o kultivaci, včetně nutnosti správně tepelně inaktivovat koňské sérum, jestliže se při selekci mutantů používá médium RPMI, lze nalézt v (19)(31)(38)(39)(40)(42).
|
Příprava kultur
|
18.
|
Buňky se pomnoží z kmenových kultur a nasadí se do kultivačního média v takové hustotě, aby kultury v suspenzích pokračovaly v exponenciálním růstu po celou dobu expozice a exprese.
|
Metabolická aktivace
|
19.
|
Při použití buněk L5178Y and TK6 by se měly použít exogenní metabolizující systémy, protože tyto buňky mají nedostatečnou endogenní metabolickou schopnost. Nejčastěji používaným systémem, který je standardně doporučován, pokud není zdůvodněno použití jiného systému, je kofaktorem dotovaná postmitochondriální frakce (S9) připravená z jater hlodavců (zpravidla potkanů) zpracovaná činidlem indukujícím enzymy, jako je Aroclor 1254 (43)(44)(45) nebo kombinace fenobarbitalu a β-naftoflavonu (46)(47)(48)(49)(50)(51). Posledně jmenovaná kombinace není v rozporu se Stockholmskou úmluvou o perzistentních organických znečišťujících látkách (52) a bylo prokázáno, že je stejně účinná jako Aroclor 1254 pro indukování oxidáz se smíšenou funkcí (45)(46)(47)(48)(49). Frakce S9 je obvykle používána v koncentracích od 1–2 % obj., avšak v konečném testovacím médiu je možné koncentraci zvýšit do 10 % obj. Volbu typu a koncentrace systému exogenní metabolické aktivace nebo metabolického induktoru, který se použije, může ovlivnit třída zkoušených chemických látek.
|
Příprava zkoušených chemických látek
|
20.
|
Pevné zkoušené chemické látky by měly být před aplikací na buňky rozpuštěny ve vhodných rozpouštědlech a popřípadě zředěny (viz odstavec 21). Kapalné látky lze přidat přímo do zkušebních systémů a/nebo je lze před použitím k ošetření zkušebních systémů zředit. Plynné a těkavé zkoušené chemické látky by se měly zkoušet za použití vhodně upravených standardních protokolů, například prostřednictvím ošetření buněk v neprodyšně uzavřených kultivačních nádobách (53)(54)(55). Měly by se používat čerstvě připravené zkoušené látky, pokud údaje o stálosti neprokazují možnost skladování.
|
ZKUŠEBNÍ PODMÍNKY
Rozpouštědla
|
21.
|
Rozpouštědlo by mělo být zvoleno tak, aby optimalizovalo rozpustnost zkoušené chemické látky, aniž by nepříznivě ovlivnilo provádění zkoušky, např. měnilo růst buněk, ovlivňovalo integritu zkoušené chemické látky, reagovalo s kultivačními nádobami, narušovalo systém metabolické aktivace. Doporučuje se pokud možno nejprve zvážit použití vodného rozpouštědla (nebo kultivačního média). Dobře zavedenými rozpouštědly jsou voda nebo dimethylsulfoxid. Podíl organických rozpouštědel by zpravidla neměl být větší než 1 % obj. a podíl vodných rozpouštědel (fyziologického roztoku nebo vody) v konečném expozičním médiu by neměl být větší než 10 % obj. Jsou-li použita jiná než osvědčená rozpouštědla (např. ethanol nebo aceton), mělo by být jejich použití podloženo údaji o jejich kompatibilitě se zkoušenými chemickými látkami, zkušebním systému a o tom, že nejsou v použité koncentraci genotoxická. Pokud takové podpůrné údaje neexistují, je důležité přidat neexponované kontroly (viz dodatek 1, Definice) prokazující, že zvolené rozpouštědlo nevyvolává žádné škodlivé nebo mutagenní účinky.
|
MĚŘENÍ CYTOTOXICITY A VOLBA EXPOZIČNÍCH KONCENTRACÍ
|
22.
|
Při stanovení nejvyšší koncentrace zkoušené chemické látky by neměly být použity koncentrace, které jsou schopny vyvolat falešnou pozitivní odpověď, jako např. koncentrace, jež vyvolávají nadměrnou cytotoxicitu (viz odstavec 28), srážení (viz odstavec 29) kultivačního média nebo zřetelné změny pH nebo osmolality (viz odstavec 8). Pokud zkoušená chemická látka v době jejího přidání způsobuje zřetelnou změnu hodnoty pH média, bylo by možné pH upravit pufrováním konečného expozičního média tak, aby nedošlo k falešným pozitivním výsledkům a aby byly zachovány vhodné kultivační podmínky.
|
|
23.
|
Volba koncentrace vychází z cytotoxicity a dalších faktorů (viz odstavce 27–30). Hodnocení cytotoxicity při předběžné zkoušce může být užitečné a může napomoci k lepšímu určení koncentrací, jež mají být použity při hlavní zkoušce, avšak předběžná zkouška není vyžadována. I v případě, že se provede předběžné hodnocení cytotoxicity, je při hlavním experimentu přesto požadováno měření cytotoxicity pro každou kulturu. Jestliže se provádí experiment ke zjištění rozsahu, měl by zahrnovat široké rozmezí koncentrací, přičemž může být ukončen buď v den 1 po expozici, nebo pokračovat expresí v den 2 a k selekci mutantů (pokud se budou použité koncentrace jevit jako vhodné).
|
|
24.
|
Cytotoxicita by měla být stanovena pro každou jednotlivou zkoušenou kulturu i kontrolní kulturu: metody pro MLA (2) a TK6 (15) jsou definovány podle mezinárodně dohodnuté praxe.
|
|
25.
|
Pro obě verze testu MLA, s použitím agaru i mikrotitračních destiček: Cytotoxicita se hodnotí podle relativního celkového růstu (RTG), který původně definoval v roce 1975 Clive a Spector (2). Toto měření zahrnuje relativní růst v suspenzi (RSG: zkoušená kultura v porovnání s kontrolou s rozpouštědlem) v průběhu expozice buněk, dobu exprese a relativní účinnost klonování (RCE: zkoušená kultura v porovnání s kontrolou s rozpouštědlem) v době, kdy jsou selektováni mutanti (2). Je třeba poznamenat, že RSG zahrnuje každou ztrátu buněk, k níž dojde ve zkoušené kultuře v průběhu expozice (viz dodatek 2, kde jsou uvedeny vzorce).
|
|
26.
|
Pro TK6: Cytotoxicita by se měla hodnotit podle relativního přežití (RS), tj. podle účinnosti klonování buněk umístěných na destičky okamžitě po expozici, upravené podle případné ztráty buněk v průběhu expozice, a to podle počtu buněk, v porovnání s negativní kontrolou (s přiřazeným přežitím 100 %) (viz dodatek 2, kde je uveden vzorec).
|
|
27.
|
Měly by se vyhodnotit nejméně čtyři zkušební koncentrace (nepočítaje v to kontroly s rozpouštědlem a pozitivní kontroly), které splňují kritéria přijatelnosti (vhodná cytotoxicita, počet buněk atd.). I když se doporučuje použít duplicitní kultury, lze pro každou zkušební koncentraci použít repliky nebo jednu exponovanou kulturu. Výsledky získané z duplicitních kultur při dané koncentraci by měly být vykazovány samostatně, ale pro účely analýzy údajů je lze sloučit (55). U zkoušených látek, jež vykazují malou cytotoxicitu nebo nevykazují žádnou cytotoxicitu, budou obvykle vhodné přibližně dvojnásobné až trojnásobné intervaly koncentrací. V případě cytotoxicity by měly zvolené koncentrace pokrývat rozpětí od koncentrace vyvolávající cytotoxicitu, jak je popsáno v odstavci 28, po koncentrace, při nichž dochází ke střední a nízké cytotoxicitě nebo nedochází k žádné cytotoxicitě. Mnohé zkoušené chemické látky vykazují strmé křivky závislosti odezvy na koncentraci, a aby bylo možné postihnout celý rozsah cytotoxicity nebo podrobně studovat závislost odpovědi na koncentraci, může být nezbytné zvolit menší rozdíly mezi jednotlivými koncentracemi a více než čtyři koncentrace, zejména v situacích, kdy je požadován opakovaný experiment (viz odstavec 70). Použití více než 4 koncentrací může být zvláště důležité, když se používá jedna kultura.
|
|
28.
|
Je-li maximální koncentrace odvozena od cytotoxicity, měla by být nejvyšší koncentrace zvolena tak, aby se dosáhlo relativního přežití mezi 20 a 10 % RTG u MLA a mezi 20 a 10 % RS u TK6 (odstavec 67).
|
|
29.
|
V případě špatně rozpustných zkoušených chemických látek, které nejsou cytotoxické při koncentracích nižších, než je jejich rozpustnost, by nejvyšší analyzovaná koncentrace měla na konci expozice zkoušené chemické látce vyvolat zákal nebo sraženinu viditelnou pouhým okem nebo pomocí inverzního mikroskopu. Dokonce i v případech, kdy k výskytu cytotoxicity dochází při koncentracích vyšších, než je rozpustnost, se doporučuje testovat pouze při jedné koncentraci vyvolávající zákal nebo viditelné sraženiny, neboť sraženina může vést k falešným účinkům. Protože testy MLA i TK6 používají kultury v suspenzi, je třeba zvláště dbát na to, aby provádění zkoušky nemohla ovlivňovat sraženina. Užitečné může být také stanovení rozpustnosti v kultivačním médiu před pokusem.
|
|
30.
|
Není-li pozorována sraženina nebo omezení cytotoxicity, měla by největší zkušební koncentrace odpovídat 10 mM, 2 mg/ml nebo 2 μl/ml, podle toho, která z uvedených hodnot je nejnižší (57) (58). Pokud u zkoušené chemické látky není stanoveno její složení, např. u látek s neznámým nebo proměnlivým složením, komplexních reakčních produktů nebo biologického materiálu [tj. chemické látky s neznámým nebo proměnlivým složením, (UVCB)], přírodních extraktů atd., bude možná muset být při nedostatečné cytotoxicitě nejvyšší koncentrace vyšší (např. 5 mg/ml), aby se zvýšila koncentrace každé ze složek. Je však třeba poznamenat, že u humánních léčivých přípravků se tyto požadavky mohou lišit (59).
|
Kontroly
|
31.
|
V okamžiku každého odběru by měly být použity souběžné negativní kontroly (viz odstavec 21) skládající se ze samotného rozpouštědla v kultivačním médiu, s nimiž se zachází stejným způsobem jako s exponovanými kulturami.
|
|
32.
|
K prokázání schopnosti laboratoře stanovit mutageny za podmínek použitého zkušebního protokolu, (případně) účinnosti exogenního systému metabolické aktivace a k prokázání adekvátní detekce malých/pozdních a velkých/časných TK mutantů jsou nutné souběžné pozitivní kontroly. Příklady pozitivních kontrol jsou uvedeny v tabulce 1 níže. V odůvodněných případech mohou být k pozitivní kontrole použity jiné látky. Jelikož zkoušky genetické toxicity savčích buněk in vitro při krátkodobé expozici (3–4 hodiny) prováděné souběžně s metabolickou aktivací a bez metabolické aktivace s použitím stejné doby trvání expozice jsou již dostatečně standardizovány, je možné použití pozitivních kontrol omezit na testování mutagenu vyžadujícího metabolickou aktivaci. V tomto případě tato reakce jediné pozitivní kontroly prokáže činnost systému metabolické aktivace i citlivost zkušebního systému. Pokud se však používá dlouhodobá expozice (tj. 24 hodin bez S9), měla by mít svou vlastní pozitivní kontrolu, protože doba trvání expozice bude odlišná než při zkoušce za použití metabolické aktivace. Každá pozitivní kontrola by měla být použita při jedné nebo více koncentracích, u nichž se očekává, že poskytnou reprodukovatelný a detekovatelný nárůst oproti pozadí, čímž se prokáže citlivost testovacího systému, a reakce by neměla být narušena cytotoxicitou přesahující limitní hodnoty stanovené v této zkušební metodě (viz odstavec 28).
|
Tabulka 1
Referenční látky doporučené pro posouzení způsobilosti laboratoře a pro výběr pozitivních kontrol
|
Kategorie
|
Látka
|
CASRN
|
|
1. Mutageny působící bez metabolické aktivace
|
|
methyl-methansulfonát
|
66-27-3
|
|
mitomycin C
|
50-07-7
|
|
4-nitrochinolin-1-oxid
|
56-57-5
|
|
2. Mutageny vyžadující metabolickou aktivaci
|
|
benzo[a]pyren
|
50-32-8
|
|
cyklofosfamid (cyklofosfamid monohydrát)
|
50-18-0 R60/6055/-19-2
|
|
7,12-dimethylbenzoanthracen
|
57-97-6
|
|
3-methylcholanthren
|
56-49-5
|
POSTUP
Expozice zkoušené chemické látce
|
33.
|
Proliferující buňky se vystaví zkoušené chemické látce jak za přítomnosti metabolického aktivačního systému, tak bez něho. Expozice by měla trvat vhodnou dobu (obvykle je postačující doba 3 až 4 hodin). Je však třeba poznamenat, že u humánních léčivých přípravků se tyto požadavky mohou lišit (59). Pro MLA, v případech, kdy krátkodobá expozice dává negativní výsledky a existují informace naznačující nutnost dlouhodobější expozice [např. analoga nukleosidů, špatně rozpustné chemické látky (5)(59)], je třeba zvážit provedení zkoušky s delší expozicí, tj. 24 hodin bez S9.
|
|
34.
|
Minimální počet buněk použitých pro každou zkušební (kontrolní a exponovanou) kulturu v každém stádiu zkoušky by měl vycházet z četnosti spontánních mutací. Obecným pravidlem je exponovat a pasážovat v každé experimentální kultuře dostatečný počet buněk, aby bylo uchováno v každé fázi zkoušky alespoň 10, ideálně však 100 spontánních mutantů (expozice, fenotypová exprese a selekce mutantů) (56).
|
|
35.
|
Pro MLA je doporučená přijatelná frekvence spontánních mutací mezi 35–140 × 10-6 (verze s agarem) a 50–170 × 10-6 (verze s mikrotitračními destičkami) (viz tabulka 2). Pro získání alespoň 10 a ideálně 100 spontánních mutantů, kteří v každé zkoušené kultuře přežijí expozici, je nezbytné exponovat alespoň 6 × 106 buněk. Exponování takového počtu buněk a zachování dostatečného počtu buněk v průběhu exprese a klonování pro selekci mutantů poskytuje dostatečný počet spontánních mutací (10 nebo více) ve všech fázích experimentu, a to i u kultur exponovaných při koncentracích, jejichž výsledkem je 90 % cytotoxicita (měřeno podle 10 % RTG) (19)(38)(39).
|
|
36.
|
U TK6 činí četnost spontánních mutací obvykle 2 až 10 × 10-6. Pro získání alespoň 10 spontánních mutantů, kteří v každé kultuře přežijí expozici, je nezbytné exponovat alespoň 20 × 106 buněk. Expozice tohoto počtu buněk zajistí dostatečný počet spontánních mutantů (10 nebo více) i v kulturách exponovaných při koncentracích, které vyvolávají při expozici 90 % cytotoxicitu (10 % relativní přežití). Kromě toho musí být v období exprese kultivován a umístěn na destičky pro výběr mutantů dostatečný počet buněk (60).
|
Doba exprese fenotypu a měření cytotoxicity a četnosti mutací
|
37.
|
Na konci expoziční doby se buňky po stanovenou dobu kultivují, aby se umožnila téměř optimální fenotypová exprese nově indukovaných mutantů, specifická pro každou buněčnou linii. U testu MLA je doba fenotypové exprese 2 dny. U testu TK6 činí doba fenotypové exprese 3–4 dny. Jestliže se použije 24hodinová expozice, začíná doba exprese po ukončení expozice.
|
|
38.
|
V průběhu doby fenotypové exprese se buňky počítají každý den. U testu MLA se denní počet buněk použije pro výpočet denního růstu v suspenzi (SG). Po dvoudenním období exprese se buňky suspendují v médiu se selekčním činidlem a bez něj za účelem stanovení počtu mutantů (destičky pro selekci), respektive účinnosti klonování (destičky pro životaschopnost). Pro test MLA existují dvě stejně přijatelné metody pro selektivní klonování mutantů; jedna s použitím měkkého agaru a druhá s použitím kapalného média v 96jamkových destičkách (19)(38)(39). Klonování v testu TK6 se provádí s použitím kapalného média v 96jamkových destičkách (16).
|
|
39.
|
Jediným doporučeným selekčním činidlem pro mutanty TK je triflurothymidin (TFT) (61).
|
|
40.
|
U testu MLA se agarové destičky a mikrotitrační destičky počítají po 10–12 dnech inkubace. U testu TK6 se po 10–14 dnech stanoví skóre časných kolonií mutantů v mikrotitračních destičkách. Za účelem obnovení pomalu rostoucích (pozdních kolonií) mutantů TK6 je třeba po spočítání časných kolonií mutantů dodat buňkám růstové médium a TFT a poté destičky inkubovat dalších 7–10 dnů (62). Viz odstavce 42 a 44, kde je uvedena diskuze o vyčíslení mutantů TK s pomalým a normálním růstem.
|
|
41.
|
Příslušné výpočty pro tyto dva testy včetně obou metod (agar a mikrodestičky) pro test MLA jsou uvedeny v dodatku 2. U testu MLA s použitím agarové metody se spočítají kolonie a počet mutantních kolonií se upraví podle účinnosti klonování, aby bylo možné vypočítat četnost mutací. U testu MLA ve verzi s mikrodestičkami a u testu TK6 se stanoví účinnost klonování u destiček pro selekci i destiček pro účinnost klonování podle Poissonova rozdělení (63). Z těchto dvou hodnot účinnosti klonování se vypočítá četnost mutací.
|
Charakterizace mutantních kolonií
|
42.
|
Je-li u testu MLA zkoušená chemická látka pozitivní (viz odstavce 62–63), charakterizace podle velikosti nebo růstu kolonií by měla být provedena alespoň na jedné ze zkušebních kultur (obvykle na kultuře s nejvyšší pozitivní koncentrací) a na negativních a pozitivních kontrolách. Je-li zkoušená látka negativní (viz odstavec 64), měla být charakterizace mutantních kolonií provedena na negativních a pozitivních kontrolách. U metody testu MLA s využitím mikrodestiček se mutanti tvořící malé kolonie definují jako kolonie pokrývající méně než 25 % průměru jamky a mutanti tvořící velké kolonie jako kolonie pokrývající více než 25 % průměru jamky. U agarové metody se na vyčíslení kolonií mutantů a stanovení jejich velikosti používá automatické počítadlo kolonií. Způsoby stanovení velikosti kolonií jsou podrobně popsány v literatuře (19)(38)(40). Aby bylo prokázáno, že studie se provádějí správně, je třeba charakterizovat kolonie na negativní a pozitivní kontrole.
|
|
43.
|
Zkoušenou chemickou látku nelze určit jako negativní, pokud nejsou v pozitivní kontrole adekvátně detekováni mutanti tvořící velké i malé kolonie. Charakterizace kolonií může sloužit k poskytnutí obecných informací o schopnosti zkoušené chemické látky způsobit bodové mutace anebo chromozomální jevy (odstavec 4).
|
|
44.
|
TK6: Normálně rostoucí a pomalu rostoucí mutanti se diferencují podle rozdílu v době inkubace (viz odstavec 40). U testu TK6 se obvykle stanoví skóre časných i pozdních mutantů pro všechny kultury včetně negativních a pozitivních kontrol. Aby bylo prokázáno, že studie se provádějí správně, je třeba charakterizovat kolonie u negativní a pozitivní kontroly. Zkoušenou chemickou látku nelze určit jako negativní, pokud nejsou v pozitivní kontrole adekvátně detekováni časní i pozdní mutanti. Charakterizace kolonií může sloužit k poskytnutí obecných informací o schopnosti zkoušené chemické látky způsobit bodové mutace anebo chromozomální jevy (odstavec 4).
|
Způsobilost laboratoře
|
45.
|
Aby bylo možné před zahájením rutinního zkoušení prokázat, zda laboratoř má s danou zkouškou dostatečné zkušenosti, měla by provést sérii experimentů s referenčními pozitivními látkami, jež působí prostřednictvím různých mechanismů (alespoň s jednou látkou účinnou s metabolickou aktivací a jednou účinnou bez metabolické aktivace, které se vyberou ze seznamu látek uvedeného v tabulce 1), a s různými negativními kontrolami (včetně neexponovaných kultur a různých rozpouštědel/vehikul). Odpovědi těchto pozitivních a negativních kontrol by měly být v souladu s odbornou literaturou. Tento požadavek se nepoužije na laboratoře, které tuto zkušenost mají, tj. které mají k dispozici databázi historických údajů, jak je definována v odstavcích 47–50. V testu MLA by hodnoty dosažené u pozitivní i negativní kontroly měly odpovídat doporučením IWGT (viz tabulka 2).
|
|
46.
|
Výběr z látek pro pozitivní kontroly (viz tabulka 1) by měl být zkoumán s krátkou i dlouhou expozicí (pokud se používají dlouhé expozice) bez metabolické aktivace a také s krátkou expozicí a s metabolickou aktivací, aby byla prokázána způsobilost pro detekci mutagenních chemických látek, stanovena účinnost systému metabolické aktivace a prokázána vhodnost podmínek pro růst buněk v průběhu expozice, fenotypovou expresi a selekci mutantů a postupů hodnocení. Rozsah koncentrací by u vybraných látek měl být zvolen tak, aby poskytl opakovatelná zvýšení účinku v závislosti na dávce nad úroveň pozadí, aby se tak prokázala citlivost a dynamické rozpětí testovacího systému.
|
Historické kontrolní údaje
|
47.
|
Laboratoř by měla stanovit:
|
—
|
historické rozmezí a distribuci pozitivních kontrol,
|
|
—
|
historické rozmezí a distribuci negativních kontrol (neexponovaných, s rozpouštědlem).
|
|
|
48.
|
Při prvním získávání údajů o historické distribuci negativních kontrol by měly být výsledky souběžných negativních kontrol v souladu s publikovanými negativními kontrolními údaji. S tím, jak je do distribuce kontrol přidáváno více údajů z experimentů, měly by aktuální negativní kontroly v ideálním případě spadat do 95 % rozmezí této distribuce (64)(65).
|
|
49.
|
Nejprve by měla být vybudována historická databáze negativních kontrol dané laboratoře na základě nejméně 10 experimentů, avšak raději by měla zahrnovat nejméně 20 experimentů provedených za srovnatelných zkušebních podmínek. V laboratořích by měly být uplatňovány metody kontroly jakosti, jako např. regulační diagramy (např. C-diagramy nebo diagramy X s pruhem (65)), z nichž je patrné, jak proměnlivé jsou jejich údaje o pozitivních a negativních kontrolách a že metodika je v dané laboratoři „pod kontrolou“ (66). Další podrobnosti a doporučení jak vytvářet a používat historická data lze najít v literatuře (64).
|
|
50.
|
Údaje o negativních kontrolách by měly zahrnovat četnost mutací z jedné kultury nebo raději z replikovaných kultur, jak je popsáno v odstavci 27. Výsledky souběžných negativních kontrol by v ideálním případě měly být uvnitř 95 % kontrolních limitů distribuce v historické databázi negativních kontrol laboratoře. Pokud jsou údaje z negativních kontrol mimo 95 % rozmezí, může být přijatelné je zahrnout do dosavadní distribuce kontrol, pokud tyto údaje nejsou mimořádně odlehlé, je to důkaz, že testovací systém je „pod kontrolou“ (viz odstavec 49), a důkaz, že nedošlo k technické nebo lidské chybě.
|
|
51.
|
Jakékoli změny zkušebního protokolu by měly být zvažovány z hlediska jejich souladu se stávajícími historickými kontrolními databázemi laboratoře. Jakýkoli větší rozdíl ve zkušebním protokolu by měl vést k zavedení nové historické kontrolní databáze.
|
ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV
Vyjádření výsledků
|
52.
|
Prezentace údajů pro test MLA i TK6 by měla zahrnovat údaje za exponované i kontrolní kultury potřebné pro výpočet cytotoxicity (RTG, respektive RS) a četnosti mutací, jak je popsáno níže.
|
|
53.
|
U testu MLA by měly být uvedeny údaje za jednotlivé kultury pro RSG, RTG, účinnost klonování v době selekce mutantů a počet mutantních kolonií (u agarové verze) nebo počet prázdných jamek (u mikrodestičkové verze). Četnost mutantů by měla být vyjádřena jako počet mutantních buněk na milion přeživších buněk. Je-li odpověď pozitivní, měly by se uvést četnosti mutací tvořících malé a velké kolonie (anebo procento z celkové četnosti mutací) u alespoň jedné koncentrace zkoušené chemické látky (obvykle látky s nejvyšší pozitivní koncentrací) a negativní a pozitivní kontroly. V případě negativní odpovědi by se měla uvést četnost mutací tvořících malé a velké kolonie pro negativní a pozitivní kontrolu.
|
|
54.
|
U testu TK6 by měly být uvedeny údaje za jednotlivé kultury pro RS, účinnost klonování v době selekce mutantů a počet prázdných jamek u časných a pozdních mutací. Četnost mutací (MF) se vyjadřuje jako počet mutantních buněk na počet přeživších buněk a měla by zahrnovat celkovou MF i MF (anebo procento z celkové MF) časných a pozdních mutantů.
|
Kritéria přijatelnosti
|
55.
|
Pro test MLA i test TK6 by měla být splněna tato kritéria před stanovením celkových výsledků za konkrétní zkoušenou chemickou látku:
|
—
|
Test se provedl za dvou experimentálních uspořádání (krátká expozice s metabolickou aktivací a bez ní – viz odstavec 33), pokud jedno z nich nevedlo k pozitivním výsledkům.
|
|
—
|
Měl by být analyzovatelný adekvátní počet buněk a koncentrací (viz odstavce 27, 34–36).
|
|
—
|
Kritéria pro výběr nejvyšší koncentrace jsou v souladu s kritérii popsanými v odstavcích 28–30.
|
|
Kritéria přijatelnosti pro negativní a pozitivní kontroly
|
56.
|
Na základě analýzy expertní pracovní skupiny IWGT pro MLA zahrnující velké množství údajů o MLA byl dosažen mezinárodní konsensus ohledně specifických kritérií přijatelnosti pro MLA (1)(2)(3)(4)(5). Proto tato zkušební metoda poskytuje konkrétní doporučení pro určení přijatelnosti negativních a pozitivních kontrol a pro hodnocení výsledků jednotlivých látek v testu MLA. Test TK6 má mnohem menší databázi a nebyl hodnocen pracovní skupinou.
|
|
57.
|
V testu MLA by se mělo u každého experimentu hodnotit, zda neexponovaná kontrola / kontrola s rozpouštědlem splňuje kritéria přijatelnosti pracovní skupiny IWGT pro MLA ((4) a níže uvedená tabulka) pro: 1) četnost mutací (nezapomeňte, že přijatelné MF podle IWGT jsou jiné pro verzi testu MLA s agarem a verzi s mikrodestičkami), 2) účinnost klonování (CE) v době selekce mutantů a 3) růst v suspenzi (SG) pro kontrolu s rozpouštědlem (viz dodatek 2, kde jsou uvedeny vzorce).
Tabulka 2
Kritéria přijatelnosti pro MLA
|
Parametr
|
Metoda s použitím měkkého agaru
|
Metoda s použitím mikrodestiček
|
|
Četnost mutantů
|
35–140 × 10-6
|
50–170 × 10-6
|
|
Účinnost klonování
|
65–120 %
|
65–120 %
|
|
Růst v suspenzi
|
8–32násobek (3–4 hodiny expozice) 32–180násobek (24 hodin expozice, je-li prováděna)
|
8–32násobek (3–4 hodiny expozice) 32–180násobek (24 hodin expozice, je-li prováděna)
|
|
|
58.
|
V testu MLA by se u každé zkoušky mělo hodnotit také, zda pozitivní kontrola/y splňuje/í alespoň jedno z těchto dvou kritérií přijatelnosti stanovených pracovní skupinou IWGT:
|
—
|
Pozitivní kontrola by měla prokázat absolutní nárůst celkové četnosti mutací, to znamená zvýšení nad spontánní četnost mutací na pozadí [indukovaná četnost mutací (IMF)] alespoň 300 × 10-6. Alespoň 40 % IMF by se mělo projevit v četnosti mutací tvořících malé kolonie.
|
|
—
|
Pozitivní kontrola má zvýšenou četnost mutací tvořících malé kolonie alespoň 150 × 10-6 nad MF pozorovanou u souběžné neexponované kontroly / kontroly s rozpouštědlem (IMF tvořících malé kolonie 150 × 10-6).
|
|
|
59.
|
U testu TK6 bude zkouška přijatelná, jestliže se souběžná negativní kontrola považuje za přijatelnou pro přidání do databáze historických negativních kontrol laboratoře, jak je popsáno v odstavcích 48–49. Kromě toho by souběžné pozitivní kontroly (viz odstavec 32) měly vyvolat odpovědi, které jsou slučitelné s odpověďmi uvedenými v historické databázi pozitivních kontrol, a měly by vyvolat statisticky významný nárůst v porovnání se souběžnou negativní kontrolou.
|
|
60.
|
U obou testů by horní mez cytotoxicity pozorovaná v kultuře pozitivní kontroly měla být stejná jako v experimentálních kulturách. To znamená, že poměr RTG/RS by neměl být menší než 10 %. K prokázání, že kritéria přijatelnosti byla u pozitivní kontroly splněna, postačí použít jednu koncentraci (nebo jednu z koncentrací kultur pozitivní kontroly, jestliže je použita více než jedna koncentrace). Dále musí být četnost mutací pozitivní kontroly v rámci přijatelného rozmezí stanoveného pro laboratoř.
|
Hodnocení a interpretace výsledků
|
61.
|
Pro test MLA vykonala významnou práci v oblasti biologické relevance a kritérií pro pozitivní odpověď expertní pracovní skupina IWGT pro myší lymfom (4). Proto tato zkušební metoda poskytuje konkrétní doporučení pro interpretaci výsledků zkoušené chemické látky v testu MLA (viz odstavce 62–64). Test TK6 má mnohem menší databázi a nebyl hodnocen pracovní skupinou. Proto jsou doporučení pro interpretaci dat v testu TK6 uvedena obecněji (viz odstavce 65–66). Další doporučení se vztahují na oba testy (viz odstavce 67–71).
|
MLA
|
62.
|
Doporučuje se, aby způsob definování pozitivní a negativní odpovědi zajišťoval, že zvýšená četnost mutací bude biologicky relevantní. Místo statistické analýzy, která se obvykle používá u jiných testů, vychází z použití předem definované indukované frekvence mutací (tj. zvýšení frekvence mutací nad souběžnou kontrolu), určeného globálního hodnotícího faktoru (GEF), který vychází z analýzy distribuce dat o četnosti mutací u negativních kontrol od zúčastněných laboratoří (4). Pro agarovou verzi testu MLA je faktor GEF 90 × 10-6 a pro mikrodestičkovou verzi testu MLA je faktor GEF 126 × 10-6.
|
|
63.
|
Za předpokladu, že všechna kritéria přijatelnosti byla splněna, lze zkoušenou chemickou látku považovat za jasně pozitivní, pokud v některém ze zkoumaných experimentálních uspořádání (viz odstavec 33), přesahuje zvýšení četnosti mutací nad souběžné pozadí faktor GEF a toto zvýšení souvisí s koncentrací (např. s použitím trend testu). Pak se má za to, že zkoušená chemická látka je schopna vyvolávat mutaci v tomto testovacím systému.
|
|
64.
|
Za předpokladu, že byla splněna všechna kritéria přijatelnosti, lze zkoušenou chemickou látku považovat za jasně negativní, pokud za všech zkoumaných experimentálních uspořádání (viz odstavec 33), nedochází k žádné odpovědi v závislosti na koncentraci nebo četnost mutací, jestliže se zvýšila, nepřevyšuje faktor GEF. Pak se má za to, že zkoušená chemická látka není schopna vyvolávat mutace v tomto testovacím systému.
|
TK6
|
65.
|
Za předpokladu, že všechna kritéria přijatelnosti byla splněna, lze zkoušenou chemickou látku považovat za jasně pozitivní, pokud v některém ze zkoumaných experimentálních uspořádání (viz odstavec 33):
|
—
|
alespoň jedna ze zkušebních koncentrací vykazuje statisticky významný nárůst v porovnání se souběžnou negativní kontrolou,
|
|
—
|
hodnocení pomocí vhodného trend-testu ukáže závislost tohoto nárůstu na koncentraci (viz odstavec 33),
|
|
—
|
všechny výsledky jsou mimo distribuci historických údajů o negativních kontrolách (např. mimo 95 % rozmezí dle Poissonova rozdělení; viz odstavec 48).
|
Jsou-li splněna všechna tato kritéria, má se za to, že zkoušená chemická látka je schopna vyvolávat mutace v tomto testovacím systému. Doporučení ohledně nejvhodnějších statistických metod lze najít v literatuře (66)(67).
|
|
66.
|
Za předpokladu, že byla splněna všechna kritéria přijatelnosti, lze zkoušenou chemickou látku považovat za jasně negativní, pokud za všech zkoumaných experimentálních uspořádání (viz odstavec 33):
|
—
|
žádná ze zkušebních koncentrací nevykazuje statisticky významný nárůst v porovnání se souběžně provedenou negativní kontrolou,
|
|
—
|
hodnocení pomocí vhodné zkoušky trendu ukáže, že nedochází k nárůstu v závislosti na koncentraci,
|
|
—
|
všechny výsledky spadají do rámce distribuce dosavadních údajů o negativních kontrolách (např. 95 % rozmezí dle Poissonova rozdělení, viz odstavec 48).
|
Pak se má za to, že zkoušená chemická látka není schopna vyvolávat mutace v tomto testovacím systému.
|
Pro MLA i TK6:
|
67.
|
Je-li maximální koncentrace odvozena od cytotoxicity, měla by být nejvyšší koncentrace zvolena tak, aby se dosáhlo mezi 20 a 10 % RTG/RS. Podle konsensu je třeba dbát na to, aby byly interpretovány pouze ty pozitivní výsledky, které jsou vykazovány mezi 20 a 10 % RTG/RS, přičemž výsledek se nepovažuje za pozitivní, jestliže ke zvýšení četnosti dotací došlo pouze při 10 % RTG/RS nebo nižším poměru (pokud byl hodnocen) (2)(59).
|
|
68.
|
Existují některé okolnosti, za nichž mohou pomoci další informace určit, že zkoušená chemická látka není mutagenní, když žádná kultura nevykazuje hodnotu RTG mezi 10–20 % RTG/RS. Jde o tyto situace: 1) V sérii datových bodů v rozmezí od 100 % do 20 % RTG/RS není žádný důkaz o mutagenitě (např. žádná závislost odpovědi na dávce, žádné četnosti mutací nad četnost pozorovanou při souběžné negativní kontrole nebo v historických rozsazích pozadí atd.) a alespoň jeden datový bod je mezi 20 % a 25 % RTG/RS. 2) V sérii datových bodů od 100 % do 25 % RTG/RS není žádný důkaz o mutagenitě (např. žádná závislost odpovědi na dávce, žádné četnosti mutací nad četnost pozorovanou při souběžné negativní kontrole nebo v historických rozsazích pozadí atd.) a také jeden datový bod je mírně pod 10 % RTG/RS. V obou těchto situacích lze zkoušenou chemickou látku posoudit jako negativní.
|
|
69.
|
Ověření jasně pozitivního či jasně negativního účinku není nutné.
|
|
70.
|
V případech, kdy odpověď není ani jasně negativní, ani jasně pozitivní, jak je popsáno výše, nebo s cílem napomoci při stanovení biologického významu výsledku by měly být údaje vyhodnoceny na základě odborného posouzení a/nebo dalším šetřením. Užitečné by mohlo být provedení opakovaného experimentu, případně za použití upravených zkušebních podmínek [např. rozsahu koncentrací, aby se zvýšila pravděpodobnost dosažení datových bodů v rozmezí 10–20 % RTG/RS, použití jiných podmínek metabolické aktivace (např. koncentrace S9 nebo původ S9) a délky expozice].
|
|
71.
|
Ve vzácných případech ani po dalším šetření nebude možné na základě daného souboru údajů dospět k závěru o pozitivitě či negativitě výsledku. Proto by měl být učiněn závěr, že odpověď na zkoumanou chemickou látku je neurčitá (interpretováno jako stejná pravděpodobnost pozitivní nebo negativní odpovědi).
|
ZÁVĚREČNÁ ZPRÁVA
|
72.
|
Závěrečná zpráva by měla obsahovat tyto informace:
|
|
Zkoušená chemická látka:
|
—
|
zdroj, číslo šarže a je-li k dispozici, datum použitelnosti,
|
|
—
|
stabilita zkoušené chemické látky samotné, je-li známa,
|
|
—
|
rozpustnost a stabilita zkoušené chemické látky v rozpouštědle, jsou-li známy,
|
|
—
|
měření pH, případně osmolality a sraženiny v kultivačním médiu, do něhož byla přidána zkoušená chemická látka.
|
|
|
Jednosložková látka:
|
—
|
fyzický vzhled, rozpustnost ve vodě a další relevantní fyzikálně-chemické vlastnosti,
|
|
—
|
chemická identifikace, např. název podle IUPAC nebo CAS, číslo CAS, kód SMILES nebo InChI, strukturní vzorec, čistota, případně chemická identita nečistot, je-li to prakticky možné, atd.
|
|
|
|
Vícesložková látka, látka s neznámým nebo proměnlivým složením, komplexní reakční produkt nebo biologický materiál a směsi:
|
—
|
charakterizované pokud možno chemickou identitou (viz výše), kvantitativním výskytem a relevantními fyzikálně-chemickými vlastnostmi složek.
|
|
|
|
|
Rozpouštědlo:
|
—
|
zdůvodnění volby rozpouštědla,
|
|
—
|
percentuální podíl rozpouštědla v konečném kultivačním médiu.
|
|
|
|
Buňky:
|
|
Pro základní laboratorní kultury:
|
—
|
druh a zdroj buněk a historie ve zkušební laboratoři,
|
|
—
|
vlastnosti karyotypu a/nebo modální hodnota počtu chromozomů,
|
|
—
|
metody udržování buněčných kultur,
|
|
—
|
nepřítomnost mykoplasmat,
|
|
—
|
doby zdvojnásobení buněk.
|
|
|
|
|
Zkušební podmínky:
|
—
|
zdůvodnění volby koncentrací a počtu buněčných kultur, včetně např. údajů o cytotoxicitě a mezích rozpustnosti,
|
|
—
|
složení média, koncentrace CO2, vlhkost,
|
|
—
|
koncentrace zkoušené chemické látky vyjádřená jako konečná koncentrace v kultivačním médiu (např. v μg nebo mg/ml nebo mM kultivačního média),
|
|
—
|
koncentrace (a/nebo objem) rozpouštědla a zkoumané chemické látky přidaných do kultivačního média,
|
|
—
|
hustota buněk během expozice;
|
|
—
|
typ a složení použitého metabolického aktivačního systému (zdroj S9, metoda přípravy směsi S9, koncentrace nebo objem směsi S9 a S9 v konečném kultivačním médiu, kontroly jakosti S9),
|
|
—
|
látky použité v pozitivních a negativních kontrolách, konečné koncentrace pro jednotlivé podmínky expozice,
|
|
—
|
délka doby exprese (případně včetně počtu nasazených buněk, subkultur a výměny média),
|
|
—
|
identifikace selekčního činidla a jeho koncentrace,
|
|
—
|
u testu MLA by měla být uvedena použitá verze (agar nebo mikrodestička),
|
|
—
|
kritéria přijatelnosti zkoušek,
|
|
—
|
metody použité k počítání životaschopných a mutantních buněk,
|
|
—
|
metody použité pro měření cytotoxicity,
|
|
—
|
jakékoli doplňkové informace týkající se cytotoxicity a použité metody,
|
|
—
|
délka inkubace po umístění na destičky,
|
|
—
|
definice kolonií podle velikosti a typu (případně včetně kritérií pro „malé“ a „velké“ kolonie),
|
|
—
|
kritéria klasifikace studie na pozitivní, negativní nebo neurčitou,
|
|
—
|
metody použité ke stanovení pH, osmolality, je-li test proveden, a v příslušných případech sraženiny.
|
|
|
|
Výsledky:
|
—
|
počet exponovaných buněk a počet subkultivovaných buněk v každé kultuře,
|
|
—
|
parametry toxicity (RTG pro MLA a RS pro TK6),
|
|
—
|
známky srážení a čas jejich stanovení,
|
|
—
|
počet buněk kultivovaných v selektivním a neselektivním médiu,
|
|
—
|
počet kolonií v neselektivním médiu a počet rezistentních kolonií v selektivním médiu a související četnosti mutací,
|
|
—
|
stanovení velikosti kolonií u negativních a pozitivních kontrol a v případě, že je zkoušená chemická látka pozitivní, alespoň jedna koncentrace a související četnosti mutací,
|
|
—
|
závislost účinku na koncentraci, pokud je to možné,
|
|
—
|
údaje o souběžných negativních kontrolách (rozpouštědlo) a pozitivních kontrolách (koncentrace a rozpouštědla),
|
|
—
|
dosavadní údaje o negativní (rozpouštědlo) a pozitivní kontrole (koncentrace a rozpouštědla) s rozmezími, středními hodnotami a směrodatnými odchylkami, počet zkoušek, z nichž vychází historické kontroly,
|
|
—
|
statistické analýzy (pro jednotlivé kultury, případně spojené repliky) a případné p hodnoty; a pro test MLA hodnocení GEF.
|
|
|
LITERATURA
|
(1)
|
Moore, M.M., Honma, M. Clements, J. (Rapporteur), Awogi, T., Bolcsfoldi, G., Cole, J., Gollapudi, B., Harrington-Brock, K., Mitchell, A., Muster, W., Myhr, B., O'Donovan, M., Ouldelhkim, M-C., San, R., Shimada, H. and Stankowski, L.F. Jr. (2000). Mouse Lymphoma Thymidine Kinase Locus (TK) Gene Mutation Assay: International Workshop on Genotoxicity Test Procedures (IWGTP) Workgroup Report, Environ. Mol. Mutagen., 35 (3): 185-190.
|
|
(2)
|
Moore, M.M., Honma, M., Clements, J., Harrington-Brock, K., Awogi, T., Bolcsfoldi, G., Cifone, M., Collard, D., Fellows, M., Flanders, K., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Kraycer, J., McEnaney, S., Muster, W., Myhr, B., O’Donovan, M., Oliver, Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Shimada, H. and Stankowski, L.F. Jr. (2002). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: Follow-Up International Workshop on Genotoxicity Test Procedures, New Orleans, Louisiana, (April 2000), Environ. Mol. Mutagen., 40 (4): 292-299.
|
|
(3)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Cifone, M., Delongchamp, R., Fellows, M., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Muster, W., Myhr, B., O’Donovan, M., Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A., Wakuri, S. and Yoshimura, I. (2003). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: International Workshop (Plymouth, UK) on Genotoxicity Test Procedures Workgroup Report, Mutation Res., 540: 127-140.
|
|
(4)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Delongchamp, R., Durward, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Lloyd, M., Majeska, J., Myhr, B., O’Donovan, M., Omori, T., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A.K., Van Goethem, F., Wakuri, S. and Yoshimura, I. (2006). Mouse Lymphoma Thymidine Kinase Gene Mutation Assay: Follow-Up Meeting of the International Workshop on Genotoxicity Tests – Aberdeen, Scotland, 2003 – Assay Acceptance Criteria, Positive Controls, and Data Evaluation, Environ. Mol. Mutagen., 47 (1): 1-5.
|
|
(5)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Clay, P., Doppalapudi, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Muster, W., Pant, K., Kidd, D.A., Lorge, E., Lloyd, M., Myhr, B., O’Donovan, M., Riach, C., Stankowski, L.F. Jr., Thakur A.K. and Van Goethem, F. (2007). Mouse Lymphoma Thymidine Kinase Mutation Assay: Meeting of the International Workshop on Genotoxicity Testing, San Francisco, 2005, Recommendations for 24-h Treatment, Mutation. Res., 627 (1): 36-40.
|
|
(6)
|
OECD (2016). Overview of the set of OECD Genetic Toxicology Test Guidelines and updates performed in 2014-2015. ENV Publications. Series on Testing and Assessment, No. Series on Testing and Assessment, No 234, OECD, Paris.
|
|
(7)
|
Fellows M.D., Luker, T., Cooper, A. and O'Donovan, M.R. (2012). Unusual Structure-Genotoxicity Relationship in Mouse Lymphoma Cells Observed with a Series of Kinase Inhibitors. Mutation, Res., 746 (1): 21-28.
|
|
(8)
|
Honma, M., Momose, M., Sakamoto, H., Sofuni, T. and Hayashi, M. (2001). Spindol Poisons Induce Allelic Loss in Mouse Lymphoma Cells Through Mitotic Non-Disjunction. Mutation Res., 493 (1-2): 101-114.
|
|
(9)
|
Wang, J., Sawyer, J.R., Chen, L., Chen, T., Honma, M., Mei, N. and Moore, M.M. (2009). The Mouse Lymphoma Assay Detects Recombination, Deletion, and Aneuploidy, Toxicol. Sci.., 109 (1): 96-105.
|
|
(10)
|
Applegate, M.L., Moore, M.M., Broder, C.B., Burrell, A., and Hozier, J.C. (1990). Molecular Dissection of Mutations at the Heterozygous Thymidine Kinase Locus in Mouse Lymphoma Cells. Proc. National. Academy. Sci. USA, 87 (1): 51-55.
|
|
(11)
|
Hozier, J., Sawyer, J., Moore, M., Howard, B. and Clive, D. (1981). Cytogenetic Analysis of the L5178Y/TK+/- Leads to TK-/- Mouse Lymphoma Mutagenesis Assay System, Mutation Res., 84 (1): 169-181.
|
|
(12)
|
Hozier, J., Sawyer, J., Clive, D. and Moore, M.M. (1985). Chromosome 11 Aberrations in Small Colony L5178Y TK-/- Mutants Early in their Clonal History, Mutation Res., 147 (5): 237-242.
|
|
(13)
|
Moore, M.M., Clive, D., Hozier, J.C., Howard, B.E., Batson, A.G., Turner, N.T. and Sawyer, J. (1985). Analysis of Trifluorothymidine-Resistant (TFTr) Mutants of L5178Y/TK+/- Mouse Lymphoma Cells. Mutation Res., 151 (1): 161-174.
|
|
(14)
|
Liber H.L., Call K.M. and Little J.B. (1987). Molecular and Biochemical Analyses of Spontaneous and X-Ray-Induced Mutants in Human Lymphoblastoid Cells. Mutation Res., 178 (1): 143-153.
|
|
(15)
|
Li C.Y., Yandell D.W. and Little J.B. (1992). Molecular Mechanisms of Spontaneous and Induced Loss of Heterozygosity in Human Cells In Vitro. Somat. Cell Mol. Genet., 18 (1): 77-87.
|
|
(16)
|
Honma M., Hayashi M. and Sofuni T. (1997). Cytotoxic and Mutagenic Responses to X-Rays and Chemical Mutagens in Normal and P53-Mutated Human Lymphoblastoid Cells. Mutation. Res., 374 (1): 89-98.
|
|
(17)
|
Honma, M., Momose, M., Tanabe, H., Sakamoto, H., Yu, Y., Little, J.B., Sofuni, T. and Hayashi, M. (2000). Requirement of Wild-Type P53 Protein for Maintenance of Chromosomal Integrity. Mol. Carcinogen., 28 (4): 203-14.
|
|
(18)
|
Amundson S.A. and Liber H.L. (1992). A Comparison of Induced Mutation at Homologous Alleles of the TK Locus in Human Cells. II. Molecular Analysis of Mutants. Mutation Res., 267 (1): 89-95.
|
|
(19)
|
Schisler M.R., Moore M.M. and Gollapudi B.B. (2013). In Vitro Mouse Lymphoma (L5178Y TK+/- -3.7.2C) Forward Mutation Assay. In Protocols in Genotoxicity Assessment A. Dhawan and M. Bajpayee (Eds.), Springer Protocols, Humana Press: 27-50.
|
|
(20)
|
Long L.H., Kirkland D., Whitwell J. and Halliwell B. (2007). Different Cytotoxic and Clastogenic Effects of Epigallocatechin Gallate in Various Cell-Culture Media Due to Variable Rates of its Oxidation in the Culture Medium, Mutation Res., 634 (1-2): 177-183.
|
|
(21)
|
Nesslany, F., Simar-Meintieres, S., Watzinger, M., Talahari, I. and Marzin, D. (2008). Characterization of the Genotoxicity of Nitrilotriacetic Acid. Environ. Mol. Mutagen., 49 (6): 439-452.
|
|
(22)
|
Brusick D. (1986). Genotoxic Effects in Cultured Mammalian Cells Produced by Low pH Treatment Conditions and Increased Ion Concentrations. Environ. Mutagen., 8 (6): 879-886.
|
|
(23)
|
Morita, T., Nagaki, T., Fukuda, I. and Okumura, K., (1992). Clastogenicity of Low pH to Various Cultured Mammalian Cells. Mutation Res., 268 (2): 297-305.
|
|
(24)
|
Scott, D., Galloway, S.M., Marshall, R.R., Ishidate, M.Jr, Brusick, D., Ashby, J. and Myhr, B.C. (1991). Genotoxicity Under Extreme Culture Conditions. A report from ICPEMC Task Group 9. Mutation Res., 257: 147-204.
|
|
(25)
|
Wang J., Heflich R.H. and Moore M.M. (2007). A Method to Distinguish Between the De Novo Induction of Thymidine Kinase Mutants and the Selection of Pre-Existing Thymidine Kinase Mutants in the Mouse Lymphoma Assay. Mutation Res., 626 (1-2): 185-190.
|
|
(26)
|
Fischer, G.A. (1958). Studies on the Culture of Leukemic Cells In Vitro. Ann. N.Y. Academy Sci., 76: 673-680.
|
|
(27)
|
Clive, D., Johnson, K.O., Spector, J.F.S., Batson, A.G. and Brown, M.M.M. (1979). Validation and Characterization of the L5178Y/TK+/– Mouse Lymphoma Mutagen Assay System. Mutation Res., 59(1): 61-108.
|
|
(28)
|
Sawyer, J., Moore, M.M., Clive, D. and Hozier, J. (1985). Cytogenetic Characterization of the L5178Y TK+/- 3.7.2C Mouse Lymphoma Cell Line, Mutation Res., 147 (5): 243-253.
|
|
(29)
|
Sawyer J.R., Moore M.M. and Hozier J.C. (1989). High-Resolution Cytogenetic Characterization of the L5178Y TK+/- Mouse Lymphoma Cell Line, Mutation Res., 214 (2): 181-193.
|
|
(30)
|
Sawyer, J.R., Binz, R.L., Wang, J. and Moore, M.M. (2006). Multicolor Spectral Karyotyping of the L5178Y TK+/--3.7.2C Mouse Lymphoma Cell Line, Environ. Mol. Mutagen., 47 (2): 127-131.
|
|
(31)
|
Fellows, M.D., McDermott, A., Clare, K.R., Doherty, A. and Aardema, M.J. (2014). The Spectral Karyotype of L5178Y TK+/- Mouse Lymphoma Cells Clone 3.7.2C and Factors Affecting Mutant Frequency at the Thymidine Kinase (TK) Locus in the Microtitre Mouse Lymphoma Assay, Environ. Mol. Mutagen., 55 (1): 35-42.
|
|
(32)
|
Storer, R.D., Jraynak, A.R., McKelvey, T.W., Elia, M.C., Goodrow, T.L. and DeLuca, J.G. (1997). The Mouse Lymphoma L5178Y TK+/- Cell Line is Heterozygous for a Codon 170 Mutation in the P53 Tumor Suppressor Gene. Mutation. Res., 373 (2): 157-165.
|
|
(33)
|
Clark L.S., Harrington-Brock, K., Wang, J., Sargent, L., Lowry, D., Reynolds, S.H. and Moore, M.M. (2004). Loss of P53 Heterozygosity is not Responsible for the Small Colony Thymidine Kinase Mutant Phenotype in L5178Y Mouse Lymphoma Cells. Mutagen., 19 (4): 263-268.
|
|
(34)
|
Skopek T.R., Liber, H.L., Penman, B.W. and Thilly, W.G. (1978). Isolation of a Human Lymphoblastoid Line Heterozygous at the Thymidine Kinase Locus: Possibility for a Rapid Human Cell Mutation Assay. Biochem. Biophys. Res. Commun., 84 (2): 411-416.
|
|
(35)
|
Honma M. (2005). Generation of Loss of Heterozygosity and its Dependency on P53 Status in Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 45, 2-3. 162-176.
|
|
(36)
|
Xia, F., Wang, X., Wang, Y.H., Tsang, N.M., Yandell, D.W., Kelsey, K.T. and Liber, H.L. (1995). Altered P53 Status Correlates with Differences in Sensitivity to Radiation-Induced Mutation and Apoptosis in Two Closely Related Human Lymphoblast Lines. Cancer. Res., 55 (1): 12-15.
|
|
(37)
|
Lorge, E., M. Moore, J. Clements, M. O Donovan, M. Honma, A. Kohara, J. van Benthem, S. Galloway, M.J. Armstrong, V. Thybaud, B. Gollapudi, M. Aardema, J. Kim, A. Sutter, D.J. Kirkland (2015). Standardized Cell Sources and Recommendations for Good Cell Culture Practices in Genotoxicity Testing. (Manuscript in preparation).
|
|
(38)
|
Lloyd M. and Kidd D. (2012). The Mouse Lymphoma Assay. Springer Protocols: Methods in Molecular Biology 817, Genetic Toxicology Principles and Methods, ed. Parry and Parry, Humana Press. ISBN, 978-1-61779-420-9, 35-54.
|
|
(39)
|
Mei N., Guo X. and Moore M.M. (2014). Methods for Using the Mouse Lymphoma Assay to Screen for Chemical Mutagenicity and Photo-Mutagenicity. In: Optimization in Drug Discover: In Vitro Methods: Yan Z and Caldwell(Eds) , 2nd Edition, GW; Humana Press, Totowa, NJ.
|
|
(40)
|
Liber H.L. and Thilly W.G. (1982). Mutation Assay at the Thymidine Kinase Locus in Diploidhuman Lymphoblasts. Mutation Res., 94 (2): 467-485.
|
|
(41)
|
Coecke, S., Balls, M., Bowe, G., Davis, J., Gstraunthaler, G., Hartung, T., Hay, R., Merten, OW., Price, A., Schechtman, L., Stacey, G. and Stokes, W. (2005). Guidance on Good Cell Culture Practice. A Report of the Second ECVAM Task Force on Good Cell Culture Practice. ATLA, 33 (3): 261-287.
|
|
(42)
|
Moore M.M. and Howard B.E. (1982). Quantitation of Small Colony Trifluorothymidine-Resistant Mutants of L5178Y/TK+/- Mouse Lymphoma Cells in RPMI-1640 Medium, Mutation Res., 104 (4-5): 287-294.
|
|
(43)
|
Ames B.N., McCann J. and Yamasaki E. (1975). Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian-Microsome Mutagenicity Test. Mutation Res., 31 (6): 347-364.
|
|
(44)
|
Maron D.M. and Ames B.N. (1983). Revised Methods for the Salmonella Mutagenicity Test. Mutation Res., 113 (3-4): 173-215.
|
|
(45)
|
Natarajan, A.T., Tates, A.D, Van Buul, P.P.W., Meijers, M. and De Vogel, N. (1976). Cytogenetic Effects of Mutagens/Carcinogens After Activation in a Microsomal System In Vitro, I. Induction of Chromosomal Aberrations and Sister Chromatid Exchanges by Diethylnitrosamine (DEN) and Dimethylnitrosamine (DMN) in CHO Cells in the Presence of Rat-Liver Microsomes. Mutation Res., 37 (1): 83-90.
|
|
(46)
|
Matsuoka A., Hayashi M. and Ishidate M. Jr. (1979). Chromosomal Aberration Tests on 29 Chemicals Combined with S9 Mix In Vitro. Mutation Res., 66 (3): 277-290.
|
|
(47)
|
Ong T.M., et al. (1980). Differential Effects of Cytochrome P450-Inducers on Promutagen Activation Capabilities and Enzymatic Activities of S-9 from Rat Liver, J. Environ. Pathol. Toxicol., 4 (1): 55-65
|
|
(48)
|
Elliott, B.M., Combes, R.D., Elcombe, C.R., Gatehouse, D.G., Gibson, G.G., Mackay, J.M. and Wolf, R.C. (1992). Report of UK Environmental Mutagen Society Working Party. Alternatives to Aroclor 1254-Induced S9 in In Vitro Genotoxicity Assays. Mutagen., 7 (3): 175-177.
|
|
(49)
|
Matsushima, T., Sawamura, M., Hara, K. and Sugimura, T. (1976). A Safe Substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems. In: In Vitro Metabolic Activation in Mutagenesis Testing. de Serres F.J., et al. (Eds, Elsevier, North-Holland, pp. 85-88.
|
|
(50)
|
Galloway S.M., et al. (1994). Report from Working Group on In Vitro Tests for Chromosomal Aberrations. Mutation Res., 312 (3): 241-261.
|
|
(51)
|
Johnson T.E., Umbenhauer D.R. and Galloway S.M. (1996). Human Liver S-9 Metabolic Activation: Proficiency in Cytogenetic Assays and Comparison with Phenobarbital/Beta-Naphthoflavone or Aroclor 1254 Induced Rat S-9, Environ. Mol. Mutagen., 28 (1): 51-59.
|
|
(52)
|
UNEP (2001). Stockholm Convention on Persistent Organic Pollutants, United Nations Environment Programme (UNEP).
|
|
(53)
|
Krahn D.F., Barsky F.C. and McCooey K.T. (1982). CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids. In: Genotoxic Effects of Airborne Agents Tice R.R., Costa D.L.and Schaich K.M. (Eds.). New York, Plenum, pp. 91–103.
|
|
(54)
|
Zamora, P.O., Benson, J.M., Li, A.P. and Brooks, A.L. (1983). Evaluation of an Exposure System Using Cells Grown on Collagen Gels for Detecting Highly Volatile Mutagens in the CHO/HGPRT Mutation Assay. Environ. Mutagen., 5 (6): 795-801.
|
|
(55)
|
Asakura M., Sasaki T., Sugiyama T., Arito H., Fukushima, S. and Matsushima, T. (2008). An Improved System for Exposure of Cultured Mammalian Cells to Gaseous Compounds in the Chromosomal Aberration Assay. Mutation Res., 652 (2): 122-130.
|
|
(56)
|
Arlett C.F., et al. (1989). Mammalian Cell Gene Mutation Assays Based upon Colony Formation. V: Statistical Evaluation of Mutagenicity Test Data, Kirkland, D.J. (Ed.), CambridgeUniversity Press, pp. 66-101.
|
|
(57)
|
Morita T., Honma M. and Morikawa K. (2012). Effect of Reducing the Top Concentration Used in the In Vitro Chromosomal Aberration Test in CHL Cells on the Evaluation of Industrial Chemical Genotoxicity. Mutation Res., 741 (1-2): 32-56.
|
|
(58)
|
Brookmire L., Chen J.J. and Levy D.D. (2013). Evaluation of the Highest Concentrations Used in the In Vitro Chromosome Aberrations Assay. Environ. Mol. Mutagen., 54 (1): s. 36-43.
|
|
(59)
|
USFDA (2012). International Conference on Harmonisation (ICH) Guidance S2 (R1) on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended For Human Use. Dostupné na webových stránkách: [https://www.federalregister.gov/a/2012-13774].
|
|
(60)
|
Honma M. and Hayashi M. (2011). Comparison of In Vitro Micronucleus and Gene Mutation Assay Results for P53-Competent Versus P53-Deficient Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 52 (5): 373-384.
|
|
(61)
|
Moore-Brown, M.M., Clive, D., Howard, B.E., Batson, A.G. and Johnson, K.O. (1981). The Utilization of Trifluorothymidine (TFT) to Select for Thymidine Kinase-Deficient (TK-/-) Mutants from L5178Y/TK+/- Mouse Lymphoma Cells, Mutation Res., 85 (5): 363-378.
|
|
(62)
|
Liber H.L., Yandell D.W. and Little J.B. (1989). A Comparison of Mutation Induction at the TK and HRPT Loci in Human Lymphoblastoid Cells; Quantitative Differences are Due to an Additional Class of Mutations at the Autosomal TK locus. Mutation Res., 216 (1): 9-17.
|
|
(63)
|
Furth E.E., Thilly, W.G., Penman, B.W., Liber, H.L. and Rand, W.M. (1981). Quantitative Assay for Mutation in Diploid Human Lymphoblasts Using Microtiter Plates. Chem. Biochem., 110 (1): 1-8.
|
|
(64)
|
Hayashi, M, Dearfield, K., Kasper, P., Lovell, D., Martus, H. J. and Thybaud, V. (2011). Compilation and Use of Genetic Toxicity Historical Control Data, Mutation Res., 723 (2): 87-90.
|
|
(65)
|
Ryan T.P. (2000). Statistical Methods for Quality Improvement. John Wiley and Sons, New York 2nd Edition.
|
|
(66)
|
OECD (2014). Statistical analysis supporting the revision of the genotoxicity Test Guidelines. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 199.), Organisation for Economic Cooperation and Development, Paris.
|
|
(67)
|
Fleiss J.L., Levin B. and Paik M.C. (2003). Statistical Methods for Rates and Proportions, Third Edition, New York: John Wiley & Sons.
|
Dodatek 1
DEFINICE
Aneugen
: Jakákoli chemická látka nebo proces, který v interakci se složkami mitotického a meiotického cyklu buněčného dělení vede k aneuploidii v buňkách nebo organismech.
Aneuploidie
: Jakákoli odchylka od normálního diploidního (nebo haploidního) počtu chromozomů o jeden nebo více chromozomů, avšak nikoli o celou sadu (nebo více sad) chromozomů (polyploidie).
Mutageny substituce párů bází
: Chemické látky, které způsobují substituci jednoho nebo více párů bází v DNA.
Chemická látka
: Látka nebo směs.
Účinnost klonování
: Procento buněk kultivovaných v nízké hustotě, které jsou schopny se rozrůst do kolonií, jež mohou být spočítány.
Klastogen
: Jakákoli chemická látka nebo proces, které způsobují strukturní chromozomové aberace v populacích buněk nebo organismů.
Cytotoxicita
: U zkoušek prováděných podle této zkušební metody se cytotoxicita vyjadřuje jako snížení relativního celkového růstu (RTG) nebo relativního přežití (RS) v testu MLA, respektive TK6.
Přímá mutace
: Genová mutace výchozího typu mutované formy, která způsobuje změnu nebo ztrátu enzymatické aktivity nebo funkce kódovaných bílkovin.
Posunové mutageny
: Chemické látky, které způsobují adici nebo deleci jednoho nebo více párů bází v molekule DNA.
Genotoxický
: Obecný termín zahrnující všechny typy poškození DNA nebo chromozomů včetně jejich rozlámání, přestavby aduktů, mutací, chromozomálních aberací a aneuploidie. Ne všechny druhy genotoxických účinků způsobují mutace nebo stálé poškození chromozomů.
Mitotická rekombinace
: Rekombinace mezi homologními chromatidami v průběhu mitózy, v jejímž důsledku může dojít k indukci dvojných zlomů vláken DNA nebo ke ztrátě heterozygotnosti.
Mutagenní
: Produkující dědičné změny sekvence (sekvencí) párů bází DNA v genech nebo struktury chromozomů (chromozomové aberace).
Četnost mutací (MF)
: Počet pozorovaných mutantních buněk dělený počtem životaschopných buněk.
Doba exprese fenotypu
: Doba po expozici, během níž v genomu vznikne genetická změna a všechny dříve existující genové produkty vymizí do té míry, že se změní fenotypová vlastnost.
Relativní přežití
: Relativní přežití se u TK6 používá jako míra cytotoxicity související s expozicí. Je to relativní účinnost klonování (CE) buněk umístěných na destičky okamžitě po jejich expozici, upravená podle případné ztráty buněk v průběhu expozice v porovnání s účinností klonování u negativní kontroly.
Relativní růst v suspenzi (RSG)
: V testu MLA se relativní celkový dvoudenní růst zkoušené kultury v suspenzi porovná s celkovým dvoudenním růstem negativní kontroly/kontroly s rozpouštědlem v suspenzi (Clive a Spector, 1975). RSG by měl zahrnovat relativní růst zkoušené kultury v porovnání s negativní kontrolou/kontrolou s rozpouštědlem v průběhu expozice.
Relativní celkový růst (RTG)
: Relativní celkový růst se u MLA používá jako míra cytotoxicity související s expozicí. Je to míra relativního (ke kontrole s vehikulem) růstu zkoušených kultur v průběhu expozice, ve fázi dvoudenní exprese a ve fázi selekce mutantních klonů. RSG každé zkoušené kultury se vynásobí relativní účinností klonování zkoušené kultury v době selekce mutantů a vyjadřuje se ve vztahu k účinnosti klonování negativní kontroly/kontroly s rozpouštědlem (Clive a Spector, 1975).
Jaterní frakce S9
: Supernatant homogenátu jater po odstředění při 9 000g, tj. surový jaterní extrakt
Směs S9
: Směs jaterní frakce S9 a kofaktorů nezbytných pro metabolickou aktivaci enzymů.
Růst v suspenzi (SG)
: Násobné zvýšení počtu buněk v průběhu expozice a fáze exprese v testu MLA. SG se vypočítá vynásobením násobného zvýšení v den 1 násobným zvýšením v den 2 u krátkodobé (3 nebo 4 hodiny) expozice. Jestliže se použije 24hodinová expozice, je SG násobné zvýšení v průběhu 24hodinové expozice vynásobené násobným zvýšením v dny exprese 1 a 2.
Kontrola s rozpouštědlem
: Obecný termín k označení kontrolních kultur, ke kterým se přidává pouze rozpouštědlo používané k rozpuštění zkoušené chemické látky.
Zkoušená chemická látka
: Jakákoli látka nebo směs zkoušená touto zkušební metodou.
Neexponované kontroly
: Neexponované/neošetřené kontroly jsou kultury, které se neexponují (tj. ani chemické látce, ani rozpouštědlu), ale zpracovávají se stejným způsobem jako kultury, k nimž se přidává zkoušená chemická látka.
Dodatek 2
VZORCE
CYTOTOXICITA
Pro obě verze testu MLA (s použitím agaru i mikrotitračních destiček)
Cytotoxicita je definována jako relativní celkový růst (RTG), který zahrnuje relativní růst v suspenzi (RSG) v průběhu dvoudenní exprese a relativní účinnost klonování (RCE) dosaženou v době selekce mutantů. RTG, RSG a RCE jsou vyjádřeny v procentech.
Výpočet RSG: Růst v suspenzi jedna (SG1) je rychlost růstu mezi dnem 0 a dnem 1 (koncentrace buněk v den 1 / koncentrace buněk v den 0) a růst v suspenzi dva (SG2) je rychlost růstu mezi dnem 1 a dnem 2 (koncentrace buněk v den 2 / koncentrace buněk v den 1). RSG je celkový růst SG (SG1 x SG2) pro exponovanou kulturu v porovnání s neexponovanou kulturou / kulturou s rozpouštědlem. To znamená: RSG = [SG1(test) x SG2(test)] / [SG1(control) x SG2(control)] SG1 se vypočítá z počáteční koncentrace buněk použité na začátku expozice buněk. Zahrnuje to případnou diferenciální cytotoxicitu, k níž dochází ve zkoušené kultuře (kulturách) v průběhu expozice buněk.
RCE je relativní účinnost klonování zkoušené kultury v porovnání s relativní účinností klonování neexponované kontroly / kontroly s rozpouštědlem zjištěné v době selekce mutantů.
Relativní celkový růst (RTG):
RTG = RSG x RCE
TK6
RELATIVNÍ PŘEŽITÍ (RS):
Cytotoxicita se hodnotí podle relativního přežití, tj. účinnosti klonování (CE) buněk umístěných na destičky okamžitě po expozici, upravené podle případné ztráty buněk v průběhu expozice v porovnání s účinností klonování u negativních kontrol (s přiřazeným přežitím 100 %). Korekci ztráty buněk v průběhu expozice lze vypočítat jako:
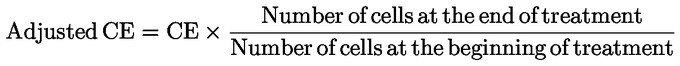
Relativní přežití u kultury exponované zkoušené chemické látce se vypočítá jako:
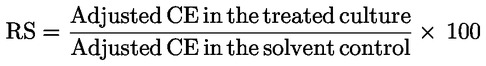
ČETNOST MUTACÍ PRO MLA I TK6
Četnost mutací (MF) je účinnost klonování kolonií mutací v selektivním médiu (CEM) upraveno podle účinnosti klonování v neselektivním médiu v době selekce mutací (CEV). To znamená MF = CEM / CEV. Výpočet těchto dvou účinností klonování je popsán níže pro metodu klonování s použitím agaru a metodu s použitím mikrodestiček.
Verze testu MLA s použitím agaru:
Ve verzi testu MLA s použitím měkkého agaru se počet kolonií na destičce pro selekci mutantů (CM) a počet kolonií na neselektované destičce nebo destičce pro účinnost klonování (životaschopný počet) (CV) zjistí přímým spočítáním klonů. Když se umístí 600 buněk pro účinnost klonování (CE) na destičky pro selekci mutantů (CEM) a neselektované destičky nebo destičky pro účinnost klonování (životaschopný počet) (CEV) a 3 x 106 buněk se použije pro selekci mutantů,
CEM = CM / (3 x 106) = (CM / 3) x 10-6
CEV = CV / 600
Verze testu MLA a TK6 s použitím mikrodestiček:
Ve verzi testu MLA s použitím mikrodestiček se hodnoty CM a CV určí jako součin celkového počtu mikrodestiček (TW) a pravděpodobného počtu kolonií na jamku (P) na mikrotitračních destičkách.
CM = PM x TWM
CV = PV x TWV
Od nulového výrazu Poissonova rozdělení (Furth et al., 1981) je P dáno vztahem
P = – ln (EW / TW)
kde EW jsou prázdné jamky a TW je celkový počet jamek. Proto,
CEM = CM / TM = (PM x TWM) / TM
CEV = CV / TV = (PV x TWV) / TV
Ve verzi testu MLA s použitím mikrodestiček se četnosti mutací tvořících malé a velké kolonie vypočítají stejným způsobem s použitím příslušného počtu prázdných jamek pro malé a velké kolonie.
V testu TK6 vycházejí četnosti mutací tvořících malé a velké kolonie z časně a pozdně se objevujících mutantů.
B.68 ZKUŠEBNÍ METODA VYUŽÍVAJÍCÍ KRÁTKODOBOU EXPOZICI IN VITRO PRO IDENTIFIKACI i) CHEMICKÝCH LÁTEK VYVOLÁVAJÍCÍCH VÁŽNÉ POŠKOZENÍ OČÍ A ii) CHEMICKÝCH LÁTEK, KTERÉ NENÍ NUTNÉ KLASIFIKOVAT JAKO LÁTKY MAJÍCÍ DRÁŽDIVÉ ÚČINKY NA OČI NEBO VYVOLÁVAJÍCÍ VÁŽNÉ POŠKOZENÍ OČÍ
ÚVOD
|
1.
|
Tato zkušební metoda je rovnocenná Pokynu OECD pro zkoušení (TG) č. 491 (2017). Zkušební metoda využívající krátkodobou expozici (STE) je metoda in vitro, kterou lze použít za určitých okolností a se specifickými omezeními pro klasifikaci a označování nebezpečnosti chemických látek (látek a směsí), které vyvolávají vážné poškození očí, i chemických látek, které není nutné klasifikovat jako látky vyvolávající vážné poškození očí nebo mající dráždivé účinky na oči, jak je definuje Globálně harmonizovaný systém klasifikace a označování chemických látek (GHS) Organizace spojených národů (OSN) (1) a nařízení Evropské unie (EU) č. 1272/2008 o klasifikaci, označování a balení látek a směsí (nařízení CLP) (67).
|
|
2.
|
Po řadu let byl potenciál nebezpečnosti chemických látek pro oči posuzován primárně pomocí oční zkoušky in vivo na králících (zkušební metoda B.5 (8), ekvivalentní zkušební metodice OECD č. 405). Obecně se uznává, že v dohledné budoucnosti nebude moci žádný alternativní test in vitro plně nahradit oční zkoušku na králících in vivo, pokud jde o předpověď celé škály závažného poškození očí / dráždivosti u různých tříd chemických látek. Oční zkouška na králících by však mohla být nahrazena strategickou kombinací alternativních zkušebních metod použitých v rámci strategie (odstupňovaného) zkoušení (2). Pro zkoušení chemických látek, u nichž lze na základě stávajících informací očekávat, že budou mít vysoký potenciál dráždivosti nebo že budou vyvolávat závažné poškození očí, je navržen přístup shora dolů. Naopak přístup zdola nahoru je navržen pro zkoušení chemických látek, u nichž lze na základě stávajících informací očekávat, že nebudou působit takové podráždění očí, aby to vyžadovalo klasifikaci. Ačkoli není zkušební metoda STE považována za úplnou náhradu očního testu in vivo na králících, je vhodná pro použití jako součást odstupňované strategie zkoušení pro klasifikaci a označování pro regulační účely, například v rámci přístupu shora dolů / zdola nahoru, aby bylo možné určit bez dalšího zkoušení i) chemické látky vyvolávající závažné poškození očí (kategorie 1 podle GHS OSN/CLP) a ii) chemické látky (s výjimkou vysoce těkavých látek a všech pevných chemických látek jiných než povrchově aktivních látek), které nevyžadují klasifikaci jako látky dráždivé pro oči nebo vyvolávající vážné poškození očí (kategorie 2 podle GHS OSN/CLP) (1)(2). U chemické látky, o níž se na základě použití zkušební metody STE nepředpokládá, že vyvolává vážné poškození očí (kategorie 1 podle GHS OSN/CLP) ani není klasifikována jako „bez kategorie podle GHS OSN/CLP“ (nevyvolává vážné poškození očí ani podráždění očí), by však pro stanovení konečné klasifikace bylo nutné další zkoušení. Použití zkušební metody STE v rámci postupu „zdola nahoru“ podle jiných klasifikačních systémů než GHS OSN/CLP by mělo být předem konzultováno s příslušnými regulačními orgány. Výběr nejvhodnější zkušební metody a její použití je třeba posuzovat v souvislosti s Pokynem OECD k integrovanému přístupu ke zkoušení a posuzování vážného poškození očí a podráždění očí (14).
|
|
3.
|
Účelem této zkušební metody je popsat postupy používané k hodnocení potenciálních nebezpečných účinků zkoušené chemické látky na oči podle její schopností vyvolávat toxicitu při použití zkušební metody s krátkodobou expozicí. Cytotoxický účinek chemických látek na epitelové buňky rohovky je důležitým způsobem účinku (MOA) vedoucím k poškození epitelu rohovky a podráždění očí. Životaschopnost buněk se ve zkušební metodě STE posuzuje kvantitativním měřením po extrakci soli modrého formazanu z buněk produkovaného živými buňkami enzymatickou konverzí vitálního barviva MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-difenyltetrazolium-bromid), které se také nazývá bromthiazolová modř (3). Zjištěná životaschopnost buněk se porovná s kontrolou s rozpouštědlem (relativní životaschopnost) a použije se na odhad potenciální nebezpečnosti zkoušené chemické látky pro oči. Zkoušená chemická látka je klasifikována jako kategorie 1 podle systému GHS OSN/CLP, když je životaschopnost buněk při koncentraci 5 % i 0,05 % menší nebo rovna (≤) 70 %. Naopak, u zkoušené chemické látky lze předpokládat klasifikaci bez kategorie podle systému GHS OSN/CLP, když je životaschopnost buněk při koncentraci 5 % i 0,05 % větší než (>) 70 %.
|
|
4.
|
Termín „zkoušená chemická látka“ se v této zkušební metodě používá k označení toho, co se zkouší, a netýká se použitelnosti zkušební metody STE ke zkoušení látek a/nebo směsí. Definice jsou uvedeny v dodatku.
|
VÝCHOZÍ ÚVAHY A OMEZENÍ
|
5.
|
Tato zkušební metoda je založena na protokolu, který vyvinula společnost Kao Corporation (4) a který byl validován ve dvou různých studiích: jednu studii provedl validační výbor Japonské společnosti pro alternativy experimentů na zvířatech (JSAAE) (5) a druhou japonské středisko pro validaci alternativních metod (JaCVAM) (6). Vzájemné hodnocení provedl NICEATM/ICCVAM na základě zpráv o validační studii a všeobecných dokumentů o kontrole této zkušební metody (7).
|
|
6.
|
Když byla zkušební metoda STE použita pro zjišťování chemických látek (látek a směsí) vyvolávajících vážné poškození očí (kategorie 1 GHS OSN/CLP (1), vykázala data zjištěná u 125 chemických látek (včetně látek i směsí) celkovou přesnost 83 % (104/125), míru falešné pozitivity 1 % (1/86) a míru falešné negativity 51 % (20/39) ve srovnání se zkouškou in vivo na očích králíků (7). Zjištěná míra falešné negativity není v této souvislosti kriticky důležitá, protože všechny zkoušené chemické látky, které indukují životaschopnost buněk ≤ 70 % při 5 % koncentraci a > 70 % při 0,05 % koncentraci by byly následně zkoušeny dalšími náležitě validovanými zkouškami in vitro nebo jako poslední možnost oční zkouškou in vivo na králících, v závislosti na požadavcích právních předpisů a v souladu se strategií postupného zkoušení a podle analýzy průkaznosti výsledků, která je v současnosti doporučena (1)(8). Zkoušeny byly hlavně jednosložkové látky, ačkoli určité omezené množství údajů existuje i o zkoušení směsí. Zkušební metoda je nicméně technicky použitelná i na zkoušení vícesložkových látek a směsí. Před použitím této zkušební metody ke zkoušení směsi za účelem získání údajů pro zamýšlené použití v právních předpisech by však mělo být zváženo, zda tato metoda může pro tento účel poskytnout přiměřené výsledky, a pokud ano, proč je tomu tak. Takové posouzení není nutné, pokud existuje právní požadavek na zkoušení dané směsi. Když byla zkušební metoda STE použita na zjištění chemických látek klasifikovaných jako kategorie 1 GHS OSN, nevykazovala žádné jiné konkrétní nedostatky. Zkoušející by mohli zvážit použití této zkušební metody na všechny zkoušené chemické látky, přičemž životaschopnost buněk ≤ 70 % při koncentraci 5 % i 0,05 % by měla být považována za odpověď indukující vážné poškození očí, které je třeba klasifikovat v kategorii 1 GHS OSN/CLP bez dalšího zkoušení.
|
|
7.
|
Když byla zkušební metoda STE použita pro zjišťování chemických látek (látek a směsí) nevyžadujících klasifikaci jako látky vyvolávající podráždění očí a závažné poškození očí (tj. bez kategorie GHS OSN/CLP), vykázala data zjištěná u 130 chemických látek (včetně látek i směsí) celkovou přesnost 85 % (110/130), míru falešné negativity 12 % (9/73) a míru falešné pozitivity 19 % (11/57) ve srovnání se zkouškou in vivo na očích králíků (7). Pokud se z datového souboru vyloučí vysoce těkavé látky a pevné látky jiné než povrchově aktivní látky, celková přesnost se zlepší na 90 % (92/102), míra falešné negativity na 2 % (1/54) a míra falešné pozitivity na 19 % (9/48) (7). Potenciálním nedostatkem zkušební metody STE použité pro zjištění zkoušených chemických látek, které nevyžadují klasifikaci jako látky vyvolávající podráždění očí a závažné poškození očí (bez kategorie GHS OSN/CLP), tedy je vysoká míra falešné negativity u i) vysoce těkavých látek s tlakem výparů nad 6 kPa a ii) pevných chemických látek (látek a směsí) jiných než povrchově aktivních látek a směsí složených pouze z povrchově aktivních látek. Tyto chemické látky jsou ze sféry použitelnosti zkušební metody STE vyloučeny (7).
|
|
8.
|
Kromě chemických látek uvedených v odstavcích 6 a 7 obsahuje datový soubor generovaný pomocí zkušební metody STE také interní údaje o 40 směsích, které při porovnání s Draizovou oční zkouškou in vivo vykázaly přesnost 88 % (35/40), míru falešné pozitivity 50 % (5/10) a míru falešné negativity 0 % (0/30) při predikci směsí, které nevyžadují klasifikaci podle klasifikačních systémů GHS OSN/CLP (9). Zkušební metoda STE tedy může být použita pro zjištění směsí klasifikovaných jako směsi bez kategorie GHS OSN/CLP při použití přístupu zdola nahoru s výjimkou pevných směsí jiných než směsí složených pouze z povrchově aktivních látek navíc k jejímu omezení u pevných látek. Dále směsi obsahující látky s tenzí par vyšší než 6 kPa by měly být hodnoceny opatrně, aby nedošlo k případnému podcenění, přičemž každý případ by měl být individuálně odůvodněn.
|
|
9.
|
Zkušební metodu STE nelze použít pro identifikaci zkoušených chemických látek klasifikovaných jako látky kategorie 2 GHS OSN/CLP nebo kategorie 2A GHS OSN (podráždění očí) nebo kategorie 2B GHS OSN (mírné podráždění očí), a to vzhledem k značnému počtu chemických látek kategorie 1 GHS OSN/CLP podhodnoceně klasifikovaných v kategorii 2, 2A nebo 2B a látek bez kategorie GHS OSN/CLP nadhodnoceně klasifikovaných v kategorii 2, 2A nebo 2B (7). Pro tento účel může být nezbytné další zkoušení s použitím jiné vhodné metody.
|
|
10.
|
Zkušební metoda STE je vhodná pro zkoušené chemické látky, které jsou rozpuštěny nebo rovnoměrně suspendovány po dobu alespoň 5 minut ve fyziologickém roztoku, 5 % dimethylsulfoxidu (DMSO) ve fyziologickém roztoku nebo v minerálním oleji. Zkušební metoda STE není vhodná pro zkoušené chemické látky, které jsou nerozpustné nebo které nelze rovnoměrně suspendovat po dobu alespoň 5 minut ve fyziologickém roztoku, 5 % DMSO ve fyziologickém roztoku nebo v minerálním oleji. Použití minerálního oleje ve zkušební metodě STE je možné vzhledem ke krátkodobé expozici. Zkušební metoda STE je tedy vhodná pro predikci potenciálu nebezpečnosti zkoušených chemických látek nerozpustných ve vodě pro oči (např. mastné alkoholy nebo ketony s dlouhým řetězcem), pokud jsou mísitelné alespoň s jedním ze tří výše uvedených navrhovaných rozpouštědel (4).
|
|
11.
|
Termín „zkoušená chemická látka“ se v této zkušební metodě používá k označení toho, co se zkouší (68), a netýká se použitelnosti zkušební metody STE ke zkoušení látek a/nebo směsí.
|
PRINCIP ZKOUŠKY
|
12.
|
Zkušební metoda STE je zkouška in vitro založená na cytotoxicitě, která se provádí na konfluentní monovrstvě buněk rohovky králíka (Statens Seruminstitut Rabbit Cornea – SIRC), které se kultivují na 96jamkové polykarbonátové mikrotitrační destičce (4). Po pětiminutové expozici zkoušené chemické látce se pomocí zkoušky MTT kvantitativně změří cytotoxicita jako relativní životaschopnost buněk SIRC (4). Snížená životaschopnost buněk se použije na predikci potenciálních nežádoucích účinků vedoucích k poškození oka.
|
|
13.
|
Bylo hlášeno, že 80 % roztoku nakapaného do oka králíka se vyloučí přes spojivkový vak do tří až čtyř minut, zatímco více než 80 % roztoku nakapaného do lidského oka se vyloučí do jedné až dvou minut (10). Zkušební metoda STE se pokouší aproximovat tyto doby expozice a využít cytotoxicitu jako sledovanou vlastnost pro posouzení rozsahu poškození buněk SIRC po pětiminutové expozici zkoušené chemické látce.
|
PROKÁZÁNÍ ZPŮSOBILOSTI
|
14.
|
Dříve, než začnou laboratoře rutinně používat zkoušku STE popsanou v této zkušební metodě, měly by prokázat svou odbornou způsobilost správným určením klasifikace jedenácti vhodných látek doporučených v tabulce 1. Tyto látky byly vybrány tak, aby představovaly celý rozsah odpovědí u závažného poškození očí nebo podráždění očí na základě výsledků oční zkoušky in vivo na králících (TG 405) a klasifikačního systému GHS OSN (1). Dalšími kritérii výběru bylo, aby látky byly komerčně dostupné, aby k nim byly k dispozici kvalitní referenční údaje in vivo a aby existovaly kvalitní údaje in vitro ze zkoušky STE (3). V situacích, kdy některá z uvedených látek není k dispozici, lze v odůvodněných případech použít jinou látku, u níž jsou k dispozici odpovídající referenční údaje o použití in vivo a in vitro, pokud budou použita stejná kritéria jako kritéria zde popsaná.
Tabulka 1
Seznam vhodných látek
|
Látka
|
CASRN
|
Třída chemických látek (69)
|
Skupenství
|
Kat. GHS OSN/CLP in vivo
(70)
|
Rozpouštědlo ve zkoušce STE
|
Kat. GHS OSN/CLP
|
|
benzalkoniumchlorid (10 %, vodný)
|
8 001 -54-5
|
oniová sloučenina
|
kapalina
|
kategorie 1
|
fyziologický roztok
|
kategorie 1
|
|
triton X-100 (100 %)
|
9 002 -93-1
|
ether
|
kapalina
|
kategorie 1
|
fyziologický roztok
|
kategorie 1
|
|
kyselá červeň 92
|
18 472 -87-2
|
heterocyklická sloučenina; sloučenina bromu; sloučenina chloru
|
pevná látka
|
kategorie 1
|
fyziologický roztok
|
kategorie 1
|
|
hydroxid sodný
|
1 310 -73-2
|
zásada; anorganická chemická látka
|
pevná látka
|
kategorie 1 (71)
|
fyziologický roztok
|
kategorie 1
|
|
butyrolakton
|
96-48-0
|
lakton; heterocyklická sloučenina
|
kapalina
|
kategorie 2A (kategorie 2 v CLP)
|
fyziologický roztok
|
nelze předpovědět
|
|
oktan-1-ol
|
111-87-5
|
alkohol
|
kapalina
|
kategorie 2A/B (72)(kategorie 2 v CLP)
|
minerální olej
|
nelze předpovědět
|
|
cyklopentanol
|
96-41-3
|
alkohol; uhlovodík, cyklický
|
kapalina
|
kategorie 2A/B (73) (kategorie 2 v CLP)
|
fyziologický roztok
|
nelze předpovědět
|
|
2-ethoxyethyl-acetát
|
111-15-9
|
alkohol; ether
|
kapalina
|
bez kategorie
|
fyziologický roztok
|
bez kategorie
|
|
dodekan
|
112-40-3
|
uhlovodík, acyklický
|
kapalina
|
bez kategorie
|
minerální olej
|
bez kategorie
|
|
methyl(isobutyl)keton
|
108-10-1
|
keton
|
kapalina
|
bez kategorie
|
minerální olej
|
bez kategorie
|
|
1,1-dimethylguanidin-sulfát
|
598-65-2
|
amidin; sloučenina síry
|
pevná látka
|
bez kategorie
|
fyziologický roztok
|
bez kategorie
|
|
Zkratky: CAS RN = registrační číslo CAS (Chemical Abstracts Service)
|
|
POSTUP
Příprava monovrstvy buněk
|
15.
|
Zkušební metoda STE se používá s buněčnou linií z rohovky králíka SIRC. Doporučuje se, aby byly buňky SIRC pořízeny z kvalifikované buněčné banky, jako je například Americká sbírka typových kultur CCL60.
|
|
16.
|
Buňky SIRC se kultivují při teplotě 37 oC ve zvlhčené atmosféře s 5 % CO2 v kultivační lahvi obsahující kultivační médium, které sestává z Eaglova minimálního esenciálního média (MEM) doplněného 10 % fetálního bovinního séra (FBS), 2 mM L-glutaminu, 50–100 jednotkami/ml penicilinu a 50–100 μg/ml streptomycinu. Buňky, které v kultivační lahvi splynuly, je třeba rozdělit pomocí roztoku trypsinu a kyseliny ethylendiamintetraoctové, s použitím nebo bez použití stěrky. Před použitím k rutinním zkouškám se buňky nechají rozmnožit (např. 2 až 3 pasáže) v kultivační láhvi, přičemž by neměly být pasážovány více než 25krát od rozmrazení.
|
|
17.
|
Buňky připravené k použití ke zkoušce STE se poté upraví na vhodnou hustotou a nasadí do 96jamkových destiček. Doporučená hustota nasazování buněk je 6,0 × 103 buněk na jamku, když jsou buňky používány čtyři dny po nasazení, nebo 3,0 × 103 buněk na jamku, když jsou buňky používány pět dnů po nasazení, v 200 μl kultivačního média. Buňky použité ke zkoušce STE, které jsou nasazeny do kultivačního média ve vhodné hustotě, dosáhnou konfluence v době zkoušení více než 80 %, tj. čtyři nebo pět dnů od nasazení.
|
Aplikace zkoušených chemických látek a kontrolních látek
|
18.
|
Rozpouštědlem první volby pro ředění nebo rozpuštění chemických látek je fyziologický roztok. Jestliže zkoušená chemická látka vykazuje nízkou rozpustnost nebo ji nelze rozpustit či rovnoměrně suspendovat po dobu alespoň pěti minut ve fyziologickém roztoku, použije se jako rozpouštědlo druhé volby 5 % DMSO (CAS#67-68-5) ve fyziologickém roztoku. Pro zkoušené chemické látky, které nelze rozpustit či rovnoměrně suspendovat po dobu alespoň pěti minut ve fyziologickém roztoku ani v 5 % DMSO ve fyziologickém roztoku, se jako rozpouštědlo třetí volby použije minerální olej (CAS#8042-47-5).
|
|
19.
|
Zkoušené chemické látky se rozpustí nebo rovnoměrně suspendují ve zvoleném rozpouštědle na koncentraci 5 % (hmotnostních) a poté se dále ředí postupným desetinásobným ředěním na koncentraci 0,5 % a 0,05 %. Každá zkoušená chemická látka se zkouší v koncentraci 5 % i 0,05 %. Buňky kultivované v 96jamkové mikrotitrační destičce se exponují roztoku (nebo suspenzi) zkoušené chemické látky v množství 200 μl/jamku v koncentraci 5 % nebo 0,05 %, a to po dobu pěti minut při pokojové teplotě. Zkoušené chemické látky (jednosložkové látky nebo vícesložkové látky či směsi) se považují za čisté látky a ředí se nebo suspendují podle této metody bez ohledu na čistotu.
|
|
20.
|
Kultivační médium popsané v odstavci 16 se používá jako kontrola s médiem na každé destičce a při každém opakování. Kromě toho se na každé destičce a při každém opakování buňky exponují také vzorkům kontroly s rozpouštědlem. Bylo potvrzeno, že rozpouštědla uvedená v odstavci 18 nemají žádné nežádoucí účinky na životaschopnost buněk SIRC.
|
|
21.
|
Ve zkušební metodě STE se na každé destičce a při každém opakování používá jako pozitivní kontrola 0,01 % laurylsíran sodný (SLS) ve fyziologickém roztoku. Aby bylo možné vypočítat životaschopnost buněk u pozitivní kontroly, musí každá destička při každém opakování zahrnovat také kontrolu s rozpouštědlem ve fyziologickém roztoku.
|
|
22.
|
Aby bylo možné stanovit kompenzaci pro optickou hustotu, je třeba provést slepou zkoušku, přičemž tato slepá zkouška se provádí na jamkách obsahujících pouze fosfátový pufr, ale žádné kalcium a magnézium (PBS-) nebo buňky.
|
|
23.
|
Každý vzorek (zkoušená chemická látka v koncentraci 5 % a 0,05 %, kontrola s médiem, kontrola s rozpouštědlem a pozitivní kontrola) je třeba zkoušet při každém opakování třikrát exponováním buněk 200 μl příslušné zkoušené nebo kontrolní látky po dobu pěti minut při pokojové teplotě.
|
|
24.
|
Srovnávací látky jsou účelné pro hodnocení potenciálu podráždění očí neznámými chemickými látkami nebo specifickými třídami chemických látek nebo produktů nebo pro hodnocení relativního potenciálu podráždění látkou s dráždivými účinky na oči ve specifickém rozmezí dráždivých reakcí.
|
Měření životaschopnosti buněk
|
25.
|
Po expozici se buňky dvakrát promyjí 200 μl PBS a přidá se 200 μl roztoku MTT (0,5 mg MTT/ml kultivačního média). Po dvouhodinové reakční době v inkubátoru (37 °C, 5 % CO2) se roztok MTT dekantuje, formazan MTT se extrahuje přidáním 200 μl 0,04 N kyseliny chlorovodíkové v isopropanolu po dobu 60 minut v temnu při pokojové teplotě a pomocí čtečky destiček se změří absorbance roztoku formazanu MTT při 570 nm. K interferenci zkoušených chemických látek s analýzou MTT (barvivy nebo přímými redukčními činidly MTT) dochází pouze, když se ve zkušebním systému ponechá po oplachu po expozici významné množství zkoušené chemické látky, což platí pro 3D tkáně rekonstruované lidské rohovky nebo rekonstruované lidské epidermis, ale neplatí pro 2D buněčné kultury používané pro zkušební metodu STE.
|
Interpretace výsledků a predikční model
|
26.
|
Poté se použijí hodnoty optické hustoty (OD) dosažené pro každou zkoušenou chemickou látku k výpočtu relativní životaschopnosti buněk vzhledem ke kontrole s rozpouštědlem, která je stanovena na úroveň 100 %. Relativní životaschopnost buněk se vyjadřuje jako procento a zjistí se vydělením optické hustoty zkoušené chemické látky optickou hustotou kontroly s rozpouštědlem po odečtení optické hustoty slepého pokusu od obou hodnot.
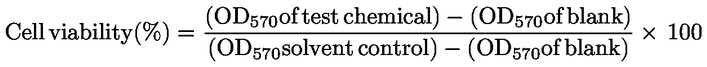
Podobně se relativní životaschopnost buněk každé kontroly s rozpouštědlem vyjadřuje jako procento a zjistí se vydělením optické hustoty každé kontroly s rozpouštědlem optickou hustotou kontroly s médiem po odečtení optické hustoty slepého pokusu od obou hodnot.
|
|
27.
|
Měla by být provedena tři nezávislá opakování, přičemž každé zahrnuje tři replikované jamky (tj. n = 9). Aritmetický průměr tří jamek pro každou zkoušenou chemickou látku a kontrolu s rozpouštědlem při každém nezávislém opakování se použije pro výpočet aritmetického průměru relativní životaschopnosti buněk. Ze tří nezávislých opakování se vypočítá konečný aritmetický průměr životaschopnosti buněk.
|
|
28.
|
Dále jsou uvedeny hraniční hodnoty životaschopnosti buněk pro identifikaci zkoušených chemických látek vyvolávajících vážné poškození očí (kategorie 1 GHS OSN/CLP) a zkoušených chemických látek, které není nutné klasifikovat jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí (bez kategorie GHS OSN/CLP).
|
Tabulka 2
Prediktivní model zkušební metody STE
|
Životaschopnost buněk
|
Klasifikace GHS OSN/CLP
|
Použitelnost
|
|
při 5 %
|
při 0,05 %
|
|
> 70 %
|
> 70 %
|
bez kategorie
|
látky a směsi, s výjimkou: i) vysoce těkavých látek s tlakem výparů nad 6 kPa (74) a ii) pevných chemických látek (látek a směsí) jiných než povrchově aktivních látek a směsí složených pouze z povrchově aktivních látek
|
|
≤ 70 %
|
> 70 %
|
nelze předpovědět
|
nepoužije se
|
|
≤ 70 %
|
≤ 70 %
|
kategorie 1
|
látky a směsi (75)
|
Kritéria přijatelnosti
|
29.
|
Výsledky zkoušky se považují za přijatelné, když jsou splněna všechna následující kritéria:
|
a)
|
Optická hustota kontroly s médiem (exponované kultivačnímu médiu) by měla být po odečtení optické hustoty slepého pokusu 0,3 nebo vyšší.
|
|
b)
|
Životaschopnost kontroly s rozpouštědlem by měla být 80 % nebo vyšší ve vztahu ke kontrole s médiem. Pokud se při každém opakování používají kontroly s více rozpouštědly, každá z těchto kontrol by měla vykazovat životaschopnost buněk větší než 80 %, aby byly vhodné pro chemické látky zkoušené těmito rozpouštědly.
|
|
c)
|
Životaschopnost buněk zjištěná při pozitivní kontrole (0,01 % SLS) by měla být mezi dvěma směrodatnými odchylkami od historické střední hodnoty. Horní a dolní mez přijatelnosti pro pozitivní kontrolu je třeba často aktualizovat, tj. každé tři měsíce nebo pokaždé, když se přijatelná zkouška vykonává v laboratořích, kde se zkoušky nedělají často (tj. méně než jednou měsíčně). Pokud laboratoř neprovedla dostatečný počet experimentů, aby mohla stanovit statisticky robustní distribuci pozitivních kontrol, je přijatelné, aby byla použita horní a dolní mez přijatelnosti stanovená subjektem, který metodu vyvinul, tj. mezi 21,1 % a 62,3 % podle historických dat laboratoře, přičemž v průběhu prvních rutinních zkoušek se stanoví interní distribuce.
|
|
d)
|
Směrodatná odchylka konečné životaschopnosti buněk odvozená ze tří nezávislých opakování by měla být menší než 15 % pro koncentraci 5 % i 0,05 % zkoušené chemické látky.
Není-li jedno nebo několik těchto kritérií splněno, měly by se výsledky zlikvidovat a provést další tři nezávislá opakování.
|
|
ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV
Údaje
|
30.
|
Vykazují se údaje za každou jednotlivou jamku (např. hodnoty životaschopnosti buněk) z každého opakování, jakož i celková střední hodnota, směrodatná odchylka a klasifikace.
|
ZÁVĚREČNÁ ZPRÁVA
|
31.
|
Závěrečná zpráva by měla obsahovat tyto informace:
|
|
Zkoušené chemické látky a látky pro kontrolu
|
—
|
Jednosložková látka: chemická identifikace, např. název (názvy) IUPAC nebo CAS, registrační číslo (čísla) CAS, kód SMILES nebo InChI, strukturní vzorec a/nebo jiné identifikátory,
|
|
—
|
vícesložková látka, UVCB a směs: charakterizovaná v co možná největší míře např. chemickou identitou (viz výše), čistotou, kvantitativním výskytem a důležitými fyzikálně-chemickými vlastnostmi (viz výše) složek, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
—
|
skupenství, těkavost, pH, logP, molekulová hmotnost, chemická třída a další důležité fyzikálně-chemické vlastnosti podstatné pro provedení studie, a to v rozsahu, v jakém jsou tyto informace dostupné,
|
|
—
|
čistota, případně chemická identita nečistot, je-li to prakticky proveditelné atd.,
|
|
—
|
případné ošetření před zkoušením (např. zahřívání, mletí),
|
|
—
|
podmínky skladování a stabilita, v rozsahu, v jakém jsou tyto informace dostupné.
|
|
|
|
Podmínky a postupy zkušební metody
|
—
|
Jméno a adresa zadavatele, zkušebního zařízení a vedoucího studie,
|
|
—
|
popis použité zkušební metody,
|
|
—
|
použitá buněčná linie, její zdroj, počet pasáží a konfluence buněk použitých ke zkoušení,
|
|
—
|
podrobnosti o použitém zkušebním postupu,
|
|
—
|
počet opakování a použitých replik,
|
|
—
|
koncentrace zkoušených chemických látek (pokud se liší od doporučených),
|
|
—
|
zdůvodnění volby rozpouštědla pro každou zkoušenou chemickou látku,
|
|
—
|
délka expozice zkoušené chemické látce (pokud se liší od doporučené),
|
|
—
|
popis všech úprav zkušebního postupu,
|
|
—
|
popis použitých hodnotících kritérií a kritérií rozhodování,
|
|
—
|
odkaz na dosavadní střední hodnoty pozitivních kontrol a směrodatnou odchylku (SD):
|
|
—
|
prokázání způsobilosti laboratoře k provádění této zkušební metody (např. zkoušením vhodných látek) nebo prokázání reprodukovatelného provedení této zkušební metody v průběhu doby.
|
|
|
|
Výsledky
|
—
|
Pro každou zkoušenou chemickou látku a kontrolní látku a pro každou zkoušenou koncentraci je třeba v tabulkové formě uvést jednotlivé hodnoty optické hustoty každé replikované jamky, aritmetický průměr hodnot optické hustoty pro každé nezávislé opakování, % životaschopnosti buněk pro každé nezávislé opakování a konečný aritmetický průměr % životaschopnosti buněk a směrodatnou odchylku ze tří opakování,
|
|
—
|
výsledky z kontroly s médiem, kontroly s rozpouštědlem a pozitivní kontroly prokazující vhodná kritéria přijatelnosti studie,
|
|
—
|
popis ostatních pozorovaných účinků,
|
|
—
|
celková odvozená klasifikace s odkazem na použitý predikční model / kritéria rozhodování.
|
|
|
LITERATURA
|
(1)
|
United Nations UN (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Fifth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. Dostupné na webových stránkách: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(2)
|
Scott L, et al. (2010). A proposed Eye Irritation Testing Strategy to Reduce and Replace in vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. Chem. In Vitro 24, 1-9.
|
|
(3)
|
Mosmann T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to 7 Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(4)
|
Takahashi Y, et al. (2008). Development of the Short Time Exposure (STE) Test: an In Vitro Eye Irritation Test Using SIRC Cells. Toxicol. Chem. In Vitro 22,760-770.
|
|
(5)
|
Sakaguchi H, et al. (2011). Validation Study of the Short Time Exposure (STE) Test to Assess the Eye Irritation Potential of Chemicals. Toxicol. Chem. In Vitro 25,796-809.
|
|
(6)
|
Kojima H, et al. (2013). Second-Phase Validation of Short Time Exposure Tests for Assessment of Eye Irritation Potency of Chemicals. Toxicol. Chem. In Vitro 27, pp.1855-1869.
|
|
(7)
|
ICCVAM (2013). Short Time Exposure (STE) Test Method Summary Review Document, NIH. Dostupné na webových stránkách: [http://www.ntp.niehs.nih.gov/iccvam/docs/ocutox_docs/STE-SRD-NICEATM-508.pdf].
|
|
(8)
|
Kapitola B.5 této přílohy: Akutní dráždivé/leptavé účinky na oči.
|
|
(9)
|
Saito K, et al. (2015). Predictive Performance of the Short Time Exposure Test for Identifying Eye Irritation Potential of Chemical Mixtures.
|
|
(10)
|
Mikkelson TJ, Chrai SS and Robinson JR. (1973). Altered Bioavailability of Drugs in the Eye Due to Drug-Protein Interaction. J. Pharm. Sci.1648-1653.
|
|
(11)
|
ECETOC (1998). Eye Irritation Reference Chemicals Data Bank. Technical Report (No 48. (2)), Brussels, Belgium.
|
|
(12)
|
Gautheron P, et al. (1992). Bovine Corneal Opacity and Permeability Test: an In Vitro Assay of Ocular Irritancy. Fundam Appl Toxicol. 18, 442-449.
|
|
(13)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental Health and Safety Publications, Series on Testing and Assessment (No 34). Organisation for Economic Cooperation and Development, Paris.
|
|
(14)
|
OECD (2017). Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Environmental Health and Safety Publications, Series on Testing and Assessment (No 263). Organisation for Economic Cooperation and Development, Paris.
|
Dodatek
DEFINICE
Přesnost
: Přesnost shody mezi výsledky zkušební metody a přijatými referenčními hodnotami. Je to míra funkčnosti zkušební metody a jeden z aspektů její relevantnosti. Tento pojem se často používá místo pojmu soulad a rozumí se jím podíl správných výsledků zkušební metody (13).
Srovnávací látka
: Látka použitá jako norma pro srovnání se zkoušenou látkou. Srovnávací látka by měla mít tyto vlastnosti: i) nesporný a spolehlivý zdroj (nesporné a spolehlivé zdroje); ii) strukturální a funkční podobnost s třídou zkoušených látek; iii) známé fyzikální/chemické vlastnosti; iv) podpůrné údaje o známých účincích; a v) známou sílu v rozsahu žádoucí reakce.
Přístup „zdola nahoru“
: Přístup zkoušení po krocích používaný u zkoušené chemické látky, u níž se předpokládá, že ji není nutné klasifikovat jako látku mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí, který začíná odlišením chemických látek, u nichž není nutná klasifikace (negativní výsledek), od ostatních chemických látek (pozitivní výsledek).
Chemická látka
: Látka nebo směs.
Podráždění očí
: Vyvolání změny na oku po aplikaci zkoušené chemické látky na přední plochu oka, která je plně vratná do 21 dní od aplikace. Tento pojem je zaměnitelný s výrazy „vratné účinky na oko“ a kategorie 2 GHS OSN/CLP.
Míra falešné negativity
: Podíl všech pozitivních chemických látek falešně zjištěných zkušební metodou jako negativní. Je to jeden z ukazatelů výsledku zkušební metody.
Míra falešné pozitivity
: Podíl všech negativních chemických látek falešně zjištěných zkušební metodou jako pozitivní. Je to jeden z ukazatelů výsledku zkušební metody.
Nebezpečnost
: Základní charakteristika látky nebo situace, která má potenciál vyvolat nepříznivé účinky, jsou-li organismus, systém nebo (sub)populace vystaveny této látce.
Kontrola s médiem
: Neošetřená replika, která obsahuje všechny složky zkušebního systému. Tento vzorek se zpracovává se vzorky zkoušené chemické látky a jinými kontrolními vzorky, aby se zjistilo, zda rozpouštědlo vzájemně reaguje se zkušebním systémem.
Směs
: Směs nebo roztok složený ze dvou nebo více látek.
Jednosložková látka
: Látka definovaná svým kvantitativním složením, v níž je přítomna jedna hlavní složka v míře alespoň 80 % hmot.
MTT
: 3–(4,5-dimethylthiazol-2-yl)-2,5-difenyl-2H-tetrazolium-bromid; thiazolylová modř tetrazolium-bromid.
Vícesložková látka
: Látka definovaná svým kvantitativním složením, v níž je přítomna více než jedna hlavní složka v koncentraci ≥ 10 % hmot. a < 80 % hmot. Vícesložková látka je výsledkem výrobního procesu. Rozdíl mezi směsí a vícesložkovou látkou spočívá v tom, že směs se získá smísením dvou nebo více látek bez chemické reakce. Vícesložková látka je výsledkem chemické reakce.
OD
: Optická hustota.
Pozitivní kontrola
: Replika obsahující všechny složky zkušebního systému a látku, o níž je známo, že způsobuje pozitivní reakci. Aby se zajistilo, že proměnlivost v reakci na pozitivní kontrolu v čase bude možné vyhodnotit, rozsah pozitivní reakce by neměl být nadměrný.
Relevantnost
: Popis vztahu zkoušky k účinku, který je předmětem zájmu, a také toho, zda je zkouška smysluplná a použitelná pro konkrétní účel. Vyjadřuje, do jaké míry zkouška správně měří nebo predikuje biologický účinek, který je předmětem zájmu. Relevantnost zahrnuje také posouzení přesnosti (shody) zkušební metody (10).
Spolehlivost
: Měření rozsahu, v jakém může být zkušební metoda časem reprodukovatelná v laboratořích a mezi laboratořemi, když se provádí s použitím stejného protokolu. Hodnotí se to výpočtem laboratorní a mezilaboratorní reprodukovatelnosti a laboratorní opakovatelnosti (13).
Citlivost
: Podíl všech pozitivních/aktivních chemických látek, které jsou na základě zkoušky správně klasifikovány. Je měřítkem přesnosti zkušební metody, která poskytuje kategorické výsledky, a je důležitým prvkem při hodnocení relevantnosti zkušební metody (10).
Vážné poškození očí
: Tvorba poškození tkání v oku nebo závažné fyzikální slábnutí vidění po aplikaci zkoušené látky na přední plochu oka, které není plně vratné do 21 dní od aplikace. Tento pojem je zaměnitelný s výrazy „nevratné účinky na oči“ a kategorie 1 GHS OSN/CLP.
Kontrola s rozpouštědlem/vehikulem
: Neošetřený vzorek obsahující všechny složky zkušebního systému, včetně rozpouštědla nebo vehikula, který se zpracovává s exponovanými zkoušenými chemickými látkami a jinými kontrolními vzorky, aby se zjistila základní reakce vzorků zkoušených se zkoušenou chemickou látkou rozpuštěnou ve stejném rozpouštědle nebo vehikulu. Pokud se vzorek zkouší souběžnou kontrolou s médiem, ukazuje také, zda rozpouštědlo nebo vehikulum vzájemně reaguje se zkušebním systémem.
Specifičnost
: Podíl všech negativních/neaktivních chemických látek, které jsou na základě zkušební metody správně klasifikovány. Je měřítkem přesnosti zkušební metody, která poskytuje kategorické výsledky, a je důležitým prvkem při hodnocení relevantnosti zkušební metody (13).
Látka
: Chemický prvek a jeho sloučeniny v přírodním stavu nebo získané výrobním postupem, včetně jakýchkoliv aditiv potřebných pro zachování stability výrobku a všech nečistot pocházejících z použitého postupu, ale s vyloučením všech rozpouštědel, která je možno oddělit bez ovlivnění stability dané látky nebo změny jejího složení.
Smáčedlo
: Nazývané též povrchově aktivní látka, je to chemická látka, jako např. detergent, která je schopna snížit povrchové napětí kapaliny a umožnit jí vytvářet pěnu nebo pronikat do pevných látek; je též známa jako surfaktant.
Zkoušená chemická látka
: Jakákoli látka nebo směs zkoušená pomocí této zkušební metody.
Strategie stupňovitého zkoušení
: Strategie postupného zkoušení, kde se všechny existující informace o zkoušené chemické látce přezkoumávají ve stanoveném pořadí s použitím postupu váhy důkazů na každém stupni, aby se zjistilo, zda je k dispozici dost informací pro rozhodnutí o klasifikaci nebezpečnosti, než se postoupí do dalšího stupně. Pokud potenciál dráždivých účinků zkoušené chemické látky lze určit na základě existujících informací, další zkoušení není nutné. Jestliže potenciál dráždivých účinků zkoušené látky nelze určit na základě existujících informací, použije se stupňovitý postupný postup zkoušení na zvířatech po krocích, dokud se nestanoví jednoznačná klasifikace.
Přístup „shora dolů“
: Přístup zkoušení po krocích používaný u zkoušených chemických látek, u nichž se předpokládá, že způsobují vážné poškození očí, který začíná odlišením chemických látek vyvolávajících vážné poškození očí (pozitivní výsledek) od jiných chemických látek (negativní výsledek).
Globálně harmonizovaný systém klasifikace a označování chemických látek Organizace spojených národů (GHS OSN)
: Systém navrhující klasifikaci látek a směsí podle standardizovaných typů a úrovní fyzikálních, zdravotních a environmentálních nebezpečí a řešící příslušné komunikační prvky, jako jsou piktogramy, signální slova, výroky o nebezpečí, výroky o bezpečnostních opatřeních a bezpečnostní datové listy, které obsahují informace o jejich nežádoucích účincích s ohledem na ochranu lidí (včetně zaměstnavatelů, pracovníků, přepravců, spotřebitelů a osob reagujících na havárie) a životního prostředí (1).
Kategorie 1 GHS OSN/CLP
: Viz „vážné poškození očí“.
Kategorie 2 GHS OSN/CLP
: Viz „dráždivé účinky na oči“.
Bez kategorie GHS OSN/CLP
: Chemické látky, které nesplňují požadavky na klasifikaci v kategorii 1 nebo 2 GHS OSN/CLP (nebo 2A nebo 2B GHS OSN).
UVCB
: látky s neznámým nebo proměnlivým složením, komplexní reakční produkty nebo biologický materiál.
B.69 ZKUŠEBNÍ METODA S POUŽITÍM REKONSTRUOVANÉHO EPITELU PODOBNÉHO LIDSKÉ ROHOVCE (RhCE) PRO STANOVENÍ CHEMICKÝCH LÁTEK, KTERÉ NEVYŽADUJÍ KLASIFIKACI A OZNAČENÍ JAKO LÁTKY MAJÍCÍ DRÁŽDIVÉ ÚČINKY NA OČI NEBO VYVOLÁVAJÍCÍ VÁŽNÉ POŠKOZENÍ OČÍ
ÚVOD
|
1.
|
Tato zkušební metoda je rovnocenná Pokynu OECD pro zkoušení (TG) č. 492 (2017). Vážné poškození očí znamená vznik poškození tkáně oka nebo závažné zhoršení zraku po aplikaci zkoušené chemické látky na přední plochu oka, které není plně vratné do 21 dnů po aplikaci, jak je definováno v globálně harmonizovaném systému klasifikace a označování chemických látek (GHS) OSN) (1) a v nařízení Evropského unie (EU) č. 1272/2008 o klasifikaci, označování a balení látek a směsí (CLP) (76). Rovněž podle GHS OSN a CLP znamená podráždění očí vznik změn na oku po aplikaci zkoušené chemické látky na přední plochu oka, které jsou plně vratné do 21 dní od aplikace. Zkoušené chemické látky vyvolávající vážné poškození oka jsou klasifikovány jako látky kategorie 1 GHS OSN a CLP, zatímco zkoušené chemické látky vyvolávající podráždění oka jsou klasifikovány jako kategorie 2 GHS OSN a CLP. Zkoušené chemické látky, které nejsou klasifikovány jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí, jsou definovány jako chemické látky, které nesplňují požadavky pro klasifikaci v kategorii 1 nebo 2 (2A nebo 2B) GHS OSN a CLP, tj. označují se jako chemické látky bez kategorie GHS OSN a CLP.
|
|
2.
|
Posuzování vážného poškození oka / podráždění oka bylo obvykle prováděno na pokusných zvířatech (zkušební metoda B.5 (2)). Výběr nejvhodnější zkušební metody a její použití je třeba posuzovat v souvislosti s Pokynem OECD k integrovanému přístupu ke zkoušení a posuzování vážného poškození očí a podráždění očí (39).
|
|
3.
|
Tato zkušební metoda popisuje postup in vitro, který umožňuje určení chemických látek (látek a směsí), které nevyžadují klasifikaci a označení jako látky vyvolávající podráždění oka nebo vážné poškození oka podle systému GHS OSN a nařízení CLP. Využívá rekonstruovaného epitelu podobného lidské rohovce (RhCE), který věrně napodobuje histologické, morfologické, biochemické a fyziologické vlastnosti epitelu lidské rohovky. Byly validovány čtyři další zkušební metody in vitro, které jsou považovány za vědecky platné a byly přijaty jako zkušební metody B.47 (3), B.48 (4), B.61 (5) a B.68 (6) pro zkoumání účinků na lidské zdraví, a to vážného poškození oka / podráždění oka.
|
|
4.
|
Tato zkušební metoda zahrnuje dvě validované zkoušky využívající komerčně dostupné modely RhCE. Validační studie k posouzení podráždění oka / vážného poškození oka byly provedeny (7)(8)(9)(10)(11)(12)(13) s použitím zkoušek na podráždění oka (EIT) EpiOcular™ a SkinEthic™ Human Corneal Epithelium (HCE). Každá z těchto zkoušek, které se v dalším textu nazývají validované referenční metody – VRM 1 respektive VRM2, využívá jako zkušební systém komerčně dostupné tkáňové konstrukce RhCE. Na základě těchto validačních studií a jejich nezávislého vzájemného hodnocení (9)(12) byl vyvozen závěr, že zkoušky na podráždění oka EpiOcular™ EIT a SkinEthic™ HCE dokážou správně určit chemické látky (látky i směsi), které nevyžadují klasifikaci a označení jako látky vyvolávající podráždění oka nebo vážné poškození oka podle GHS OSN, a tyto zkoušky byly doporučeny jako vědecky platné pro tento účel (13).
|
|
5.
|
V současnosti se obecně uznává, že v dohledné budoucnosti nebude moci žádný alternativní test in vitro plně nahradit Draizeovu oční zkoušku in vivo (2)(14), pokud jde o předpověď celé škály závažného poškození očí/dráždivosti u různých tříd chemických látek. Draizeova oční zkouška (15) by však mohla být zcela nahrazena strategickými kombinacemi několika alternativních zkušebních metod v rámci strategií (odstupňovaného) zkoušení, jako je přístup zdola nahoru / shora dolů. Postup „zdola nahoru“ (15) má být použit, pokud se na základě existujících informací očekává, že chemická látka nevyvolává tak intenzivní podráždění oka, aby vyžadovala klasifikaci, zatímco postup „shora dolů“ (15) se má použít, jestliže se na základě existujících informací předpokládá, že chemická látka vyvolává závažné poškození oka. Zkoušky na podráždění oka EpiOcular™ EIT a SkinEthic™ HCE se doporučují použít k identifikaci chemických látek, které není nutné klasifikovat jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí podle systému GHS OSN (bez kategorie), bez dalšího zkoušení, a to v rámci strategie zkoušení, jako je přístup zdola nahoru, jak navrhuje Scott et al., například jako první krok v přístupu zdola nahoru nebo jako jeden z posledních kroků v přístupu shora dolů (15). Zkoušky na podráždění oka EpiOcular™ EIT a SkinEthic™ HCE však nejsou určeny pro rozlišení látek kategorie 1 GHS OS/CLP (vážné poškození oka) od látek kategorie 2 GHS OS/CLP (podráždění oka). Toto rozlišení bude třeba řešit dalším stupněm strategie zkoušení (15). Zkoušená chemická látka, která je pomocí zkoušek na podráždění oka EpiOcular™ EIT nebo SkinEthic™ HCE určena jako látka vyžadující klasifikaci z důvodu podráždění oka / vážného poškození oka, tedy bude vyžadovat další zkoušení (in vitro anebo in vivo), aby bylo možné stanovit konečný závěr (bez kategorie, kategorie 2 nebo kategorie 1 GHS OSN/CLP), a to pomocí např. zkušební metody B.47, B.48, B.61 nebo B.68.
|
|
6.
|
Účelem této zkušební metody je popsat postup používaný k hodnocení potenciálních nebezpečných účinků zkoušené chemické látky na oči podle její schopností vyvolávat toxicitu v tkáňové konstrukci RhCE, měřenou analýzou MTT (16) (viz odstavec 21). Životaschopnost tkáně RhCE po expozici zkoušené chemické látce se určuje porovnáním s tkáněmi ošetřenými negativní kontrolní látkou (% životaschopnosti) a poté se používá k predikci potenciální nebezpečnosti zkoušené chemické látky pro oko.
|
|
7.
|
K dispozici jsou standardy funkčnosti (17), které usnadní validaci nových nebo pozměněných zkoušek in vitro využívajících RhCE, obdobných zkouškám na podráždění oka EpiOcular™ EIT a SkinEthic™ HCE, v souladu se zásadami pokynu OECD č. 34 (18), a které umožní včas provést změnu pokynu OECD pro zkoušení č. 492, aby do něj mohly být zahrnuty. Vzájemné uznávání údajů bude podle postupu dohodnutého v rámci OECD zaručeno pouze u zkušebních metod validovaných podle těchto standardů funkčnosti, pokud tyto zkoušky byly podrobeny revizi a pokud je OECD zahrnula do odpovídajícího pokynu ke zkoušení.
|
DEFINICE
|
8.
|
Definice jsou uvedeny v dodatku 1.
|
VÝCHOZÍ ÚVAHY A OMEZENÍ
|
9.
|
Tato zkušební metoda vychází z komerčních trojrozměrných tkáňových konstrukcí RhCE, které se vyrábějí s využitím primárních lidských epidermálních keratinocytů (tj. EpiOcular™ OCL-200), nebo imortalizovaných lidských buněk epitelu rohovky (tj. SkinEthic™ HCE/S). Tkáňové konstrukce EpiOcular™ OCL-200 a SkinEthic™ HCE/S RhCE jsou podobné trojrozměrné struktuře rohovkového epitelu in vivo a vyrábějí se s použitím buněk zájmových druhů (19)(20). Tyto zkoušky navíc přímo měří cytotoxicitu v důsledku průniku chemické látky přes rohovku a vznik poškození buněk a tkání; cytotoxická odpověď poté určuje celkový výsledek vážného poškození oka / podráždění oka in vivo. K poškození buněk může dojít několika způsoby účinku (viz odstavec 20), ale cytotoxicita hraje důležitou, pokud ne primární, mechanistickou roli při určování celkové odpovědi na působení chemické látky ve formě poškození oka / podráždění oka, které se projevuje in vivo zejména zákalem rohovky, iritidou, zarudnutím spojivek anebo chemózou spojivek bez ohledu na fyzikálně chemické procesy, které jsou příčinou poškození tkáně.
|
|
10.
|
Ve validační studii k této zkušební metodě byla zkoušena široká škála chemických látek zahrnujících nejrůznější chemické druhy, chemické třídy, molekulové hmotnosti, logP, chemické struktury atd. Validační databáze zkoušky na podráždění oka EpiOcular™ obsahovala celkem 113 chemických látek zahrnujících 95 různých organických funkčních skupin podle analýzy OECD QSAR (8). Většina těchto chemických látek představovala jednosložkové látky, ale do studie bylo zařazeno také několik vícesložkových látek (včetně 3 homopolymerů, 5 kopolymerů a 10 kvazipolymerů). Z hlediska skupenství a kategorií GHS OSN/CLP bylo 113 zkoušených chemických látek rozděleno takto: 13 kapalin kategorie 1, 15 pevných látek kategorie 1, 6 kapalin kategorie 2A, 10 pevných látek kategorie 2A, 7 kapalin kategorie 2B, 7 pevných látek kategorie 2B, 27 kapalin bez kategorie a 28 pevných látek bez kategorie (8). Validační databáze zkoušky na podráždění oka SkinEthic™ HCE obsahovala celkem 200 chemických látek zahrnujících 165 různých organických funkčních skupin (8)(10)(11). Většina těchto chemických látek představovala jednosložkové látky, ale do studie bylo zařazeno také několik vícesložkových látek (včetně 10 polymerů). Z hlediska skupenství a kategorií GHS OSN/CLP bylo 200 zkoušených chemických látek rozděleno takto: 27 kapalin kategorie 1, 24 pevných látek kategorie 1, 19 kapalin kategorie 2A, 10 pevných látek kategorie 2A, 9 kapalin kategorie 2B, 7 pevných látek kategorie 2B, 50 kapalin bez kategorie a 53 pevných látek bez kategorie (10)(11).
|
|
11.
|
Tato zkušební metoda je použitelná pro látky a směsi a pro pevné látky, kapaliny, polotuhé látky a vosky. Kapaliny mohou být vodné či nevodné; pevné látky mohou být ve vodě rozpustné či nerozpustné. Pevné látky by měly být pokud možno před aplikací rozemlety na jemný prášek; žádná další předběžná úprava vzorku není vyžadována. Plyny a aerosoly nebyly ve validační studii hodnoceny. Ačkoli není jejich zkoušení postupem za použití techniky RhCE teoreticky vyloučeno, současná zkušební metoda zkoušení plynů a aerosolů neumožňuje.
|
|
12.
|
Zkoušené chemické látky absorbující světlo ve stejném pásmu jako formazan MTT (přirozeně nebo po expozici) a zkoušené chemické látky schopné přímé redukce vitálního barviva MTT (na formazan MTT) mohou interferovat s měřením životaschopnosti tkání, proto je třeba pro korekce použít upravené kontroly. Druh upravených kontrol, které mohou být potřebné, se bude lišit v závislosti na druhu interference vyvolané zkoušenou chemickou látkou a na postupu použitém pro kvantifikaci formazanu MTT (viz odstavce 36–42).
|
|
13.
|
Výsledky získané v předběžné validační studii (21)(22) a v plné validační studii (8)(10)(11) prokázaly, že obě zkoušky na podráždění oka EpiOcular™ EIT i SkinEthic™ HCE jsou přenosné do laboratoří, které doposud tyto analýzy neprováděly, a také že jsou reprodukovatelné v rámci laboratoře i mezi laboratořemi. Podle těchto studií je míra reprodukovatelnosti z hlediska shody předpovědí, které lze od zkoušky na podráždění oka EpiOcular™ očekávat na základě údajů o 113 chemických látkách, řádově 95 % v rámci laboratoří a 93 % mezi laboratořemi. Míra reprodukovatelnosti z hlediska shody předpovědí, které lze očekávat od zkoušky na podráždění oka SkinEthic™ HCE na základě údajů o 120 chemických látkách, je řádově 92 % v rámci laboratoří a 95 % mezi laboratořemi.
|
|
14.
|
Zkoušku na podráždění oka EpiOcular™ lze použít k identifikaci chemických látek, které není nutné klasifikovat jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí podle klasifikačního systému GHS OSN a CLP. Podle údajů získaných ve validační studii (8) má zkouška na podráždění oka EpiOcular™ celkovou přesnost 80 % (podle 112 chemických látek), citlivost 96 % (podle 57 chemických látek), míru falešné negativity 4 % (podle 57 chemických látek), specifičnost 63 % (podle 55 chemických látek) a míru falešné pozitivity 37 % (podle 55 chemických látek) při porovnání s údaji z referenční oční zkoušky in vivo na králících (zkušební metoda B.5) (2)(14) klasifikovaných podle klasifikačního systému GHS OSN a CLP. Studie, v níž bylo zkoušeno 97 kapalných agrochemických přípravků ve zkoušce na podráždění oka EpiOcular™, prokázala podobnou výkonnost zkušební metody u tohoto druhu směsí, jaká byla získána ve validační studii (23). 97 přípravků bylo rozděleno takto: 21 kategorie 1, 19 kategorie 2A, 14 kategorie 2B a 43 bez kategorie, klasifikováno podle klasifikačního systému GHS OSN na základě údajů z referenční oční zkoušky in vivo na králících (zkušební metoda B.5) (2)(14). Byla dosažena celková přesnost 82 % (podle 97 přípravků), citlivost 91 % (podle 54 přípravků), míra falešné negativity 9 % (podle 54 přípravků), specifičnost 72 % (podle 43 přípravků) a míra falešné pozitivity 28 % (podle 43 přípravků) (23).
|
|
15.
|
Zkoušku na podráždění očí SkinEthic™ HCE lze použít k identifikaci chemických látek, které není nutné klasifikovat jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí podle klasifikačního systému GHS OSN a CLP. Podle údajů získaných ve validační studii (10)(11) má zkouška na podráždění oka SkinEthic™ HCE celkovou přesnost 84 % (podle 200 chemických látek), citlivost 95 % (podle 97 chemických látek), míru falešné negativity 5 % (podle 97 chemických látek), specifičnost 72 % (podle 103 chemických látek) a míru falešné pozitivity 28 % (podle 103 chemických látek) při porovnání s údaji z referenční oční zkoušky in vivo na králících (zkušební metoda B.5) (2)(14) klasifikovaných podle klasifikačního systému GHS OSN a CLP.
|
|
16.
|
Míry falešné negativity zjištěné u obou zkoušek RhCE u látek nebo směsí spadají do 12 % celkové pravděpodobnosti, že chemické látky jsou při opakovaných testech v Draizově oční zkoušce in vivo identifikovány jako kategorie 2 podle GHS OSN a CLP nebo bez kategorie podle GHS OSN a CLP; je to způsobeno inherentní variabilitou metody v rámci zkoušky (24). Míry falešné pozitivity zjištěné u obou zkušebních metod RhCE u látek nebo směsí nejsou u této zkušební metody zásadně důležité, neboť všechny zkoušené chemické látky, které vykazují životaschopnost tkáně rovnou nebo nižší než stanovené mezní hodnoty (viz odstavec 44), budou vyžadovat další zkoušení pomocí jiných zkušebních metod in vitro nebo jako poslední možnost zkoušku na králících, v závislosti na požadavcích právních předpisů, s využitím strategie postupného zkoušení v rámci přístupu založeného na průkazných důkazech. Tyto zkušební metody lze použít pro všechny druhy chemických látek, přičemž negativní výsledek by měl být uznán pro neklasifikování chemické látky jako látky mající dráždivé účinky na oči a vyvolávající vážné poškození očí (bez kategorie GHS OSN a CLP). Použití zkoušek na podráždění oka EpiOcular™ a SkinEthic™ HCE podle jiných klasifikačních systémů než GHS OSN/CLP by mělo být předem konzultováno s příslušnými regulačními orgány.
|
|
17.
|
Omezení této zkušební metody spočívá v tom, že neumožňuje rozlišit mezi podrážděním oka / vratnými účinky na oko (kategorie 2) a vážným poškozením oka / nevratnými účinky na oko (kategorie 1) podle definice GHS OSN a CLP, ani mezi látkami vyvolávajícími podráždění oka (volitelná kategorie 2A) a látkami vyvolávajícími mírné podráždění oka (volitelná kategorie 2B) podle definice GHS OSN (1). Pro tyto účely je nutné další zkoušení s použitím jiných zkušebních metod in vitro.
|
|
18.
|
Termín „zkoušená chemická látka“ se v této zkušební metodě používá k označení toho, co se zkouší, (77) a netýká se použitelnosti zkušební metody RhCE ke zkoušení látek a/nebo směsí.
|
PRINCIP ZKOUŠKY
|
19.
|
Zkoušená chemická látka se aplikuje lokálně na minimálně dvě trojrozměrné konstrukce tkáně RhCE a po expozici a inkubační době po ošetření se změří životaschopnost tkáně. Tkáně RhCE jsou rekonstruovány z primárních lidských epidermálních keratinocytů nebo lidských imortalizovaných buněk epitelu rohovky, které byly kultivovány po několik dnů, aby vytvořily stratifikovaný, vysoce diferencovaný dlaždicový epitel morfologicky podobný epitelu, který se nachází v lidské rohovce. Tkáňová konstrukce EpiOcular™ RhCE sestává alespoň ze 3 životaschopných vrstev buněk a nekeratinizovaného povrchu vykazujícího strukturu podobnou rohovce, která je analogická struktuře in vivo. Tkáňová konstrukce SkinEthic™ HCE RhCE sestává alespoň ze 4 životaschopných vrstev buněk, včetně cylindrických bazálních buněk, přechodových křídlových buněk a povrchových dlaždicových buněk, podobných buňkám normálního lidského epitelu rohovky (20) (26).
|
|
20.
|
Chemicky vyvolané vážné poškození očí / podráždění očí, které se projevuje in vivo zejména zákalem rohovky, iritidou, zarudnutím spojivek anebo chemózou spojivek, je výsledkem kaskády událostí, která začíná proniknutím chemické látky přes rohovku anebo spojivku a vznikem poškození buněk. K poškození buněk může dojít několika způsoby účinku, včetně: lýzy buněčné membrány (např. působením povrchově aktivních látek, organických rozpouštědel); koagulace makromolekul (zejména bílkovin) (např. působením povrchově aktivních látek, organických rozpouštědel, alkalických látek a kyselin); saponifikace lipidů (např. působením alkalických látek); a alkylace nebo jiných kovalentních interakcí s makromolekulami (např. působením bělidel, peroxidů a alkylačních látek) (15)(27)(28). Bylo však prokázáno, že cytotoxicita hraje důležitou, pokud ne hlavní, mechanistickou roli při určování celkové odpovědi na působení chemické látky ve formě poškození oka / podráždění oka bez ohledu na fyzikálně chemické procesy, které jsou příčinou poškození tkáně (29)(30). Kromě toho potenciál chemické látky vyvolat vážné poškození oka / podráždění oka je v zásadě určen rozsahem počátečního poranění (31), které koreluje s rozsahem odumření buněk (29) a s rozsahem následných odpovědí a konečných výsledků (32). Slabě dráždivé látky tak obvykle zasáhnou pouze povrchový epitel rohovky, mírně a středně dráždivé látky poškozují zásadně epitel a povrchové vrstvy stromatu a silně dráždivé látky poškozují epitel, hluboké vrstvy stromatu a někdy i endotel rohovky (30)(33). Měření životaschopnosti tkáňové konstrukce RhCE po lokální expozici zkoušené chemické látce za účelem určení chemických látek, které nevyžadují klasifikaci jako látky vyvolávající vážné poškození oka / podráždění oka (bez kategorie podle GHS OSN a CLP), vychází z předpokladu, že všechny chemické látky vyvolávající vážné poškození oka nebo podráždění oka vyvolají cytotoxicitu v epitelu rohovky anebo ve spojivce.
|
|
21.
|
Životaschopnost tkáně RhCE se klasicky měří enzymatickou přeměnou vitálního barviva MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-difenyl-2H-tetrazolium-bromid; thiazolylová modř tetrazolium-bromid; číslo CAS 298–93-1] vitálníni buňkami tkáně na sůl modrého formazanu MTT, jehož množství se změří po extrakci z tkání (16). Chemické látky, které nevyžadují klasifikaci a označení podle GHS OSN / CLP (bez kategorie), se určují jako látky, které nesnižují životaschopnost tkáně pod stanovenou mez (tj. životaschopnost tkáně > 60 % ve zkouškách EpiOcular™ EIT a SkinEthic™ HCE EITL (78), nebo > 50 % ve zkouškách na dráždění oka SkinEthic™ HCE EITS (79)) (viz odstavec 44).
|
PROKÁZÁNÍ ZPŮSOBILOSTI
|
22.
|
Dříve než začnou laboratoře rutinně používat zkoušky RhCE pro regulační účely, měly by prokázat svou odbornou způsobilost správnou predikcí patnácti vhodných chemických látek uvedených v tabulce 1. Tyto chemické látky byly vybrány z chemických látek použitých ve validačních studiích validovaných referenčních metod (8)(10)(11). Výběr zahrnuje pokud možno chemické látky, které: i) zahrnují různá skupenství; ii) celý rozsah odpovědí in vivo na vážné poškození očí / podráždění očí na základě kvalitních výsledků získaných v referenční oční zkoušce in vivo na králících (zkušební metoda B.5) (2)(14) a klasifikačního systému GHS OSN (tj. kategorie 1, 2A, 2B nebo bez kategorie) (1) a klasifikačního systému CLP (tj. kategorie 1, 2 nebo bez kategorie); iii) zahrnují různé faktory in vivo důležité pro klasifikaci (24)(25); iv) jsou charakteristické pro chemické třídy použité ve validační studii (8)(10)(11); v) zahrnují správné a široké zastoupení organických funkčních skupin (8)(10)(11); vi) mají chemické struktury, které jsou dobře definovány (8)(10)(11); vii) jsou zabarvené anebo přímo redukují MTT; viii) mají reprodukovatelné výsledky ve zkušebních metodách RhCE v průběhu validace; ix) byly správně predikovány zkušebními metodami RhCE v průběhu validace; x) zahrnují celý rozsah odpovědí in vitro na základě kvalitních údajů ze zkušebních metod RhCE (0 až 100 % životaschopnost); xi) jsou komerčně dostupné; a xii) nejsou spojeny s příliš vysokými pořizovacími náklady a/nebo náklady na likvidaci odpadu. V situacích, kdy uvedená chemická látka není k dispozici nebo ji nelze použít z jiných oprávněných důvodů, může být použita jiná chemická látka splňující výše popsaná kritéria, např. některá z chemických látek použitých při validaci validované referenční metody. Tyto změny by však měly být odůvodněny.
Tabulka 1
Seznam vhodných chemických látek
|
Chemický název
|
CASRN
|
Organická funkční skupina (80)
|
Skupenství
|
Životaschopnost VRM1 (%) (81)
|
Životaschopnost VRM2 (%) (82) Předpověď VRM
|
Redukční činidlo MTT
|
Barevná interf.
|
Kategorie in vivo 1 (83)
|
|
Methylthioglykolát
|
|
|
2 365 -48-2
|
ester kyseliny karboxylové; thioalkohol
|
K
|
10,9 ± 6,4
|
5,5 ± 7,4
|
nelze předpovědět
|
A (silně)
|
N
|
Hydroxyethyl-akrylát
|
|
818-61-1
|
akrylát;
|
alkohol
|
K
|
7,5 ± 4,7 (84)
|
1,6 ± 1,0
|
nelze předpovědět
|
N
|
N
|
|
2,5-dimethyl-2,5-hexandiol
|
110-03-2
|
alkohol
|
PL
|
2,3 ± 0,2
|
0,2 ± 0,1
|
nelze předpovědět
|
N
|
N
|
|
Šťavelan sodný
|
62-76-0
|
kyselina oxokarboxylová
|
S
|
29,0 ± 1,2
|
5,3 ± 4,1
|
nelze předpovědět
|
N
|
N
|
|
Kategorie in vivo 2A (83)
|
|
|
2,4,11,13-tetraazatetradekan-diimidamid, N,N″-bis(4-chlorofenyl)-3,12-diimino-, di-D-glukonát (20 %, vodný) (85)
|
18 472 -51-0
|
aromatický heterocyklický halogenid; arylhalogenid; dihydroxylová skupina; guanidin
|
K
|
4,0 ± 1,1
|
1,3 ± 0,6
|
nelze předpovědět
|
N
|
A (slabě)
|
|
Benzoan sodný
|
532-32-1
|
aryl; karboxylová kyselina
|
PL
|
3,5 ± 2,6
|
0,6 ± 0,1
|
nelze předpovědět
|
N
|
N
|
|
Kategorie in vivo 2B (83)
|
|
|
Diethyl-toluamid
|
134-62-3
|
benzamid
|
K
|
15,6 ± 6,3
|
2,8 ± 0,9
|
nelze předpovědět
|
N
|
N
|
|
2,2-dimethyl-3-methylenebicyklo [2.2.1] heptan
|
79-92-5
|
alkan, rozvětvený s terciárním uhlíkem; alken; bicykloheptan; můstky propojené cyklické uhlovodíky; cykloalkan
|
PL
|
4,7 ± 1,5
|
15,8 ± 1,1
|
nelze předpovědět
|
N
|
N
|
|
Bez kategorie in vivo
(83)
|
|
|
1-ethyl-3-methylimidazol ethylsufát
|
3425 73 -75-5
|
alkoxy; amonná sůl; aryl; imidazol; síran
|
K
|
79,9 ± 6,4
|
79,4 ± 6,2
|
bez kat.
|
N
|
N
|
|
Dikaprylylether
|
629-82-3
|
alkoxy; ether
|
K
|
97,8 ± 4,3
|
95,2 ± 3,0
|
bez kat.
|
N
|
N
|
|
Piperonylbutoxid
|
51-03-6
|
alkoxy; benzodioxol; benzyl; ether
|
K
|
104,2 ± 4,2
|
96,5 ± 3,5
|
bez kat.
|
N
|
N
|
|
Polyethylenglykol (PEG-40) hydrogenovaný ricinový olej
|
61788-85-0
|
acylal; alkohol; allyl; ether
|
viskózní
|
77,6 ± 5,4
|
89,1 ± 2,9
|
bez kat.
|
N
|
N
|
|
1-(4-chlorfenyl)-3-(3,4-dichlorfenyl) močovina
|
101-20-2
|
aromatický heterocyklický halogenid; arylhalogenid; deriváty močoviny
|
PL
|
106,7 ± 5,3
|
101,9 ± 6,6
|
bez kat.
|
N
|
N
|
|
2,2′-methylen-bis-[6-(2H-benzotriazol-2-yl)-4-(1,1,3,3-tetramethylbutyl)fenol]
|
1035 97 -45-1
|
alkan, rozvětvený s kvartérním uhlíkem; kondenzovaná karbocyklická aromatická látka; kondenzované nasycené heterocykly; prekurzorové chinoidní sloučeniny; terc-butanol
|
PL
|
102,7 ± 13,4
|
97,7 ± 5,6
|
bez kat.
|
N
|
N
|
|
Tetrafluoroboritan draselný
|
14 075 -53-7
|
anorganická sůl
|
S
|
88,6 ± 3,3
|
92,9 ± 5,1
|
bez kat.
|
N
|
N
|
|
Zkratky:
CASRN = Chemical Abstracts Service Registry Number (registrační číslo CAS); GHS OSN = Globálně harmonizovaný systém klasifikace a označování chemických látek Organizace spojených národů (1); VRM1 = validovaná referenční metoda, EpiOcular™ EIT; VRM2 = validovaná referenční metoda, SkinEthic™ HCE EIT; Barevná interf. = měření barevné interference se standardní absorbancí (optická hustota (OD)) formazanu MTT.
|
|
|
23.
|
V rámci zkoušení způsobilosti se doporučuje, aby uživatelé po obdržení modelu rekonstruované lidské epidermis ověřili bariérové vlastnosti tkání uvedené výrobcem tkáňové konstrukce RhCE (viz odstavce 25, 27 a 30). To je zvláště důležité v případě, že se tkáně zasílají na dlouhé vzdálenosti nebo že jejich přeprava trvá dlouhou dobu. Pokud je zkouška úspěšně zavedena a je získána a prokázána způsobilost správně ji používat, není nutno běžně provádět takové ověřování. Při běžném používání zkoušky se však doporučuje i nadále v pravidelných intervalech bariérové vlastnosti posuzovat.
|
POSTUP
|
24.
|
Zkoušky, které v současné době zahrnuje tato zkušební metoda, jsou vědecky platné zkoušky na podráždění oka EpiOcular™ EIT a SkinEthic™ HCET (9)(12)(13), nazývané validovaná referenční metoda (VRM1, respektive VRM2). K dispozici jsou standardní pracovní postupy pro zkušební metody RhCE, které by měly být používány pro provádění a využívání těchto zkušebních metod v laboratoři (34)(35). V následujících odstavcích a v dodatku 2 je uveden popis hlavních složek a postupů zkoušek RhCE.
|
SLOŽKY ZKUŠEBNÍ METODY RHCE
Obecné podmínky
|
25.
|
Na rekonstrukci trojrozměrné epitelové tkáně podobné rohovce, která by se měla skládat z postupně stratifikovaných, nikoli však kornifikovaných buněk, se používají příslušné lidské derivované buňky. Tkáňová konstrukce RhCE se připravuje ve vložkách s porézní syntetickou membránou, přes kterou mohou do buněk procházet živiny. V rekonstruovaném epitelu podobném rohovce by mělo být přítomno několik vrstev životaschopných, nekeratinizovaných epitelových buněk. Tkáňová konstrukce RhCE by měla mít epitelový povrch v přímém kontaktu se vzduchem, aby byla možná přímá lokální expozice zkoušených chemických látek způsobem podobným tomu, jak by byl epitel rohovky exponován in vivo. Tkáňová konstrukce RhCE by měla tvořit funkční bariéru s dostatečnou robustností, aby odolala rychlému průniku cytotoxických referenčních látek, např. přípravku Triton X-100 nebo dodecylsulfátu sodného (SDS). Bariérová funkce by měla být prokázána a může být hodnocena stanovením doby expozice potřebné ke snížení životaschopnosti tkáně o 50 % (ET50) po aplikaci referenční látky v pevně stanovené koncentraci (např. 100 μl 0,3 % (obj.) přípravku Triton X-100) nebo v koncentraci, při níž referenční látka snižuje životaschopnost tkání o 50 % (IC50) po pevně stanovené době expozice (např. 30minutová expozice 50 μl SDS) (viz odstavec 30). Zadržovací vlastnosti tkáňové konstrukce RhCE by měly zabránit průchodu zkoušené chemické látky kolem okraje životaschopné tkáně, což by mohlo vést k nesprávnému modelování expozice rohovky. Lidské derivované buňky použité k vytvoření tkáňové konstrukce RhCE by neměly být kontaminovány bakteriemi, viry, mykoplazmaty ani houbovými mikroorganismy. Dodavatel by měl kontrolovat sterilitu tkáně rekonstrukce, zda není kontaminována houbami a bakteriemi.
|
Funkční podmínky
Životaschopnost
|
26.
|
K určení míry životaschopnosti se používá zkouška MTT (16). Vitální buňky tkáňové konstrukce RhCE redukují vitální barvivo MTT na modrou sraženinu formazanu, která je poté z tkáně extrahována pomocí isopropylalkoholu (nebo podobného rozpouštědla). Extrahovaný formazan MTT může být kvantifikován pomocí standardního měření absorbance (optické hustoty (OD)) nebo spektrofotometrickým postupem HPLC/UPLC (36). Optická hustota samotného extrakčního rozpouštědla by měla být dostatečně malá, tzn. OD < 0,1. Uživatelé tkáňové konstrukce RhCE by měli zajistit, aby každá použitá šarže tkáňové konstrukce RhCE splňovala stanovená kritéria pro negativní kontrolu. Rozsahy přijatelnosti pro hodnoty OD negativní kontroly pro validované referenční metody jsou uvedeny v tabulce 2. Uživatel spektrofotometrie HPLC/UPLC by měl jako kritérium přijatelnosti pro negativní kontrolu používat rozsahy OD negativní kontroly uvedené v tabulce 2. V závěrečné zprávě je třeba doložit, že tkáně ošetřené negativní kontrolní látkou jsou stabilně kultivovány (poskytují podobné naměřené hodnoty životaschopnosti tkáně) po dobu trvání zkušební doby expozice. Podobný postup by měl používat výrobce tkání v rámci řízení kvality při propouštění šarží tkání, ale v tomto případě mohou platit jiná kritéria přijatelnosti než kritéria uvedená v tabulce 2. Rozsah přijatelnosti (horní a dolní mez) hodnot OD negativní kontroly (za podmínek zkušební metody QC) stanoví subjekt, který model RhCE vyvinul nebo dodává.
|
Tabulka 2
Rozsahy přijatelnosti pro hodnoty OD negativní kontroly (pro uživatele testu)
|
Zkouška
|
Dolní mez přijatelnosti
|
Horní mez přijatelnosti
|
|
EpiOcular™ EIT (OCL-200) – VRM1 (protokoly pro kapaliny i pevné látky)
|
> 0,8 (86)
|
< 2,5
|
|
SkinEthic™ HCE EIT (HCE/S) – VRM2 (protokoly pro kapaliny i pevné látky)
|
> 1,0
|
≤ 2,5
|
Bariérová funkce
|
27.
|
Tkáňová konstrukce RhCE by měla být dostatečně silná a robustní, aby odolávala rychlému průniku cytotoxických referenčních látek, podle odhadu na základě hodnoty ET50 (např. Triton X-100) nebo IC50 (SDS) (tabulka 3). Bariérovou funkci každé šarže použité tkáňové konstrukce RhCE by měl prokázat vývojář/prodejce modelu RhCE při dodávce tkání koncovému uživateli (viz odstavec 30).
|
Morfologie
|
28.
|
Histologické vyšetření tkáňové konstrukce RhCE by mělo prokázat strukturu podobnou epitelu lidské rohovky (včetně alespoň 3 vrstev životaschopných epitelových buněk a nekeratinizovaného povrchu). Pro validované referenční metody stanovil vhodnou morfologii vývojář/dodavatel, a proto ji uživatel zkušební metody nemusí znovu prokazovat při každé použité šarži tkáně.
|
Reprodukovatelnost
|
29.
|
Výsledky zkušební metody za použití pozitivních a negativních kontrol by měly prokázat reprodukovatelnost v průběhu času.
|
Kontrola kvality
|
30.
|
Tkáňová konstrukce RhCE by měla být použita pouze v případě, že subjekt, který ji vyvinul nebo dodává, prokáže, že každá použitá šarže tkáňové konstrukce RhCE splňuje stanovená produkční kritéria pro uvolnění do oběhu, přičemž mezi nejvýznamnější z nich patří kritéria životaschopnosti (odstavec 26) a bariérové funkce (odstavec 27). Rozsah přijatelnosti (horní a dolní mez) pro bariérové funkce měřené podle hodnoty ET50 nebo IC50 (viz odstavce 25 a 26) stanoví vývojář/dodavatel tkáňové konstrukce RhCE. Rozsah přijatelnosti hodnot ET50 a IC50 použitý jako kritérium řízení kvality pro propouštění šarží u vývojáře/dodavatele tkáňových konstrukcí RhCE (použitých ve validovaných referenčních metodách) je uveden v tabulce 3. Údaje prokazující shodu se všemi výrobními kritérií pro propuštění by měl vývojář/dodavatel tkáňové konstrukce RhCE poskytnout uživatelům zkušební metody, aby je mohli uvést v závěrečné zprávě. Pro spolehlivou predikci chemických látek, které nevyžadují klasifikaci a označení jako látky vyvolávající podráždění oka nebo vážné poškození oka podle systému GHS OSN/CLP, lze akceptovat pouze výsledky získané u tkání splňujících všechna tato výrobní kritéria pro propuštění.
|
Tabulka 3
Kritérium kontroly kvality pro uvolnění šarže do oběhu
|
Zkouška
|
Dolní mez přijatelnosti
|
Horní mez přijatelnosti
|
|
EpiOcular™ EIT (OCL-200) – VRM1 (100 μl 0,3 % (obj.) Tritonu X-100)
|
ET50 = 12,2 min
|
ET50 = 37,5 min
|
|
SkinEthic™ HCE EIT (HCE/S) – VRM2 (30minutová expozice 50 μl SDS)
|
IC50 = 1 mg/ml
|
IC50 = 3,2 mg/ml
|
Aplikace zkoušené chemické látky a kontrolních látek
|
31.
|
Pro každou zkoušenou látku a každou kontrolní látku by se při každém provedení zkoušky měly použít alespoň dva tkáňové replikáty. Používají se dva různé zkušební protokoly, jeden pro kapalné zkoušené chemické látky a jeden pro pevné zkoušené chemické látky (34)(35). Před aplikací zkoušených chemických látek u obou metod a protokolů by měl být povrch tkáňové konstrukce zvlhčen Dulbeccovým fosfátovým pufrem bez vápníku a hořčíku (DPBS bez Ca2+/Mg2+), aby se napodobily vlhké podmínky lidského oka. Ošetření tkání se zahajuje expozicí zkoušené chemické látce (látkám) a kontrolní látkám. U obou zkušebních protokolů v obou validovaných referenčních metodách je zapotřebí nanést dostatečné množství zkoušené chemické látky nebo kontrolní látky, aby rovnoměrně pokryla povrch epitelu, a současně se vyhnout nekonečné dávce (viz odstavce 32 a 33) (dodatek 2).
|
|
32.
|
Se zkoušenými chemickými látkami, které lze pipetovat při teplotě 37 oC nebo při nižších teplotách (v případě potřeby pomocí objemové pipety), se ve validovaných referenčních metodách nakládá jako s kapalinami, jinak by se s nimi mělo nakládat jako s pevnými látkami (viz odstavec 33). Ve validovaných referenčních metodách se kapalná zkoušená chemická látka rovnoměrně rozprostře na povrch tkáně (tj. aplikace minimálně 60 μl/cm2) (viz dodatek 2, (33)(34)). V maximální možné míře by měly být vyloučeny kapilární jevy (účinky povrchového napětí), k nimž může dojít v důsledku malých objemů aplikovaných na vložku (na povrch tkáně), aby bylo zaručeno správné dávkování na tkáň. Tkáně ošetřené kapalnými zkoušenými chemickými látkami se inkubují po dobu 30 minut při standardních kultivačních podmínkách (37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % relativní vlhkost). Na konci expoziční doby je nutno kapalnou zkoušenou chemickou látku a kontrolní látky pečlivě odstranit z povrchu tkáně důkladným opláchnutím pomocí DPBS bez Ca2+/Mg2+ při pokojové teplotě. Po opláchnutí následuje poexpoziční ponoření do čerstvého média při pokojové teplotě (aby se odstranila veškerá zkoušená chemická látka absorbovaná do tkáně) po stanovenou dobu, která se liší v závislosti na použité validované referenční metodě. Pouze u VMR1 platí poexpoziční inkubační doba v čerstvém médiu při standardních kultivačních podmínkách před provedením analýzy MTT (viz dodatek 2, (34)(35)).
|
|
33.
|
Se zkoušenými chemickými látkami, které nelze pipetovat při teplotách do 37 oC, se ve validovaných referenčních metodách zachází jako s pevnými látkami. Množství aplikované zkoušené chemické látky by mělo být dostatečné, aby pokrylo celý povrch tkáně, tj. mělo by být aplikováno minimálně 60 mg/cm2 (dodatek 2). Pevné látky by se měly pokud možno zkoušet ve formě jemného prášku. Tkáně ošetřené pevnými zkoušenými chemickými látkami se inkubují po stanovenou dobu (v závislosti na použité validované referenční metodě) při standardních kultivačních podmínkách (viz dodatek 2, (34)(35)). Na konci expoziční doby je nutno pevnou zkoušenou chemickou látku a kontrolní látky pečlivě odstranit z povrchu tkáně důkladným opláchnutím pomocí DPBS bez Ca2+/Mg2+ při pokojové teplotě. Po opláchnutí následuje poexpoziční ponoření do čerstvého média při pokojové teplotě (aby se odstranila veškerá zkoušená chemická látka absorbovaná do tkáně) po stanovenou dobu, která se liší v závislosti na použité validované referenční metodě, a poexpoziční inkubace v čerstvém médiu při standardních kultivačních podmínkách před provedením analýzy MTT (viz dodatek 2, (34)(35)).
|
|
34.
|
Do každého provedení zkoušky by se měly zařadit souběžné negativní a pozitivní kontroly, aby se prokázalo, že životaschopnost (stanovená při negativní kontrole) a citlivost (stanovená při pozitivní kontrole) tkání jsou v rámci rozpětí přijatelnosti definovaného podle dosavadních hodnot. Souběžná negativní kontrola poskytuje také výchozí hodnotu (100 % životaschopnost tkáně) pro výpočet relativní percentuální životaschopnosti tkání ošetřených zkoušenou chemickou látkou (%Viabilitytest). Doporučenou látkou pro pozitivní kontrolu, která se má používat u těchto validovaných referenčních metod, je čistý methyl-acetát (CAS č. 79-20-9, komerčně dostupný např. od společnosti Sigma-Aldrich, kat. č. 45997; kapalný). Doporučenými látkami pro negativní kontrolu, které se mají používat u VRM1 a VRM2, je ultračistá H2O, respektive DPBS bez Ca2+/Mg2+. Tyto kontrolní látky byly použity ve validačních studiích validovaných referenčních metod a většina dosavadních údajů se týká právě těchto látek. Použití vhodných alternativních látek pro pozitivní nebo negativní kontrolu by mělo být adekvátně vědecky odůvodněno. Negativní a pozitivní kontroly by měly být zkoušeny podle stejného protokolu (protokolů), jako byly protokoly použité pro zkoušené chemické látky zařazené do provedení zkoušky (tj. pro kapaliny anebo pevné látky). Po jejich aplikaci by měla následovat experimentální expozice, oplach, poexpoziční ponoření, případně poexpoziční inkubace, jak je popsáno pro kontroly prováděné souběžně u kapalných zkoušených chemických látek (viz odstavec 32) nebo pro kontroly prováděné souběžně u pevných zkoušených chemických látek (viz odstavec 33) před provedením analýzy MTT (viz odstavec 35) (34)(35). Postačí jedna sada negativních a pozitivních kontrol pro všechny zkoušené chemické látky stejného skupenství (kapaliny nebo pevné látky) zařazené do stejného provedení zkoušky.
|
Měření životaschopnosti tkání
|
35.
|
Zkouška MTT je standardizovaná kvantitativní metoda (16), která by se měla používat k měření životaschopnosti tkání podle této zkušební metody. Je slučitelná s použitím trojrozměrné tkáňové konstrukce. Analýza MTT se provádí okamžitě po poexpoziční inkubační době. Ve validovaných referenčních metodách se vzorek tkáňové konstrukce RhCE vloží do 0,3 ml roztoku MTT v koncentraci 1 mg/ml na dobu 180 ± 15 min při standardních kultivačních podmínkách. Životaschopné buňky tkáňové konstrukce RhCE redukují vitální barvivo MTT na modrou sraženinu formazanu MTT. Vysrážený modrý formazan MTT se poté z tkáně extrahuje pomocí příslušného množství isopropylalkoholu (nebo podobného rozpouštědla) (34)(35). Tkáně testované kapalnými zkoušenými chemickými látkami by měly být extrahovány z horní i dolní části tkání, zatímco tkáně testované pevnými zkoušenými chemickými látkami a barvenými kapalinami by měly být extrahovány pouze z dolní části tkáně (aby se minimalizovala potenciální kontaminace extrakčního roztoku isopropylalkoholu některou zkoušenou chemickou látkou, jež mohla zůstat v tkáni). Tkáně testované kapalnými zkoušenými chemickými látkami, které nelze snadno vymýt, mohou být také extrahovány pouze z dolní části tkáně. Se souběžně testovanými negativními a pozitivními kontrolními látkami se zachází podobně jako se zkoušenou chemickou látkou. Extrahovaný formazan MTT může být kvantifikován pomocí standardního měření absorbance (OD) při vlnové délce 570 nm za použití filtru o pásmové propustnosti nejvýše ± 30 nm nebo spektrofotometrickým postupem HPLC/UPLC (viz odstavce 42) (11)(36).
|
|
36.
|
Optické vlastnosti zkoušené látky nebo její chemické působení na MTT mohou měření formazanu MTT ovlivnit, což vede k nepravdivému odhadu životaschopnosti tkáně. Zkoušené chemické látky mohou interferovat s měřením formazanu MTT, a to přímou redukcí MTT na modrý formazan MTT, anebo barevnou interferencí, jestliže zkoušená chemická látka absorbuje, přirozeně nebo v důsledku experimentálních postupů, ve stejném rozsahu OD jako formazan MTT (tj. cca 570 nm). Před testováním by měly být provedeny předběžné zkoušky, aby bylo možné identifikovat potenciální přímá redukční činidla MTT anebo barevně interferující chemické látky, a k detekci a korekci potenciální interference těchto zkoušených chemických látek by měly být provedeny dodatečné kontroly (viz odstavce 37–41). To je zvláště důležité v případě, že se určitá zkoušená chemická látka opláchnutím z tkáňové konstrukce RhCE zcela neodstraní nebo že pronikne do epitelu podobného rohovce, takže je přítomna v tkáňových konstrukcích, když se provádí analýza MTT. U zkoušených chemických látek absorbujících světlo ve stejném pásmu jako formazan MTT (přirozeně nebo po expozici), které nejsou kompatibilní se standardním měřením absorbance (OD) formazanu MTT vzhledem k příliš velké interferenci, tj. silná absorbance při vlnové délce 570 ± 30 nm, může být na měření formazanu MTT použit HPLC/UPLC spektrofotometrický postup (viz odstavce 41 a 42) (11)(36). Podrobný popis, jak detekovat a korigovat přímou redukci MTT a interference způsobené barvivy, je k dispozici ve standardních prováděcích postupech validovaných referenčních metod (34)(35). V dodatku III, respektive IV jsou také uvedeny grafy ilustrující identifikaci a nakládání s přímými redukčními činidly MTT anebo barevně interferujícími chemickými látkami pro VRM1 a VRM2.
|
|
37.
|
Aby bylo možné identifikovat potenciální interferenci zkoušených chemických látek absorbujících světlo ve stejném pásmu jako formazan MTT (přirozeně nebo po expozici) a rozhodnout, zda jsou nutné další kontroly, zkoušená chemická látka se přidá do vody anebo isopropylalkoholu a inkubuje se po vhodnou dobu při pokojové teplotě (viz dodatek 2, (34)(35)). Jestliže zkoušená chemická látka ve vodě anebo isopropylalkoholu absorbuje dostatečně světlo v pásmu 570 ± 20 nm u VRM1 (viz dodatek 3) nebo jestliže po smíchání zkoušené chemické látky s vodou vznikne zbarvený roztok u VRM2 (viz dodatek 4), považuje se zkoušená chemická látka za interferující se standardním měřením absorbance (OD) formazanu MTT a je třeba provést další kontroly s barvivy, případně použít spektrofotometrický postup HPLC/UPLC, přičemž v tomto případě nejsou tyto kontroly nutné (viz odstavce 41 a 42 a dodatky III a IV) (34)(35). Při provádění standardního měření absorbance (OD) by měla být každá interferující zkoušená chemická látka aplikována na alespoň dva životaschopné tkáňové replikáty, které se podrobí celému zkušebnímu postupu, ale v průběhu inkubačního kroku MTT se inkubují v médiu místo roztoku MTT za účelem vytvoření kontroly nespecifického zbarvení v živých tkáních (NSCliving) (34)(35). Vzhledem k inherentní biologické variabilitě živých tkání je třeba kontrolu NSCliving provádět současně se zkoušením zbarvené chemické látky a v případě většího počtu zkoušek je třeba u každé zkoušky (při každém provedení zkoušky) provést nezávislou kontrolu NSCliving. Skutečná životaschopnost tkáně se vypočte jako: percentuální životaschopnost tkáně získaná z živých tkání vystavených působení interferující zkoušené chemické látky a inkubovaných v roztoku MTT (% Viabilitytest) minus percentuální nespecifické zbarvení získané z živých tkání vystavených působení interferující zkoušené chemické látky a inkubovaných v roztoku bez MTT, prováděno současně s korigovanou zkouškou (% NSCliving), tj. skutečná životaschopnost tkáně = [%Viabilitytest]– [%NSCliving].
|
|
38.
|
Aby bylo možné identifikovat přímá redukční činidla MTT, je třeba každou zkoušenou chemickou látku přidávat do čerstvě připraveného roztoku MTT. Do roztoku MTT se přidá vhodné množství zkoušené chemické látky a směs se inkubuje přibližně po dobu přibližně 3 hodin při standardních kultivačních podmínkách (viz dodatky III a IV) (34)(35). Jestliže směs MTT obsahující zkoušenou chemickou látku (nebo suspenzi u nerozpustných zkoušených chemických látek) zmodrá/zfialoví, je zkoušená chemická látka považována za přímé redukční činidlo MTT, přičemž je třeba provést další funkční kontrolu na neživotaschopných tkáňových konstrukcích RhCE nezávisle na použití standardního měření absorbance (OD) nebo spektrofotometrického postupu HPLC/UPLC. Tato další funkční kontrola využívá neživé tkáně, které mají pouze zbytkovou metabolickou aktivitu, ale absorbují a udržují v sobě zkoušenou chemickou látku podobně jako životaschopné tkáně. Neživé tkáně u VRM1 se připravují expozicí nízké teplotě („usmrcené zmrazením“). Neživé tkáně u VRM2 se připravují dlouhodobou inkubací (např. minimálně 24 ± 1 hodina) ve vodě s následným uložením při nízké teplotě („usmrcené vodou“). Každá zkoušená chemická látka redukující MTT se aplikuje na alespoň dva neživé tkáňové replikáty, které se podrobí celému zkušebnímu postupu, aby došlo ke kontrole na nespecifickou redukci MTT (NSMTT (34)(35). Postačí jedna kontrola NSMTT na zkoušenou chemickou látku bez ohledu na počet provedených nezávislých zkoušek/experimentů. Skutečná životaschopnost tkáně se vypočte jako: percentuální životaschopnost tkáně získaná z živých tkání vystavených působení redukčního činidla MTT (%Viabilitytest) minus percentuální nespecifická redukce MTT získaná z neživých tkání vystavených stejnému redukčnímu činidlu MTT, počítáno ve vztahu k negativní kontrole prováděné současně s korigovanou zkouškou (% NSMTT), tj. skutečná životaschopnost tkáně = [%Viabilitytest] – [%NSMTT].
|
|
39.
|
Zkoušené chemické látky, u nichž se zjistí, že působí barevné interference (viz odstavec 37) i že přímo redukují MTT (viz odstavec 38), budou při provádění standardního měření absorbance (OD) také vyžadovat třetí skupinu kontrol vedle kontrol NSMTT a NSCliving popsaných v předchozích odstavcích. Obvykle je tomu tak u tmavě zbarvených zkoušených chemických látek absorbujících světlo v rozsahu 570 ± 30 nm (např. modré, fialové, černé), protože jejich vlastní barva narušuje posouzení jejich schopnosti přímo redukovat MTT, jak je popsáno v odstavci 38. Je tedy nutné použít standardně kontroly NSMTT společně s kontrolami NSCliving. Zkoušené chemické látky, u nichž je provedena kontrola NSMTT i NSCliving, mohou být absorbovány a zachyceny v živých i neživých tkáních. Proto v tomto případě kontrola NSMTT musí nejen korigovat potenciální přímou redukci MTT způsobenou zkoušenou chemickou látkou, ale také barevnou interferenci vznikající z absorpce a retence zkoušené chemické látky v neživých tkáních. Mohlo by to vést k dvojí korekci barevné interference, protože kontrola NSCliving již koriguje barevnou interferenci vznikající z absorpce a retence zkoušené chemické látky v živých tkáních. Aby byla vyloučena možná dvojí korekce barevné interference, je třeba provést třetí kontrolu na nespecifické zbarvení u neživých tkání (NSCkilled) (viz dodatky III a IV) (34)(35). Při této další kontrole se zkoušená chemická látka aplikuje na alespoň dva neživé tkáňové replikáty, které se podrobí celé zkoušce, ale v průběhu inkubačního kroku MTT se inkubují v médiu místo roztoku MTT. Postačí jedna kontrola NSCkilled na zkoušenou chemickou látku bez ohledu na počet provedených nezávislých zkoušek/experimentů, ale tuto kontrolu je třeba provést současně s kontrolou NSMTT a se stejnou šarží tkáně. Skutečná životaschopnost tkáně se vypočte jako: percentuální životaschopnost tkáně získaná z živých tkání vystavených působení zkoušené chemické látky (%Viabilitytest) minus % NSMTT minus % NSCliving plus percentuální nespecifické zbarvení získané z neživých tkání vystavených působení interferující zkoušené chemické látky a inkubovaných v roztoku bez MTT, počítáno ve vztahu k negativní kontrole provedené současně s korigovanou zkouškou (% NSCkilled), tj. skutečná životaschopnost tkáně = [%Viabilitytest] – [%NSMTT] – [%NSCliving] + [%NSCkilled].
|
|
40.
|
Je důležité uvést, že nespecifická redukce MTT a nespecifické barevné interference mohou zvyšovat OD (při provádění standardních měření absorbance) tkáňového extraktu nad rozsah linearity spektrofotometru a že nespecifická redukce MTT může také zvyšovat plochu píku formazanu MTT (při provádění HPLC/UPLC-spektrofotometrických měření) tkáňového extraktu nad rozsah linearity spektrofotometru. Proto je důležité, aby každá laboratoř stanovila OD / rozsah linearity plochy píku svého spektrofotometru s použitím např. formazanu MTT (CAS č. 57360-69-7), který je komerčně dostupný např. od společnosti Sigma-Aldrich (kat. č. M2003), než zahájí testování zkoušených chemických látek pro regulační účely.
|
|
41.
|
Standardní měření absorbance (OD) pomocí spektrofotometru je vhodné pro posouzení zkoušených chemických látek, které přímo redukují MTT a působí barevné interference, když interference pozorované při měření formazanu MTT nejsou příliš silné (tzn. optická hustota tkáňových extraktů zjištěná za použití zkoušené chemické látky bez korekce přímé redukce MTT anebo barevné interference je v lineárním rozsahu spektrofotometru). Avšak výsledky u zkoušených chemických látek, které dávají % NSMTT anebo % NSCliving ≥ 60 % (protokol VRM1 a VRM2 pro kapaliny) nebo ≥ 50 % (protokol VRM2 pro pevné látky) negativní kontroly, je třeba posuzovat opatrně, neboť je to stanovená hraniční hodnota používaná ve validovaných referenčních metodách na odlišení klasifikovaných chemických látek od neklasifikovaných (viz odstavec 44). Standardní absorbanci (OD) však nelze měřit, když je interference s měřením formazanu MTT příliš silná (tj. vede k nesprávným hodnotám OD u zkoušených tkáňových extraktů spadajících mimo lineární rozsah spektrofotometru). Zabarvené zkoušené chemické látky nebo zkoušené chemické látky, které se zabarví při kontaktu s vodou nebo isopropylalkoholem a které interferují příliš silně se standardním měřením absorbance (OD) formazanu MTT, mohou být přesto hodnoceny pomocí HPLC/UPLC spektrofotometrie (viz dodatky III a IV). Je tomu tak proto, že systém HPLC/UPLC umožňuje oddělit formazan MTT od chemické látky před její kvantifikací (36). Z tohoto důvodu se při použití HPLC/UPLC spektrofotometrie nepožadují kontroly NSCliving ani NSCkilled, a to nezávisle na zkoušené chemické látce. Kontroly NSMTT by se však měly použít, pokud existuje podezření, že zkoušená chemická látka přímo redukuje MTT (podle postupu popsaného v odstavci 38). Kontroly NSMTT by se také měly použít u zkoušených chemických látek majících barvu (vlastní barvu nebo zbarvení ve vodě), která znesnadňuje posouzení jejich schopnosti přímé redukce MTT, jak je popsáno v odstavci 38. Při použití HPLC/UPLC spektrofotometrie na měření formazanu MTT se percentuální životaschopnost tkáně vypočítá jako percentuální plocha píku formazanu MTT získaná z živých tkání vystavených působení zkoušené chemické látky ve vztahu k píku formazanu MTT získanému při souběžné negativní kontrole. U zkoušených chemických látek, které dokážou přímo redukovat MTT, se skutečná životaschopnost tkáně vypočítá jako: % Viabilitytest minus % NSMTT, jak je popsáno v poslední větě odstavce 38. Konečně je třeba uvést, že přímá redukční činidla MTT nebo přímá redukční činidla MTT, jež jsou také barevně interferující, která zůstanou v tkáních po expozici a redukují MTT tak silně, že to vede k takovým hodnotám optické hustoty (při využití standardního měření OD) nebo plochám píků (při použití UPLC/HPLC spektrofotometrie) zkoušených tkáňových extraktů, které spadají mimo rozsah linearity spektrofotometrie, nelze posuzovat zkušebními metodami RhCE, i když lze očekávat, že k tomu dochází velmi vzácně.
|
|
42.
|
HPLC/UPLC spektrofotometrie může být použita u všech druhů zkoušených chemických látek (barevných, nebarevných, látek redukujících MTT i látek, které MTT neredukují) pro měření formazanu MTT (11)(36). Vzhledem k různým spektrofotometrickým systémům HPLC/UPLC není možné, aby každý uživatel vytvořil přesně stejné systémové podmínky. Proto je třeba prokázat způsobilost spektrofotometrického systému HPLC/UPLC před jeho použitím pro kvantifikaci formazanu MTT z tkáňových extraktů splněním kritérií přijatelnosti pro skupinu standardních parametrů způsobilosti vycházejících z parametrů popsaných v pokynu amerického Úřadu pro potraviny a léčiva pro průmysl k validaci bioanalytických metod (36)(38). Tyto klíčové parametry a jejich kritéria přijatelnosti jsou uvedeny v dodatku 5. Po splnění kritérií přijatelnosti definovaných v dodatku 5 je spektrofotometrický systém HPLC/UPLC považován za způsobilý a připravený na měření formazanu MTT za experimentálních podmínek popsaných v této zkušební metodě.
|
Kritéria přijatelnosti
|
43.
|
U každého provedení zkoušky s použitím šarží tkáně RhCE, které splňují požadavky na kvalitu (viz odstavec 30), by tkáně ošetřené látkou pro negativní kontrolu měly vykazovat hodnoty OD odrážející kvalitu tkání, která vznikla po všech přepravních a přejímacích krocích a po všech procesech podle protokolu zkoušky, a neměly by být mimo doposud stanové meze popsané v tabulce 2 (viz odstavec 26). Podobně tkáně ošetřené látkou pro pozitivní kontrolu, tj. methylacetátem, by měly vykazovat střední životaschopnost tkáně < 50 % ve vztahu k negativní kontrole ve VRM1 u protokolu pro kapaliny i protokolu pro pevné látky a ≤ 30 % (protokol pro kapaliny) nebo ≤ 20 % (protokol pro pevné látky) ve vztahu k negativní kontrole ve VRM2, a odrážet tak schopnost tkání reagovat na dráždivou zkoušenou chemickou látku za podmínek zkušební metody (34)(35). Variabilita mezi tkáňovými replikáty u zkoušených chemických látek anebo kontrolních látek by měla spadat do přijatelných limitů (tj. rozdíl životaschopnosti mezi dvěma tkáňovými replikáty by měl být menší než 20 % a směrodatná odchylka (SD) mezi třemi tkáňovými replikáty by neměla přesahovat 18 %). Jestliže je negativní kontrola nebo pozitivní kontrola zařazená do zkoušky mimo přijatelné rozsahy, je toto provedení zkoušky považováno za „nezpůsobilé“ a zkoušku je třeba opakovat. Jestliže je variabilita mezi tkáňovými replikáty zkoušené chemické látky mimo přijatelný rozsah, je třeba zkoušku považovat za „nezpůsobilou“ a zkoušenou chemickou látku je třeba testovat znovu.
|
Interpretace výsledků a predikční model
|
44.
|
Hodnoty OD / plochy píku zjištěné u replikovaných tkáňových extraktů pro každou zkoušenou chemickou látku se použijí k výpočtu střední percentuální životaschopnosti tkání (průměru z tkáňových replikátů) normalizované podle negativní kontroly, která je stanovena na 100 %. Percentuální hraniční hodnoty životaschopnosti tkání pro určení zkoušených chemických látek, které není nutné klasifikovat jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí (bez kategorie GHS OSN/CLP), jsou uvedeny v tabulce 4. Výsledky by tedy měly být interpretovány takto:
|
—
|
Zkoušená chemická látka je určena jako látka nevyžadující klasifikaci a označení podle systému GHS OSN a CLP (bez kategorie), jestliže je střední percentuální životaschopnost tkáně po expozici a poexpoziční inkubaci větší než (>) stanovená mezní hodnota percentuální životaschopnosti tkáně, jak je uvedeno v tabulce 4. V tomto případě není vyžadováno žádné další zkoušení jinými zkušebními metodami.
|
|
—
|
Jestliže je střední percentuální životaschopnost tkáně po expozici a poexpoziční inkubaci menší než nebo rovna (≤) stanovené mezní hodnotě percentuální životaschopnosti tkáně, nelze provést žádnou predikci, jak je uvedeno v tabulce 4. V tomto případě bude nutné další zkoušení jinými zkušebními metodami, protože zkušební metody RhCE vykazují určitý počet falešně pozitivních výsledků (viz odstavce 14–15) a nedokážou rozlišit mezi kategoriemi 1 a 2 podle GHS OSN a CLP (viz odstavec 17).
|
Tabulka 4
Predikční modely podle klasifikace GHS OSN a CLP
|
Validovaná referenční metoda
|
Bez kategorie
|
Nelze předpovědět
|
|
VRM 1 – EpiOcular™ EIT (pro oba protokoly)
|
střední životaschopnost tkáně > 60 %
|
střední životaschopnost tkáně ≤ 60 %
|
|
VRM 2 – SkinEthic™ HCE EIT (pro protokoly pro kapaliny)
|
střední životaschopnost tkáně > 60 %
|
střední životaschopnost tkáně ≤ 60 %
|
|
VRM2 – SkinEthic™ HCE EIT (pro protokol pro pevné látky)
|
střední životaschopnost tkáně > 50 %
|
střední životaschopnost tkáně ≤ 50 %
|
|
|
45.
|
U zkoušené chemické látky s jednoznačným výsledkem by měla stačit jedna zkouška za použití alespoň dvou tkáňových replikátů. Avšak v případě hraničních výsledků, jako jsou například nesouhlasné výsledky opakovaných měření a/nebo průměrná hodnota životaschopnosti ve výši 60 ± 5 % (VRM1 a VRM2 pro protokol pro kapaliny) nebo 50 ± 5 % (VRM2 pro protokol pro pevné látky), by se měla zvážit druhá zkouška, jakož i třetí zkouška v případě nesouhlasných výsledků mezi prvními dvěma zkouškami.
|
|
46.
|
Pokud je to vhodné a zdůvodněné, lze pro konkrétní druhy směsí uvažovat různé mezní hodnoty percentuální životaschopnosti tkání rozlišující klasifikované zkoušené chemické látky od neklasifikovaných, aby se zvýšila celková účinnost zkušební metody pro tyto druhy směsí (viz odstavec 14). Srovnávací chemické látky mohou být užitečné pro hodnocení potenciálu vážného poškození očí / podráždění očí neznámými zkoušenými chemickými látkami nebo třídami produktů nebo pro hodnocení potenciálu relativní toxicity klasifikované chemické látky pro oči ve specifickém rozmezí pozitivních odpovědí.
|
ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV
Údaje
|
47.
|
Údaje získané ve zkoušce za použití jednotlivých tkáňových replikátů (např. hodnoty OD / plochy píku formazanu MTT a vypočítané údaje percentuální životaschopnosti tkání za zkoušenou chemickou látku a kontrolní látky a konečná predikce zkušební metody RhCE) by se měly vykazovat ve formě tabulky za každou zkoušenou chemickou látku, popřípadě včetně údajů z opakovaných zkoušek. Kromě toho by se za každou jednotlivou zkoušenou chemickou látku a kontrolní látku měla uvést střední percentuální životaschopnost tkáně a rozdíl životaschopnosti mezi dvěma tkáňovými replikáty (jestliže n = 2 replikované tkáně) nebo SD (jestliže n ≥ 3 replikované tkáně). Za každou zkoušenou chemickou látku je třeba uvést všechny pozorované interference zkoušené chemické látky s měřením formazanu MTT ve formě přímé redukce MTT anebo barevné interference.
|
ZÁVĚREČNÁ ZPRÁVA
|
48.
|
Závěrečná zpráva by měla obsahovat tyto informace:
Zkoušená chemická látka
|
|
Jednosložková látka:
|
—
|
Chemická identifikace, např. název (názvy) IUPAC nebo CAS, číslo (čísla) CAS, kód SMILES nebo InChI, strukturní vzorec a/nebo jiné identifikátory,
|
|
—
|
skupenství, těkavost, pH, logP, molekulová hmotnost, chemická třída a další důležité fyzikálně-chemické vlastnosti podstatné pro provedení studie, a to v rozsahu, v jakém jsou tyto informace dostupné,
|
|
—
|
čistota, případně chemická identita nečistot, je-li to prakticky proveditelné atd.,
|
|
—
|
případné ošetření před zkoušením (např. zahřívání, mletí),
|
|
—
|
podmínky skladování a stabilita, v rozsahu, v jakém jsou tyto informace dostupné.
|
|
|
|
Vícesložková látka, UVCB a směs:
|
—
|
Charakterizovaná v co možná největší míře např. chemickou identitou (viz výše), čistotou, kvantitativním výskytem a důležitými fyzikálně-chemickými vlastnostmi (viz výše) složek, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
—
|
skupenství a další důležité fyzikálně-chemické vlastnosti podstatné pro provedení studie, a to v rozsahu, v jakém jsou tyto informace dostupné,
|
|
—
|
čistota, případně chemická identita nečistot, je-li to prakticky proveditelné atd.,
|
|
—
|
případné ošetření před zkoušením (např. zahřívání, mletí),
|
|
—
|
podmínky skladování a stabilita, v rozsahu, v jakém jsou tyto informace dostupné.
|
|
Látky pozitivní a negativní kontroly
|
—
|
Chemická identifikace, např. název (názvy) IUPAC nebo CAS, číslo (čísla) CAS, kód SMILES nebo InChI, strukturní vzorec a/nebo jiné identifikátory,
|
|
—
|
skupenství, těkavost, molekulová hmotnost, chemická třída a další důležité fyzikálně-chemické vlastnosti podstatné pro provedení studie, a to v rozsahu, v jakém jsou tyto informace dostupné,
|
|
—
|
čistota, případně chemická identita nečistot, je-li to prakticky proveditelné atd.,
|
|
—
|
případné ošetření před zkoušením (např. zahřívání, mletí),
|
|
—
|
podmínky skladování a stabilita, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
—
|
zdůvodnění použití jiné negativní kontroly než ultračisté H2O, případně DPBS bez Ca2
+/Mg2
+,
|
|
—
|
zdůvodnění případného použití jiné pozitivní kontroly než čistého methyl-acetátu,
|
|
—
|
odkaz na dosavadní výsledky pozitivních a negativních kontrol prokazující vhodnost kritérií přijatelnosti provedení.
|
Informace týkající se zadavatele a zkušebního zařízení
|
—
|
Jméno a adresa zadavatele, zkušebního zařízení a vedoucího studie.
|
|
—
|
Použitý model tkáňové konstrukce RhCE a protokol (případně s uvedením zdůvodnění jejich volby)
|
Podmínky zkušební metody
|
—
|
Použitá tkáňová konstrukce RhCE, včetně čísla šarže,
|
|
—
|
(případně) vlnová délka a pásmová propustnost použitá pro kvantifikaci formazanu MTT a rozsah linearity měřicího zařízení (např. spektrofotometru),
|
|
—
|
popis metody použité pro kvantifikaci formazanu MTT,
|
|
—
|
případně popis použitého HPLC/UPLC spektrofotometrického systému,
|
|
—
|
úplné podklady ke konkrétní použité tkáňové konstrukci RhCE včetně jejího chování při zkoušce. Ty by měly zejména zahrnovat:
|
i)
|
řízení kvality životaschopnosti (dodavatel);
|
|
ii)
|
životnost za podmínek zkušební metody (dodavatel);
|
|
iii)
|
řízení kvality bariérové funkce;
|
|
iv)
|
morfologii, je-li k dispozici;
|
|
v)
|
reprodukovatelnost a prediktivní schopnost;
|
|
vi)
|
jiné údaje o řízení kvality tkáňové konstrukce RhCE, jsou-li k dispozici,
|
|
|
—
|
odkaz na dosavadní údaje tkáňové konstrukce RhCE. Měl by zejména zahrnovat: přijatelnost údajů z kontrol kvality s odkazem na dosavadní údaje o šaržích,
|
|
—
|
prohlášení, že zkoušející zařízení prokázalo způsobilost při používání zkušební metody před rutinním použitím otestováním vhodných chemických látek.
|
Kritéria přijatelnosti provedení zkoušky
|
—
|
Střední hodnoty pozitivních a negativních kontrol a rozpětí přijatelnosti založené na porovnání s historickými údaji,
|
|
—
|
přijatelná variabilita mezi tkáňovými replikáty pro pozitivní a negativní kontroly,
|
|
—
|
přijatelná variabilita mezi tkáňovými replikáty pro zkoušenou chemickou látku.
|
Postup zkoušky
|
—
|
Podrobnosti použitého zkušebního postupu,
|
|
—
|
dávky zkoušené chemické látky a použitých kontrolních látek,
|
|
—
|
doba a teplota expozice, doba poexpozičního ponoření a (případně) doba poexpoziční inkubace,
|
|
—
|
popis všech úprav zkušebního postupu,
|
|
—
|
přehled kontrol použitých pro látky, které přímo redukují MTT, případně pro zbarvující zkoušené chemické látky,
|
|
—
|
počet tkáňových replikátů použitých na jednu zkoušenou chemickou látku a kontroly (pozitivní kontrola, negativní kontrola a NSMTT, případně NSCliving a NSCkilled).
|
Výsledky
|
—
|
Tabulka s údaji o jednotlivých zkoušených chemických látkách a kontrolách pro každé provedení zkoušky (případně včetně opakovaných experimentů) a každé měření replikátů včetně hodnoty OD nebo plochy píku formazanu MTT, percentuální životaschopnosti tkáně, střední percentuální životaschopnosti tkáně, rozdílu mezi replikáty nebo směrodatnými odchylkami a konečné predikce,
|
|
—
|
případně výsledky kontrol použitých pro přímá redukční činidla MTT anebo zbarvené zkoušené chemické látky včetně hodnot OD nebo plochy píku formazanu MTT, %NSMTT, %NSCliving, %NSCkilled, rozdílu mezi tkáňovými replikáty nebo směrodatnými odchylkami, konečné korigované percentuální životaschopnosti tkáně a konečné predikce,
|
|
—
|
výsledky získané u zkoušených chemických látek a kontrolních látek ve vztahu k definovaným kritériím přijatelnosti provedení zkoušky,
|
|
—
|
popis ostatních pozorovaných účinků, např. zbarvení tkání zbarvenou zkoušenou chemickou látkou.
|
Diskuse o výsledcích
Závěr
|
LITERATURA
|
(1)
|
UN (2015). United Nations Globally Harmonized System of Classification and Labelling of Chemicals (GHS). ST/SG/AC.10/30/Rev.6, Sixth Revised Edition, New York and Geneva: United Nations. Dostupné na webových stránkách: http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(2)
|
Kapitola B.5 této přílohy, Akutní dráždivé/leptavé účinky na oči.
|
|
(3)
|
Kapitola B.47 této přílohy, Zkušební metoda pro zákal a propustnost rohovky u skotu pro zjišťování i) chemických látek vyvolávajících vážné poškození očí a ii) chemických látek, které není nutné klasifikovat jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí.
|
|
(4)
|
Kapitola B.48 této přílohy, Zkušební metoda odděleného kuřecího oka pro zjišťování i) chemických látek vyvolávajících vážné poškození očí a ii) chemických látek, které není nutné klasifikovat.
|
|
(5)
|
Kapitola B.61 této přílohy, Zkušební metoda průniku fluoresceinu ke zjišťování látek s leptavými a silně dráždivými účinky na oči.
|
|
(6)
|
Kapitola B.68 této přílohy, Zkušební metoda využívající krátkodobou expozici in vitro pro zjišťování i) chemických látek vyvolávajících vážné poškození očí a ii) chemických látek, které není nutné klasifikovat jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí.
|
|
(7)
|
Freeman, S.J., Alépée N., Barroso, J., Cole, T., Compagnoni, A., Rubingh, C., Eskes, C., Lammers, J., McNamee, P., Pfannenbecker, U., Zuang, V. (2010). Prospective Validation Study of Reconstructed Human Tissue Models for Eye Irritation Testing. ALTEX 27, Special Issue 2010,261-266.
|
|
(8)
|
EC EURL-ECVAM (2014). The EURL ECVAM - Cosmetics Europe prospective validation study of Reconstructed human Cornea-like Epithelium (RhCE)-based test methods for identifying chemicals not requiring classification and labelling for serious eye damage/eye irritation: Validation Study Report. EUR 28125 EN; doi:10.2787/41680. Dostupné na webových stránkách: http://publications.jrc.ec.europa.eu/repository/handle/JRC100280.
|
|
(9)
|
EURL ECVAM Science Advisory Committee (2014). ESAC Opinion on the EURL ECVAM Eye Irritation Validation Study (EIVS) on EpiOcular™ EIT and SkinEthic™ HCE and a related Cosmetics Europe study on HPLC/UPLC-spectrophotometry as an alternative endpoint detection system for MTT-formazan. ESAC Opinion No 2014-03 of 17 November 2014; EUR 28 173 EN; doi: LX 4610,2787/043697. Dostupné na webových stránkách: http://publications.jrc.ec.europa.eu/repository/handle/JRC103702.
|
|
(10)
|
Alépée, N., Leblanc, V., Adriaens, E., Grandidier, M.H., Lelièvre, D, Meloni, M., Nardelli, L., Roper, C.S, Santirocco, E., Toner, F., Van Rompay, A., Vinall, J., Cotovio, J. (2016). Multi-laboratory validation of SkinEthic HCE test method for testing serious eye damage/eye irritation using liquid chemicals. Toxicol. In Vitro 31, 43-53.
|
|
(11)
|
Alépée, N., Adriaens, E., Grandidier, M.H., Meloni, M., Nardelli, L., Vinall, C.J., Toner, F., Roper, C.S, Van Rompay, A.R., Leblanc, V., Cotovio, J. (2016). Multi-laboratory evaluation of SkinEthic HCE test method for testing serious eye damage/eye irritation using solid chemicals and overall performance of the test method with regard to solid and liquid chemicals testing. Toxicol. Chem. In Vitro 34, 55-70.
|
|
(12)
|
EURL ECVAM Science Advisory Committee (2016). ESAC Opinion on the SkinEthic™ Human Corneal Epithelium (HCE) Eye Irritation Test (EIT). ESAC Opinion No 2016-02 of 24 June 2016; EUR 28175 EN; doi: LX 4610,2787/390390. Dostupné na webových stránkách: http://publications.jrc.ec.europa.eu/repository/handle/JRC103704.
|
|
(13)
|
EC EURL-ECVAM (2016). Recommendation on the Use of the Reconstructed human Cornea-like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Serious Eye Damage/Eye Irritation According to UN GHS. (Návrh).
|
|
(14)
|
Draize, J.H., Woodard, G., Calvery, H.O. (1944). Methods for the Study of Irritation and Toxicity of Substances Applied Topically to the Skin and Mucous Membranes. Journal of Pharmacol. and Exp. Therapeutics 82, 377-390.
|
|
(15)
|
Scott, L., Eskes, C., Hoffmann, S., Adriaens, E., Alépée, N., Bufo, M., Clothier, R., Facchini, D., Faller, C., Guest, R., Harbell, J., Hartung, T., Kamp, H., Le Varlet, B., Meloni, M., McNamee, P., Osborne, R., Pape, W., Pfannenbecker, U., Prinsen, M., Seaman, C., Spielman, H., Stokes, W., Trouba, K., Van den Berghe, C., Van Goethem, F., Vassallo, M., Vinardell, P., Zuang, V. (2010). A Proposed Eye Irritation Testing Strategy to Reduce and Replace In Vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. In Vitro 24, 1-9.
|
|
(16)
|
Mosmann, T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(17)
|
OECD (2016). Series on Testing and Assessment No 216: Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Reconstructed Human Cornea-Like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Eye Irritation or Serious Eye Damage, Based on the Validated Reference Methods EpiOcular™ EIT and SkinEthic™ HCE EIT described in TG 492. Organisation for Economic Cooperation and Development, Paris.
|
|
(18)
|
OECD (2005). Series on Testing and Assessment No 34: Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Organisation for Economic Cooperation and Development, Paris.
|
|
(19)
|
Kaluzhny, Y., Kandárová, H., Hayden, P., Kubilus, J., d’Argembeau-Thornton, L., Klausner, M. (2011). Development of the EpiOcular™ Eye Irritation Test for Hazard Identification and Labelling of Eye Irritating Chemicals in Response to the Requirements of the EU Cosmetics Directive and REACH Legislation. Altern. Lab. Anim. 39, 339-364.
|
|
(20)
|
Nguyen, D.H., Beuerman, R.W., De Wever, B., Rosdy, M. (2003). Three-dimensional construct of the human corneal epithelium for in vitro toxicology. In: Salem, H., Katz, S.A. (Eds), Alternative Toxicological Methods, CRC Press, pp. 147-159.
|
|
(21)
|
Pfannenbecker, U., Bessou-Touya, S., Faller, C., Harbell, J., Jacob, T., Raabe, H., Tailhardat, M., Alépée, N., De Smedt, A., De Wever, B., Jones, P., Kaluzhny, Y., Le Varlet, B., McNamee, P., Marrec-Fairley, M., Van Goethem, F. (2013). Cosmetics Europe multi-laboratory pre-validation of the EpiOcular™ reconstituted Human Tissue Test Method for the Prediction of Eye Irritation. Toxicol. Chem. In Vitro 27, 619-626.
|
|
(22)
|
Alépée, N., Bessou-Touya, S., Cotovio, J., de Smedt, A., de Wever, B., Faller, C., Jones, P., Le Varlet, B., Marrec-Fairley, M., Pfannenbecker, U., Tailhardat, M., van Goethem, F., McNamee, P. (2013). Cosmetics Europe Multi-Laboratory Pre-Validation of the SkinEthic™ Reconstituted Human Corneal Epithelium Test Method for the Prediction of Eye Irritation. Toxicol. Chem. In Vitro 27, 1476-1488.
|
|
(23)
|
Kolle, S.N., Moreno, M.C.R., Mayer, W., van Cott, A., van Ravenzwaay, B., Landsiedel, R. (2015). The EpiOcular™ Eye Irritation Test is the Method of Choice for In Vitro Eye Irritation Testing of Agrochemical Formulations: Correlation Analysis of EpiOcular™ Eye Irritation Test and BCOP Test Data to UN GHS, US EPA and Brazil ANIVSA Classifications. Altern. Lab. Anim. 43, 1-18.
|
|
(24)
|
Adriaens, E., Barroso, J., Eskes, C., Hoffmann, S., McNamee, P., Alépée, N., Bessou-Touya, S., De Smedt, A., De Wever, B., Pfannenbecker, U., Tailhardat, M., Zuang, V. (2014). Retrospective Analysis of the Draize Test for Serious Eye Damage/Eye Irritation: Importance of Understanding the In Vivo Endpoints Under UN GHS/EU CLP for the Development and Evaluation of In Vitro Test Methods. Arch. Toxicol. 88, 701-723.
|
|
(25)
|
Barroso, J., Pfannenbecker, U., Adriaens, E., Alépée, N., Cluzel, M., De Smedt, A., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Templier, M., McNamee, P. (2017). Cosmetics Europe compilation of historical serious eye damage/eye irritation in vivo data analysed by drivers of classification to support the selection of chemicals for development and evaluation of alternative methods/strategies: the Draize eye test Reference Database (DRD). Arch. Toxicol. 91, 521-547.
|
|
(26)
|
Meloni, M., De Servi, B., Marasco, D., Del Prete, S. (2011). Molecular mechanism of ocular surface damage: Application to an in vitro dry eye model on human corneal epithelium. Molecular Vision 17, 113-126.
|
|
(27)
|
Hackett, R.B., McDonald, T.O. (1991). Eye Irritation. In Advances in Modern Toxicology: Dermatoxicology Marzulli F.N.and Maibach H.I. (Eds.), 4th Edition, pp. 749–815. Washington, DC, USA: Hemisphere Publishing Corporation.
|
|
(28)
|
Fox, D.A., Boyes, W.K. (2008). Toxic Responses of the Ocular and Visual System. In Cassaret and Doull’s Toxicology: The Basic Science of Poisons Klaassen C.D.(Ed.), 7th Edition, pp. 665–697. Withby, ON, Canada: McGraw-Hill Ryerson.
|
|
(29)
|
Jester, J.V., Li, H.F., Petroll, W.M., Parker, R.D., Cavanagh, H.D., Carr, G.J., Smith, B., Maurer, J.K. (1998). Area and Depth of Surfactant Induced Corneal Injury Correlates with Cell Death. Invest. Ophthalmol. Vis. Sci. 39, 922-936.
|
|
(30)
|
Maurer, J.K., Parker, R.D., Jester, J.V. (2002). Extent of Corneal Injury as the Mechanistic Basis for Ocular Irritation: Key Findings and Recommendations for the Development of Alternative Assays. Reg. Tox. Pharmacol. 36, 106-117.
|
|
(31)
|
Jester, J.V., Li, L., Molai, A., Maurer, J.K. (2001). Extent of Corneal Injury as a Mechanistic Basis for Alternative Eye Irritation Tests. Toxicol. Chem. In Vitro 15, 115–130.
|
|
(32)
|
Jester, J.V., Petroll, W.M., Bean, J., Parker, R.D., Carr, G.J., Cavanagh, H.D., Maurer, J.K. (1998). Area and Depth of Surfactant-Induced Corneal Injury Predicts Extent of Subsequent Ocular Responses. Invest. Ophthalmol. Vis. Sci. 39, 2610-2625.
|
|
(33)
|
Jester, J.V. (2006). Extent of Corneal Injury as a Biomarker for Hazard Assessment and the Development of Alternative Models to the Draize Rabbit Eye Test. Cutan. Ocul. Toxicol. 25, 41-54.
|
|
(34)
|
EpiOcular™ EIT SOP, Version 8 (March 05, 2013). EpiOcular™ EIT for the Prediction of Acute Ocular Irritation of Chemicals. Dostupné na webových stránkách: [https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index].
|
|
(35)
|
SkinEthic™ HCE EIT SOP, Version 1. (July 20, 2015). SkinEthic™ HCE Eye Irritation Test (EITL for Liquids, EITS for Solids) for the Prediction of Acute Ocular Irritation of Chemicals. Dostupné na webových stránkách: https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index
|
|
(36)
|
Alépée, N., Barroso, J., De Smedt, A., De Wever, B., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Pfannenbecker, U., Tailhardat, M., Templier, M., McNamee, P. (2015). Use of HPLC/UPLC-Spectrophotometry for Detection of Formazan in In Vitro Reconstructed Human Tissue (RhT)-Based Test Methods Employing the MTT-Reduction Assay to Expand their Applicability to Strongly Coloured Test Chemicals. Toxicol. In Vitro 29, 741-761.
|
|
(37)
|
Kaluzhny, Y., Kandárová, H., Handa, Y., DeLuca, J., Truong, T., Hunter, A., Kearney, P., d’Argembeau-Thor135nton, L., Klausner, M. (2015). EpiOcular™ Eye Irritation Test (EIT) for Hazard Identification and Labeling of Eye Irritating Chemicals: Protocol Optimization for Solid Materials and Extended Shipment Times. Altern. Lab Anim. 43, 101-127.
|
|
(38)
|
US FDA (2001). Guidance for Industry: Bioanalytical Method Validation. U.S. Department of Health and Human Services, Food and Drug Administration. May 2001. Dostupné na webových stránkách: http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf.
|
|
(39)
|
OECD (2017). Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Series on Testing and Assessment No. 263. ENV Publications, Organisation for Economic Cooperation and Development, Paris.
|
Dodatek 1
DEFINICE
Přesnost
: Přesnost shody mezi výsledky zkušební metody a přijatými referenčními hodnotami. Je to míra funkčnosti zkušební metody a jeden z aspektů její „relevantnosti“. Tento pojem se často používá místo pojmu „soulad“ a rozumí se jím podíl správných výsledků zkušební metody (18).
Srovnávací chemická látka
: Látka použitá jako norma pro srovnání se zkoušenou látkou. Srovnávací látka by měla mít tyto vlastnosti: i) neměnný a spolehlivý zdroj (zdroje) pro její určení a charakterizaci; ii) podobnost se strukturou, funkcí anebo chemickou či produktovou třídou se zkoušenou chemickou látkou (látkami); iii) známé fyzikálněchemické vlastnosti; iv) podklady ke známým účinkům; a v) známé účinky v rozsahu žádoucí reakce.
Přístup „zdola nahoru“
: Postupný přístup používaný u zkoušených chemických látek, u nichž se předpokládá, že je není nutné klasifikovat a označovat jako látky mající dráždivé účinky na oči nebo vyvolávající vážné poškození očí, který začíná odlišením chemických látek, u nichž není nutná klasifikace a označení (negativní výsledek), od ostatních chemických látek (pozitivní výsledek).
Chemická látka
: Látka nebo směs.
Soulad
: Viz „přesnost“.
Rohovka
: Průhledná přední část oční bulvy, která zahrnuje duhovku a zornici a propouští světlo dovnitř oka.
CV
: Variační koeficient.
Dev
: Odchylka.
EIT
: Zkouška dráždivosti pro oči.
EURL-ECVAM
: Referenční laboratoř EU pro alternativy ke zkouškám na zvířatech.
Podráždění očí
: Vyvolání změn na oku po aplikaci zkoušené chemické látky na přední plochu oka, které jsou plně vratné do 21 dní od aplikace. Tento pojem je zaměnitelný s výrazy „vratné účinky na oko“ a „kategorie 2 GHS OSN/CLP“.
ET50
: Doba expozice potřebné k tomu, aby se životaschopnost tkáně snížila o 50 % po aplikaci referenční chemické látky v pevně stanovené koncentraci.
Míra falešné negativity
: Podíl všech pozitivních látek falešně zjištěných zkušební metodou jako negativní. Je to jeden z ukazatelů výsledku zkušební metody.
Míra falešné pozitivity
: Podíl všech negativních látek falešně zjištěných zkušební metodou jako pozitivní. Je to jeden z ukazatelů výsledku zkušební metody.
Nebezpečnost
: Základní charakteristika látky nebo situace, která má potenciál vyvolat nepříznivé účinky, jsou-li organismus, systém nebo (sub)populace vystaveny této látce.
HCE
: SkinEthic™ Human Corneal Epithelium (lidský rohovkový epitel).
HPLC
: Vysokoúčinná kapalinová chromatografie
IC50
: Koncentrace, při níž referenční chemická látka snižuje životaschopnost tkání o 50 % po pevně stanovené době expozice (např. 30minutová expozice SDS).
Nekonečná dávka
: Množství zkušební látky nanesené na tkáňovou konstrukci RhCE, které přesahuje množství potřebné k úplnému a rovnoměrnému pokrytí epitelového povrchu.
Nevratné účinky na oči
: Viz „vážné poškození očí“.
LLOQ
: Dolní mez kvantifikace.
logP
: Logaritmus rozdělovacího koeficientu oktanol/voda
Směs
: Směs nebo roztok složený ze dvou nebo více látek.
Jednosložková látka
: Látka definovaná svým kvantitativním složením, v níž je přítomna jedna hlavní složka v míře alespoň 80 % hmot.
Vícesložková látka
: Látka definovaná svým kvantitativním složením, v níž je přítomna více než jedna hlavní složka v koncentraci ≥ 10 % hmot. a < 80 % hmot. Vícesložková látka je výsledkem výrobního procesu. Rozdíl mezi směsí a vícesložkovou látkou spočívá v tom, že směs se získá smísením dvou nebo více látek bez chemické reakce. Vícesložková látka je výsledkem chemické reakce.
MTT
: 3-(4,5-dimethylthiazol-2-yl)-2,5-difenyl-2H-tetrazolium-bromid; thiazolylová modř tetrazolium-bromid.
Negativní kontrola
: Vzorek obsahující všechny složky zkušebního systému a látku, o níž je známo, že nezpůsobuje pozitivní odpověď ve zkušebním systému. Tento vzorek se zpracovává se vzorky zkoušené chemické látky a jinými kontrolními vzorky a používá se na stanovení 100 % životaschopnosti.
Neklasifikované
: Chemické látky, které nejsou klasifikované jako látky mající dráždivé účinky na oči (kategorie 2 GHS OSN/CLP, kategorie 2A nebo 2B GHS OSN) nebo jako látky vyvolávající vážné poškození očí (kategorie 1 GHS OSN/CLP). Tento pojem je zaměnitelný s pojmem „bez kategorie GHS OSN/CLP“.
NSCkilled
: Nespecifická barva u neživých tkání.
NSCliving
: Nespecifická barva u živých tkání.
NSMTT
: Nespecifická redukce MTT.
OD
: Optická hustota.
Standardy funkčnosti
: Standardy založené na validované referenční metodě, která je považována za vědecky platnou, jež slouží jako základ pro hodnocení srovnatelnosti navrhované zkušební metody, která je mechanicky a funkčně podobná. Zahrnují se: i) zásadní složky zkušební metody; ii) minimální seznam srovnávacích látek vybraných z chemických látek používaných k prokázání přijatelné funkčnosti validované referenční metody a iii) srovnatelné hladiny přesnosti a spolehlivosti založené na hodnotách získaných u validované referenční metody, jež by navrhovaná zkušební metoda měla prokázat při vyhodnocení za použití minimálního seznamu srovnávacích látek (18).
Pozitivní kontrola
: Vzorek obsahující všechny složky zkušebního systému a látku, o níž je známo, že způsobuje pozitivní odpověď ve zkušebním systému. Tento vzorek se zpracovává se vzorky zkoušené chemické látky a jinými kontrolními vzorky. Aby se zajistilo, že proměnlivost v reakci na pozitivní kontrolu v čase bude možné vyhodnotit, rozsah pozitivní reakce by neměl být nadměrný.
Relevantnost
: Popis vztahu zkoušky k účinku, který je předmětem zájmu, a také toho, zda je zkouška smysluplná a použitelná pro konkrétní účel. Vyjadřuje, do jaké míry zkouška správně měří nebo predikuje biologický účinek, který je předmětem zájmu. Relevantnost zahrnuje také posouzení přesnosti (shody) zkušební metody (18).
Spolehlivost
: Měření rozsahu, v jakém může být zkušební metoda časem reprodukovatelná v laboratořích a mezi laboratořemi, když se provádí s použitím stejného protokolu. Hodnotí se to výpočtem laboratorní a mezilaboratorní reprodukovatelnosti a laboratorní opakovatelnosti (18).
Náhradní zkouška
: Zkouška, která je koncipována tak, aby nahradila běžně používanou a uznávanou zkoušku, která slouží k určení nebezpečnosti a/nebo posouzení rizika a u níž je zjištěno, že poskytuje stejnou nebo lepší úroveň ochrany zdraví lidí nebo zvířat či případně ochrany životního prostředí ve srovnání s uznávanou zkouškou ve všech možných zkušebních situacích a pro všechny zkoušené chemické látky (18).
Reprodukovatelnost
: Shoda mezi výsledky získanými při opakovaném zkoušení téže zkoušené chemické látky za použití stejného zkušebního protokolu (viz „spolehlivost“) (18).
Vratné účinky na oči
: Viz „dráždivé účinky na oči“.
RhCE
: Rekonstruovaný epitel podobný lidské rohovce.
Provedení zkoušky
: Jedna či více zkoušených chemických látek se zkouší současně s negativní kontrolou a s pozitivní kontrolou.
SD
: Směrodatná odchylka.
Citlivost
: Podíl všech pozitivních/aktivních zkoušených látek, které jsou na základě zkoušky správně klasifikovány. Je měřítkem přesnosti zkušební metody, která poskytuje kategorické výsledky, a je důležitým prvkem při hodnocení relevantnosti zkušební metody (18).
Vážné poškození očí
: Tvorba poškození tkání v oku nebo závažné fyzikální slábnutí vidění po aplikaci zkoušené látky na přední plochu oka, které není plně vratné do 21 dní od aplikace. Tento pojem je zaměnitelný s výrazy „nevratné účinky na oči“ a „kategorie 1 GHS OSN a CLP“.
Standardní pracovní postupy (SOP)
: Formální písemné postupy, které podrobně popisují, jak se mají vykonávat konkrétní rutinní laboratorní operace a operace při konkrétních zkouškách. Jsou vyžadovány podle správné laboratorní praxe.
Specifičnost
: Podíl všech negativních/neaktivních zkoušených látek, které jsou na základě zkoušky správně klasifikovány. Je měřítkem přesnosti zkušební metody, která poskytuje kategorické výsledky, a je důležitým prvkem při hodnocení relevantnosti zkušební metody (18).
Látka
: Chemický prvek a jeho sloučeniny v přírodním stavu nebo získané výrobním postupem, včetně jakýchkoliv aditiv potřebných pro zachování stability výrobku a všech nečistot pocházejících z použitého postupu, ale s vyloučením všech rozpouštědel, která je možno oddělit bez ovlivnění stability dané látky nebo změny jejího složení.
Zkouška
: Jedna zkoušená chemická látka, která byla souběžně zkoušena na minimálně dvou tkáňových replikátech příslušným standardním pracovním postupem.
Životaschopnost tkáně
: Parametr, kterým se měří celková aktivita buněčné populace v rekonstruované tkáni jako její schopnost redukovat vitální barvivo MMT a který v závislosti na měřeném jevu a na použitém uspořádání zkoušky koreluje s celkovým počtem a/nebo vitalitou živých buněk.
Přístup „shora dolů“
: Postupný přístup používaný u chemických látek, u nichž se předpokládá, že způsobují vážné poškození očí, který začíná odlišením chemických látek vyvolávajících vážné poškození očí (pozitivní výsledek) od jiných chemických látek (negativní výsledek).
Zkoušená chemická látka
: Jakákoli látka nebo směs zkoušená touto zkušební metodou.
Strategie postupného zkoušení
: Strategie zkoušení po krocích, při které se zkušební metody využívají postupně. Všechny existující informace o zkoušené chemické látce se přezkoumávají na každém stupni s použitím hodnocení závažnosti důkazů, aby se zjistilo, zda jsou k dispozici dostačující informace pro rozhodnutí o klasifikaci nebezpečnosti, než se postoupí do dalšího stupně strategie. Pokud potenciál/intenzitu nebezpečnosti zkoušené chemické látky lze určit na základě existujících informací na daném stupni, další zkoušení není nutné (18).
ULOQ
: Horní mez kvantifikace.
Globálně harmonizovaný systém klasifikace a označování chemických látek Organizace spojených národů (GHS OSN)
: Systém navrhující klasifikaci chemických látek (látek a směsí) podle standardizovaných typů a úrovní fyzikálních, zdravotních a environmentálních nebezpečí a řešící příslušné komunikační prvky, jako jsou piktogramy, signální slova, výroky o nebezpečí, výroky o bezpečnostních opatřeních a bezpečnostní datové listy, které obsahují informace o jejich nežádoucích účincích s ohledem na ochranu lidí (včetně zaměstnavatelů, zaměstnanců, přepravců, spotřebitelů a osob reagujících na havárie) a životního prostředí (1).
Kategorie 1 podle GHS OSN/CLP
: Viz „vážné poškození očí“.
Kategorie 2 podle GHS OSN/CLP
: Viz „dráždivé účinky na oči“.
Bez kategorie GHS OSN/CLP
: Chemické látky, které nesplňují požadavky na klasifikaci v kategorii 1 nebo 2 GHS OSN/CLP (2A nebo 2B GHS OSN). Tento pojem je zaměnitelný s výrazem „neklasifikované“.
UPLC
: Vysokoúčinná kapalinová chromatografie.
UVCB
: Látky s neznámým nebo proměnlivým složením, komplexní reakční produkty nebo biologický materiál.
Validovaná zkušební metoda
: Zkušební metoda, u níž se má za to, že je dostatečně relevantní a spolehlivá pro určitý účel, a která je založena na vědecky spolehlivých zásadách. Zkušební metoda není nikdy validovaná v absolutním smyslu, ale pouze pro určitý konkrétní účel (18).
Validovaná zkušební metoda
: Zkušební metoda, u níž byly provedeny validační studie k určení její relevantnosti (včetně přesnosti) a spolehlivosti pro konkrétní účel. Je důležité si uvědomit, že validovaná zkušební metoda nemusí mít dostatečnou funkčnost, pokud jde o přesnost a spolehlivost, aby mohla být shledána přijatelnou pro navrhovaný účel (18).
VRM
: Validovaná referenční metoda.
VRM1
: EpiOcular™ EIT se nazývá validovaná referenční metoda 1.
VRM2
: SkinEthic™ HCE EIT se nazývá validovaná referenční metoda 2.
Váha důkazů
: Postup zvažování silných a slabých stránek různých informací v dosahování a podpoře závěru týkajícího se potenciálu nebezpečnosti zkoušené látky.
Dodatek 2
HLAVNÍ SLOŽKY ZKOUŠEK RHCE VALIDOVANÝCH PRO STANOVENÍ CHEMICKÝCH LÁTEK, KTERÉ NEVYŽADUJÍ KLASIFIKACI A OZNAČENÍ JAKO LÁTKY MAJÍCÍ DRÁŽDIVÉ ÚČINKY NA OČI NEBO VYVOLÁVAJÍCÍ VÁŽNÉ POŠKOZENÍ OČÍ
|
Složky zkoušky
|
EpiOcular™ EIT
(VRM 1)
|
SkinEthic™ HCE EIT
(VRM 2)
|
|
Protokoly
|
Kapaliny
(pipetovatelné při 37 ± 1°C nebo nižších teplotách po dobu 15 min)
|
Pevné látky
(nepipetovatelné)
|
Kapaliny a viskózní látky
(pipetovatelné)
|
Pevné látky
(nepipetovatelné)
|
|
Povrch modelu
|
0,6 cm2
|
0,6 cm2
|
0,5 cm2
|
0,5 cm2
|
|
Počet tkáňových replikátů
|
nejméně 2
|
nejméně 2
|
nejméně 2
|
nejméně 2
|
|
Předběžná kontrola na barevnou interferenci
|
50 μl + 1 ml H2O po dobu 60 min. při 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH (nezbarvené zkoušené chemické látky) nebo 50 μl + 2 ml isopropylalkoholu míchaného po dobu 2–3 hod. při pokojové teplotě (zbarvené zkoušené chemické látky)
|
 jestliže je OD zkoušené chemické látky při vlnové délce 570 ± 20 nm po odečtení OD isopropylalkoholu nebo vody > 0,08 (což odpovídá přibližně 5 % střední OD negativní kontroly), je třeba provést upravené kontroly na živé tkáni. jestliže je OD zkoušené chemické látky při vlnové délce 570 ± 20 nm po odečtení OD isopropylalkoholu nebo vody > 0,08 (což odpovídá přibližně 5 % střední OD negativní kontroly), je třeba provést upravené kontroly na živé tkáni.
|
|
50 mg + 1 ml H2O po dobu 60 min. při 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH (nezbarvené zkoušené chemické látky)
a/nebo
50 mg + 2 ml isopropylalkoholu míchaného po dobu 2–3 hod. při pokojové teplotě (zbarvené i nezbarvené zkoušené chemické látky)
|
 jestliže je OD zkoušené chemické látky při vlnové délce 570 ± 20 nm po odečtení OD isopropylalkoholu nebo vody > 0,08 (což odpovídá přibližně 5 % střední OD negativní kontroly), je třeba provést upravené kontroly na živé tkáni. jestliže je OD zkoušené chemické látky při vlnové délce 570 ± 20 nm po odečtení OD isopropylalkoholu nebo vody > 0,08 (což odpovídá přibližně 5 % střední OD negativní kontroly), je třeba provést upravené kontroly na živé tkáni.
|
|
10 μl + 90 μl H2O mícháno po dobu 30 ± 2 min. při pokojové teplotě (RT, 18–28 °C)
|
 jestliže se zkoušená chemická látka zbarví, je třeba provést upravené kontroly na živé tkáni jestliže se zkoušená chemická látka zbarví, je třeba provést upravené kontroly na živé tkáni
|
|
10 mg + 90 μl H2O mícháno po dobu 30 ± 2 min. při pokojové teplotě
|
 jestliže se zkoušená chemická látka zbarví, je třeba provést upravené kontroly na živé tkáni jestliže se zkoušená chemická látka zbarví, je třeba provést upravené kontroly na živé tkáni
|
|
|
Předběžná kontrola na přímou redukci MTT
|
50 μl + 1 ml MTT 1 mg/ml roztoku po dobu 180 ± 15 min při 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
 jestliže roztok zmodrá/zfialoví, je třeba provést upravené kontroly na tkáni usmrcené zmrazením jestliže roztok zmodrá/zfialoví, je třeba provést upravené kontroly na tkáni usmrcené zmrazením
(jako negativní kontrola se používá 50 μl sterilní deionizované vody v roztoku MTT)
|
|
50 mg + 1 ml MTT 1 mg/ml roztoku po dobu 180 ± 15 min při 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
 jestliže roztok zmodrá/zfialoví, je třeba provést upravené kontroly na tkáni usmrcené zmrazením jestliže roztok zmodrá/zfialoví, je třeba provést upravené kontroly na tkáni usmrcené zmrazením
(jako negativní kontrola se používá 50 μl sterilní deionizované vody v roztoku MTT)
|
|
30 μl + 300 μl MTT 1 mg/ml roztoku po dobu 180 ± 15 min při 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
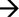 jestliže roztok zmodrá/zfialoví, je třeba provést upravené kontroly na tkáni usmrcené vodou jestliže roztok zmodrá/zfialoví, je třeba provést upravené kontroly na tkáni usmrcené vodou
(jako negativní kontrola se používá 30 μl sterilní deionizované vody v roztoku MTT)
|
|
30 mg + 300 μl MTT 1 mg/ml roztoku po dobu 180 ± 15 min při 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
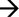 jestliže roztok zmodrá/zfialoví, je třeba provést upravené kontroly na tkáni usmrcené vodou jestliže roztok zmodrá/zfialoví, je třeba provést upravené kontroly na tkáni usmrcené vodou
(jako negativní kontrola se používá 30 μl sterilní deionizované vody v roztoku MTT)
|
|
|
Předběžné ošetření
|
20 μl Ca2+/Mg2+bez DPBS
po dobu 30 ± 2 min při 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH, chráněno před světlem.
|
20 μl Ca2+/Mg2+ – bez DPBS
po dobu 30±2 min při 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH, chráněno před světlem.
|
-
|
-
|
|
Experimentální dávky a aplikace
|
50 μl (83,3 μl/cm2)
|
50 mg (83,3 mg/cm2) pomocí kalibrovaného nástroje (např. zarovnaná kalibrovaná odměrka s obsahem 50 mg chloridu sodného).
|
10 μl Ca2+/Mg2+ bez DPBS + 30 ± 2 μl (60 μl/cm2)
U viskózních látek použijte nylonovou síťku
|
30 μl Ca2+/Mg2+bez DPBS + 30 ± 2 mg (60 mg/cm2)
|
|
Doba a teplota expozice
|
30 min (± 2 min)
v kultivačním médiu
při 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
6 hodin (± 0,25 hod.)
v kultivačním médiu
při 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
30 min (± 2 min)
v kultivačním médiu
při 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
4 hodiny (± 0,1 hod.)
v kultivačním médiu
při 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
|
Propláchnutí při pokojové teplotě
|
3krát ve 100 ml DPBS bez Ca2+/Mg2+
|
3krát ve 100 ml DPBS bez Ca2+/Mg2+
|
20 ml DPBS bez Ca2+/Mg2+
|
25 ml DPBS bez Ca2+/Mg2+
|
|
Ponoření po expozici
|
12 min (± 2 min) při pokojové teplotě v kultivačním médiu
|
25 min (± 2 min) při pokojové teplotě v kultivačním médiu
|
30 min (± 2 min) při 37 oC, 5 % CO2, 95 % RH v kultivačním médiu
|
30 min (± 2 min) při pokojové teplotě v kultivačním médiu
|
|
Inkubace po expozici
|
120 min (± 15 min) v kultivačním médiu při 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
18 hod. (± 0,25 hod.) v kultivačním médiu při 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
žádná
|
18 hod. (± 0,5 hod.) v kultivačním médiu při 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
|
Negativní kontrola
|
50 μl H2O
Zkoušeno současně
|
50 μl H2O
Zkoušeno současně
|
DPBS bez 30 ± 2μl Ca2+/Mg2
Zkoušeno současně
|
DPBS bez 30 ± 2μl Ca2+/Mg2
Zkoušeno současně
|
|
Pozitivní kontrola
|
50 μl methylacetátu
Zkoušeno současně
|
50 μl methylacetátu
Zkoušeno současně
|
30 ± 2μl methylacetátu
Zkoušeno současně
|
30 ± 2μl methylacetátu
Zkoušeno současně
|
|
Roztok MTT
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
|
Doba inkubace MTT a teplota
|
180 min (± 15 min) při 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
180 min (± 15 min) při 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
180 min (± 15 min) při 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
180 min (± 15 min) při 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH
|
|
Extrakční rozpouštědlo
|
2 ml isopropylalkoholu
(extrakce z horní a dolní části vložky propíchnutím tkáně)
|
2 ml isopropylalkoholu
(extrakce z horní a dolní části vložky propíchnutím tkáně)
|
1,5 ml isopropylalkoholu
(extrakce z horní a dolní části vložky)
|
1,5 ml isopropylalkoholu
(extrakce z dolní části vložky)
|
|
Doba a teplota extrakce
|
2–3 hod. s protřepáváním (~120 ot./min.) při pokojové teplotě nebo přes noc při 4–10°C
|
2–3 hod. s protřepáváním (~120 ot./min.) při pokojové teplotě nebo přes noc při 4–10°C
|
4 hod. s protřepáváním (~120 ot./min.) při pokojové teplotě nebo alespoň přes noc bez protřepávání při 4–10°C
|
Alespoň 2 hod. s protřepáváním (~120 rpm) při pokojové teplotě
|
|
Odečet OD
|
570 nm (550–590 nm)
bez referenčního filtru
|
570 nm (550–590 nm)
bez referenčního filtru
|
570 nm (540–600 nm)
bez referenčního filtru
|
570 nm (540–600 nm)
bez referenčního filtru
|
|
Kontrola kvality tkání
|
Expozice za použití 100 μl 0,3 % (obj.) přípravku Triton X-100
12,2 min ≤ ET50 ≤ 37,5 min
|
Expozice za použití 100 μl 0,3 % (obj.) přípravku Triton X-100
12,2 min ≤ ET50 ≤ 37,5 min
|
30 min. expozice za použití SDS (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,5 mg/ml
|
30 min. expozice za použití SDS (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,2 mg/ml
|
|
Kritéria přijatelnosti
|
|
1.
|
Střední hodnota OD tkáňových replikátů vystavených působení negativní kontroly by měla být > 0,8 a < 2,5.
|
|
2.
|
Střední životaschopnost tkáňových replikátů vystavených působení pozitivní kontroly po dobu 30 minut, vyjádřeno jako % negativní kontroly, by měla být < 50 %.
|
|
3.
|
Rozdíl životaschopnosti mezi dvěma tkáňovými replikáty by měl být menší než 20 %.
|
|
|
1.
|
Střední hodnota OD tkáňových replikátů vystavených působení negativní kontroly by měla být > 0,8 a < 2,5.
|
|
2.
|
Střední životaschopnost tkáňových replikátů vystavených působení pozitivní kontroly po dobu 6 hodin, vyjádřeno jako % negativní kontroly, by měla být < 50 %.
|
|
3.
|
Rozdíl životaschopnosti mezi dvěma tkáňovými replikáty by měl být menší než 20 %.
|
|
|
1.
|
Střední hodnota OD tkáňových replikátů vystavených působení negativní kontroly by měla být > 1,0 a ≤ 2,5.
|
|
2.
|
Střední životaschopnost tkáňových replikátů vystavených působení pozitivní kontroly po dobu 30 minut, vyjádřeno jako % negativní kontroly, by měla být ≤ 30 %.
|
|
3.
|
Rozdíl životaschopnosti mezi dvěma tkáňovými replikáty by měl být menší než 20 %.
|
|
|
1.
|
Střední hodnota OD tkáňových replikátů vystavených působení negativní kontroly by měla být > 1,0 a ≤ 2,5.
|
|
2.
|
Střední životaschopnost tkáňových replikátů vystavených působení pozitivní kontroly po dobu 4 hodin, vyjádřeno jako % negativní kontroly, by měla být ≤ 20 %.
|
|
3.
|
Rozdíl životaschopnosti mezi dvěma tkáňovými replikáty by měl být menší než 20 %.
|
|
Dodatek 3
GRAF ILUSTRUJÍCÍ IDENTIFIKACI A NAKLÁDÁNÍ S PŘÍMÝMI REDUKČNÍMI ČINIDLY MTT ANEBO BAREVNĚ INTERFERUJÍCÍMI CHEMICKÝMI LÁTKAMI NA ZÁKLADĚ STANDARDNÍHO PRACOVNÍHO POSTUPU PRO VRM1
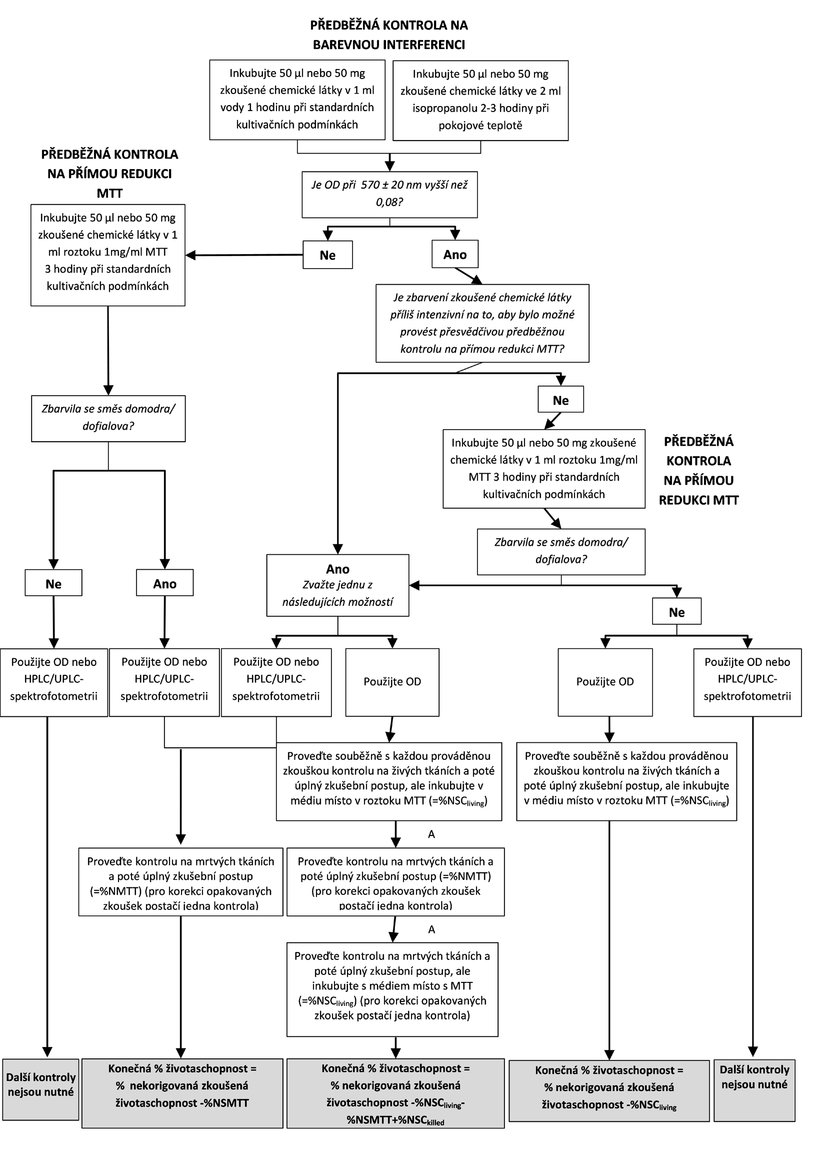
Dodatek 4
GRAF ILUSTRUJÍCÍ IDENTIFIKACI A NAKLÁDÁNÍ S PŘÍMÝMI REDUKČNÍMI ČINIDLY MTT ANEBO BAREVNĚ INTERFERUJÍCÍMI CHEMICKÝMI LÁTKAMI NA ZÁKLADĚ STANDARDNÍHO PRACOVNÍHO POSTUPU PRO VRM2
Dodatek 5
KLÍČOVÉ PARAMETRY A KRITÉRIA PŘIJATELNOSTI PRO ZPŮSOBILOST SPEKTROFOTOMETRICKÉHO SYSTÉMU HPLC/UPLC PRO MĚŘENÍ FORMAZANU MTT EXTRAHOVANÉHO Z TKÁŇOVÝCH KONSTRUKCÍ RHCE
|
Parametr
|
Protokol vycházející z pokynu FDA (36)(38)
|
Kritéria přijatelnosti
|
|
Selektivita
|
Analýza isopropylalkoholu, slepý pokus s živou tkání (isopropylalkoholový extrakt z tkáňových konstrukcí RhCE bez jakékoli expozice), slepý pokus s neživou tkání (isopropylalkoholový extrakt z tkáňových konstrukcí RhCE bez jakékoli expozice), a analýza barviva (např. methylenová modř)
|
plochainterference ≤ 20 % plochyLLOQ
(87)
|
|
Přesnost
|
Kontroly kvality (tj. formazan MTT při 1,6 μg/ml, 16 μg/ml a 160 μg/ml) v isopropylalkoholu (n = 5)
|
CV ≤ 15 % nebo ≤ 20 % pro LLOQ
|
|
Přesnost
|
Kontroly kvality v isopropylalkoholu (n = 5)
|
%Dev ≤ 15 % nebo ≤ 20 % pro LLOQ
|
|
Vlivy matrice
|
Kontroly kvality ve slepém pokusu s živou tkání (n = 5)
|
85 % ≤ % matricového jevu ≤ 115 %
|
|
Únik
|
Analýza isopropylalkoholu po standardu ULOQ (88)
|
plochainterference ≤ 20 % plochyLLOQ
|
|
Reprodukovatelnost (v rámci dne)
|
3 nezávislé kalibrační křivky (vycházející z 6 postupných třetinových ředění formazanu MTT v isopropylalkoholu s počátkem při ULOQ, tj. 200 μg/ml);
Kontroly kvality v isopropylalkoholu (n = 5)
|
Kalibrační křivky: %Dev ≤ 15 % nebo ≤ 20 % pro LLOQ
Kontroly kvality: %Dev ≤ 15 % a CV ≤ 15 %
|
|
Reprodukovatelnost (v rámci dne)
|
Den 1: 1 kalibrační křivka a kontroly kvality v isopropylalkoholu (n = 3)
Den 2: 1 kalibrační křivka a kontroly kvality v isopropylalkoholu (n = 3)
Den 3: 1 kalibrační křivka a kontroly kvality v isopropylalkoholu (n = 3)
|
|
Krátkodobá stabilita formazanu MTT v tkáňovém extraktu RhCE
|
Kontroly kvality ve slepém pokusu s živou tkání (n = 3) analyzované v den přípravy a po 24 hodinách uložení při pokojové teplotě
|
%Dev ≤ 15 %
|
|
Dlouhodobá stabilita formazanu MTT v tkáňovém extraktu RhCE, v případě potřeby
|
Kontroly kvality ve slepém pokusu s živou tkání (n = 3) analyzované v den přípravy a po Několika dnech uložení při –20°C
|
%Dev ≤ 15 %
|
B.70 ZKOUŠKA IN VITRO S VYUŽITÍM REKOMBINANTNÍCH ESTROGENOVÝCH RECEPTORŮ NA DETEKCI CHEMICKÝCH LÁTEK S VAZEBNOU AFINITOU NA ER
VŠEOBECNÝ ÚVOD
Pokyn OECD vycházející z funkční způsobilosti
|
1.
|
Tato zkušební metoda odpovídá pokynu OECD pro zkoušení (TG) 493 (2015). Pokyn č. 493 je pokyn vycházející z funkční způsobilosti (PBTG), který popisuje metodiku lidských rekombinantních analýz in vitro na detekci látek s vazebnou afinitou na estrogenové receptory (analýzy vazby hrER). Skládá se ze dvou mechanisticky a funkčně podobných analýz pro zjištění látek, které se vážou na estrogenový receptor (tj. ERα), a měl by usnadnit vypracování nových podobných nebo modifikovaných analýz podle zásad pro validaci stanovených v dokumentu OECD o validaci a mezinárodním uznání nových nebo aktualizovaných zkušebních metod pro posuzování nebezpečnosti (1). Plně validovanými referenčními zkušebními metodami (dodatek 2 a dodatek 3), které představují základ pro tuto metodiku vycházející z funkční způsobilosti, jsou:
|
—
|
Freyberger-Wilsonova (FW) in vitro analýza vazby na estrogenové receptory (ER) s využitím celého lidského rekombinantního ERα (2) a
|
|
—
|
in vitro analýza vazby na estrogenové receptory s využitím lidského rekombinantního proteinu s ligand vázající doménou Ústavu pro hodnocení a výzkum chemických látek (CERI) (2).
|
K dispozici jsou standardy funkčnosti (PS) (3), které usnadňují vývoj a validaci podobných zkušebních metod pro stejné riziko a umožňují včasnou změnu pokynu PBTG 493, aby mohly být do aktualizovaného pokynu přidány nové podobné analýzy. Podobné analýzy však budou přidány až po kontrole a dohodě s OECD, že splňují standardy funkčnosti. Analýzy zařazené do pokynu 493 mohou být použity libovolně pro řešení požadavků členských zemí OECD na výsledky zkoušek na vazbu na estrogenové receptory při využití vzájemného uznávání údajů podle postupu dohodnutého v rámci OECD.
|
Obecné informace a principy analýz zahrnutých do této zkušební metody
|
2.
|
V roce 1998 zahájila OECD činnost vysoké priority, jejíž podstatou je revize stávajících a vytvoření nových pokynů pro screening a zkoušení potenciálních endokrinních disruptorů. V roce 2012 byl revidován koncepční rámec OECD pro zkoušky a hodnocení potenciálních endokrinních disruptorů. Původní i revidovaný koncepční rámec je uveden v přílohách standardizovaných pokynů pro zkoušení a posuzování endokrinních disruptorů (Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption) (4). Koncepční rámec je tvořen pěti úrovněmi, které odpovídají různým úrovním biologické komplexnosti. Analýzy vazby na ER popsané v této zkušební metodě představují úroveň 2, která zahrnuje „analýzy in vivo poskytující údaje o vybraných endokrinních mechanismech/drahách“. Tato zkušební metoda je v testech vazby ligandu na receptor in vitro určena na zjištění ligandů na lidský estrogenový receptor alfa (ERα).
|
|
3.
|
Relevance testu vazby ER in vitro pro biologické funkce byla jasně prokázána. Testy vazby ER jsou určeny k identifikaci chemických látek, které mají potenciál narušit dráhu estrogenových hormonů a v posledních dvou desetiletích se široce používají pro charakterizaci distribuce ER v tkáních, jakož i pro identifikaci agonistů/antagonistů ER. Tyto testy odrážejí interakci mezi ligandem a receptorem, což je první krok estrogenové signální dráhy, který je zásadně důležitý pro reprodukční funkci všech obratlovců.
|
|
4.
|
Interakce estrogenů s estrogenovými receptory může ovlivnit transkripci estrogenem kontrolovaných genů a indukovat negenomové účinky, což může vést k indukci nebo inhibici buněčných procesů, včetně procesů nezbytných pro buněčnou proliferaci, normální vývoj plodu a reprodukční funkci (5)(6)(7). Narušení normálních estrogenních systémů může spustit nežádoucí účinky v oblasti normálního vývoje (ontogeneze), reprodukčního zdraví a správné funkce reprodukčního systému. Nesprávná signalizace ER může vést k takovým účinkům, jako je zvýšené riziko hormonálně dependentního karcinomu, narušení plodnosti a změny růstu a vývoje plodu (8).
|
|
5.
|
Analýzy vazby in vitro vycházejí z přímé interakce látky s vazebným místem ligandu na specifický receptor, který reguluje transkripci genu. Hlavní složka testu vazby lidského rekombinantního estrogenového receptoru alfa (hrERα) měří schopnost radioizotopově značeného ligandu ([3H]17β-estradiol) vázat se na ER v přítomnosti zvyšujících se koncentrací zkoušené chemické látky (tj. kompetitor). Zkoušené chemické látky, které mají vysokou afinitu na ER, konkurují radioizotopově značenému ligandu při nižší koncentraci v porovnání s chemickými látkami s nižší afinitou na tento receptor. Tato analýza sestává ze dvou hlavních složek: saturačního vazebného experimentu za účelem charakterizace parametrů interakce mezi receptorem a ligandem a dokumentace specifičnosti ER, po němž následuje kompetitivní vazebný experiment, který charakterizuje kompetici mezi zkoušenou chemickou látkou a radioizotopově značeným ligandem ve vazbě na ER.
|
|
6.
|
Validační studie vazebných analýz CERI a FW prokázaly jejich relevanci a spolehlivost pro zamýšlený účel (2).
|
|
7.
|
Definice a zkratky používané v této zkušební metodě jsou popsány v dodatku 1.
|
Platnost a omezení testů vazby ligandu na receptor
|
8.
|
Tyto analýzy jsou navrženy pro účely screeningu a stanovení priorit, ale mohou také poskytovat informace o molekulární iniciační události (MIE), které lze použít při analýze průkaznosti výsledků. Týkají se chemické vazby na doménu ERα vázající ligand v systému in vitro. Výsledky proto nelze přímo extrapolovat na komplexní signalizaci a regulaci intaktního endokrinního systému in vivo.
|
|
9.
|
Vazba přirozeného ligandu, 17β-estradiolu, je prvním krokem v sérii molekulárních událostí, který aktivuje transkripci cílových genů a nakonec vrcholí fyziologickou změnou (9). Vazba na doménu ERα vázající ligand se tedy považuje za jeden z klíčových mechanismů narušení endokrinního systému (ED) mediovaného ER, i když existují i jiné mechanismy, jejichž prostřednictvím může k tomuto narušení dojít, včetně i) interakcí s místy ERα jinými, než je kapsa vázající ligand, ii) interakcí s jinými receptory relevantními pro estrogenovou signalizaci, ERβ a estrogenovým receptorem spřaženým s G-proteinem, jinými receptory a enzymatickými systémy v endokrinním systému, iii) syntézy hormonů, iv) metabolické aktivace anebo inaktivace hormonů, v) distribuce hormonů do cílových tkání a vi) clearance hormonů z těla. Žádná z analýz podle této zkušební metody se nezabývá těmito způsoby účinku.
|
|
10.
|
Tato zkušební metoda se zabývá schopností látek vázat se na lidský ERα, ale nerozlišuje mezi agonisty a antagonisty ERα. Tyto analýzy neřeší další události v řetězci, jako je genová transkripce nebo fyziologické změny. Vzhledem k tomu, že při validaci byly použity pouze jednotlivé jednosložkové látky, nebyla řešena použitelnost na zkoušené směsi. Analýzy jsou však teoreticky použitelné i na zkoušení vícesložkových látek a směsí. Před použitím této zkušební metody ke zkoušení směsi s cílem získat údaje pro zamýšlené regulační účely by mělo být zváženo, zda tato metoda může pro tento účel poskytnout přiměřené výsledky, a pokud ano, proč je tomu tak. Takové úvahy nejsou nutné, pokud existuje právní požadavek na zkoušení dané směsi.
|
|
11.
|
Receptorové systémy bez buněk nemají žádnou vnitřní metabolickou kapacitu a nebyly validovány v kombinaci s metabolickými enzymatickými systémy. Mohlo by však být možné začlenit metabolickou aktivitu do plánu studie, ale vyžadovalo by to další práci v oblasti validace.
|
|
12.
|
Chemické látky, které by mohly protein (tj. receptorový protein) denaturovat, jako jsou povrchově aktivní látky nebo chemické látky, které mohou změnit pH zkušebního pufru, nesmí být testovány nebo mohou být testovány pouze při koncentracích, které tyto interakce vylučují. Jinak je rozsah koncentrací, které mohou být zkoušeny v testech zkoušené chemické látky, omezen na její rozpustnost ve zkušebním pufru.
|
|
13.
|
V tabulce 1 jsou pro informaci uvedeny výsledky testu 24 látek, které byly zkoušeny oběma plně validovanými analýzami popsanými v této zkušební metodě. Z těchto látek je na základě publikovaných zpráv, včetně analýz in vitro na transkripční aktivaci ER anebo uterotrofní analýzy, 17 klasifikováno jako ER vázající látky a 6 jako ER nevázající látky (9)(10)(11)(12)(13)(14)(15). Pokud jde o údaje uvedené v tabulce 1, byla mezi oběma analýzami u klasifikace všech látek do 10-4 M téměř 100 % shoda, přičemž každá látka byla správně klasifikována jako ER vázající nebo nevázající. Doplňkové informace o této skupině látek i o dalších látkách zkoušených v analýzách vazby ER v průběhu validačních studií jsou uvedeny ve standardech funkčnosti pro analýzu vazby hrER (3), dodatek 2 (tabulky 1, 2 a 3).
Tabulka 1
Klasifikace látek vázajících a nevázajících se na ER při zkoušení pomocí analýz vazby na hrER FW a CERI v porovnání s očekávanou odpovědí
|
|
Název látky
|
CASRN
|
Předpokládaná odpověď
|
Analýza FW
|
Analýza CERI
|
MESH Chemická třída
|
Třída produktu
|
|
|
Koncentrace Rozsah (M)
|
Klasifikace
|
Koncentrace Rozsah (M)
|
Klasifikace
|
|
1
|
17β-estradiol
|
50-28-2
|
váže se
|
1 × 10-11 – 1 × 10-6
|
váže se
|
1 × 10-11 – 1 × 10-6
|
váže se
|
Steroid
|
Léčivý přípravek, veterinární látka
|
|
2
|
Norethynodrel
|
68-23-5 Org.
|
váže se
|
3 × 10-9 – 30 × 10-4
|
váže se
|
3 × 10-9 – 30 × 10-4
|
váže se
|
Steroid
|
Léčivý přípravek, veterinární látka
|
|
3
|
Norethindron
|
68-22-4
|
váže se
|
3 × 10-9 – 30 × 10-4
|
váže se
|
3 × 10-9 – 30 × 10-4
|
váže se
|
Steroid
|
Léčivý přípravek, veterinární látka
|
|
4
|
Di-n-butylftalát
|
84-74-2
|
neváže se (*5)
|
1 × 10-10 – 1 × 10-4
|
neváže se (*6)
(*7)
|
1 × 10-10 – 1 × 10-4
|
neváže se (*6)
(*7)
|
Uhlovodík (cyklický), ester
|
Plastifikátor, chemický meziprodukt
|
|
5
|
DES
|
56-53-1
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
Uhlovodík (cyklický), fenol
|
Léčivý přípravek, veterinární látka
|
|
6
|
17α-ethynylestradiol
|
57-63-6
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
Steroid
|
Léčivý přípravek, veterinární látka
|
|
7
|
Meso-Hexestrol
|
84-16-2
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
Uhlovodík (cyklický), fenol
|
Léčivý přípravek, veterinární látka
|
|
8
|
Genistein
|
446-72-0
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
Uhlovodík (heterocyklický), flavonoid
|
Přírodní produkt
|
|
9
|
Equol
|
531-95-3
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
Fytoestrogenový metabolit
|
Přírodní produkt
|
|
10
|
Butylparaben (n butyl-4-hydroxybenzoan)
|
94-26-8
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
Paraben
|
Konzervant
|
|
11
|
Nonylfenol (směs)
|
84852-15-3
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
Alkylfenol
|
Meziprodukt
|
|
12
|
o,p’-DDT
|
789-02-6
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
Organochlor
|
Insekticid
|
|
13
|
Kortikosteron
|
50-22-6
|
neváže se (*5)
|
1 × 10-10 – 1 × 10-4
|
neváže se
|
1 × 10-10 – 1 × 10-4
|
neváže se
|
Steroid
|
Přírodní produkt
|
|
14
|
Zearalenon
|
17924-92-4 Org.
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
Uhlovodík (heterocyklický), lakton
|
Přírodní produkt
|
|
15
|
Tamoxifen
|
10540-29-1
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
Uhlovodík (cyklický)
|
Léčivý přípravek, veterinární látka
|
|
16
|
5α-dihydrotestosteron
|
521-18-6
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
Steroid, nefenolový
|
Přírodní produkt
|
|
17
|
Bisfenol A
|
80-05-7
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
Fenol
|
Chemický meziprodukt
|
|
18
|
4-n-heptylphenol
|
1987-50-4
|
váže se
|
1 × 10-10 – 1 × 10-3
|
neurčité (6)
|
1 × 10-10 – 1 × 10-3
|
váže se
|
Alkylfenol
|
Meziprodukt
|
|
19
|
Kepon (Chlordecon)
|
143-50-0
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
Uhlovodík (halogenovaný)
|
Pesticid
|
|
20
|
Benzo(a)anthracen
|
56-55-3
|
neváže se
|
1 × 10-10 – 1 × 10-3
|
neváže se (7)
|
1 × 10-10 – 1 × 10-3
|
neváže se (7)
|
Aromatický uhlovodík
|
Meziprodukt
|
|
21
|
Enterolakton
|
78473-71-9
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
1 × 10-10 – 1 × 10-3
|
váže se
|
Fytoestrogen
|
Přírodní produkt
|
|
22
|
Progesteron
|
57-83-0
|
neváže se (*5)
|
1 × 10-10 – 1 × 10-4
|
neváže se
|
1 × 10-10 – 1 × 10-4
|
neváže se
|
Steroid
|
Přírodní produkt
|
|
23
|
Oktyltriethoxysilan
|
2943-75-1
|
neváže se
|
1 × 10-10 – 1 × 10-3
|
neváže se
|
1 × 10-10 – 1 × 10-3
|
neváže se
|
Silan
|
Povrchový modifikátor
|
|
24
|
Atrazin
|
1912-24-9
|
neváže se (*5)
|
1 × 10-10 – 1 × 10-4
|
neváže se
|
1 × 10-10 – 1 × 10-4
|
neváže se
|
Heterocyklická sloučenina
|
Herbicid
|
|
SLOŽKY VAZEBNÉ ANALÝZY hrER
Zásadní složky zkoušky
|
14.
|
Tato zkušební metoda platí pro analýzy používající receptor ER a vhodný silný ligand na receptor, který může být použit jako marker/stopovací látka pro analýzu a se zvyšujícími se koncentracemi zkoušené chemické látky může být vytěsněn. Vazebné analýzy sestávají z těchto dvou hlavních složek: 1) saturační vazby a 2) kompetitivní vazby. Saturační vazebná analýza se používá na potvrzení specifičnosti a aktivity receptorových přípravků, zatímco kompetitivní vazebný experiment se používá na vyhodnocení schopnosti zkoušené chemické látky vázat se na hrER.
|
Kontroly
|
15.
|
Je třeba popsat východisko pro navrhované souběžné referenční zkoušky s estrogenem a kontroly. Souběžné kontroly (rozpouštědlo (vehikulum), pozitivní (vazba na ER; silná a slabá afinita), negativní (neváže se)) slouží podle potřeby jako indikace, že analýza je za zkušebních podmínek funkční a poskytuje základ pro porovnání jednotlivých experimentů; obvykle jsou součástí kritérií přijatelnosti pro daný experiment (1). Při každém provedení zkoušky by měly být na jedné destičce použity celé křivky koncentrace pro referenční estrogen a kontroly (tj. slabá vazba a neváže se). Všechny ostatní destičky by měly obsahovat: 1) vysokou (přibližně úplné vytěsnění radioizotopově značeného ligandu) a střední (přibližně IC50) koncentraci každého E2 a látky se slabou vazbou, a to třikrát; 2) kontrolu s rozpouštědlem a látkou s nespecifickou vazbou, a to vždy třikrát.
|
Standardní postupy kontroly kvality
|
16.
|
Standardní postupy řízení kvality by měly být prováděny tak, jak je popsáno pro každou analýzu, aby byly zajištěny aktivní receptory a správné koncentrace chemické látky a aby bylo zajištěno, že toleranční meze zůstanou u vícečetných replikátů stabilní a bude zachována schopnost podávat očekávané odpovědi vazby na ER v průběhu času.
|
Prokázání odborné způsobilosti laboratoře
|
17.
|
Před zkoušením neznámých chemických látek pomocí některé z analýz v rámci této zkušební metody by měla každá laboratoř prokázat odbornou způsobilost v používání analýzy provedením saturačních zkoušek na potvrzení specifičnosti a aktivity přípravku ER a kompetitivních vazebných zkoušek s referenčním estrogenem a kontrolami (s látkou se slabou vazbou a látkou, která se neváže). Laboratoř by si měla vytvořit historickou databázi s výsledky za referenční estrogen a kontroly získanými z 3–5 nezávislých experimentů provedených v různé dny. Tyto experimenty budou základem pro referenční estrogen a historické kontroly pro tuto laboratoř a budou použity jako částečné hodnocení přijatelnosti analýzy pro budoucí zkoušky.
|
|
18.
|
Odpovědi zkušebního systému budou potvrzeny také zkoušením vhodných látek uvedených v tabulce 2. Seznam vhodných látek představuje podmnožinu referenčních látek uvedených ve standardech funkčnosti pro analýzy vazby na ER (3). Tyto látky jsou komerčně dostupné, představují třídy chemických látek běžně souvisejících s vazebnou aktivitou na ER a vykazují vhodný rozsah síly očekávané od látek, které se vážou na ER (tj. silné až slabé), a látek, které se na ER nevážou (tj. negativní). U každé látky pro prokázání způsobilosti by měly zkoušené koncentrace zahrnovat rozmezí uvedené v tabulce 2. Pro každou látku by se měly provést alespoň tři experimenty a výsledky by měly být v souladu s očekávanou chemickou aktivitou. Každý experiment by se měl provádět nezávisle (tj. s čerstvými roztoky zředěného receptoru, chemickými látkami a reagenciemi), a to se třemi opakováními u každé koncentrace. Odborná způsobilost je prokázána správnou klasifikací (pozitivní/negativní) každé vhodné látky. Zkoušení odborné způsobilosti by měl provést každý technik, který se analýzy učí.
Tabulka 2
Seznam kontrolních látek a látek pro prokázání způsobilosti pro kompetitivní vazebné analýzy hrER
(89)
|
Č.
|
Název látky
|
CASRN (90)
|
Předpokládaná odpověď (91)
(92)
|
Rozpětí zkušebních koncentrací (M)
|
Třída chemických látek MeSH (93)
|
Třída produktu (94)
|
|
Kontroly (referenční estrogen, slabá vazba, neváže se)
|
|
1
|
17-εστραδιολ
|
50-28-2
|
váže se
|
1 × 10-11 – 1 × 10-6
|
Steroid
|
Léčivý přípravek, veterinární látka
|
|
2
|
Norethynodrel (nebo) Norethindron
|
68-23-5 (or) 68-22-4
|
váže se
|
3 × 10-9 – 30 × 10-6
|
Steroid
|
Léčivý přípravek, veterinární látka
|
|
3
|
Oktyltriethoxysilan
|
2943-75-1
|
neváže se
|
1 × 10-10 – 1 × 10-3
|
Silan
|
Povrchový modifikátor
|
|
Vhodné látky (94)
|
|
4
|
Diethylstilbestrol
|
56-53-1
|
váže se
|
1 × 10-11 – 1 × 10-6
|
Uhlovodík (cyklický), fenol
|
Léčivý přípravek, veterinární látka
|
|
5
|
17α-ethynylestradiol
|
57-63-6
|
váže se
|
1 × 10-11 – 1 × 10-6
|
Steroid
|
Léčivý přípravek, veterinární látka
|
|
6
|
meso-Hexestrol
|
84-16-2
|
váže se
|
1 × 10-11 – 1 × 10-6
|
Uhlovodík (cyklický), fenol
|
Léčivý přípravek, veterinární látka
|
|
7
|
Tamoxifen
|
10540-29-1
|
váže se
|
1 × 10-11 – 1 × 10-6
|
Uhlovodík (cyklický)
|
Léčivý přípravek, veterinární látka
|
|
8
|
Genistein
|
446-72-0
|
váže se
|
1 × 10-10 – 1 × 10-3
|
Heterocyklická sloučenina, flavonoid
|
Přírodní produkt
|
|
9
|
Bisfenol A
|
80-05-7
|
váže se
|
1 × 10-10 – 1 × 10-3
|
Fenol
|
Chemický meziprodukt
|
|
10
|
Zearalonone
|
17924-92-4
|
váže se
|
1 × 10-11 – 1 × 10-3
|
Heterocyklická sloučenina, lakton
|
Přírodní produkt
|
|
11
|
Butylparaben
|
94-26-8
|
váže se
|
1 × 10-11 – 1 × 10-3
|
Kyselina karboxylová, fenol
|
Konzervant
|
|
12
|
Atrazin
|
1912-24-9
|
neváže se
|
1 × 10-11 – 1 × 10-6
|
Heterocyklická sloučenina
|
Herbicid
|
|
13
|
Di-n-butylftalát (DBP) (95)
|
84-74-2
|
neváže se (96)
|
1 × 10-10 – 1 × 10-4
|
Uhlovodík (cyklický), ester
|
Plastifikátor, chemický meziprodukt
|
|
14
|
Kortikosteron
|
50-22-6
|
neváže se
|
1 × 10-11 – 1 × 10-4
|
Steroid
|
Přírodní produkt
|
|
Zkoušení rozpustnosti a stanovení rozsahu koncentrace pro zkoušené chemické látky
|
19.
|
Měla by být provedena předběžná zkouška, aby bylo možné stanovit mez rozpustnosti pro každou zkoušenou chemickou látku a zjistit vhodný rozsah koncentrace, jenž má být použit při provádění zkoušky. Mez rozpustnosti každé zkoušené chemické látky se nejprve stanoví v rozpouštědle a dále potvrdí za zkušebních podmínek. Konečná koncentrace zkoušená při analýze by neměla překročit 1 nm. Zkoušení za účelem zjištění rozsahu sestává z kontroly s rozpouštědlem společně s osmi log-sériovými ředěními, počínaje maximální přijatelnou koncentrací (např. 1 mM nebo menší, na základě meze rozpustnosti), přičemž je třeba zaznamenat přítomnost zákalu nebo sraženiny. Při druhém a třetím experimentu by se měla podle potřeby upravit koncentrace, aby lépe charakterizovala křivku závislosti odpovědi na koncentraci.
|
Kritéria přijatelnosti provedení zkoušky
|
20.
|
Přijetí nebo odmítnutí provedené zkoušky vychází z hodnocení výsledků získaných pro referenční estrogen a kontrolu použitou pro každý experiment. Za prvé, na destičce 1 by měly celé křivky koncentrace pro referenční kontroly z každého experimentu odpovídat mírám funkčnosti u parametrů vynesené křivky (např. IC50 a Hillův koeficient) podle výsledků uvedených pro příslušné protokoly pro analýzy CERI a FW (dodatek 2 a 3) a dosavadních kontrolních dat z laboratoře provádějící zkoušku. Všechny kontroly (referenční estrogen, slabá vazba a neváže se) by měly být u každého experimentu správně klasifikovány. Za druhé, u kontrol na všech dalších destičkách je třeba posuzovat, zda odpovídají destičce 1. Mělo by být použito dostatečné rozmezí koncentrací, aby byl jednoznačně definován vrchol kompetitivní vazebné křivky. Variabilita mezi replikáty při každé koncentraci zkoušené chemické látky i mezi třemi nezávislými provedeními zkoušky by měla být přiměřená a vědecky obhajitelná. Schopnost konzistentního provedení analýzy by měla být prokázána vytvořením a údržbou historické databáze referenčního estrogenu a kontrol. Jako měřítko vnitrolaboratorní reprodukovatelnosti mohou být použity směrodatné odchylky (SD) nebo variační koeficienty (CV) pro střední hodnoty parametrů stanovené křivky referenčního estrogenu a slabé vazby z vícečetných experimentů. Při posuzování výsledků kontrol z každého provedení zkoušky i výsledků za každou zkoušenou chemickou látku je třeba postupovat s odborným úsudkem.
Kromě toho by měly být splněny tyto zásady, pokud jde o kritéria přijatelnosti:
|
—
|
Údaje by měly postačovat na kvantitativní posouzení vazby na ER.
|
|
—
|
Zkoušené koncentrace by měly zůstat v rozmezí rozpustnosti zkoušené chemické látky.
|
|
Analýza údajů
|
21.
|
Stanovený postup analýzy údajů o nasycené a kompetitivní vazbě by měl odpovídat hlavním zásadám pro charakterizaci interakcí mezi receptorem a ligandem. Údaje o nasycené vazbě se obvykle analyzují promocí nelineárního regresního modelu, který zohledňuje celkovou a nespecifickou vazbu. Při určování Bmax a Kd může být nutná korekce úbytku ligandu (např. Swillens, 1995 (19)). Data z kompetitivních vazebných analýz se obvykle transformují (např. percentuální specifická vazba a koncentrace zkoušené chemické látky (log M)). Odhady log (IC50) za každou zkoušenou chemickou látku se stanoví pomocí vhodného softwaru pro nelineární vynesení křivky, aby byly vhodné pro čtyřparametrickou Hillovu rovnici. Po první analýze je třeba stanovit parametry vynesené křivky a vizuálně zkontrolovat, jak dobře vazebná data odpovídají generované kompetitivní vazebné křivce. V některých případech může být nutná další analýza, aby byl dosažen nejlepší tvar křivky (např. omezení horní anebo dolní části křivky, použití pravidla 10 %, viz dodatek 4 a odkaz 2 (oddíl III.A.2).
|
|
22.
|
Splnění kritérií přijatelnosti (odstavec 20) ukazuje, že systém funguje správně, ale nezaručuje to, že konkrétní provedení zkoušky podá přesné údaje. Opakování správných výsledků první zkoušky je nejlepším důkazem, že údaje byly přesné.
|
Obecná kritéria interpretace údajů
|
23.
|
V současné době neexistuje žádná univerzálně akceptovaná metoda pro interpretaci údajů o vazbě na ER. Kvalitativní (např. váže se / neváže se) anebo kvantitativní (např. log iC50, relativní vazebná afinita (RBA) atd.) posouzení aktivity zprostředkované hrER by však mělo vycházet z empirických údajů a řádného vědeckého úsudku.
|
ZÁVĚREČNÁ ZPRÁVA
|
24.
|
Závěrečná zpráva by měla obsahovat tyto informace:
|
|
Kontrola / referenční látka / zkoušená chemická látka
|
—
|
zdroj, číslo šarže a je-li k dispozici, datum použitelnosti,
|
|
—
|
stabilita zkoušené chemické látky samotné, je-li známa,
|
|
—
|
rozpustnost a stabilita zkoušené chemické látky v rozpouštědle, jsou-li známy,
|
|
—
|
měření pH, osmolality a sraženiny v kultivačním médiu, do něhož byla podle potřeby přidána zkoušená chemická látka.
|
|
|
|
Jednosložková látka:
|
—
|
fyzický vzhled, rozpustnost ve vodě a další relevantní fyzikálně-chemické vlastnosti,
|
|
—
|
chemická identifikace, např. název IUPAC nebo CAS, číslo CAS, SMILES nebo kód InChI, strukturní vzorec, čistota, případně chemická identita nečistot, je-li to prakticky proveditelné atd.
|
|
|
|
Vícesložková látka, látka s neznámým nebo proměnlivým složením, komplexní reakční produkt nebo biologický materiál a směsi:
|
—
|
charakterizované pokud možno chemickou identitou (viz výše), kvantitativním výskytem a relevantními fyzikálně-chemickými vlastnostmi složek.
|
|
|
|
Rozpouštědlo/vehikulum:
|
—
|
charakterizace (povaha, dodavatel a šarže),
|
|
—
|
zdůvodnění volby rozpouštědla/vehikula,
|
|
—
|
rozpustnost a stabilita zkoušené chemické látky v rozpouštědle/vehikulu, jsou-li známy,
|
|
|
|
Receptory:
|
—
|
zdroj receptoru (dodavatel, katalogové číslo, šarže, druh receptoru, koncentrace aktivního receptoru uvedená dodavatelem, certifikace dodavatele),
|
|
—
|
charakterizace receptorů (včetně výsledků nasycené vazby): Kd, Bmax,
|
|
—
|
radioizotopově značený ligand:
|
|
—
|
dodavatel, katalogové číslo, šarže, měrná aktivita.
|
|
|
|
Zkušební podmínky:
|
—
|
meze rozpustnosti za podmínek zkoušky,
|
|
—
|
složení vazebného pufru,
|
|
—
|
koncentrace stopovací látky (tj. radioizotopově značený ligand),
|
|
—
|
koncentrace zkoušené chemické látky,
|
|
—
|
procento vehikula v konečné analýze,
|
|
—
|
teplota a doba inkubace,
|
|
—
|
způsob separace vázané/volné látky,
|
|
—
|
pozitivní a negativní kontroly / referenční látky,
|
|
—
|
kritéria klasifikace zkoušky na pozitivní, negativní nebo dvojznačnou.
|
|
|
|
Kontrola přijatelnosti:
|
—
|
skutečné hodnoty IC50 a hodnoty Hillova koeficientu pro souběžné pozitivní kontroly / referenční látky.
|
|
|
|
Výsledky:
|
—
|
naměřená data a data o vázaných/volných látkách,
|
|
—
|
případně kontrola potvrzení denaturace,
|
|
—
|
pokud existuje, nejnižší účinná koncentrace (LEC),
|
|
—
|
hodnoty RBA a IC50 podle potřeby,
|
|
—
|
závislost účinku na koncentraci, pokud je to možné,
|
|
—
|
případné statistické analýzy společně s měřítkem chyby a spolehlivosti (např. SEM, SD, CV nebo 95 % CI) a popis, jak byly tyto hodnoty získány.
|
|
|
|
Diskuse o výsledcích:
|
—
|
uplatnění pravidla 10 %.
|
|
Závěr
|
LITERATURA
|
(1)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental Health and Safety Publications, Series on Testing and Assessment, (No 34), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
OECD (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 226), Organisation for Economic Cooperation and Development, Paris.
|
|
(3)
|
OECD (2015). Performance Standards for Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 222), Organisation for Economic Cooperation and Development, Paris.
|
|
(4)
|
OECD (2012). Guidance Document on Standardized Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environmental Health and Safety Publications, Series on Testing and Assessment, (No 150), Organisation for Economic Cooperation and Development, Paris.
|
|
(5)
|
Cavailles V. (2002). Estrogens and Receptors: an Evolving Concept, Climacteric, 5 Suppl 2: p. 20-6.
|
|
(6)
|
Welboren W.J., et al. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and how are they Regulated? Endocr. Relat. Cancer., 16(4): p. 1073-89.
|
|
(7)
|
Younes M. and Honma N. (2011). Estrogen Receptor Beta, Arch. Pathol. Lab. Med., 135(1): 63-6.
|
|
(8)
|
Diamanti-Kandarakis et al. (2009). Endocrine-Disrupting Chemicals: an Endocrine Society Sci. Statement, Endo Rev 30(4):293-342.
|
|
(9)
|
ICCVAM (2002). Background Review Document. Current Status of Test Methods for Detecting Endocrine Disruptors: In Vitro Estrogen Receptor Binding Assays. (NIH Publication No 03-4504). National Institute of Environmental Health Sciences, Research Triangle Park, NC.
|
|
(10)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(11)
|
ICCVAM (2006). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(12)
|
Akahori Y. et al. (2008). Relationship Between the Results of In Vitro Receptor Binding Assay to Human Estrogen Receptor Alpha and In Vivo Uterotrophic Assay: Comparative Study with 65 Selected Chemicals, Toxicol. In Vitro, 22(1): 225-231.
|
|
(13)
|
OECD (2007). Additional Data Supporting the Test Guideline on the Uterotrophic Bioassay in Rodents, Environment, Health and Safety Publications, Series on Testing and Assessment (No 67), Organisation for Economic Cooperation and Development, Paris.
|
|
(14)
|
Takeyoshi, M. (2006). Draft Report of Pre-validation and Inter-laboratory Validation For Stably Transfected Transcriptional Activation (TA) Assay to Detect Estrogenic Activity - The Human Estrogen Receptor Alpha Mediated Reporter Gene Assay Using hER-HeLa-9903 Cell Line, Chemicals Evaluation and Research Institute (CERI): Japan. p. 1-188.
|
|
(15)
|
Yamasaki, K; Noda, S; Imatanaka, N; Yakabe, Y. (2004). Comparative Study of the Uterotrophic Potency of 14 Chemicals in a Uterotrophic Assay and their Receptor-Binding Affinity, Toxicol. Letters, 146: 111-120.
|
|
(16)
|
Kummer V; Maskova, J; Zraly, Z; Neca, J; Simeckova, P; Vondracek, J; Machala, M. (2008). Estrogenic Activity of Environmental Polycyclic Aromatic Hydrocarbons in Uterus of Immature Wistar Rats. Toxicol. Letters, 180: 213-221.
|
|
(17)
|
Gozgit, JM; Nestor, KM; Fasco, MJ; Pentecost, BT; Arcaro, KF. (2004). Differential Action of Polycyclic Aromatic Hydrocarbons on Endogenous Estrogen-Responsive Genes and on a Transfected Estrogen-Responsive Reporter in MCF-7 Cells. Toxicol. and Applied Pharmacol., 196: 58-67.
|
|
(18)
|
Santodonato, J. (1997). Review of the Estrogenic and Antiestrogenic Activity of Polycyclic Aromatic Hydrocarbons: Relationship to Carcinogenicity. Chemosphere, 34: 835-848.
|
|
(19)
|
Swillens S (1995). Interpretation of Binding Curves Obtained with High Receptor Concentrations: Practical Aid for Computer Analysis, Mol Pharmacol 47(6):1197-1203.
|
Dodatek 1
DEFINICE A ZKRATKY
Pravidlo 10 %
: Možnost vyloučit z analýz datové body, pokud je průměr replikátů pro procento specifické vazby [3H]17β-estradiolu o 10 % nebo více nad hodnotu pozorovanou u střední hodnoty při nižší koncentraci (viz dodatek 4).
Kritéria přijatelnosti
: Minimální pravidla pro výkonnost experimentálních kontrol a referenčních standardů. Aby se experiment mohl považovat za platný, měla by být splněna všechna kritéria přijatelnosti.
Přesnost (soulad)
: Přesnost shody mezi výsledky analýzy a přijatými referenčními hodnotami. Je to měřítko funkčnosti analýzy a jeden z aspektů relevance. Tento pojem se často používá místo pojmu „soulad“ a rozumí se jím podíl správných výsledků analýzy (1).
CF
: Koncepční rámec OECD pro zkoušky a hodnocení endokrinních distruptorů.
Chemická látka
: Látka nebo směs.
CV
: Variační koeficient.
E2
: 17β-estradiol.
ED
: Narušení endokrinní činnosti.
hERα
: Lidský estrogenový receptor alfa.
ER
: Estrogenový receptor.
Estrogenní aktivita
: Schopnost chemické látky napodobit schopnost 17β-estradiolu vázat se na estrogenové receptory. Touto zkušební metodou lze detekovat vazbu na hERα.
IC
50
: Poloviční maximální účinná koncentrace inhibiční zkoušené chemické látky.
ICCVAM
: Meziagenturní koordinační výbor pro validaci alternativních metod.
Mezilaboratorní reprodukovatelnost
: Míra, v jaké mohou různé kvalifikované laboratoře za použití stejného protokolu a při testování stejných látek získat kvalitativně a kvantitativně podobné výsledky. Mezilaboratorní reprodukovatelnost se určuje během předvalidačních a validačních postupů a ukazuje, do jaké míry lze analýzu úspěšně přenášet mezi různými laboratořemi (1).
Vnitrolaboratorní reprodukovatelnost
: Určení míry, v jaké může kvalifikovaný personál ve stejné laboratoři jindy úspěšně dosáhnout stejných výsledků za použití určitého protokolu. Označuje se také jako „reprodukovatelnost v rámci laboratoře“ (1).
LEC
: Nejnižší účinná koncentrace je nejnižší koncentrace zkoušené chemické látky, která dává odpověď (tj. nejnižší koncentrace zkoušené chemické látky, při níž je násobná indukce statisticky odlišná od souběžné kontroly vehikulem).
Zkouška typu „me-too“
: Hovorový výraz pro analýzu, která je strukturálně a funkčně podobná validované a uznávané referenční zkušební metodě. Používá se namísto podobné zkušební metody.
PBTG
: Pokyn vycházející z funkční způsobilosti.
Standardy funkčnosti
: Standardy založené na validované zkušební metodě, které slouží jako základ pro hodnocení srovnatelnosti navrhované analýzy, která je mechanicky a funkčně podobná. Zahrnují 1) zásadní složky analýzy; 2) minimální seznam srovnávacích látek vybraných z chemických látek používaných k prokázání přijatelné funkčnosti validované referenční metody; a 3) srovnatelné hladiny přesnosti a spolehlivosti založené na hodnotách získaných u validované zkušební metody, jež by navrhovaná analýza měla prokázat při vyhodnocení za použití minimálního seznamu srovnávacích látek (1).
Vhodné látky
: Podsoubor referenčních látek zařazených do standardů funkčnosti, které mohou laboratoře použít k prokázání způsobilosti k provádění standardizované analýzy. Kritéria výběru u těchto látek obvykle zahrnují to, že reprezentují celou škálu odpovědí, jsou komerčně dostupné a jsou k nim dostupné kvalitní referenční údaje.
Způsobilost
: Prokázaná schopnost řádně provést analýzu před zkoušením neznámých látek.
Referenční estrogen
: 17ß-estradiol (E2, CAS 50-28-2).
Referenční zkušební metody
: Analýzy, z nichž vychází pokyn PBTG č. 493.
RBA
: Relativní vazebná afinita. RBA látky se vypočítá jako procento log (IC50) pro látku vzhledem k log (IC50) pro 17β-estradiol.
Relevantnost
: Popis vztahu analýzy k účinku, který je předmětem zájmu, a také toho, zda je analýza smysluplná a použitelná pro konkrétní účel. Vyjadřuje, do jaké míry analýza správně měří nebo predikuje biologický účinek, který je předmětem zájmu. Relevantnost zahrnuje také posouzení přesnosti (shody) analýzy (1).
Spolehlivost
: Míra rozsahu, v jakém může být analýza časem reprodukovatelná v laboratořích a mezi laboratořemi, když se provádí s použitím stejného protokolu. Vyhodnocuje se na základě výpočtu vnitrolaboratorní a mezilaboratorní reprodukovatelnosti.
SD
: Směrodatná odchylka.
Zkoušená chemická látka
: Jakákoli látka nebo směs zkoušená touto zkušební metodou.
Validovaná zkušební metoda
: Analýza, u níž byly provedeny validační studie k určení její relevantnosti (včetně přesnosti) a spolehlivosti pro konkrétní účel. Je důležité si uvědomit, že validovaná zkušební metoda nemusí mít dostatečnou funkčnost, pokud jde o přesnost a spolehlivost, aby mohla být shledána přijatelnou pro navrhovaný účel (1).
Validace
: Proces, při němž je stanovena spolehlivost a relevance konkrétního přístupu, metody, analýzy, procesu nebo hodnocení pro definovaný účel (1).
Dodatek 2
FREYBERGER-WILSONOVA (FW) IN VITRO SATURAČNÍ ANALÝZA A ANALÝZA KOMPETITIVNÍ VAZBY NA ESTROGENOVÝ RECEPTOR (ERΑ) S VYUŽITÍM CELÉ DÉLKY LIDSKÉHO REKOMBINANTNÍHO ERΑ
VÝCHOZÍ ÚVAHY A OMEZENÍ (VIZ TAKÉ VŠEOBECNÝ ÚVOD)
|
1.
|
Tato in vitro analýza saturace a kompetitivní vazby na estrogenový receptor (ERα) používají celou délku lidského receptoru ERαα(hrERα), který je vytvořen a izolován z buněk hmyzu infikovaných bakulovirem. Protokol, který vypracovali Freyberger a Wilson, prošel mezinárodní validační studií v několika laboratořích (2), která prokázala jeho relevanci a spolehlivost pro zamýšlený účel analýzy.
|
|
2.
|
Tato analýza je screeningovým postupem pro určení látek, které se mohou vázat na hrERα v celé délce. Používá se na určení schopnosti zkoušené chemické látky konkurovat 17β-estradiolu ve vazbě na hrERα. Výsledky kvantitativní analýzy mohou zahrnovat IC50 (míra koncentrace zkoušené chemické látky potřebná na vytěsnění poloviny [3H]-17β-estradiolu z hrERα) a relativní vazebné afinity zkoušených chemických látek pro hrERα v porovnání s 17β-estradiol. Pro účely chemického screeningu mohou přijatelné výsledky kvalitativní analýzy zahrnovat klasifikace zkoušených chemických látek jako látek, které se vážou na hrERα, látek, které se na něj nevážou, nebo látek neurčitých, a to na základě kritérií popsaných u vazebných křivek.
|
|
3.
|
Analýza používá radioaktivní ligand, což vyžaduje, aby laboratoř měla povolení nakládat s radioaktivními materiály. Při používání radioizotopů a nebezpečných chemických látek musí být dodržovány předpisy a postupy stanovené vnitrostátními právními předpisy.
|
|
4.
|
Před použitím této analýzy pro regulační účely je třeba přečíst si části „VŠEOBECNÝ ÚVOD“ a „SLOŽKY VAZEBNÉ ANALÝZY hrER“. Definice a zkratky používané v této zkušební metodě jsou popsány v dodatku 1.
|
PRINCIPY ANALÝZY (VIZ TAKÉ VŠEOBECNÝ ÚVOD)
|
5.
|
Test vazby na hrERα měří schopnost radioizotopově značeného ligandu ([3H]17β-estradiol) vázat se na ER v přítomnosti zvyšujících se koncentrací zkoušené chemické látky (tj. kompetitor). Zkoušené chemické látky, které mají vysokou afinitu na ER, konkurují radioizotopově značenému ligandu při nižší koncentraci v porovnání s chemickými látkami s nižší afinitou na tento receptor.
|
|
6.
|
Tato analýza sestává ze dvou hlavních složek: saturačního vazebného experimentu za účelem charakterizace parametrů interakce mezi receptorem a ligandem a dokumentace specifičnosti ER, po němž následuje kompetitivní vazebný experiment, který charakterizuje kompetici mezi zkoušenou chemickou látkou a radioizotopově značeným ligandem ve vazbě na ER.
|
|
7.
|
Účelem saturačního vazebného experimentu je charakterizovat konkrétní šarži receptorů z hlediska vazebné afinity a počtu při přípravě na kompetitivní vazebný experiment. Saturační vazebný experiment měří za rovnovážných podmínek afinitu pevně stanovené koncentrace estrogenového receptoru na jeho přirozený ligand (představovaný disociační konstantou Kd) a koncentraci míst aktivního receptoru (Bmax).
|
|
8.
|
Kompetitivní vazebný experiment měří afinitu látky konkurovat [3H]17β-estradiolu ve vazbě na ER. Afinita je kvantifikována koncentrací zkoušené chemické látky, která za rovnovážného stavu inhibuje 50 % specifické vazby [3H]17β-estradiolu (nazývá se „50 % inhibiční koncentrace“ nebo IC50). Může se hodnotit také pomocí relativní vazebné afinity (RBA, vzhledem k IC50 estradiolu měřené samostatně při stejném provedení zkoušky). Kompetitivní vazebný experiment měří vazbu [3H]17β-estradiolu při pevně stanovené koncentraci v přítomnosti širokého rozmezí (osm řádů) koncentrací zkoušené chemické látky. Data se poté vynesou pokud možno do určité formy Hillovy rovnice (Hill, 1910), která popisuje vytěsnění radioligandu kompetiční látkou s jedním vazebným místem. Rozsah vytěsnění radioizotopově značeného estradiolu v rovnovážném stavu se používá na charakterizaci zkoušené chemické látky jako látky, která se váže, která se neváže nebo která generuje neurčitou odpověď.
|
POSTUP
Prokázání funkční způsobilosti proteinu hrERα
|
9.
|
Před rutinním prováděním saturačních a kompetitivních vazebných analýz by mělo být u každé nové šarže hrERα prokázáno, že v laboratoři, v níž bude používán, funguje správně. K prokázání způsobilosti by se měl použít proces zahrnující dva kroky. Jsou to tyto kroky:
|
—
|
Proveďte saturační vazebnou analýzu [3H]-17β-estradiolu na prokázání specifičnosti a saturace hrERα. V nelineární regresní analýze těchto dat (např. BioSoft; McPherson, 1985; Motulsky, 1995) a na následném Scatchardově grafu by měla být dokumentována vazebná afinita hrERα vůči [3H]-17β-estradiolu (Kd) a počet receptorů (Bmax) pro každou šarži hrERα.
|
|
—
|
Proveďte kompetitivní vazebnou analýzu pomocí kontrolních látek (referenčního estrogenu (17β-estradiol)), látky se slabou vazbou (např. norethynodrel nebo norethindron) a látky, která se neváže (oktyltriethoxysilan, OTES). Každá laboratoř by si měla vytvořit historickou databázi dokumentující konzistentnost hodnot IC50 a dalších relevantních hodnot pro referenční estrogen a látku se slabou vazbou u experimentů a různých šarží hrERα. Parametry kompetitivních vazebných křivek pro kontrolní látky by měly být v rozmezí 95 % konfidenčního intervalu (viz tabulka 1), který byl stanoven pomocí dat z laboratoří, jež se účastnily validační studie pro tuto analýzu (2).
|
|
Tabulka 1
Kritéria výkonnosti vypracovaná pro referenční estrogen a látku se slabou vazbou, vazebná analýza FW hrER
|
Látka
|
Parametr
|
Střední hodnota (8)
|
Směrodatná odchylka (n)
|
95 % konfidenční intervaly (9)
|
|
Dolní mez
|
Horní mez
|
|
17β-estradiol
|
Horní (%)
|
100,44
|
10,84 (67)
|
97,8
|
103,1
|
|
Dolní (%)
|
0,29
|
1,25 (67)
|
–0,01
|
0,60
|
|
Hillův koeficient
|
–1,06
|
0,20 (67)
|
–1,11
|
–1,02
|
|
logIC50 (M)
|
–8,92 (10)
|
0,18 (67)
|
–8,97
|
–8,88
|
|
Norethynodrel
|
Horní (%)
|
99,42
|
8,90 (68)
|
97,27
|
101,60
|
|
Dolní (%)
|
2,02
|
3,42 (68)
|
1,19
|
2,84
|
|
Hillův koeficient
|
–1,01
|
0,38 (68)
|
–1,10
|
–0,92
|
|
log IC50 (M)
|
–6,39
|
0,27 (68)
|
–6,46
|
–6,33
|
|
Norethindrone (10)
|
Horní (%)
|
96,14
|
8,44 (27)
|
92,80
|
99,48
|
|
Dolní (%)
|
2,38
|
5,02 (27)
|
0,40
|
4,37
|
|
Hillův koeficient
|
–1,41
|
0,32 (27)
|
–1,53
|
-1,28
|
|
logIC50(M)
|
–5,73
|
0,27 (27)
|
–5,84
|
–5,62
|
Prokázání odborné způsobilosti laboratoře
|
10.
|
Viz odstavce 17 a 18 a tabulka 2 v části „SLOŽKY VAZEBNÉ ANALÝZY hrER“ této zkušební metody. Každá analýza (saturační a kompetitivní vazby) by měla zahrnovat tři nezávislé zkoušky (tzn. s čerstvě naředěným receptorem, chemickými látkami a reagenciemi) provedené v různé dny, přičemž každé provedení by mělo obsahovat tři opakování.
|
Určení koncentrace receptoru (hrERα)
|
11.
|
Koncentrace aktivního receptoru se poněkud liší podle šarže a podmínek uchovávání. Z tohoto důvodu je třeba stanovit koncentraci aktivního receptoru přijatého od dodavatele. Získá se tím vhodná koncentrace aktivního receptoru v době provedení zkoušky.
|
|
12.
|
Za podmínek odpovídajících kompetitivní vazbě (tj. 1 nM [3H]-estradiol) by se měly nominální koncentrace receptoru 0,25, 0,5, 0,75 a 1 nM inkubovat v nepřítomnosti (celková vazba) a v přítomnosti (nespecifická vazba) 1 μM neznačeného estradiolu. Specifická vazba vypočítaná jako rozdíl celkové a nespecifické vazby se nanese proti nominální koncentraci receptoru. Koncentrace receptoru, která dává hodnoty specifické vazby odpovídající 20 % přidaného radioizotopového značení, souvisí s odpovídající nominální koncentrací receptoru a tato koncentrace receptoru se používá pro saturační a kompetitivní vazebné experimenty. Často bude tuto podmínku splňovat konečná koncentrace hrER 0,5 nM.
|
|
13.
|
Jestliže nelze opakovaně splnit 20 % kritérium, je třeba zkontrolovat, zda uspořádání experimentu neobsahuje možné chyby. Nedosažení 20 % kritéria může ukazovat, že v rekombinantní šarži je příliš málo aktivního receptoru, a potom je třeba zvážit použití jiné šarže receptoru.
|
Saturační analýza
|
14.
|
Hodnotí se osm zvyšujících se koncentrací [3H]17β-estradiolu v trojím provedení, a to za těchto tří podmínek (viz tabulka 2):
|
—
|
Za nepřítomnosti neznačeného 17β-estradiolu a v přítomnosti ER. Je to stanovení celkové vazby měřením radioaktivity v jamkách, v nichž je pouze [3H]17β-estradiol.
|
|
—
|
V přítomnosti tisícinásobně zvýšené koncentrace neznačeného 17β-estradiolu nad značený 17β-estradiol a v přítomnosti ER. Záměrem této podmínky je saturovat aktivní vazebná místa neznačeným 17β-estradiolem a změřením radioaktivity v jamkách stanovit nespecifickou vazbu. Případný zbývající radioaktivní estradiol, který se může vázat na receptor, se považuje za navázaný na nespecifické místo, neboť neradioaktivní estradiol by měl být v tak vysoké koncentraci, aby se navázal na všechna dostupná specifická místa na receptoru.
|
|
—
|
Za nepřítomnosti neznačeného 17β-estradiolu a nepřítomnosti ER (stanovení celkové radioaktivity)
|
|
Příprava roztoků [3H]-17β-estradiolu a neznačeného 17β-estradiolu
|
15.
|
Zředěné roztoky [3H]-17β-estradiolu se připravují přidáním zkušebního pufru do 12 nM zásobního roztoku [3H]-17β-estradiolu, aby se získaly koncentrace v počátečním rozsahu od 0,12 nM do 12 nM. Přidáním 40 μl těchto roztoků do příslušných jamek 96jamkové mikrotitrační destičky (v konečném objemu 160 μl) se získají konečné zkušební koncentrace v rozsahu od 0,03 do 3,0 nM. Příprava zkušebního pufru, zásobního roztoku [3H]-17β-estradiolu a zředěných roztoků a stanovení koncentrací je podrobně popsáno v protokolu FW (2).
|
|
16.
|
Zředěné etanolové roztoky 17β-estradiolu se připravují přidáním zkušebního pufru, aby bylo dosaženo osmi zvyšujících se koncentrací v počátečním rozsahu od 0,06 μM do 6 μM. Přidáním 80 μl těchto roztoků do příslušných jamek 96jamkové mikrotitrační destičky (v konečném objemu 160 μl) se získají konečné zkušební koncentrace v rozsahu od 0,03 μM do 3,0 μM. Konečná koncentrace neznačeného 17β-estradiolu v jednotlivých jamkách nespecifické vazebné analýzy by měla být tisícinásobkem koncentrace značeného [3H]-17β- estradiolu. Příprava roztoků neznačeného 17β-estradiolu je podrobně popsána v protokolu FW (2).
|
|
17.
|
Měla by se použít taková nominální koncentrace receptoru, která dává specifickou vazbu 20 ± 5 % (viz odstavce 12–13). Roztok hrERα je třeba připravovat bezprostředně před použitím.
|
|
18.
|
96jamkové mikrotitrační destičky se připraví tak, jak je zobrazeno v tabulce 2, a to s třemi replikáty na každou koncentraci. Příklad uspořádání koncentrací a objemů [3H]-17β-estradiolu, neznačeného 17β-estradiolu, pufru a receptoru je uveden v dodatku 2.2.
|
Tabulka 2
Uspořádání mikrotitrační destičky při saturační vazebné analýze
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3H] E2 + ER
|
0,06 nM [3H] E2 + ER
|
0,08 nM [3H] E2 + ER
|
0,10 nM [3 H] E2 + ER
|
Celková vazba (rozpouštědlo)
|
|
B
|
0,30 nM [3H] E2 + ER
|
0,60 nM [3H] E2 + ER
|
1,0 nM [3H] E2 + ER
|
3,0 nM [3H] E2 + ER
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3H] E2 + ER + 0,03 μM E2
|
0,06 nM [3H] E2 + ER + 0,06 μM E2
|
0,08 nM [3H] E2 + ER + 0,08 μM E2
|
0,10 nM [3H] E2 + ER
+ 0,10 μM E2
|
Nespecifická vazba
|
|
E
|
0,30 nM [3H] E2 + ER + 0,30 μM E2
|
0,60 nM [3H] E2 + ER + 0,60 μM E2
|
1,0 nM [
3
H] E2 + ER
+ 1,0 μM E2
|
3,0 nM [3H] E2 + ER + 3,0 μM E2
|
|
F
|
|
|
|
|
|
|
G
|
|
|
|
|
|
|
H
|
|
|
|
|
|
[3H] E2:: [3H]-17β-estradiol
ER:: estrogenový receptor
E2:: neznačený 17β-estradiol
|
|
19.
|
Experimentální mikrotitrační destičky se inkubují při 2–8 oC po dobou 16 až 20 hodin a v průběhu inkubace se umístí na otáčecí podložku.
|
Měření [3H]-17β-estradiolu navázaného na hrERα
|
20.
|
[3H]-17β-estradiol navázaný na hrERα se separuje z volného [3H]-17β-estradiolu přidáním 80 μl studené suspenze DCC do každé jamky, mikrotitrační destičky se protřepávají po dobu 10 minut a odstřeďují po dobu 10 minut při 2 500 ot./min. Aby se v průběhu tohoto procesu minimalizovala disociace vázaného [3H]-17β-estradiolu z hrERα, je velmi důležité, aby byly pufry i experimentální jamky udržovány při teplotě mezi 2 a 8 oC a aby byl každý krok proveden rychle. Pro efektivní a rychlé zpracování destiček je nezbytná třepačka na mikrotitrační destičky.
|
|
21.
|
Poté se velmi opatrně odebere 50 μl supernatantu obsahujícího [3H]-17β-estradiol navázaný na hrERα, aby nedošlo ke kontaminaci jamek dotykem DCC, a umístí se na druhou mikrotitrační destičku.
|
|
22.
|
Pak se do každé jamky přidá 200 μl scintilační kapaliny, schopné přeměnit kinetickou energii jaderných emisí na světelnou energii (A1–B12 a D1 až E12). Jamky G1–H12 (označené jako celkové dpm) představují sériové naředěné roztoky [3H]-17β-estradiolu (40 μl), které je třeba přemístit přímo do scintilační kapaliny v jamkách měřicí destičky, jak je vyznačeno v tabulce 3, tj. tyto jamky obsahují pouze 200 μl scintilační kapaliny a příslušně naředěný roztok [3H]-17β-estradiolu. Tato měření ukazují, kolik [3H]-17β-estradiolu v dpm bylo přidáno do každé sady jamek pro celkovou vazbu a nespecifickou vazbu.
|
Tabulka 3
Uspořádání mikrotitrační destičky při saturační vazebné analýze, měření radioaktivity
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3H] E2 + ER
|
0,06 nM [3H] E2 + ER
|
0,08 nM [3H] E2 + ER
|
0,10 nM [3 H] E2 + ER
|
Celková vazba (rozpouštědlo)
|
|
B
|
0,30 nM [3H] E2 + ER
|
0,60 nM [3H] E2 + ER
|
1,0 nM [3H] E2 + ER
|
3,0 nM [3H] E2 + ER
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3H] E2 + ER + 0,03 μM E2
|
0,06 nM [3H] E2 + ER + 0,06 μM E2
|
0,08 nM [3H] E2 + ER + 0,08 μM E2
|
0,10 nM [3H] E2 + ER
+ 0,10 μM E2
|
Nespecifická vazba
|
|
E
|
0,30 nM [3H] E2 + ER + 0,30 μM E2
|
0,60 nM [3H] E2 + ER + 0,60 μM E2
|
1,0 nM [
3
H] E2 + ER
+ 1,0 μM E2
|
3,0 nM [3H] E2 + ER + 3,0 μM E2
|
|
F
|
|
|
|
|
|
|
G
|
0,03 nM [3H] E2 (dpm celkem)
|
0,06 nM [3H] E2
|
0,08 nM [3H] E2
|
0,10 nM [3H] E2
|
Celkem dpm (*8)
|
|
H
|
0,30 nM [3H] E2
|
0,60 nM [3H] E2
|
1,0 nM [3H] E2
|
3,0 nM [3H] E2
|
|
[3H] E2:: [3H]-17β-estradiol
ER:: estrogenový receptor
E2:: neznačený 17β-estradiol
dpm:: rozpady za minutu
|
|
23.
|
Měření by mělo být zahájeno se zpožděním alespoň 2 hodiny a doba počítání by měla být 40 minut na jamku. Ke stanovení dpm/jamku s korekcí na zhášení se používá scintilační spektrometr mikrotitračních destiček. Není-li k dispozici scintilační spektrometr pro mikrotitrační destičky, mohou se vzorky alternativně měřit klasickým spektrometrem. Za těchto podmínek může být zváženo zkrácení doby počítání.
|
Kompetitivní vazebná analýza
|
24.
|
Kompetitivní vazebná analýza měří vazbu jedné koncentrace [3H]17β-estradiolu v přítomnosti zvyšujících se koncentrací zkoušené chemické látky. Při každé koncentraci by se měly v jednom provedení zkoušky použít tři souběžné replikáty. Kromě toho by se měla pro každou zkoušenou chemickou látku provést tři nesouběžná provedení zkoušky. Analýza se provádí v jedné nebo několika 96jamkových mikrotitračních destičkách.
|
Kontrolys
|
25.
|
Při provádění analýzy by měly být do každého experimentu zařazeny souběžné testy s rozpouštědlem a kontroly (tj. referenční estrogen, slabá vazba a neváže se). Při každém provedení zkoušky by měly být na jedné destičce použity celé křivky koncentrace pro referenční estrogen a kontroly (tj. slabá vazba a neváže se). Všechny ostatní destičky by měly obsahovat i) vysokou (maximální vytěsnění) a střední (přibližně IC50) koncentraci každého E2 a látky se slabou vazbou, a to třikrát; ii) kontrolu s rozpouštědlem a látkou s nespecifickou vazbou, a to vždy alespoň třikrát. Postupy pro přípravu experimentálního pufru, kontrol, [3H]-17β-estradiolu, hrERα a roztoků zkoušených chemických látek jsou popsány v odkazu 2 (příloha K, viz protokol pro analýzu FW).
|
Kontrola s rozpouštědlem:
|
26.
|
Kontrola s rozpouštědlem ukazuje, že rozpouštědlo nereaguje se zkušebním systémem, a měří také celkovou vazbu (TB). Nejvhodnějším rozpouštědlem je ethanol. Jestliže nejvyšší koncentrace zkoušené chemické látky není rozpustná v ethanolu, může být alternativně použit DMSO. Koncentrace ethanolu nebo DMSO, je-li použit, v jamkách pro konečnou analýzu je 1,5 % a nesmí překročit 2 %.
|
Kontrola s pufrem:
|
27.
|
Kontrola s pufrem (BC) by neměla obsahovat rozpouštědlo ani zkoušenou chemickou látku, ale všechny ostatní složky analýzy. Výsledky kontroly s pufrem se porovnají s kontrolou s rozpouštědlem za účelem ověření, že použité rozpouštědlo neovlivňuje zkušební systém.
|
Látka se silnou vazbou (referenční estrogen)
|
28.
|
17β-estradiol (CAS 50-28-2) je endogenní ligand a váže se s vysokou afinitou na ER, podtyp alfa. Pro každou kompetitivní vazebnou analýzu hrERα by měla být vypracována standardní křivka s použitím neznačeného 17β-estradiolu, aby bylo možné posoudit variabilitu při provádění analýzy v čase ve stejné laboratoři. Připraví se osm roztoků neznačeného 17β-estradiolu v ethanolu, přičemž koncentrace v jamkách jsou v rozmezí 100 nM–10 pM (od –7[logM] do –11[logM]), a to v těchto rozestupech: (–7[logM], –8[logM], –8,5[logM], –9[logM], –9,5[logM], –10[logM], –11[logM]). Nejvyšší koncentrace neznačeného 17β-estradiolu (1 μM) slouží také jako indikátor nespecifické vazby. Tato koncentrace je v tabulce 4 odlišena označením „NSB“, ačkoliv je také součástí standardní křivky.
|
Látka se slabou vazbou
|
29.
|
Měla by být zařazena látka se slabou vazbou (norethynodrel (CAS 68-23-5) nebo norethindron (CAS 68-22-4)), aby se prokázala citlivost každého experimentu a aby se umožnilo posouzení variability při provádění analýzy v čase. Připraví se osm roztoků látky se slabou vazbou v ethanolu, přičemž koncentrace v jamkách jsou v rozmezí od 3 nM do 30 μM (od –8,5[logM] do –4,5[logM]), a to v těchto rozestupech: –4,5[logM], –5[logM], –5,5[logM], –6[logM], –6,5[logM], –7[logM], –7,5[logM], –8,5[logM].
|
Látka, která se neváže
|
30.
|
Jako negativní kontrola (látka, která se neváže) se používá oktyltriethoxysilan (OTES, CAS 2 943-75-1). Poskytuje jistotu, že analýza, tak jak je prováděna, dokáže detekovat, když se zkoušené chemické látky nenavážou na hrERα. Připraví se osm roztoků látky, která se neváže, v ethanolu, přičemž koncentrace v jamkách jsou v rozmezí od 0,1 nM do 1 000 μM (od –10[logM] do –3[logM]), a to v logaritmických přírůstcích. Jako alternativní kontrolní látka, která se neváže, může být použit di-n-butylftalát (DBP). Bylo prokázáno, že jeho maximální rozpustnost je–4[logM].
|
Koncentrace hrERα
|
31.
|
Mělo by se použít takové množství receptoru, které dává specifickou vazbu 20 ± 5 % 1 nM radioligandu (viz odstavce 12–13 v dodatku 2). Roztok hrERα je třeba připravovat bezprostředně před použitím.
|
[3H]-17β-estradiol
|
32.
|
Koncentrace [3H]-17β-estradiolu v jamkách by měla být 1,0 nM.
|
Zkoušené chemické látky
|
33.
|
V první řadě je třeba provést zkoušku rozpustnosti, aby bylo možné stanovit mez rozpustnosti pro každou zkoušenou chemickou látku a zjistit vhodný rozsah koncentrace, jenž má být použit při provádění zkušebního protokolu. Mez rozpustnosti každé zkoušené chemické látky se nejprve stanoví v rozpouštědle a dále potvrdí za zkušebních podmínek. Konečná koncentrace zkoušená při analýze by neměla překročit 1 mM. Zkoušení za účelem zjištění rozsahu sestává z kontroly s rozpouštědlem společně s 8 log-sériovými ředěními, počínaje maximální přijatelnou koncentrací (např. 1 mM nebo menší, na základě meze rozpustnosti), přičemž je třeba zaznamenat přítomnost zákalu nebo sraženiny (viz také odstavec 35). Zkoušená chemická látka by měla být testována s použitím 8 logaritmických křivek koncentrací v rozestupech definovaných předchozí zkouškou ke stanovení rozsahu. Při druhém a třetím experimentu by se měla podle potřeby upravit koncentrace, aby lépe charakterizovala křivku závislosti odpovědi na koncentraci.
|
|
34.
|
Zkoušená chemická látka se naředí ve vhodném rozpouštědle (viz odstavec 26 v dodatku 2). Jestliže nejvyšší koncentrace zkoušené chemické látky není rozpustná v ethanolu ani v DMSO a přidání většího množství rozpouštědla by způsobilo, že koncentrace rozpouštědla v poslední zkumavce bude větší než přijatelná mez, může se nejvyšší koncentrace snížit na nejbližší nižší koncentraci. V tomto případě může být na dolní konec série koncentrací přidána další koncentrace. Ostatní koncentrace v sérii by měly zůstat beze změny.
|
|
35.
|
Roztoky zkoušené chemické látky je třeba po přidání do jamky pečlivě sledovat, neboť zkoušená chemická látka se může po přidání do jamky srážet. Data za všechny jamky, které obsahují sraženinu, by měla být vyloučena z vynesené křivky a měl by být uveden důvod pro vyloučení těchto dat.
|
|
36.
|
Jestliže již existují informace z jiných zdrojů, které uvádějí log(IC50) zkoušené chemické látky, může být vhodné stanovit geometrické rozestupy ředění (tj. 0,5 log jednotky kolem předpokládané hodnoty log(IC50)). Konečný výsledek by měl odrážet dostatečný rozsah koncentrací na každé straně log(IC50), včetně „horní“ a „dolní“, aby vazebná křivka mohla být adekvátně charakterizována.
|
Uspořádání zkušebních destiček
|
37.
|
Připraví se označené mikrotitrační destičky pro šestinásobné inkubace s kódy pro kontrolu s rozpouštědlem, nejvyšší koncentraci referenčního estrogenu, který slouží také jako indikátor nespecifické vazby (NSB), a kontrolu s pufrem a pro trojnásobné inkubace s kódy pro každou z osmi koncentrací kontrolní látky, která se neváže (oktyltriethoxysilan), sedm nižších koncentrací pro referenční estrogen, osm úrovní koncentrace látky se slabou vazbou a osm koncentrací každé zkoušené chemické látky (TC). Příklad diagramu destičky pro celé koncentrační křivky pro referenční estrogen a kontrolu je uveden níže v tabulce 4. Pro zkoušené chemické látky se použijí další mikrotitrační destičky, které by měly obsahovat kontroly, tj. 1) vysokou (maximální vytěsnění) a střední (přibližně IC50) koncentraci každého E2 a látky se slabou vazbou, a to třikrát; 2) kontrolu s rozpouštědlem a látkou s nespecifickou vazbou, a to vždy šestkrát (tabulka 5). Příklad postupu uspořádání mikrotitrační destičky pro kompetitivní analýzu s použitím tří neznámých zkoušených chemických látek je uveden v dodatku 2.3. Koncentrace uvedené v tabulkách 4 a 5 jsou konečné koncentrace analýzy. Maximální koncentrace pro E2 by měla být 1 × 10-7 M a pro látku se slabou vazbou by měla být použita nejvyšší koncentrace pro látku se slabou vazbou použitá na destičce 1. Koncentraci IC50 musí stanovit laboratoř na základě své dosavadní kontrolní databáze. Lze očekávat, že tato hodnota bude podobná hodnotě pozorované ve validačních studiích (viz tabulka 1).
|
Tabulka 4
Uspořádání mikrotitrační destičky při kompetitivní vazebné analýze, celé křivky koncentrace pro referenční estrogen a kontroly (destička 1)
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (pouze rozpouštědlo)
|
TB (pouze rozpouštědlo)
|
NSB
|
NSB
|
|
B
|
E2 (1 × 10-7)
|
E2 (1 × 10-8)
|
E2 (1 × 10-8,5)
|
E2 (1 × 10-9)
|
|
C
|
E2 (1 × 10-9,5)
|
E2 (1 × 10-10)
|
E2 (1 × 10-11)
|
Slepý pokus (*9)
|
|
D
|
NE (1 × 10-4.5)
|
NE (1 × 10-5)
|
NE (1 × 10-5,5)
|
NE (1 × 10-6)
|
|
E
|
NE (1 × 10-6,5)
|
NE (1 × 10-7)
|
NE (1 × 10-7,5)
|
NE (1 × 10-8,5)
|
|
F
|
OTES (1 × 10-3)
|
OTES (1 × 10-4)
|
OTES (1 × 10-5)
|
OTES (1×10-6)
|
|
G
|
OTES (1 × 10-7)
|
OTES (1 × 10-8)
|
OTES (1 × 10-9)
|
OTES (1 × 10-10)
|
|
H
|
Slepý pokus (pro radioaktivní) (*10)
|
Slepý pokus (pro radioaktivní) (*10)
|
Kontrola s pufrem
|
Kontrola s pufrem
|
|
V tomto příkladu je látkou se slabou vazbou norethinodrel (NE)
|
Tabulka 5
Uspořádání mikrotitrační destičky při analýze kompetitivní vazby, celé křivky koncentrace pro zkoušené chemické látky a kontroly
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (pouze rozpouštědlo)
|
TB (pouze rozpouštědlo)
|
NSB
|
NSB
|
|
B
|
TC1 (1 × 10-3)
|
TC1 (1 × 10-4)
|
TC1 (1 × 10-5)
|
TC1 (1 × 10-6)
|
|
C
|
TC1 (1 × 10-7)
|
TC1 (1 × 10-8)
|
TC1 (1 × 10-9)
|
TC1 (1 × 10-10)
|
|
D
|
TC2 (1 × 10-3)
|
TC2 (1 × 10-4)
|
TC2 (1 × 10-5)
|
TC2 (1 × 10-6)
|
|
E
|
TC2 (1 × 10-7)
|
TC2 (1 × 10-8)
|
TC2 (1 × 10-9)
|
TC2 (1 × 10-10)
|
|
F
|
TC3 (1 × 10-3)
|
TC3 (1 × 10-4)
|
TC3 (1 × 10-5)
|
TC3 (1×10-6)
|
|
G
|
TC3 (1 × 10-7)
|
TC3 (1 × 10-8)
|
TC3 (1 × 10-9)
|
TC3 (1 × 10-10)
|
|
H
|
NE (IC50)
|
NE (1 × 10-4,5)
|
E2 (IC50)
|
E2 (1 × 10-7)
|
|
V tomto příkladu je látkou se slabou vazbou norethinodrel (NE)
|
Dokončení analýzy kompetitivní vazby
|
38.
|
Jak je uvedeno v tabulce 6, přidá se do jamek 80 μl kontroly s rozpouštědlem, kontroly s pufrem, referenční estrogen, látka se slabou vazbou, látka, která se neváže, a zkušební chemické látky připravené ve zkušebním pufru. Poté se do každé jamky přidá 40 μl roztoku 4 nM [3H]-17β-estradiolu. Po mírném otáčení po dobu 10 až 15 minut při teplotě od 2 oC do 8 oC se přidá 40 μl roztoku hrERα. Experimentální mikrotitrační destičky se inkubují při 2 oC až 8 oC po dobou 16 až 20 hodin a v průběhu inkubace se umístí na otáčecí podložku.
|
Tabulka 6
Objem zkušebních složek pro kompetitivní vazebnou analýzu hrER, mikrotitrační destičky
|
Objem (μl)
|
Složka
|
|
80
|
Neznačený 17β-estradiol, norethynodrel, OTES, zkoušené chemické látky, rozpouštědlo nebo pufr
|
|
40
|
roztok 4 nM [3H]-17β-estradiolu
|
|
40
|
roztok hrERα, určená koncentrace
|
|
160
|
Celkový objem v každé jamce
|
|
39.
|
Poté se provede kvantifikace [3H]-17β-estradiolu navázaného na hrERα po separaci [3H]-17β-estradiolu navázaného na hrERα z volného [3H]-17β-estradiolu přidáním 80 μl studené suspenze DCC do každé jamky, jak je popsáno v odstavcích 20–23 týkajících se saturační vazebné analýzy.
|
|
40.
|
Jamky H1–6 (označené v tabulce 4 jako slepý pokus (pro radioaktivní látku)) představují dpm [3H]-značeného-estradiolu v 40 μl. Alikvotní množství z 40 μl se přidá přímo do scintilační kapaliny v jamkách H1–H6.
|
Kritéria přijatelnosti
Saturační vazebná analýza
|
41.
|
Křivka specifické vazby by měla dosáhnout plató s použitím zvyšujících se koncentrací [3H]-17β-estradiolu, což ukazuje saturaci hrERα ligandem.
|
|
42.
|
Specifická vazba při 1 nM [3H]-17β-estradiolu by měla být v přijatelném rozmezí 15 % až 25 % průměrné naměřené celkové radioaktivity přidané v průběhu jednotlivých provedení zkoušek. Občasná mírná vybočení mimo toto rozmezí jsou přijatelná, ale pokud jsou všechna provedení zkoušky mimo toto rozmezí nebo pokud je konkrétní provedení zkoušky výrazně mimo toto rozmezí, je třeba upravit koncentraci proteinu a saturační analýzu zopakovat.
|
|
43.
|
Data by měla vytvořit lineární Scatchardův graf.
|
|
44.
|
Nespecifická vazba by neměla být nadměrná. Hodnota nespecifické vazby by měla být typicky < 35 % celkové vazby. Tento poměr však může při měření velmi nízkých dpm u nejnižší koncentrace radioizotopově značeného zkoušeného 17β-estradiolu občas uvedený limit překročit.
|
Analýza kompetitivní vazby
|
45.
|
Zvyšující se koncentrace neznačeného 17β-estradiolu by měly vytěsnit [3H]-17β- estradiol z receptoru způsobem odpovídajícím jednomístné kompetitivní vazbě.
|
|
46.
|
Hodnota IC50 pro referenční estrogen (tj. 17β-estradiol) by se měla přibližně rovnat molární koncentraci [3H]-17β-estradiolu plus Kd určený ze saturační vazebné analýzy.
|
|
47.
|
Celková specifická vazba by měla být soustavně v přijatelném rozmezí 20 ± 5 %, když je průměrná naměřená koncentrace celkové radioaktivity přidaná do každé jamky 1 nM ve všech provedeních zkoušky. Občasná mírná vybočení mimo toto rozmezí jsou přijatelná, ale pokud jsou všechna provedení zkoušky mimo toto rozmezí nebo pokud je konkrétní provedení zkoušky výrazně mimo toto rozmezí, je třeba upravit koncentraci proteinu.
|
|
48.
|
Rozpouštědlo by nemělo měnit citlivost ani reprodukovatelnost analýzy. Výsledky kontroly s rozpouštědlem (jamky TB) se porovnají s kontrolou s pufrem za účelem ověření, že použité rozpouštědlo neovlivňuje zkušební systém. Jestliže rozpouštědlo nemá na analýzu žádný vliv, měly by být výsledky TB a kontroly s rozpouštědlem srovnatelné.
|
|
49.
|
Látka, která se neváže, by neměla vytěsnit více než 25 % [3H]-17β-estradiolu z hrERα při zkoušení do 10-3 M (OTES) nebo 10-4 M (DBP).
|
|
50.
|
Pro referenční estrogen a dvě látky se slabou vazbou (např. norethynodrel, norethindron) byla vypracována kritéria funkčnosti s použitím údajů z validační studie vazebné analýzy FW hrER (příloha N v odkazu 2). Jsou uvedeny 95 % konfidenční intervaly pro střední hodnotu (n) +/– SD za všechna kontrolní provedení zkoušky ve všech laboratořích účastnících se ve validační studii. Pro referenční estrogen a látky se slabou vazbou a pro log10RBA látek se slabou vazbou vzhledem k referenčnímu estrogenu byly vypočteny 95 % konfidenční intervaly pro parametry křivky (tj. horní, dolní, Hillův koeficient, logIC50), které jsou uvedeny jako kritéria funkčnosti pro pozitivní kontroly. V tabulce 1 jsou uvedeny očekávané rozsahy parametrů křivky, které mohou být použity jako kritéria funkčnosti. V praxi se může rozsah IC50 poněkud odlišovat v závislosti na Kd receptorového přípravku a koncentraci ligandu.
|
|
51.
|
Pro parametry křivky pro zkoušené chemické látky nebyla vypracována žádná kritéria funkčnosti, protože stávajících potenciálních zkoušených chemických látek existuje velké množství a značně se liší jejich potenciální afinity a výsledky (např. celá křivka, částečná křivka, mimo křivku). Při posuzování výsledků z každého provedení zkoušky za zkoušenou chemickou látku je třeba postupovat s odborným úsudkem. Mělo by být použito dostatečné rozmezí koncentrací, aby byl jednoznačně definován vrchol (např. 90–100 % vazby) kompetitivní křivky. Variabilita mezi replikáty při každé koncentraci zkoušené chemické látky i mezi 3 zkouškami neprováděnými souběžně by měla být přiměřená a vědecky obhajitelná. Kontroly z každého provedení zkoušky by se u zkoušené chemické látky měly blížit mírám funkčnosti uvedeným pro tuto analýzu FW a měly by odpovídat dosavadním kontrolním údajům z každé příslušné laboratoře.
|
ANALÝZA ÚDAJŮ
Saturační vazebná analýza
|
52.
|
Měří se celková i nespecifická vazba. Z těchto hodnot se specifická vazba zvyšujících se koncentrací [3H]-17β-estradiolu za rovnovážného stavu vypočítá odečtením nespecifické vazby od celkové. Graf specifické vazby proti koncentraci [3H]-17β-estradiolu by měl dosáhnout plató u maximální specifické vazby indikující saturaci hrERα [3H]-17β-estradiolem. Kromě toho by analýza dat měla dokumentovat vazbu [3H]-17β-estradiolu na jedno vazebné místo s vysokou afinitou. Na křivce saturační vazby by měla být zobrazena nespecifická, celková a specifická vazba. Další rozbor těchto údajů by měl být proveden pomocí nelineární regresní analýzy (např. BioSoft; McPherson, 1985; Motulsky, 1995) s konečným zobrazením dat ve formě Scatchardova grafu.
|
|
53.
|
Při analýze dat se určuje Bmax a Kd z údajů o celkové vazbě samotné, a to za použití předpokladu, že nespecifická vazba je lineární, není-li zdůvodněno použití jiné metody. Kromě toho by při určování nejlepších hodnot měla být použita robustní regrese, pokud není uvedeno zdůvodnění. Metoda zvolená pro robustní regresi by měla být uvedena. Při určování Bmax a Kd z údajů o saturační vazbě by měla být vždy použita korekce úbytku ligandu (např. pomocí metody podle Swillense 1995).
|
Analýza kompetitivní vazby
|
54.
|
Křivka kompetitivní vazby se vynáší jako specifická vazba [3H]-17β-estradiolu proti koncentraci (log10 jednotky) kompetitora. Koncentrace zkoušené chemické látky, která inhibuje 50 % maximální specifické vazby [3H]-17β-estradiolu, je hodnota IC50.
|
|
55.
|
Odhady log (IC50) pro pozitivní kontroly (např. referenční estrogen a látku se slabou vazbou) se stanoví pomocí vhodného softwaru pro nelineární vynesení křivky, aby byly vhodné pro čtyřparametrickou Hillovu rovnici (např. BioSoft; McPherson, 1985; Motulsky, 1995). Při vynášení těchto křivek se horní bod, dolní bod, sklon a log(IC50) obvykle ponechávají bez omezení. Při určování nejlepších hodnot by měla být použita robustní regrese, pokud není uvedeno zdůvodnění. Korekce úbytku ligandu se nepoužívá. Po úvodní analýze je třeba každou vazebnou křivku zkontrolovat, aby bylo zajištěno, že je vhodně aplikována na model. Relativní vazebná afinita (RBA) pro látku se slabou vazbou se vypočítá jako procento log (IC50) pro látku se slabou vazbou vzhledem k log (IC50) pro 17β-estradiol. Výsledky z pozitivních kontrol a z kontroly s látkou, která se neváže, se posuzují pomocí měřítek účinnosti analýzy uvedených v odstavci 45–50 v tomto dodatku 2.
|
|
56.
|
Data za všechny zkoušené chemické látky se analyzují postupně, aby bylo zajištěno, že data budou analyzována správně, a aby byla správně klasifikována každá kompetitivní vazebná křivka. Doporučuje se, aby každé provedení zkoušky u zkoušené chemické látky nejdříve prošlo standardizovanou analýzou dat, která je stejná jako analýza použitá pro kontroly s referenčním estrogenem a s látkou se slabou vazbou (viz odstavec 55 výše). Po dokončení je třeba provést technickou kontrolu parametrů vynesené křivky a vizuálně zkontrolovat, jak dobře data odpovídají generované kompetitivní vazebné křivce u každého provedení zkoušky. V průběhu této technické kontroly je pozorovaný pokles procenta specificky navázaného [3H]-17β-estradiolu v závislosti na koncentraci, nízká variabilita mezi technickými replikáty při jednotlivých koncentracích chemické látky a konzistentní parametry u tří provedení zkoušky dobrým ukazatelem, že zkouška a analýzy dat byly provedeny správně.
|
Interpretace údajů
|
57.
|
Za předpokladu, že jsou splněna všechna kritéria přijatelnosti, lze zkoušenou chemickou látku považovat za látku, která se váže na hrERα, jestliže lze vynést vazebnou křivku a nejnižší bod na křivce odpovědi v rozsahu dat je menší než 50 % (obrázek 1).
|
|
58.
|
Za předpokladu, že jsou splněna všechna kritéria přijatelnosti, lze zkoušenou chemickou látku považovat za látku, která se neváže na hrERα, jestliže:
|
—
|
lze vynést vazebnou křivku a nejnižší bod na křivce odpovědi v rozsahu dat je nad 75 % nebo
|
|
—
|
nelze vynést vazebnou křivku a nejnižší nevyhlazené průměrné procento vazby ze skupiny koncentrací v datech je nad 75 %.
|
|
|
59.
|
Zkoušené chemické látky se považují za neurčité, jestliže není splněna žádná z výše uvedených podmínek (např. nejnižší bod na vynesené křivce odpovědi je mezi 76 a 51 %).
|
Tabulka 7
Kritéria pro stanovení klasifikace zkoušené chemické látky podle kompetitivní vazebné křivky
|
Klasifikace
|
Kritéria
|
|
váže sea
|
Lze vynést vazebnou křivku.
Nejnižší bod na křivce odpovědi v rozsahu dat je menší než 50 %.
|
|
neváže seb
|
Jestliže lze vynést vazebnou křivku,
je nejnižší bod na křivce odpovědi v rozsahu dat nad 75 %.
Jestliže nelze vynést vazebnou křivku,
je nejnižší nevyhlazené průměrné procento vazby ze skupiny koncentrací v datech nad 75 %.
|
|
Neurčitác
|
Každé testovatelné provedení zkoušky, kdy se látka nechová ani jako látka, která se váže, ani jako látka, která se neváže
je nejnižší nevyhlazené průměrné procento vazby ze skupiny koncentrací v datech nad 75 %.
|
Obrázek 1
Příklady klasifikace zkoušené chemické látky s použitím kompetitivní vazebné křivky
|
60.
|
Sloučí se několik testů zkoušené chemické látky provedených v laboratoři, každému testu se přiřadí numerická hodnota a jednotlivé testy se zprůměrují, jak je znázorněno v tabulce 8. Výsledky sloučených testů v rámci každé laboratoře se porovnají s předpokládanou klasifikací jednotlivých zkoušených chemických látek.
|
Tabulka 8
Metoda pro klasifikaci zkoušené chemické látky s použitím několika testů v rámci laboratoře
|
Přiřazení hodnoty každému testu:
|
|
|
Klasifikace
|
Číselná hodnota
|
|
váže se
|
2
|
|
neurčitá
|
1
|
|
neváže se
|
0
|
|
Klasifikace průměru číselných hodnot ze všech testů:
|
|
|
Klasifikace
|
Číselná hodnota
|
|
váže se
|
průměr ≥ 1,5
|
|
neurčitá
|
0,5 ≤ průměr < 1,5
|
|
neváže se
|
průměr < 0,5
|
ZÁVĚREČNÁ ZPRÁVA
|
61.
|
Viz odstavec 24 „SLOŽKY VAZEBNÉ ANALÝZY hrER“ této zkušební metody.
|
Dodatek 2.1
SEZNAM VÝRAZŮ
[3H]E2
: 17β-estradiol značený radioizotopem tritia
DCC
: aktivní uhlí potažené dextranem
E2
: neznačený 17β-estradiol (inertní)
Zkušební pufr: 10 mM Tris, 10 mg bovinního sérového albuminu /ml, 2 mM DTT, 10 % glycerol, 0,2 mM leupeptinu, pH 7,5
hrERα
: lidský rekombinantní estrogenní receptor alfa
Replikát
: Jedna z několika jamek, které obsahují stejný obsah ve stejné koncentraci a analyzují se souběžně v rámci jednoho provedení zkoušky. V tomto protokolu se každá koncentrace zkoušené chemické látky testuje třikrát; to znamená, že existují tři replikáty, které se analyzují současně při každé koncentraci zkoušené chemické látky.
Provedení zkoušky
: Úplná sada jamek na mikrotitrační destičce analyzovaných souběžně, která poskytuje všechny informace potřebné pro charakterizaci vazby zkoušené chemické látky na hrERα (tzn. celkový [3H]-17β-estradiol přidaný do zkušební jamky, maximální vazba [3H]-17β-estradiolu na hrERα, nespecifická vazba a celková vazba při různých koncentracích zkoušené chemické látky). Provedení zkoušky může sestávat pouze z jedné jamky (tj. replikátu) na každou koncentraci, ale protože tento protokol vyžaduje trojí analyzování, sestává jedno provedení ze tří jamek na každou koncentraci. Dále tento protokol vyžaduje tři nezávislá (tj. nesouběžná) provedení na každou chemickou látku.
Dodatek 2.2
TYPICKÁ SATURAČNÍ ANALÝZA [3H]-17Β-ESTRADIOLU SE TŘEMI REPLIKOVANÝMI JAMKAMI
|
Typická saturační analýza [3H]-17β-estradiolu se třemi replikovanými jamkami
|
|
Poloha
|
Replikát
|
Kód typu jamky
|
Výchozí koncentrace radioaktivního E2 (nM)
|
Objem radioaktivního E2 (μl)
|
Konečná koncentrace radioaktivního E2 (nM)
|
Výchozí koncentrace neradioaktivního E2 (μM)
|
Objem neradioaktivního E2 (μl)
|
Konečná koncentrace neradioaktivního E2 (μM)
|
Objem pufru (μl)
|
Objem receptoru (μl)
|
Celkový objem v jamkách
|
|
A1
|
1
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A2
|
2
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A3
|
3
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A4
|
1
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A5
|
2
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A6
|
3
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A7
|
1
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A8
|
2
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A9
|
3
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A10
|
1
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A11
|
2
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A12
|
3
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B1
|
1
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B2
|
2
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B3
|
3
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B4
|
1
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B5
|
2
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B6
|
3
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B7
|
1
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B8
|
2
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B9
|
3
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B10
|
1
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B11
|
2
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B12
|
3
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
D1
|
1
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D2
|
2
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D3
|
3
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D4
|
1
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D5
|
2
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D6
|
3
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D7
|
1
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D8
|
2
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D9
|
3
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D10
|
1
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D11
|
2
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D12
|
3
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
E1
|
1
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E2
|
2
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E3
|
3
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E4
|
1
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E5
|
2
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E6
|
3
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E7
|
1
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E8
|
2
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E9
|
3
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E10
|
1
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E11
|
2
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E12
|
3
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
G1
|
1
|
radioaktivní
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G2
|
2
|
radioaktivní
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G3
|
3
|
radioaktivní
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G4
|
1
|
radioaktivní
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G5
|
2
|
radioaktivní
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G6
|
3
|
radioaktivní
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G7
|
1
|
radioaktivní
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G8
|
2
|
radioaktivní
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G9
|
3
|
radioaktivní
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G10
|
1
|
radioaktivní
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G11
|
2
|
radioaktivní
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G12
|
3
|
radioaktivní
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H1
|
1
|
radioaktivní
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H2
|
2
|
radioaktivní
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H3
|
3
|
radioaktivní
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H4
|
1
|
radioaktivní
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H5
|
2
|
radioaktivní
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H6
|
3
|
radioaktivní
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H7
|
1
|
radioaktivní
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H8
|
2
|
radioaktivní
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H9
|
3
|
radioaktivní
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H10
|
1
|
radioaktivní
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H11
|
2
|
radioaktivní
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H12
|
3
|
radioaktivní
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
Upozorňujeme, že „radioaktivní“ jamky jsou v průběhu inkubace prázdné. 40 μl se přidává až pro počítání scintilací.
|
Dodatek 2.3:
USPOŘÁDÁNÍ JAMEK PŘI KOMPETITIVNÍ VAZEBNÉ ANALÝZE
|
Mikrodestička
|
Poloha
|
Replikát
|
Typ jamky
|
Kód jamky
|
Kód koncentrace
|
Výchozí koncentrace kompetitora (M)
|
Zásobní hrER (μl)
|
Objem pufru (μl)
|
Objem stopovací látky (radioaktivní E2) (μl)
|
Objem z destičky s ředěním (μl)
|
Konečný objem (μl)
|
Konečná koncentrace kompetitora (M)
|
|
S
|
A1
|
1
|
celková vazba
|
TB
|
TB1
|
–
|
40
|
|
40
|
80
|
160
|
–
|
|
P
|
A2
|
2
|
celková vazba
|
TB
|
TB2
|
–
|
40
|
|
40
|
80
|
160
|
–
|
|
P
|
A3
|
3
|
celková vazba
|
TB
|
TB3
|
–
|
40
|
|
40
|
80
|
160
|
-
|
|
P
|
A4
|
1
|
celková vazba
|
TB
|
TB4
|
–
|
40
|
|
40
|
80
|
160
|
–
|
|
P
|
A5
|
2
|
celková vazba
|
TB
|
TB5
|
–
|
40
|
|
40
|
80
|
160
|
–
|
|
P
|
A6
|
3
|
celková vazba
|
TB
|
TB6
|
–
|
40
|
|
40
|
80
|
160
|
–
|
|
P
|
A7
|
1
|
neradioaktivní E2 (vysoká)
|
NSB
|
S0
|
2.00E-06
|
40
|
–
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
A8
|
2
|
neradioaktivní E2 (vysoká)
|
NSB
|
S0
|
2.00E-06
|
40
|
–
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
A9
|
3
|
neradioaktivní E2 (vysoká)
|
NSB
|
S0
|
2.00E-06
|
40
|
–
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
A10
|
1
|
neradioaktivní E2 (vysoká)
|
NSB
|
S0
|
2.00E-06
|
40
|
–
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
A11
|
2
|
neradioaktivní E2 (vysoká)
|
NSB
|
S0
|
2.00E-06
|
40
|
–
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
A12
|
3
|
neradioaktivní E2 (vysoká)
|
NSB
|
S0
|
2.00E-06
|
40
|
–
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
B1
|
1
|
neradioaktivní E2
|
S
|
S1
|
2.00E-07
|
40
|
–
|
40
|
80
|
160
|
1.0E-07
|
|
S
|
B2
|
2
|
neradioaktivní E2
|
S
|
S1
|
2.00E-07
|
40
|
–
|
40
|
80
|
160
|
1.0E-07
|
|
S
|
B3
|
3
|
neradioaktivní E2
|
S
|
S1
|
2.00E-07
|
40
|
–
|
40
|
80
|
160
|
1.0E-07
|
|
S
|
B4
|
1
|
neradioaktivní E2
|
S
|
S2
|
2.00E-08
|
40
|
-
|
40
|
80
|
160
|
1.0E-08
|
|
S
|
B5
|
2
|
neradioaktivní E2
|
S
|
S2
|
2.00E-08
|
40
|
–
|
40
|
80
|
160
|
1.0E-08
|
|
S
|
B6
|
3
|
neradioaktivní E2
|
S
|
S2
|
2.00E-08
|
40
|
–
|
40
|
80
|
160
|
1.0E-08
|
|
S
|
B7
|
1
|
neradioaktivní E2
|
S
|
S3
|
6.00E-09
|
40
|
–
|
40
|
80
|
160
|
3.0E-09
|
|
S
|
B8
|
2
|
neradioaktivní E2
|
S
|
S3
|
6.00E-09
|
40
|
–
|
40
|
80
|
160
|
3.0E-09
|
|
S
|
B9
|
3
|
neradioaktivní E2
|
S
|
S3
|
6.00E-09
|
40
|
–
|
40
|
80
|
160
|
3.0E-09
|
|
S
|
B10
|
1
|
neradioaktivní E2
|
S
|
S4
|
2.00E-09
|
40
|
–
|
40
|
80
|
160
|
1.0E-09
|
|
S
|
B11
|
2
|
neradioaktivní E2
|
S
|
S4
|
2.00E-09
|
40
|
–
|
40
|
80
|
160
|
1.0E-09
|
|
S
|
B12
|
3
|
neradioaktivní E2
|
S
|
S4
|
2.00E-09
|
40
|
-
|
40
|
80
|
160
|
1.0E-09
|
|
S
|
C1
|
1
|
neradioaktivní E2
|
S
|
S5
|
6.00E-10
|
40
|
-
|
40
|
80
|
160
|
3.0E-10
|
|
S
|
C2
|
2
|
neradioaktivní E2
|
S
|
S5
|
6.00E-10
|
40
|
–
|
40
|
80
|
160
|
3.0E-10
|
|
S
|
C3
|
3
|
neradioaktivní E2
|
S
|
S5
|
6.00E-10
|
40
|
–
|
40
|
80
|
160
|
3.0E-10
|
|
S
|
C4
|
1
|
neradioaktivní E2
|
S
|
S6
|
2.00E-10
|
40
|
–
|
40
|
80
|
160
|
1.0E-10
|
|
S
|
C5
|
2
|
neradioaktivní E2
|
S
|
S6
|
2.00E-10
|
40
|
–
|
40
|
80
|
160
|
1.0E-10
|
|
S
|
C6
|
3
|
neradioaktivní E2
|
S
|
S6
|
2.00E-10
|
40
|
–
|
40
|
80
|
160
|
1.0E-10
|
|
S
|
C7
|
1
|
neradioaktivní E2
|
S
|
S7
|
2.00E-11
|
40
|
–
|
40
|
80
|
160
|
1.0E-11
|
|
S
|
C8
|
2
|
neradioaktivní E2
|
S
|
S7
|
2.00E-11
|
40
|
-
|
40
|
80
|
160
|
1.0E-11
|
|
S
|
C9
|
3
|
neradioaktivní E2
|
S
|
S7
|
2.00E-11
|
40
|
-
|
40
|
80
|
160
|
1.0E-11
|
|
S
|
C10
|
1
|
slepá zkouška
|
slepá zkouška
|
B1
|
–
|
–
|
160
|
–
|
–
|
160
|
–
|
|
S
|
C11
|
2
|
slepá zkouška
|
slepá zkouška
|
B2
|
–
|
–
|
160
|
–
|
–
|
160
|
–
|
|
P
|
C12
|
3
|
slepá zkouška
|
slepá zkouška
|
B3
|
–
|
–
|
160
|
–
|
–
|
160
|
–
|
|
P
|
D1
|
1
|
norethynodrel
|
NE
|
WP1
|
6.00E-05
|
40
|
–
|
40
|
80
|
160
|
3.0E-05
|
|
S
|
D2
|
1
|
norethynodrel
|
NE
|
WP1
|
6.00E-05
|
40
|
–
|
40
|
80
|
160
|
3.0E-05
|
|
S
|
D3
|
1
|
norethynodrel
|
NE
|
WP1
|
6.00E-05
|
40
|
–
|
40
|
80
|
160
|
3.0E-05
|
|
S
|
D4
|
1
|
norethynodrel
|
NE
|
WP2
|
2.00E-05
|
40
|
–
|
40
|
80
|
160
|
1.0E-05
|
|
S
|
D5
|
1
|
norethynodrel
|
NE
|
WP2
|
2.00E-05
|
40
|
–
|
40
|
80
|
160
|
1.0E-05
|
|
S
|
D6
|
1
|
norethynodrel
|
NE
|
WP2
|
2.00E-05
|
40
|
–
|
40
|
80
|
160
|
1.0E-05
|
|
S
|
D7
|
1
|
norethynodrel
|
NE
|
WP3
|
6.00E-06
|
40
|
–
|
40
|
80
|
160
|
3.0E-06
|
|
S
|
D8
|
1
|
norethynodrel
|
NE
|
WP3
|
6.00E-06
|
40
|
–
|
40
|
80
|
160
|
3.0E-06
|
|
S
|
D9
|
1
|
norethynodrel
|
NE
|
WP3
|
6.00E-06
|
40
|
–
|
40
|
80
|
160
|
3.0E-06
|
|
S
|
D10
|
1
|
norethynodrel
|
NE
|
WP4
|
2.00E-06
|
40
|
–
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
D11
|
1
|
norethynodrel
|
NE
|
WP4
|
2.00E-06
|
40
|
–
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
D12
|
1
|
norethynodrel
|
NE
|
WP4
|
2.00E-06
|
40
|
–
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
E1
|
1
|
norethynodrel
|
NE
|
WP
|
6.00E-07
|
40
|
|
40
|
80
|
160
|
3.0E-07
|
|
S
|
E2
|
2
|
norethynodrel
|
NE
|
WP
|
6.00E-07
|
40
|
|
40
|
80
|
160
|
3.0E-07
|
|
S
|
E3
|
3
|
norethynodrel
|
NE
|
WP
|
6.00E-07
|
40
|
|
40
|
80
|
160
|
3.0E-07
|
|
S
|
E4
|
1
|
norethynodrel
|
NE
|
WP
|
2.00E-07
|
40
|
|
40
|
80
|
160
|
1.0E-07
|
|
S
|
E5
|
2
|
norethynodrel
|
NE
|
WP
|
2.00E-07
|
40
|
|
40
|
80
|
160
|
1.0E-07
|
|
S
|
E6
|
3
|
norethynodrel
|
NE
|
WP
|
2.00E-07
|
40
|
|
40
|
80
|
160
|
1.0E-07
|
|
S
|
E7
|
1
|
norethynodrel
|
NE
|
WP
|
6.00E-08
|
40
|
-
|
40
|
80
|
160
|
3.0E-08
|
|
S
|
E8
|
2
|
norethynodrel
|
NE
|
WP
|
6.00E-08
|
40
|
-
|
40
|
80
|
160
|
3.0E-08
|
|
S
|
E9
|
3
|
norethynodrel
|
NE
|
WP
|
6.00E-08
|
40
|
-
|
40
|
80
|
160
|
3.0E-08
|
|
S
|
E10
|
1
|
norethynodrel
|
NE
|
WP
|
6.00E-09
|
40
|
-
|
40
|
80
|
160
|
3.0E-09
|
|
S
|
E11
|
2
|
norethynodrel
|
NE
|
WP
|
6.00E-09
|
40
|
-
|
40
|
80
|
160
|
3.0E-09
|
|
S
|
E12
|
3
|
norethynodrel
|
NE
|
WP
|
6.00E-09
|
40
|
-
|
40
|
80
|
160
|
3.0E-09
|
|
S
|
F1
|
1
|
OTES
|
N
|
OTES
|
2.00E-03
|
40
|
-
|
40
|
80
|
160
|
1.0E-03
|
|
S
|
F2
|
2
|
OTES
|
N
|
OTES
|
2.00E-03
|
40
|
-
|
40
|
80
|
160
|
1.0E-03
|
|
S
|
F3
|
3
|
OTES
|
N
|
OTES
|
2.00E-03
|
40
|
-
|
40
|
80
|
160
|
1.0E-03
|
|
S
|
F4
|
1
|
OTES
|
N
|
OTES
|
2.00E-04
|
40
|
-
|
40
|
80
|
160
|
1.0E-04
|
|
S
|
F5
|
2
|
OTES
|
N
|
OTES
|
2.00E-04
|
40
|
-
|
40
|
80
|
160
|
1.0E-04
|
|
S
|
F6
|
3
|
OTES
|
N
|
OTES
|
2.00E-04
|
40
|
-
|
40
|
80
|
160
|
1.0E-04
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES
|
2.00E-05
|
40
|
-
|
40
|
80
|
160
|
3.0E-05
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES
|
2.00E-05
|
40
|
-
|
40
|
80
|
160
|
3.0E-05
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES
|
2.00E-05
|
40
|
-
|
40
|
80
|
160
|
3.0E-05
|
|
S
|
F10
|
1
|
OTES
|
N
|
OTES
|
2.00E-06
|
40
|
-
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
F11
|
2
|
OTES
|
N
|
OTES
|
2.00E-06
|
40
|
-
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
F12
|
3
|
OTES
|
N
|
OTES
|
2.00E-06
|
40
|
-
|
40
|
80
|
160
|
1.0E-06
|
|
S
|
G1
|
1
|
OTES
|
N
|
OTES
|
2.00E-07
|
40
|
-
|
40
|
80
|
160
|
3.0E-07
|
|
S
|
G2
|
2
|
OTES
|
N
|
OTES
|
2.00E-07
|
40
|
-
|
40
|
80
|
160
|
3.0E-07
|
|
S
|
G3
|
3
|
OTES
|
N
|
OTES
|
2.00E-07
|
40
|
-
|
40
|
80
|
160
|
3.0E-07
|
|
S
|
G4
|
1
|
OTES
|
N
|
OTES
|
2.00E-08
|
40
|
-
|
40
|
80
|
160
|
1.0E-08
|
|
S
|
G5
|
2
|
OTES
|
N
|
OTES
|
2.00E-08
|
40
|
-
|
40
|
80
|
160
|
1.0E-08
|
|
S
|
G6
|
3
|
OTES
|
N
|
OTES
|
2.00E-08
|
40
|
-
|
40
|
80
|
160
|
1.0E-08
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES
|
2.00E-09
|
40
|
-
|
40
|
80
|
160
|
1.0E-09
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES
|
2.00E-09
|
40
|
-
|
40
|
80
|
160
|
1.0E-09
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES
|
2.00E-09
|
40
|
-
|
40
|
80
|
160
|
1.0E-09
|
|
S
|
G10
|
1
|
OTES
|
N
|
OTES
|
2.00E-10
|
40
|
-
|
40
|
-
|
160
|
1.0E-10
|
|
S
|
G11
|
2
|
OTES
|
N
|
OTES
|
2.00E-10
|
40
|
-
|
40
|
-
|
160
|
1.0E-10
|
|
S
|
G12
|
3
|
OTES
|
N
|
OTES
|
2.00E-10
|
40
|
-
|
40
|
-
|
160
|
1.0E-10
|
|
S
|
H1
|
1
|
radioaktivní
|
H
|
H
|
-
|
-
|
-
|
40
|
-
|
40
|
-
|
|
P
|
H2
|
1
|
radioaktivní
|
H
|
H
|
-
|
-
|
-
|
40
|
-
|
40
|
-
|
|
S
|
H3
|
1
|
radioaktivní
|
H
|
H
|
-
|
-
|
-
|
40
|
-
|
40
|
-
|
|
S
|
H4
|
1
|
radioaktivní
|
H
|
H
|
-
|
-
|
-
|
40
|
-
|
40
|
-
|
|
S
|
H5
|
1
|
radioaktivní
|
H
|
H
|
-
|
-
|
-
|
40
|
-
|
40
|
-
|
|
S
|
H6
|
1
|
radioaktivní
|
H
|
H
|
-
|
-
|
-
|
40
|
-
|
40
|
-
|
|
S
|
H7
|
1
|
kontrola s pufrem
|
BC
|
BC
|
-
|
40
|
80
|
40
|
-
|
160
|
-
|
|
S
|
H8
|
1
|
kontrola s pufrem
|
BC
|
BC
|
-
|
40
|
80
|
40
|
-
|
160
|
-
|
|
S
|
H9
|
1
|
kontrola s pufrem
|
BC
|
BC
|
-
|
40
|
80
|
40
|
-
|
160
|
-
|
|
S
|
H10
|
1
|
kontrola s pufrem
|
BC
|
BC
|
-
|
40
|
80
|
40
|
-
|
160
|
-
|
|
S
|
H11
|
1
|
kontrola s pufrem
|
BC
|
BC
|
-
|
40
|
80
|
40
|
-
|
160
|
-
|
|
S
|
H12
|
1
|
kontrola s pufrem
|
BC
|
BC
|
-
|
40
|
80
|
40
|
-
|
160
|
-
|
Upozorňujeme, že „radioaktivní“ jamky jsou v průběhu inkubace prázdné. 40 μl se přidává až pro počítání scintilací.
|
Uspořádání jamek při kompetitivní vazebné analýze
|
|
Mikrodestička
|
Poloha
|
Replikát
|
Druh jamky
|
Kód jamky
|
Kód koncentrace
|
Výchozí koncentrace kompetitora (M)
|
Zásobní hrER (μl)
|
Objem pufru (μl)
|
Objem stopovací látky (radioaktivní E2) (μl)
|
Objem z destičky s ředěním (μl)
|
Konečný objem (μl)
|
Konečná koncentrace kompetitora (M)
|
|
P1
|
A1
|
1
|
celková vazba
|
TB
|
TBB1B1
|
-
|
40
|
-
|
40
|
80
|
160
|
-
|
|
P1
|
A2
|
2
|
celková vazba
|
TB
|
TB2
|
-
|
40
|
-
|
40
|
80
|
160
|
-
|
|
P1
|
A3
|
3
|
celková vazba
|
TB
|
TB3
|
-
|
40
|
-
|
40
|
80
|
160
|
-
|
|
P1
|
A4
|
1
|
celková vazba
|
TB
|
TB4
|
-
|
40
|
-
|
40
|
80
|
160
|
-
|
|
P1
|
A5
|
2
|
celková vazba
|
TB
|
TB5
|
-
|
40
|
-
|
40
|
80
|
160
|
-
|
|
P1
|
A6
|
3
|
celková vazba
|
TB
|
TB6
|
-
|
40
|
-
|
40
|
80
|
160
|
-
|
|
P1
|
A7
|
1
|
neradioaktivní E2 (vysoká)
|
NSB
|
S0
|
2.00E-06
|
40
|
-
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
A8
|
2
|
neradioaktivní E2 (vysoká)
|
NSB
|
S0
|
2.00E-06
|
40
|
-
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
A9
|
3
|
neradioaktivní E2 (vysoká)
|
NSB
|
S0
|
2.00E-06
|
40
|
-
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
A10
|
1
|
neradioaktivní E2 (vysoká)
|
NSB
|
S0
|
2.00E-06
|
40
|
-
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
A11
|
2
|
neradioaktivní E2 (vysoká)
|
NSB
|
S0
|
2.00E-06
|
40
|
-
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
A12
|
3
|
neradioaktivní E2 (vysoká)
|
NSB
|
S0
|
2.00E-06
|
40
|
-
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
B1
|
1
|
Zkoušená chemická látka 1
|
TC1
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
B2
|
2
|
Zkoušená chemická látka 1
|
TC1
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
B3
|
3
|
Zkoušená chemická látka 1
|
TC1
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
B4
|
1
|
Zkoušená chemická látka 1
|
TC1
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
B5
|
2
|
Zkoušená chemická látka 1
|
TC1
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
B6
|
3
|
Zkoušená chemická látka 1
|
TC1
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
B7
|
1
|
Zkoušená chemická látka 1
|
TC1
|
3
|
2.00E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
B8
|
2
|
Zkoušená chemická látka 1
|
TC1
|
3
|
2.00E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
B9
|
3
|
Zkoušená chemická látka 1
|
TC1
|
3
|
2.00E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
B10
|
1
|
Zkoušená chemická látka 1
|
TC1
|
4
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
B11
|
2
|
Zkoušená chemická látka 1
|
TC1
|
4
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
B12
|
3
|
Zkoušená chemická látka 1
|
TC1
|
4
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
C1
|
1
|
Zkoušená chemická látka 1
|
TC1
|
5
|
2.00E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
C2
|
2
|
Zkoušená chemická látka 1
|
TC1
|
5
|
2.00E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
C3
|
3
|
Zkoušená chemická látka 1
|
TC1
|
5
|
2.00E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
C4
|
1
|
Zkoušená chemická látka 1
|
TC1
|
6
|
2.00E-08
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
C5
|
2
|
Zkoušená chemická látka 1
|
TC1
|
6
|
2.00E-08
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
C6
|
3
|
Zkoušená chemická látka 1
|
TC1
|
6
|
2.00E-08
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
C7
|
1
|
Zkoušená chemická látka 1
|
TC1
|
7
|
2.00E-09
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
C8
|
2
|
Zkoušená chemická látka 1
|
TC1
|
7
|
2.00E-09
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
C9
|
3
|
Zkoušená chemická látka 1
|
TC1
|
7
|
2.00E-09
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
C10
|
1
|
Zkoušená chemická látka 1
|
TC1
|
8
|
2.00E-10
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
C11
|
2
|
Zkoušená chemická látka 1
|
TC1
|
8
|
2.00E-10
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
C12
|
3
|
Zkoušená chemická látka 1
|
TC1
|
8
|
2.00E-10
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
D1
|
1
|
Zkoušená chemická látka 2
|
TC2
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
D2
|
2
|
Zkoušená chemická látka 2
|
TC2
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
D3
|
3
|
Zkoušená chemická látka 2
|
TC2
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
D4
|
1
|
Zkoušená chemická látka 2
|
TC2
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
D5
|
2
|
Zkoušená chemická látka 2
|
TC2
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
D6
|
3
|
Zkoušená chemická látka 2
|
TC2
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
D7
|
1
|
Zkoušená chemická látka 2
|
TC2
|
3
|
2.00E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
D8
|
2
|
Zkoušená chemická látka 2
|
TC2
|
3
|
2.00E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
D9
|
3
|
Zkoušená chemická látka 2
|
TC2
|
3
|
2.00E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
D10
|
1
|
Zkoušená chemická látka 2
|
TC2
|
4
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
D11
|
2
|
Zkoušená chemická látka 2
|
TC2
|
4
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
D12
|
3
|
Zkoušená chemická látka 2
|
TC2
|
4
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
E1
|
1
|
Zkoušená chemická látka 2
|
TC2
|
5
|
2.00E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
E2
|
2
|
Zkoušená chemická látka 2
|
TC2
|
5
|
2.00E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
E3
|
3
|
Zkoušená chemická látka 2
|
TC2
|
5
|
2.00E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
E4
|
1
|
Zkoušená chemická látka 2
|
TC2
|
6
|
-
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
E5
|
2
|
Zkoušená chemická látka 2
|
TC2
|
6
|
-
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
E6
|
3
|
Zkoušená chemická látka 2
|
TC2
|
6
|
-
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
E7
|
1
|
Zkoušená chemická látka 2
|
TC2
|
7
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
E8
|
2
|
Zkoušená chemická látka 2
|
TC2
|
7
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
E9
|
3
|
Zkoušená chemická látka 2
|
TC2
|
7
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
E10
|
1
|
Zkoušená chemická látka 2
|
TC2
|
8
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
E11
|
2
|
Zkoušená chemická látka 2
|
TC2
|
8
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
E12
|
3
|
Zkoušená chemická látka 2
|
TC2
|
8
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
F1
|
1
|
Zkoušená chemická látka 3
|
TC3
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
F2
|
2
|
Zkoušená chemická látka 3
|
TC3
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
F3
|
3
|
Zkoušená chemická látka 3
|
TC3
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
F4
|
1
|
Zkoušená chemická látka 3
|
TC3
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
F5
|
2
|
Zkoušená chemická látka 3
|
TC3
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
F6
|
3
|
Zkoušená chemická látka 3
|
TC3
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
F7
|
1
|
Zkoušená chemická látka 3
|
TC3
|
3
|
2.00E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
F8
|
2
|
Zkoušená chemická látka 3
|
TC3
|
3
|
2.00E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
F9
|
3
|
Zkoušená chemická látka 3
|
TC3
|
3
|
2.00E-05
|
40
|
0
|
40
|
80
|
160
|
1.0E-05
|
|
P1
|
F10
|
1
|
Zkoušená chemická látka 3
|
TC3
|
4
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
F11
|
2
|
Zkoušená chemická látka 3
|
TC3
|
4
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
F12
|
3
|
Zkoušená chemická látka 3
|
TC3
|
4
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1.0E-06
|
|
P1
|
G1
|
1
|
Zkoušená chemická látka 3
|
TC3
|
5
|
2.00E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
G2
|
2
|
Zkoušená chemická látka 3
|
TC3
|
5
|
2.00E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
G3
|
3
|
Zkoušená chemická látka 3
|
TC3
|
5
|
2.00E-07
|
40
|
0
|
40
|
80
|
160
|
1.0E-07
|
|
P1
|
G4
|
1
|
Zkoušená chemická látka 3
|
TC3
|
6
|
2.00E-08
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
G5
|
2
|
Zkoušená chemická látka 3
|
TC3
|
6
|
2.00E-08
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
G6
|
3
|
Zkoušená chemická látka 3
|
TC3
|
6
|
2.00E-08
|
40
|
0
|
40
|
80
|
160
|
1.0E-08
|
|
P1
|
G7
|
1
|
Zkoušená chemická látka 3
|
TC3
|
7
|
2.00E-09
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
G8
|
2
|
Zkoušená chemická látka 3
|
TC3
|
7
|
2.00E-09
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
G9
|
3
|
Zkoušená chemická látka 3
|
TC3
|
7
|
2.00E-09
|
40
|
0
|
40
|
80
|
160
|
1.0E-09
|
|
P1
|
G10
|
1
|
Zkoušená chemická látka 3
|
TC3
|
8
|
2.00E-10
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
G11
|
2
|
Zkoušená chemická látka 3
|
TC3
|
8
|
2.00E-10
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
G12
|
3
|
Zkoušená chemická látka 3
|
TC3
|
8
|
2.00E-10
|
40
|
0
|
40
|
80
|
160
|
1.0E-10
|
|
P1
|
H1
|
1
|
norethynodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H2
|
2
|
norethynodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H3
|
3
|
norethynodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H4
|
1
|
norethynodrel
|
NE
|
|
1.00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H5
|
2
|
norethynodrel
|
NE
|
|
1.00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H6
|
3
|
norethynodrel
|
NE
|
|
1.00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H7
|
1
|
neradioaktivní E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H8
|
2
|
neradioaktivní E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H9
|
3
|
neradioaktivní E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H10
|
1
|
neradioaktivní E2 S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H11
|
2
|
neradioaktivní E2 S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H12
|
3
|
neradioaktivní E2 S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
Dodatek 3
ANALÝZA VAZBY NA ESTROGENOVÉ RECEPTORY IN VITROS VYUŽITÍM LIDSKÉHO REKOMBINANTNÍHO PROTEINU S LIGAND VÁZAJÍCÍ DOMÉNOU PODLE ÚSTAVU PRO HODNOCENÍ A VÝZKUM CHEMICKÝCH LÁTEK (CERI)
VÝCHOZÍ ÚVAHY A OMEZENÍ (VIZ TAKÉ VŠEOBECNÝ ÚVOD)
|
1.
|
Tato in vitro analýza saturační a kompetitivní vazby na estrogenový receptor (ERα) používá ligand vázající doménu lidského ERα (hrERα). Tato proteinová konstrukce, kterou vytvořil japonský Ústav pro hodnocení a výzkum chemických látek (CERI), existuje jako fúzní protein glutathion-S-transferáza (GST) a je exprimována v E. coli. Protokol CERI prošel mezinárodní validační studií v několika laboratořích (2), která prokázala jeho relevanci a spolehlivost pro zamýšlený účel analýzy.
|
|
2.
|
Tato analýza je screeningovým postupem pro určení látek, které se mohou vázat na hrERα. Používá se na určení schopnosti zkoušené chemické látky konkurovat 17β-estradiolu ve vazbě na hrERα-LBD. Výsledky kvantitativní analýzy mohou zahrnovat IC50 (míra koncentrace zkoušené chemické látky potřebná na vytěsnění poloviny [3H]-17β-estradiolu z hrERα) a relativní vazebné afinity zkoušených chemických látek pro hrERα v porovnání s 17β-estradiol. Pro účely chemického screeningu mohou přijatelné výsledky kvalitativní analýzy zahrnovat klasifikace zkoušených chemických látek jako látek, které se vážou na hrERα, látek, které se na něj nevážou, nebo látek neurčitých, a to na základě kritérií popsaných u vazebných křivek.
|
|
3.
|
Analýza používá radioaktivní ligand, což vyžaduje, aby laboratoř měla povolení nakládat s radioaktivními materiály. Při používání radioizotopů a nebezpečných chemických látek musí být dodržovány předpisy a postupy stanovené vnitrostátními právními předpisy.
|
|
4.
|
Před použitím této analýzy pro regulační účely je třeba přečíst si části „VŠEOBECNÝ ÚVOD“ a „SLOŽKY VAZEBNÉ ANALÝZY hrER“. Definice a zkratky používané v této zkušební metodě jsou popsány v dodatku 1.
|
PRINCIPY ANALÝZY (VIZ TAKÉ VŠEOBECNÝ ÚVOD)
|
5.
|
Test vazby na hrERα měří schopnost radioizotopově značeného ligandu ([3H]17β-estradiol) vázat se na ER v přítomnosti zvyšujících se koncentrací zkoušené chemické látky (tj. kompetitor). Zkoušené chemické látky, které mají vysokou afinitu na ER, konkurují radioizotopově značenému ligandu při nižší koncentraci v porovnání s chemickými látkami s nižší afinitou na tento receptor.
|
|
6.
|
Tato analýza sestává ze dvou hlavních složek: saturačního vazebného experimentu za účelem charakterizace parametrů interakce mezi receptorem a ligandem, po němž následuje kompetitivní vazebný experiment, který charakterizuje kompetici mezi zkoušenou chemickou látkou a radioizotopově značeným ligandem ve vazbě na ER.
|
|
7.
|
Účelem saturačního vazebného experimentu je charakterizovat konkrétní šarži receptorů z hlediska vazebné afinity a počtu při přípravě na kompetitivní vazebný experiment. Saturační vazebný experiment měří za rovnovážných podmínek afinitu pevně stanovené koncentrace estrogenového receptoru na jeho přirozený ligand (představovaný disociační konstantou Kd) a koncentraci míst aktivního receptoru (Bmax).
|
|
8.
|
Kompetitivní vazebný experiment měří afinitu látky konkurovat [3H]17β-estradiolu ve vazbě na ER. Afinita je kvantifikována koncentrací zkoušené chemické látky, která za rovnovážného stavu inhibuje 50 % specifické vazby [3H]17β-estradiolu (nazývá se „50 % inhibiční koncentrace“ nebo IC50). Může se hodnotit také pomocí relativní vazebné afinity (RBA, vzhledem k IC50 estradiolu měřené samostatně při stejném provedení zkoušky). Kompetitivní vazebný experiment měří vazbu [3H]17β-estradiolu při pevně stanovené koncentraci v přítomnosti širokého rozmezí (osm řádů) koncentrací zkoušené chemické látky. Data se poté vloží pokud možno do určité formy Hillovy rovnice (Hill, 1910), která popisuje vytěsnění radioligandu kompetiční látkou s jedním vazebným místem. Rozsah vytěsnění radioizotopově značeného estradiolu v rovnovážném stavu se používá na charakterizaci zkoušené chemické látky jako látky, která se váže, která se neváže nebo která generuje neurčitou odpověď.
|
POSTUP
Prokázání funkční způsobilosti proteinu hrERα
|
9.
|
Před rutinním prováděním saturačních a kompetitivních vazebných analýz by mělo být u každé nové šarže hrERα prokázáno, že v laboratoři, v níž bude používán, funguje správně. K prokázání způsobilosti by se měl použít proces zahrnující dva kroky. Jsou to tyto kroky:
|
—
|
Proveďte saturační vazebnou analýzu [3H]-17β-estradiolu na prokázání specifičnosti a saturace hrERα. V nelineární regresní analýze těchto dat (např. BioSoft; McPherson, 1985; Motulsky, 1995) a na následném Scatchardově grafu by měla být dokumentována vazebná afinita hrERα vůči [3H]-17β-estradiolu (Kd) a počet receptorů (Bmax) pro konkrétní šarži hrERα.
|
|
—
|
Proveďte kompetitivní vazebnou analýzu pomocí kontrolních látek (referenčního estrogenu (17β-estradiol), látky se slabou vazbou (např. norethynodrel nebo norethindron) a látky, která se neváže (oktyltriethoxysilan, OTES). Každá laboratoř by si měla vytvořit historickou databázi dokumentující konzistentnost hodnot IC50 a relevantních hodnot pro referenční estrogen a látku se slabou vazbou u experimentů a různých šarží hrERα. Dále by parametry kompetitivních vazebných křivek pro kontrolní látky měly být v rozmezí 95 % konfidenčního intervalu (viz tabulka 1), který byl stanoven pomocí dat z laboratoří, jež se účastnily validační studie pro tuto analýzu (2).
|
Tabulka 1
Kritéria výkonnosti vypracovaná pro referenční estrogen a látku se slabou vazbou, vazebná analýza CERI hrER
|
Látka
|
Parametr
|
Střední hodnota (11)
|
Směrodatná odchylka (n)
|
95 % konfidenční intervaly (12)
|
|
Dolní mez
|
Horní mez
|
|
17β-estradiol
|
Horní
|
104,74
|
13,12 (70)
|
101,6
|
107,9
|
|
Spodní strana
|
0,85
|
2,41 (70)
|
0,28
|
1,43
|
|
Hillův koeficient
|
–1,22
|
0,20 (70)
|
–1,27
|
–1,17
|
|
logIC50
|
–8,93
|
0,23 (70)
|
–8,98
|
–8,87
|
|
Norethynodrel
|
Horní
|
101,31
|
10,55 (68)
|
98,76
|
103,90
|
|
Spodní strana
|
2,39
|
5,01 (68)
|
1,18
|
3,60
|
|
Hillův koeficient
|
–1,04
|
0,21 (68)
|
–1,09
|
–0,99
|
|
LogIC50
|
–6,19
|
0,40 (68)
|
–6,29
|
–6,10
|
|
Norethindrone (13)
|
Nahoru
|
92,27
|
7,79 (23)
|
88,90
|
95,63
|
|
Spodní strana
|
16,52
|
10,59 (23)
|
11,94
|
21,10
|
|
Hillův koeficient
|
–1,18
|
0,32 (23)
|
–1,31
|
–1,04
|
|
logIC50
|
–6,01
|
0,54 (23)
|
–6,25
|
–5,78
|
|
Prokázání odborné způsobilosti laboratoře
|
10.
|
Viz odstavce 17 a 18 a tabulka 2 v části „SLOŽKY VAZEBNÉ ANALÝZY hrER“ této zkušební metody. Každá analýza (saturační a kompetitivní vazby) by měla zahrnovat tři nezávislé zkoušky (tzn. s čerstvě naředěným receptorem, chemickými látkami a reagenciemi) provedené v různé dny, přičemž každé provedení by mělo obsahovat tři opakování.
|
Určení koncentrace receptoru (hrERα)
|
11.
|
Koncentrace aktivního receptoru se poněkud liší podle šarže a podmínek uchovávání. Z tohoto důvodu je třeba stanovit koncentraci aktivního receptoru přijatého od dodavatele. Získá se tím vhodná koncentrace aktivního receptoru v době provedení zkoušky.
|
|
12.
|
Za podmínek odpovídajících kompetitivní vazbě (tj. 0,5 nM [3H]-estradiol) by se měly nominální koncentrace receptoru 0,1, 0,2, 0,4 a 0,6 nM inkubovat v nepřítomnosti (úplná vazba) a v přítomnosti (nespecifická vazba) 1 μM neznačeného estradiolu. Specifická vazba, vypočítaná jako rozdíl úplné a nespecifické vazby, se nanese proti nominální koncentraci receptoru. Koncentrace receptoru, která dává hodnoty specifické vazby odpovídající 40 % přidaného radioizotopového značení, souvisí s odpovídající koncentrací receptoru a tato koncentrace receptoru se používá pro saturační a kompetitivní vazebné experimenty. Často bude tuto podmínku splňovat konečná koncentrace hrER 0,2 nM.
|
|
13.
|
Jestliže nelze opakovaně splnit 40 % kritérium, je třeba zkontrolovat, zda uspořádání experimentu neobsahuje možné chyby. Nedosažení 40 % kritéria může ukazovat, že v rekombinantní šarži je příliš málo aktivního receptoru, a potom je třeba zvážit použití jiné šarže receptoru.
|
Saturační analýza
|
14.
|
Hodnotí se osm zvyšujících se koncentrací [3H]17β-estradiolu v trojím provedení, a to za těchto tří podmínek (viz tabulka 2):
|
a.
|
Za nepřítomnosti neznačeného 17β-estradiolu a v přítomnosti ER. Je to stanovení úplné vazby měřením radioaktivity v jamkách, v nichž je pouze [3H]17β-estradiol.
|
|
b.
|
V přítomnosti 2000násobně zvýšené koncentrace neznačeného 17β-estradiolu přes značený 17β-estradiol a v přítomnosti ER. Záměrem této podmínky je saturovat aktivní vazebná místa neznačeným 17β-estradiolem a změřením radioaktivity v jamkách stanovit nespecifickou vazbu. Případný zbývající radioaktivní estradiol, který se může vázat na receptor, se považuje za navázaný na nespecifické místo, neboť neradioaktivní estradiol by měl být v tak vysoké koncentraci, aby se navázal na všechna dostupná specifická místa na receptoru.
|
|
c.
|
Za nepřítomnosti neznačeného 17β-estradiolu a nepřítomnosti ER (stanovení celkové radioaktivity)
|
Příprava roztoků [3H]-17β-estradiolu a neznačeného 17β-estradiolu a hrERα
|
15.
|
40 nM roztok [3H]-17β-estradiolu se připraví z 1 μM zásobního roztoku [3H]-17β-estradiolu v DMSO přidáním DMSO (pro přípravu 200 nM) a zkušebního pufru při pokojové teplotě (pro přípravu 40 nM). Pomocí tohoto 40 nM roztoku se připraví série naředěného [3H]-17β-estradiolu v rozmezí od 0,313 nM do 40 nM se zkušebním pufrem při pokojové teplotě (jak je znázorněno ve sloupci 12 tabulky 2). Konečné zkušební koncentrace v rozmezí od 0,0313 do 4,0 nM se získají přidáním 10 μl těchto roztoků do příslušných jamek 96jamkové mikrotitrační destičky (viz tabulky 2 a 3). Příprava zkušebního pufru, výpočet původního zásobního roztoku [3H]-17β-estradiolu podle jeho měrné aktivity, příprava zředěných roztoků a stanovení koncentrací je podrobně popsáno v protokolu CERI (2).
|
|
16.
|
Zředěné roztoky neznačeného 17β-estradiolu se připraví z 1 nM zásobního roztoku 17β-estradiolu přidáním zkušebního pufru, aby bylo dosaženo osmi zvyšujících se koncentrací v počátečním rozsahu od 0,625 μM do 80 μM. Konečné zkušební koncentrace v rozmezí od 0,0625 do 8 μM se získají přidáním 10 μl těchto roztoků do příslušných jamek 96jamkové mikrotitrační destičky určené pro měření nespecifické vazby (viz tabulky 2 a 3). Příprava roztoků neznačeného 17β-estradiolu je podrobně popsána v protokolu CERI (2).
|
|
17.
|
Měla by se použít taková koncentrace receptoru, která dává specifickou vazbu 40±10 % (viz odstavce 12–13). Roztok hrERα se připraví s ledovým zkušebním pufrem bezprostředně před použitím, tj. až po přípravě všech jamek pro celkovou vazbou, nespecifickou vazbu a radioaktivní ligand samotný.
|
|
18.
|
96jamkové mikrotitrační destičky se připraví tak, jak je zobrazeno v tabulce 2, a to s třemi replikáty na každou koncentraci [3H]-17β-estradiolu. Přiřazení objemů [3H]-17β-estradiolu, neznačeného 17β-estradiolu, pufru a receptoru je uvedeno v tabulce 3.
Tabulka 2
Uspořádání mikrotitrační destičky při saturační vazebné analýze
|
|
1 (*11)
|
2 (*11)
|
3 (*11)
|
4 (*11)
|
5 (*11)
|
6 (*11)
|
7 (*11)
|
8 (*11)
|
9 (*11)
|
10
|
11 (*12)
|
12 (*12)
|
|
Pro měření TB
|
Pro měření NSB
|
Pro určení radioaktivního ligandu samotného
|
|
Neznačená ředění E2 pro sloupec 4–6
|
Ředění [3H]E2 pro sloupec 1–9
|
|
A
|
0,0313 nM [ 3H] E2+ ER
|
0,0313 nM [3H] E2+ 0,0625 μM E2+ ER
|
0,0313 nM
|
|
0,625 μM
|
0,313 nM
|
|
B
|
0,0625 nM [3H] E2+ ER
|
0,0625 nM [3H] E2+ 0,125 μM E2+ ER
|
0,0625 nM
|
|
1,25 μM
|
0,625 nM
|
|
C
|
30,125 nM [3H] E2+ ER
|
0,125 nM [3H] E2+ 0,25 μM E2+ ER
|
0,125 nM
|
|
2,5 μM
|
1,25 nM
|
|
D
|
0,250 nM [3H] E2+ ER
|
0,250 nM [3H] E2+ 0,5 μM E2+ ER
|
0,250 nM
|
|
5 μM
|
2,5 nM
|
|
E
|
0.50 nM [ H] E2+ ER
|
0,50 nM [3H] E2+ 1 μM E2+ ER
|
0,50 nM
|
|
10 μM
|
5 nM
|
|
F
|
1,00 nM [3H] E2+ ER
|
1,00 nM [3H] E2+ 2 μM E2+ ER
|
1,00 nM
|
|
20 μM
|
10 nM
|
|
G
|
2,00 nM [3H] E2+ ER
|
2,00 nM [3H] E2+ 4 μM E2+ ER
|
2,00 nM
|
|
40 μM
|
20 nM
|
|
H
|
4,00 nM [3H] E2+ ER
|
4,00 nM [3H] E2+ 8 μM E2+ ER
|
4,00 nM
|
|
80 μM
|
40 nM
|
|
TB:
|
-
|
celková vazba
|
|
NSB:
|
-
|
nespecifická vazba
|
|
[3H] E2:
|
-
|
[3H]17β-estradiol
|
|
E2:
|
-
|
neznačený 17β-estradiol
|
|
Tabulka 3
Objemy reagencií pro saturační mikrotitrační destičku
|
Číslo sloupce
|
1
|
2
|
3
|
4
|
5
|
6
|
7 (*13)
|
8 (*13)
|
9 (*13)
|
|
Postup přípravy
|
Jamky TB
|
Jamky NSB
|
Radioaktivní ligand samotný
|
|
Objem komponentů pro výše uvedené reakční jamky a pořadí přidávání
|
pufr
|
60 μl
|
50 μl
|
90 μl
|
|
neznačený E2 ze sloupce 11 v tabulce 2
|
–
|
10 μl
|
–
|
|
[3H]E2 z řádku 12 v tabulce 2
|
10 μl
|
10 μl
|
10 μl
|
|
hrERα
|
30 μl
|
30 μl
|
–
|
|
Celkový reakční objem
|
100 μl
|
100 μl
|
100 μl
|
|
Inkubace
|
REAKCE PO 2 HODINÁCH INKUBACE
|
Kvantifikace radioaktivity ihned po přípravě. Bez inkubace
|
|
Expozice 0,4 % DCC
|
Ano
|
Ano
|
Ne
|
|
Objem 0,4 % DCC
|
100 μl
|
100 μl
|
–
|
|
Filtrace
|
Ano
|
Ano
|
Ne
|
|
MĚŘENÍ DPM
|
|
Do scintilačního koktejlu přidán kvantifikační objem
|
100 μl (*14)
|
100 μl (*14)
|
50 μl
|
|
|
19.
|
Mikrotitrační destičky pro stanovení celkové vazby a nespecifické vazby by se měly inkubovat při pokojové teplotě (22oC až 28oC) po dobu dvou hodin.
|
|
Měření [3H]-17β-estradiolu navázaného na hrERα
|
20.
|
Po dvouhodinové inkubaci by měl být [3H]-17β-estradiol navázaný na hrERα separován od volného [3H]-17β-estradiolu přidáním 100 μl ledové suspenze 0,4 % DCC do jamek. Destičky se poté umístí na led na 10 minut a reakční směs a suspenze DCC se přefiltruje přenesením na filtr mikrotitračních destiček, aby se odstranil DCC. Poté se 100 μl filtrátu přidá do scintilační kapaliny ve scintilačních lahvičkách pro stanovení rozpadů za minutu (dpm) na lahvičku pomocí kapalinového scintilačního spektrometru.
|
|
21.
|
Jestliže není k dispozici filtr na mikrotitrační destičky, lze DCC alternativně odstranit odstředěním. Poté se velmi opatrně odebere 50 μl supernatantu obsahujícího [3H]-17β-estradiol navázaný na hrERα, aby nedošlo ke kontaminaci jamek dotykem DCC, a použije se pro počítání scintilací.
|
|
22.
|
Pro stanovení rozpadu za minutu (dpm) [3H]-17β-estradiolu přidaného do jamek se použije podmínka radioaktivního ligandu samotného. Radioaktivita se kvantifikuje ihned po přípravě. Tyto jamky se neinkubují a neexponují se suspenzi DCC, ale jejich obsah je třeba přemístit přímo do scintilační kapaliny. Tato měření ukazují, kolik [3H]-17β-estradiolu v dpm bylo přidáno do každé sady jamek pro celkovou vazbu a nespecifickou vazbu.
|
Kompetitivní vazebná analýza
|
23.
|
Kompetitivní vazebná analýza měří vazbu jedné koncentrace [3H]17β-estradiolu v přítomnosti zvyšujících se koncentrací zkoušené chemické látky. Při každé koncentraci by se měly v jednom provedení zkoušky použít tři souběžné replikáty. Kromě toho by se měla pro každou zkoušenou chemickou látku provést tři nesouběžná provedení zkoušky. Analýza se provádí v jedné nebo několika 96jamkových mikrotitračních destičkách.
|
Kontroly
|
24.
|
Při provádění analýzy by měly být do každého experimentu zařazeny souběžné testy s rozpouštědlem a kontroly (tj. referenční estrogen, slabá vazba a látka, která se neváže). Při každém provedení zkoušky by měly být na jedné destičce použity celé křivky koncentrace pro referenční estrogen a kontroly (tj. slabá vazba a neváže se). Všechny ostatní destičky by měly obsahovat i) vysokou (maximální vytěsnění, tj. přibližně úplné vytěsněni radioizotopově značeného ligandu) a střední (přibližně IC50) koncentraci E2 a látky se slabou vazbou, a to třikrát; ii) kontrolu s rozpouštědlem a látkou s nespecifickou vazbou, a to vždy třikrát. Postupy pro přípravu experimentálního pufru, [3H]-17β-estradiolu, hrERα a roztoků zkoušených chemických látek jsou podrobně popsány v protokolu CERI (2).
|
Kontrola s rozpouštědlem:
|
25.
|
Kontrola s rozpouštědlem ukazuje, že rozpouštědlo nereaguje se zkušebním systémem, a měří také úplnou vazbu (TB). Nejvhodnějším rozpouštědlem je DMSO. Jestliže nejvyšší koncentrace zkoušené chemické látky není rozpustná v DMSO, může být alternativně použit ethanol. Koncentrace DMSO v jamkách při konečné analýze by měla být 2,05 % a může se zvyšovat do 2,5 % v případě nerozpustnosti zkoušené chemické látky. Koncentrace DMSO nad 2,5 % by se neměly prožívat vzhledem k interferenci vyšších koncentrací rozpouštědla s analýzou. U zkoušených chemických látek, které nejsou rozpustné v DMSO, ale jsou rozpustné v ethanolu, může být při analýze použit bez interference maximálně 2 % ethanol.
|
Kontrola s pufrem:
|
26.
|
Kontrola s pufrem (BC) by neměla obsahovat rozpouštědlo ani zkoušenou chemickou látku, ale všechny ostatní složky analýzy. Výsledky kontroly s pufrem se porovnají s kontrolou s rozpouštědlem za účelem ověření, že použité rozpouštědlo neovlivňuje zkušební systém.
|
Látka se silnou vazbou (referenční estrogen)
|
27.
|
17β-estradiol (CAS 50-28-2) je endogenní ligand a váže se s vysokou afinitou na ER, podtyp alfa. Pro každou kompetitivní vazebnou analýzu hrERα by měla být vypracována standardní křivka s použitím neznačeného 17β-estradiolu, aby bylo možné posoudit variabilitu při provádění analýzy v čase ve stejné laboratoři. Připraví se osm roztoků neznačeného 17β-estradiolu v DMSO a zkušebním pufru, přičemž konečné koncentrace v jamkách, které budou použity pro standardní křivku, budou v těchto rozestupech: 10–6, 10–7, 10–8, 10–8,5, 10–9, 10–9,5, 10–10, 10–11 M. Nejvyšší koncentrace neznačeného 17β-estradiolu (1 μM) by měla sloužit také jako indikátor nespecifické vazby. Tato koncentrace je v tabulce 4 odlišena označením „NSB“, ačkoliv je také součástí standardní křivky.
|
Látka se slabou vazbou
|
28.
|
Měla by být zařazena látka se slabou vazbou (norethynodrel (CAS68-23-5) nebo alternativně norethindron (CAS 68-22-4)), aby se prokázala citlivost každého experimentu a aby se umožnilo posouzení variability při provádění analýzy v čase. Připraví se osm roztoků látky se slabou vazbou v DMSO a zkušebním pufru, přičemž konečné koncentrace v jamkách budou: 10–4,5, 10–5,5, 10–6, 10–6,5, 10–7, 10–7,5, 10–8 a 10–9 M.
|
Látka, která se neváže
|
29.
|
Jako negativní kontrola (látka, která se neváže) se používá oktyltriethoxysilan (OTES, CAS 2943-75-1). Poskytuje jistotu, že analýza, tak jak je prováděna, dokáže detekovat zkoušené chemické látky, které se nevážou na hrERα. Připraví se osm roztoků látky, která se neváže, v DMSO a zkušebním pufru, přičemž konečné koncentrace v jamkách budou: 10–3,10–4, 10–5, 10–6, 10–7, 10–8, 10–9, 10–10 M. Jako látka, která se neváže, může být alternativně použit di-n-butylftalát (DBP, CAS 84-72-2), ale může být zkoušen pouze do 10–4M. Bylo prokázáno, že maximální rozpustnost DBP v analýze je 10–4 M.
|
Koncentrace hrERα
|
30.
|
Mělo by se použít takové množství receptoru, které dává specifickou vazbu 40±10 % (viz odstavce 12–13 v dodatku 3). Roztok hrERα je třeba připravovat naředěním funkčního hrERα v ledovém zkušebním pufru bezprostředně před použitím.
|
[3H]-17β-estradiol
|
31.
|
Konečná koncentrace [3H]-17β-estradiolu v jamkách by měla být 0,5 nM.
|
Zkoušené chemické látky
|
32.
|
V první řadě je třeba provést zkoušku rozpustnosti, aby bylo možné stanovit mez rozpustnosti pro každou zkoušenou chemickou látku a zjistit vhodný rozsah koncentrace, jenž má být použit při provádění zkušebního protokolu. Mez rozpustnosti každé zkoušené chemické látky se nejprve stanoví v rozpouštědle a poté dále potvrdí za zkušebních podmínek. Konečná koncentrace zkoušená při analýze by neměla překročit 1 mM. Zkoušení za účelem zjištění rozsahu zahrnuje kontrolu s rozpouštědlem s alespoň 8 log-sériovými ředěními, počínaje maximální přijatelnou koncentrací (např. 1 mM nebo nižší, na základě meze rozpustnosti), přičemž je třeba zaznamenat přítomnost zákalu nebo sraženiny (viz také odstavec 35 v dodatku 3). Po stanovení rozsahu koncentrace pro zkoušení by zkoušená chemická látka měla být testována s použitím 8 logaritmických koncentrací v rozestupech vhodně definovaných předchozí zkouškou ke stanovení rozsahu. Při druhém a třetím experimentu by se měly podle potřeby dále upravit zkoušené koncentrace, aby lépe charakterizovaly křivku závislosti odpovědi na koncentraci, je-li to nezbytné.
|
|
33.
|
Zkoušená chemická látka se naředí ve vhodném rozpouštědle (viz odstavec 25 v dodatku 3). Jestliže nejvyšší koncentrace zkoušené chemické látky není rozpustná v DMSO ani v ethanolu a přidání většího množství rozpouštědla by způsobilo, že koncentrace rozpouštědla v poslední zkumavce bude větší než přijatelná mez, může se nejvyšší koncentrace snížit na nejbližší nižší koncentraci. V tomto případě může být na dolní konec série koncentrací přidána další koncentrace. Ostatní koncentrace v sérii by měly zůstat beze změny.
|
|
34.
|
Roztoky zkoušené chemické látky je třeba po přidání do jamky pečlivě sledovat, neboť zkoušená chemická látka se může po přidání do jamky srážet. Data za všechny jamky, které obsahují sraženinu, by měla být vyloučena z vynesené křivky a měl by být uveden důvod pro vyloučení těchto dat.
|
|
35.
|
Jestliže již existují informace z jiných zdrojů, které uvádějí log(IC50) zkoušené chemické látky, může být vhodné stanovit geometrické rozestupy ředění blíže kolem předpokládané hodnoty log(IC50) (tj. 0,5 log jednotky). Konečné výsledky by měly ukazovat dostatečný rozsah koncentrací na každé straně log(IC50), včetně „horní“ a „dolní“, aby vazebná křivka mohla být adekvátně charakterizována.
|
Uspořádání zkušebních destiček
|
36.
|
Připraví se označené mikrotitrační destičky s použitím šestinásobné inkubace pro kontrolu s rozpouštědlem, nejvyšší koncentraci referenčního estrogenu (E2), který slouží také jako indikátor nespecifické vazby (NSB), kontrolu s pufrem, trojnásobná inkubace pro každou z osmi koncentrací kontrolní látky, která se neváže (oktyltriethoxysilan), sedm nižších koncentrací pro referenční estrogen (E2), osm koncentrací látky se slabou vazbou (norethynodrel nebo norethindron) a osm koncentrací každé zkoušené chemické látky (TC). Příklad diagramu uspořádání destičky pro celé koncentrační křivky pro referenční estrogen a kontroly je uveden níže v tabulce 4. Pro zkoušenou chemickou látku se použijí další mikrotitrační destičky, které by měly obsahovat kontroly (tj. i) vysokou (maximální vytěsnění) a střední (přibližně IC50) koncentraci každého E2 a látky se slabou vazbou, a to třikrát; ii) kontrolu s rozpouštědlem (jako celkovou vazbu) a látkou s nespecifickou vazbou, a to vždy šestkrát (tabulka 5). Příklad postupu uspořádání mikrotitrační destičky pro kompetitivní analýzu s použitím tří neznámých zkoušených chemických látek je uveden v dodatku 3.3. Koncentrace uvedené v technologickém postupu a v tabulkách 4 a 5 jsou konečné koncentrace použité v každé jamce. Maximální koncentrace pro E2 by měla být 1×10-7 M a pro látku se slabou vazbou by měla být použita nejvyšší koncentrace pro látku se slabou vazbou použitá na destičce 1. Koncentraci IC50 musí stanovit laboratoř na základě své dosavadní kontrolní databáze. Lze očekávat, že tato hodnota bude podobná hodnotě pozorované ve validačních studiích (viz tabulka 1).
|
Tabulka 4
Uspořádání mikrotitrační destičky při kompetitivní vazebné analýze (97)
(98), celé křivky koncentrace pro referenční estrogen a kontroly (destička 1)
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
Kontrola s pufrem a pozitivní kontrola (E2)
|
Slabá pozitivní (norethynodrel)
|
Negativní kontrola (OTES)
|
TB a NSB
|
|
A
|
Slepý pokus (*15)
|
1×10–9 M
|
1×10–10 M
|
TB (kontrola s rozpouštědlem) (2,05 % DMSO)
|
|
B
|
E2 (1×10–11 M)
|
1×10–8 M
|
1×10–9 M
|
|
C
|
E2 (1×10–10 M)
|
1×10–7,5 M
|
1×10–8 M
|
NSB (10–6 M E2)
|
|
D
|
E2 (1×10–9,5 M)
|
1×10–7 M
|
1×10–7 M
|
|
E
|
E2 (1×10–9 M)
|
1×10–6,5 M
|
1×10–6 M
|
Kontrola s pufrem
|
|
F
|
E2 (1×10–8,5 M)
|
1×10–6 M
|
1×10–5 M
|
|
G
|
E2 (1×10–8 M)
|
1×10–5,5 M
|
1×10–4 M
|
Slepý pokus (pro radioaktivní) (*16)
|
|
H
|
E2 (1×10–7 M)
|
1×10–4,5 M
|
1×10–3 M
|
Tabulka 5
Uspořádání mikrotitrační destičky při kompetitivní vazebné analýze, další destičky pro zkoušené chemické látky (TC) a kontroly
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
Zkoušená chemická látka 1 (TC-1)
|
Zkoušená chemická látka 2 (TC-2)
|
Zkoušená chemická látka 3 (TC-3)
|
Kontroly
|
|
A
|
TC-1 (1×10–10 M)
|
TC-2 (1×10–10 M)
|
TC-3 (1×10–10 M)
|
E2 (1×10–7 M)
|
|
B
|
TC-1 (1×10–9 M)
|
TC-2 (1×10–9 M)
|
TC-3 (1×10–9 M)
|
E2 (IC50)
|
|
C
|
TC-1 (1×10–8 M)
|
TC-2 (1×10–8 M)
|
TC-3 (1×10–8 M)
|
NE (1×10–4.5 M)
|
|
D
|
TC-1 (1×10–7 M)
|
TC-2 (1×10–7 M)
|
TC-3 (1×10–7 M)
|
NE (IC50)
|
|
E
|
TC-1 (1×10–6 M)
|
TC-2 (1×10–6 M)
|
TC-3 (1×10–6 M)
|
NSB (10–6 M E2)
|
|
F
|
TC-1 (1×10–5 M)
|
TC-2 (1×10–5 M)
|
TC-3 (1×10–5 M)
|
|
G
|
TC-1 (1×10–4 M)
|
TC-2 (1×10–4 M)
|
TC-3 (1×10–4 M)
|
TB (kontrola s rozpouštědlem)
|
|
H
|
TC-1 (1×10–3 M)
|
TC-2 (1×10–3 M)
|
TC-3 (1×10–3 M)
|
|
V tomto příkladu je látkou se slabou vazbou norethinodrel (NE).
|
Dokončení kompetitivní vazebné analýzy
|
37.
|
S výjimkou jamek pro celkovou vazbu a slepé pokusy (pro radioaktivní látku), jak je uvedeno v tabulce 6, se do každé jamky dá 50 μl zkušebního pufru a smíchá se s 10 μl kontroly s rozpouštědlem, referenčního estrogenu (E2), látky se slabou vazbou, látky, která se neváže, a zkoušenými chemickými látkami, respektive s 10 μl roztoku 5 nM [3H]-17β-estradiolu. Poté se do každé destičky přidá 30 μl ledového roztoku receptoru a lehce promíchá. Roztok hrERα by měl být poslední přidávanou reagencií. Zkušební mikrotitrační destičky se inkubují při pokojové teplotě (22o až 28oC) po dobu 2 hodin.
Tabulka 6
Objem zkušebních složek pro kompetitivní vazebnou analýzu hrER, mikrotitrační destičky
|
Postup přípravy
|
Jiné než jamky TB
|
Jamky TB
|
Slepý pokus (pro radioaktivní látku)
|
|
Objem komponentů pro výše uvedené reakční jamky a pořadí přidávání
|
Zkušební pufr při pokojové teplotě
|
50 μl
|
60 μl
|
90 μl
|
|
Neznačený E2, látka se slabou vazbou, látka, která se neváže, rozpouštědlo a zkoušené chemické látky (*17)
|
10 μl
|
–
|
–
|
|
[3H]-17β-estradiol pro konečnou koncentraci 0,5 nM (tj. 5 nM)
|
10 μl
|
10 μl
|
10 μl
|
|
určená koncentrace hrERα (viz odstavce 12–13)
|
30 μl
|
30 μl
|
–
|
|
Celkový objem v každé jamce
|
100 μl
|
100 μl
|
100 μl
|
|
|
38.
|
Poté se provede kvantifikace [3H]-17β-estradiolu navázaného na hrERα po separaci [3H]-17β-estradiolu navázaného na hrERα z volného [3H]-17β-estradiolu přidáním 100 μl ledové suspenze DCC do každé jamky, jak je popsáno v odstavcích 21–23 dodatku 3 o saturační vazebné analýze.
|
|
39.
|
Jamky G10–12 a H10–12 (označené v tabulce 4 jako slepý pokus (pro radioaktivní látku)) představují dpm [3H]-značeného-estradiolu v 10 μl. Alikvotní množství z 10 μl se přidá přímo do scintilační kapaliny.
|
Kritéria přijatelnosti
Saturační vazebná analýza
|
40.
|
Křivka specifické vazby by měla dosáhnout plató s použitím zvyšujících se koncentrací [3H]-17β-estradiolu, což ukazuje saturaci hrERα ligandem.
|
|
41.
|
Specifická vazba při 0,5 nM [3H]-17β-estradiolu by měla být v přijatelném rozmezí 30 % až 50 % průměrné naměřené celkové radioaktivity přidané v průběhu jednotlivých provedení zkoušek. Občasná mírná vybočení mimo toto rozmezí jsou přijatelná, ale pokud jsou všechna provedení zkoušky mimo toto rozmezí nebo pokud je konkrétní provedení zkoušky výrazně mimo toto rozmezí, je třeba upravit koncentraci proteinu a saturační analýzu zopakovat.
|
|
42.
|
Data by měla vytvořit lineární Scatchardův graf.
|
|
43.
|
Nespecifická vazba by neměla být nadměrná. Hodnota nespecifické vazby by měla být typicky <35 % celkové vazby. Tento poměr však může při měření velmi nízkých dpm u nejnižší koncentrace radioizotopově značeného zkoušeného 17β-estradiolu občas uvedený limit překročit.
|
Kompetitivní vazebná analýza
|
44.
|
Zvyšující se koncentrace neznačeného 17β-estradiolu by měly vytěsnit [3H]-17β- estradiol z receptoru způsobem odpovídajícím jednomístné kompetitivní vazbě.
|
|
45.
|
Hodnota IC50 pro referenční estrogen (tj. 17β-estradiol) by se měla přibližně rovnat molární koncentraci [3H]-17β-estradiolu plus Kd určený ze saturační vazebné analýzy.
|
|
46.
|
Celková specifická vazba by měla být soustavně v přijatelném rozmezí 40 ± 10 %, když je průměrná naměřená koncentrace celkové radioaktivity přidaná do každé jamky 0,5 nM ve všech provedeních zkoušky. Občasná mírná vybočení mimo toto rozmezí jsou přijatelná, ale pokud jsou všechna provedení zkoušky mimo toto rozmezí nebo pokud je konkrétní provedení zkoušky výrazně mimo toto rozmezí, je třeba upravit koncentraci proteinu.
|
|
47.
|
Rozpouštědlo by nemělo měnit citlivost ani reprodukovatelnost analýzy. Výsledky kontroly s rozpouštědlem (jamky TB) se porovnají s kontrolou s pufrem za účelem ověření, že použité rozpouštědlo neovlivňuje zkušební systém. Jestliže rozpouštědlo nemá na analýzu žádný vliv, měly by být výsledky TB a kontroly s rozpouštědlem srovnatelné.
|
|
48.
|
Látka, která se neváže, by neměla vytěsnit více než 25 % [3H]-17β-estradiolu z hrERα při zkoušení do 10–3 M (OTES) nebo 10–4 M (DBP).
|
|
49.
|
Pro referenční estrogen a dvě látky se slabou vazbou (např. norethynodrel, norethindron) byla vypracována kritéria funkčnosti s použitím údajů z validační studie pro vazebnou analýzu CERI hrER (příloha N v odkazu 2). Jsou uvedeny 95 % konfidenční intervaly pro střední hodnotu ± SD (n) za všechna kontrolní provedení zkoušky ve čtyřech laboratořích účastnících se ve validační studii. Pro referenční estrogen a látky se slabou vazbou a pro log10RBA látek se slabou vazbou vzhledem k referenčnímu estrogenu byly vypočteny 95 % konfidenční intervaly pro parametry křivky (tj. horní, dolní, Hillův koeficient, log IC50). V tabulce 1 jsou uvedeny očekávané rozsahy parametrů křivky, které mohou být použity jako kritéria funkčnosti. V praxi se může rozsah IC50 poněkud odlišovat v závislosti na experimentálně zjištěné Kd receptorového přípravku a koncentraci ligandu použitého pro analýzu.
|
|
50.
|
Pro parametry křivky pro zkoušené chemické látky nebyla vypracována žádná kritéria funkčnosti, protože stávajících potenciálních zkoušených chemických látek existuje velké množství a značně se liší jejich potenciální afinity a výsledky (např. celá křivka, částečná křivka, chybějící křivka). Při posuzování výsledků z každého provedení zkoušky za zkoušenou chemickou látku je třeba postupovat s odborným úsudkem. Mělo by být použito dostatečné rozmezí koncentrací, aby byl jednoznačně definován vrchol (např. 90–100 % vazby) kompetitivní křivky. Variabilita mezi replikáty při každé koncentraci zkoušené chemické látky i mezi 3 zkouškami neprováděnými souběžně by měla být přiměřená a vědecky obhajitelná. Kontroly z každého provedení zkoušky by se u zkoušené chemické látky měly blížit mírám funkčnosti uvedeným pro tuto analýzu CERI a měly by odpovídat dosavadním kontrolním údajům z každé příslušné laboratoře.
|
ANALÝZA ÚDAJŮ
Saturační vazebná analýza
|
51.
|
Měří se celková i nespecifická vazba. Z těchto hodnot se specifická vazba zvyšujících se koncentrací [3H]-17β-estradiolu za rovnovážného stavu vypočítá odečtením nespecifické vazby od celkové. Graf specifické vazby proti koncentraci [3H]-17β-estradiolu by měl dosáhnout plató u maximální specifické vazby indikující saturaci hrERα [3H]-17β-estradiolem. Kromě toho by analýza dat měla dokumentovat vazbu [3H]-17β-estradiolu na jedno vazebné místo s vysokou afinitou. Na křivce saturační vazby by měla být zobrazena nespecifická, celková a specifická vazba. Další rozbor těchto údajů by měl být proveden pomocí nelineární regresní analýzy (např. BioSoft; McPherson, 1985; Motulsky, 1995) s konečným zobrazením dat ve formě Scatchardova grafu.
|
|
52.
|
Při analýze dat se určuje Bmax a Kd z údajů o celkové vazbě samotné, a to za použití předpokladu, že nespecifická vazba je lineární, není-li zdůvodněno použití jiné metody. Kromě toho by při určování nejlepších hodnot měla být použita robustní regrese, pokud není uvedeno zdůvodnění. Metoda zvolená pro robustní regresi by měla být uvedena. Při určování Bmax a Kd z údajů o saturační vazbě by měla být vždy použita korekce úbytku ligandu (např. pomocí metody podle Swillense 1995).
|
Kompetitivní vazebná analýza
|
53.
|
Křivka kompetitivní vazby se vynáší jako specifická vazba [3H]-17β-estradiolu proti koncentraci (log10 jednotky) kompetitora. Koncentrace zkoušené chemické látky, která inhibuje 50 % maximální specifické vazby [3H]-17β-estradiolu, je hodnota IC50.
|
|
54.
|
Odhady log (IC50) pro pozitivní kontroly (např. referenční estrogen a látku se slabou vazbou) se stanoví pomocí vhodného softwaru pro nelineární vynesení křivky, aby byly vhodné pro čtyřparametrickou Hillovu rovnici (např. BioSoft; McPherson, 1985; Motulsky, 1995). Při vynášení těchto křivek se horní bod, dolní bod, sklon a log(IC50) obvykle ponechávají bez omezení. Při určování nejlepších hodnot by měla být použita robustní regrese, pokud není uvedeno zdůvodnění. Korekce úbytku ligandu se nepoužívá. Po úvodní analýze je třeba každou vazebnou křivku zkontrolovat, aby bylo zajištěno, že je vhodně aplikována na model. Relativní vazebná afinita (RBA) pro látku se slabou vazbou se vypočítá jako procento log (IC50) pro látku se slabou vazbou vzhledem k log (IC50) pro 17β-estradiol. Výsledky z pozitivních kontrol a z kontroly s látkou, která se neváže, se posuzují pomocí měřítek účinnosti analýzy uvedených v odstavci 44–49 tohoto dodatku 3.
|
|
55.
|
Data za všechny zkoušené chemické látky se analyzují postupně, aby bylo zajištěno, že data budou analyzována správně, a aby byla správně klasifikována každá kompetitivní vazebná křivka. Doporučuje se, aby každé provedení zkoušky u zkoušené chemické látky nejdříve prošlo standardizovanou analýzou dat, která je stejná jako analýza použitá pro kontroly s referenčním estrogenem a s látkou se slabou vazbou (viz odstavec 54 tohoto dodatku 3). Po dokončení je třeba provést technickou kontrolu parametrů vynesené křivky a vizuálně zkontrolovat, jak dobře data odpovídají generované kompetitivní vazebné křivce u každého provedení zkoušky. V průběhu této technické kontroly je pozorovaný pokles procenta specificky navázaného [3H]-17β-estradiolu v závislosti na koncentraci, nízká variabilita mezi technickými replikáty při jednotlivých koncentracích zkoušené chemické látky a konzistentní parametry u tří provedení zkoušky dobrým ukazatelem, že zkouška a analýzy dat byly provedeny správně.
|
Interpretace údajů
|
56.
|
Za předpokladu, že jsou splněna všechna kritéria přijatelnosti, lze zkoušenou chemickou látku považovat za látku, která se váže na hrERα, jestliže lze vynést vazebnou křivku a nejnižší bod na křivce odpovědi v rozsahu dat je menší než 50 % (obrázek 1).
|
|
57.
|
Za předpokladu, že jsou splněna všechna kritéria přijatelnosti, lze zkoušenou chemickou látku považovat za látku, která se neváže na hrERα, jestliže:
|
—
|
lze vynést vazebnou křivku a nejnižší bod na křivce odpovědi v rozsahu dat je nad 75 % nebo
|
|
—
|
nelze vynést vazebnou křivku a nejnižší nevyhlazené průměrné procento vazby ze skupiny koncentrací v datech je nad 75 %.
|
|
|
58.
|
Zkoušené chemické látky se považují za neurčité, jestliže není splněna žádná z výše uvedených podmínek (např. nejnižší bod na vynesené křivce odpovědi je mezi 76–51 %).
|
Tabulka 7
Kritéria pro stanovení klasifikace zkoušené chemické látky podle kompetitivní vazebné křivky
|
Klasifikace
|
Kritéria
|
|
Látka, která se vážea
|
Lze vynést vazebnou křivku.
Nejnižší bod na křivce odpovědi v rozsahu dat je menší než 50 %.
|
|
Látka, která se nevážeb
|
Jestliže lze vynést vazebnou křivku,
je nejnižší bod na křivce odpovědi v rozsahu dat nad 75 %.
Jestliže nelze vynést vazebnou křivku,
je nejnižší nevyhlazené průměrné procento vazby ze skupiny koncentrací v datech nad 75 %.
|
|
Neurčitác
|
Každé testovatelné provedení zkoušky, kdy se látka nechová ani jako látka, která se váže, ani jako látka, která se neváže
(např. nejnižší bod na vynesené křivce odpovědi je mezi 76–51 %).
|
Obrázek 1
Příklady klasifikace zkoušené chemické látky s použitím kompetitivní vazebné křivky
|
59.
|
Sloučí se několik testů zkoušené chemické látky provedených v laboratoři, každému testu se přiřadí numerická hodnota a jednotlivé testy se zprůměrují, jak je znázorněno v tabulce 8. Výsledky sloučených testů v rámci každé laboratoře se porovnají s předpokládanou klasifikací jednotlivých zkoušených chemických látek.
|
Tabulka 8
Metoda pro klasifikaci zkoušené chemické látky s použitím několika testů v rámci laboratoře
|
Přiřazení hodnoty každému testu:
|
|
Klasifikace
|
Číselná hodnota
|
|
váže se
|
2
|
|
neurčitá
|
1
|
|
neváže se
|
0
|
|
Klasifikace průměru číselných hodnot ze všech testů:
|
|
Klasifikace
|
Číselná hodnota
|
|
váže se
|
průměr ≥ 1,5
|
|
neurčitá
|
0,5 ≤ průměr < 1,5
|
|
neváže se
|
průměr < 0,5
|
ZÁVĚREČNÁ ZPRÁVA
|
60.
|
Viz odstavce 24 „SLOŽKY VAZEBNÉ ANALÝZY hrER“ této zkušební metody.
|
Dodatek 3.1
SEZNAM VÝRAZŮ
[3H]E2
: 17β-estradiol značený radioizotopem tritia.
DCC
: Aktivní uhlí potažené dextranem.
E2
: Neznačený 17β-estradiol (inertní).
Zkušební pufr
: 10 mM Tris-HCl, pH 7,4, s obsahem 1 mM EDTA, 1mM EGTA, 1 mM NaVO3, 10 % glycerolu, 0,2 mM leupeptinu, 1 mM dithiothreitolu a 10 mg/ml bovinního sérového albuminu.
hrERα
: Lidský rekombinantní estrogenní receptor alfa (ligand vázající doména).
Replikát
: Jedna z několika jamek, které obsahují stejný obsah ve stejné koncentraci a analyzují se souběžně v rámci jednoho provedení zkoušky. V tomto protokolu se každá koncentrace zkoušené chemické látky testuje třikrát; to znamená, že existují tři replikáty, které se analyzují současně při každé koncentraci zkoušené chemické látky.
Provedení zkoušky
: Úplná sada jamek na mikrotitrační destičce analyzovaných souběžně, která poskytuje všechny informace potřebné pro charakterizaci vazby zkoušené chemické látky na hrERα (tzn. celkový [3H]-17β-estradiol přidaný do zkušební jamky, maximální vazba [3H]-17β-estradiolu na hrERα, nespecifická vazba a celková vazba při různých koncentracích zkoušené chemické látky). Provedení zkoušky může sestávat pouze z jedné jamky (tj. replikátu) na každou koncentraci, ale protože tento protokol vyžaduje trojí analyzování, sestává jedno provedení ze tří jamek na každou koncentraci. Dále tento protokol vyžaduje tři nezávislá (tj. nesouběžná) provedení na každou chemickou látku.
Dodatek 3.2
USPOŘÁDÁNÍ JAMEK PŘI KOMPETITIVNÍ VAZEBNÉ ANALÝZE
|
Mikrodestička
|
Poloha
|
Replikát
|
Druh jamky
|
Kód jamky
|
Kód koncentrace
|
Výchozí koncentrace kompetitora (M)
|
Zásobní hrER (μl)
|
Objem pufru (μl)
|
Objem stopovací látky (radioaktivní E2) (μl)
|
Objem z destičky s ředěním (μl)
|
Konečný objem (μl)
|
Konečná koncentrace kompetitora (M)
|
|
S
|
A1
|
1
|
Slepý pokus
|
BK
|
BK1
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A2
|
2
|
Slepý pokus
|
BK
|
BK2
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A3
|
3
|
Slepý pokus
|
BK
|
BK3
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
B1
|
1
|
neradioaktivní E2
|
S
|
S1
|
1.00E-10
|
30
|
50
|
10
|
10
|
100
|
1.0E-11
|
|
S
|
B2
|
2
|
neradioaktivní E2
|
S
|
S1
|
1.00E-10
|
30
|
50
|
10
|
10
|
100
|
1.0E-11
|
|
S
|
B3
|
3
|
neradioaktivní E2
|
S
|
S1
|
1.00E-10
|
30
|
50
|
10
|
10
|
100
|
1.0E-11
|
|
S
|
C1
|
1
|
neradioaktivní E2
|
S
|
S2
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
S
|
C2
|
2
|
neradioaktivní E2
|
S
|
S2
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
S
|
C3
|
3
|
neradioaktivní E2
|
S
|
S2
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
S
|
D1
|
1
|
neradioaktivní E2
|
S
|
S3
|
3.16E-09
|
30
|
50
|
10
|
10
|
100
|
3.2E-10
|
|
S
|
D2
|
2
|
neradioaktivní E2
|
S
|
S3
|
3.16E-09
|
30
|
50
|
10
|
10
|
100
|
3.2E-10
|
|
S
|
D3
|
3
|
neradioaktivní E2
|
S
|
S3
|
3.16E-09
|
30
|
50
|
10
|
10
|
100
|
3.2E-10
|
|
S
|
E1
|
1
|
neradioaktivní E2
|
S
|
S4
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
E2
|
2
|
neradioaktivní E2
|
S
|
S4
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
E3
|
3
|
neradioaktivní E2
|
S
|
S4
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
F1
|
1
|
neradioaktivní E2
|
S
|
S5
|
3.16E-08
|
30
|
50
|
10
|
10
|
100
|
3.2E-09
|
|
S
|
F2
|
2
|
neradioaktivní E2
|
S
|
S5
|
3.16E-08
|
30
|
50
|
10
|
10
|
100
|
3.2E-09
|
|
S
|
F3
|
3
|
neradioaktivní E2
|
S
|
S5
|
3.16E-08
|
30
|
50
|
10
|
10
|
100
|
3.2E-09
|
|
S
|
G1
|
1
|
neradioaktivní E2
|
S
|
S6
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
G2
|
2
|
neradioaktivní E2
|
S
|
S6
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
G3
|
3
|
neradioaktivní E2
|
S
|
S6
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
H1
|
1
|
neradioaktivní E2
|
S
|
S7
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
H2
|
2
|
neradioaktivní E2
|
S
|
S7
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
H3
|
3
|
neradioaktivní E2
|
S
|
S7
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
A4
|
1
|
norethynodrel
|
NE
|
WP1
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
A5
|
2
|
norethynodrel
|
NE
|
WP1
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
A6
|
3
|
norethynodrel
|
NE
|
WP1
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
B4
|
1
|
norethynodrel
|
NE
|
WP2
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
B5
|
2
|
norethynodrel
|
NE
|
WP2
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
B6
|
3
|
norethynodrel
|
NE
|
WP2
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
C4
|
1
|
norethynodrel
|
NE
|
WP3
|
3.16E-07
|
30
|
50
|
10
|
10
|
100
|
3.2E-08
|
|
S
|
C5
|
2
|
norethynodrel
|
NE
|
WP3
|
3.16E-07
|
30
|
50
|
10
|
10
|
100
|
3.2E-08
|
|
S
|
C6
|
3
|
norethynodrel
|
NE
|
WP3
|
3.16E-07
|
30
|
50
|
10
|
10
|
100
|
3.2E-08
|
|
S
|
D4
|
1
|
norethynodrel
|
NE
|
WP4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
D5
|
2
|
norethynodrel
|
NE
|
WP4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
D6
|
3
|
norethynodrel
|
NE
|
WP4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
E4
|
1
|
norethynodrel
|
NE
|
WP5
|
3.16E-06
|
30
|
50
|
10
|
10
|
100
|
3.2E-07
|
|
S
|
E5
|
2
|
norethynodrel
|
NE
|
WP5
|
3.16E-06
|
30
|
50
|
10
|
10
|
100
|
3.2E-07
|
|
S
|
E6
|
3
|
norethynodrel
|
NE
|
WP5
|
3.16E-06
|
30
|
50
|
10
|
10
|
100
|
3.2E-07
|
|
S
|
F4
|
1
|
norethynodrel
|
NE
|
WP6
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
F5
|
2
|
norethynodrel
|
NE
|
WP6
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
F6
|
3
|
norethynodrel
|
NE
|
WP6
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
G4
|
1
|
norethynodrel
|
NE
|
WP7
|
3.16E-05
|
30
|
50
|
10
|
10
|
100
|
3.2E-06
|
|
S
|
G5
|
2
|
norethynodrel
|
NE
|
WP7
|
3.16E-05
|
30
|
50
|
10
|
10
|
100
|
3.2E-06
|
|
S
|
G6
|
3
|
norethynodrel
|
NE
|
WP7
|
3.16E-05
|
30
|
50
|
10
|
10
|
100
|
3.2E-06
|
|
S
|
H4
|
1
|
norethynodrel
|
NE
|
WP8
|
3.16E-04
|
30
|
50
|
10
|
10
|
100
|
3.2E-05
|
|
S
|
H5
|
2
|
norethynodrel
|
NE
|
WP8
|
3.16E-04
|
30
|
50
|
10
|
10
|
100
|
3.2E-05
|
|
S
|
H6
|
3
|
norethynodrel
|
NE
|
WP8
|
3.16E-04
|
30
|
50
|
10
|
10
|
100
|
3.2E-05
|
|
S
|
A7
|
1
|
OTES
|
N
|
OTES1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
S
|
A8
|
2
|
OTES
|
N
|
OTES1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
S
|
A9
|
3
|
OTES
|
N
|
OTES1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
S
|
B7
|
1
|
OTES
|
N
|
OTES2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
B8
|
2
|
OTES
|
N
|
OTES2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
B9
|
3
|
OTES
|
N
|
OTES2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
S
|
C7
|
1
|
OTES
|
N
|
OTES3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
C8
|
2
|
OTES
|
N
|
OTES3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
C9
|
3
|
OTES
|
N
|
OTES3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
S
|
D7
|
1
|
OTES
|
N
|
OTES4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
D8
|
2
|
OTES
|
N
|
OTES4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
D9
|
3
|
OTES
|
N
|
OTES4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
S
|
E7
|
1
|
OTES
|
N
|
OTES5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
E8
|
2
|
OTES
|
N
|
OTES5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
E9
|
3
|
OTES
|
N
|
OTES5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
S
|
H7
|
1
|
OTES
|
N
|
OTES8DBP7
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
S
|
H8
|
2
|
OTES
|
N
|
OTES88
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
S
|
H9
|
3
|
OTES
|
N
|
OTES8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
S
|
A10
|
1
|
celková vazba
|
TB
|
TB1
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
|
P
|
A11
|
2
|
celková vazba
|
TB
|
TB2
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
|
P
|
A12
|
3
|
celková vazba
|
TB
|
TB3
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
|
P
|
B10
|
4
|
celková vazba
|
TB
|
TB4
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
|
S
|
B11
|
5
|
celková vazba
|
TB
|
TB5
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
|
S
|
B12
|
6
|
celková vazba
|
TB
|
TB6
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
|
S
|
C10
|
1
|
neradioaktivní E2 (vysoká)
|
NSB
|
S1
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
C11
|
2
|
neradioaktivní E2 (vysoká)
|
NSB
|
S2
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
C12
|
3
|
neradioaktivní E2 (vysoká)
|
NSB
|
S3
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
D10
|
4
|
neradioaktivní E2 (vysoká)
|
NSB
|
S4
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
D11
|
5
|
neradioaktivní E2 (vysoká)
|
NSB
|
S5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
D12
|
6
|
neradioaktivní E2 (vysoká)
|
NSB
|
S6
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
S
|
E10
|
1
|
Kontrola s pufrem
|
BC
|
BC1
|
-
|
-
|
100
|
-
|
-
|
100
|
-
|
|
S
|
E11
|
2
|
Kontrola s pufrem
|
BC
|
BC2
|
-
|
-
|
100
|
-
|
-
|
100
|
-
|
|
S
|
E12
|
3
|
Kontrola s pufrem
|
BC
|
BC3
|
-
|
-
|
100
|
-
|
-
|
100
|
-
|
|
S
|
F10
|
4
|
Kontrola s pufrem
|
BC
|
BC4
|
-
|
-
|
100
|
-
|
-
|
100
|
-
|
|
S
|
F11
|
5
|
Kontrola s pufrem
|
BC
|
BC5
|
-
|
-
|
100
|
-
|
-
|
100
|
-
|
|
S
|
F12
|
6
|
Kontrola s pufrem
|
BC
|
BC6
|
-
|
-
|
100
|
-
|
-
|
100
|
-
|
|
S
|
G10 (*18)
|
1
|
Slepý pokus (pro radioaktivní látku)
|
radioaktivní
|
H1
|
-
|
90
|
-
|
10
|
-
|
100
|
-
|
|
S
|
G11 (*18)
|
2
|
Slepý pokus (pro radioaktivní látku)
|
radioaktivní
|
H2
|
-
|
90
|
-
|
10
|
-
|
100
|
-
|
|
P
|
G12 (*18)
|
3
|
Slepý pokus (pro radioaktivní látku)
|
radioaktivní
|
H3
|
-
|
90
|
-
|
10
|
-
|
100
|
-
|
|
S
|
H10 (*18)
|
4
|
Slepý pokus (pro radioaktivní látku)
|
radioaktivní
|
H4
|
-
|
90
|
-
|
10
|
-
|
100
|
-
|
|
S
|
H11 (*18)
|
5
|
Slepý pokus (pro radioaktivní látku)
|
radioaktivní
|
H5
|
-
|
90
|
-
|
10
|
-
|
100
|
-
|
|
S
|
H12
|
6
|
Slepý pokus (pro radioaktivní látku)
|
radioaktivní
|
H6
|
-
|
90
|
-
|
10
|
-
|
100
|
-
|
|
P1
|
A1
|
1
|
Neznámá 1
|
U1
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
A2
|
2
|
Neznámá 1
|
U1
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
A3
|
3
|
Neznámá 1
|
U1
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
B1
|
1
|
Neznámá 1
|
U1
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
B2
|
2
|
Neznámá 1
|
U1
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
B3
|
3
|
Neznámá 1
|
U1
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
C1
|
1
|
Neznámá 1
|
U1
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
C2
|
2
|
Neznámá 1
|
U1
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
C3
|
3
|
Neznámá 1
|
U1
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
D1
|
1
|
Neznámá 1
|
U1
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
D2
|
2
|
Neznámá 1
|
U1
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
D3
|
3
|
Neznámá 1
|
U1
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
E1
|
1
|
Neznámá 1
|
U1
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E2
|
2
|
Neznámá 1
|
U1
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E3
|
3
|
Neznámá 1
|
U1
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
F1
|
1
|
Neznámá 1
|
U1
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
F2
|
2
|
Neznámá 1
|
U1
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
F3
|
3
|
Neznámá 1
|
U1
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
G1
|
1
|
Neznámá 1
|
U1
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
G2
|
2
|
Neznámá 1
|
U1
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
G3
|
3
|
Neznámá 1
|
U1
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
H1
|
1
|
Neznámá 1
|
U1
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
H2
|
2
|
Neznámá 1
|
U1
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
H3
|
3
|
Neznámá 1
|
U1
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
A4
|
1
|
Neznámá 2
|
U2
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
A5
|
2
|
Neznámá 2
|
U2
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
A6
|
3
|
Neznámá 2
|
U2
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
B4
|
1
|
Neznámá 2
|
U2
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
B5
|
2
|
Neznámá 2
|
U2
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
B6
|
3
|
Neznámá 2
|
U2
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
C4
|
1
|
Neznámá 2
|
U2
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
C5
|
2
|
Neznámá 2
|
U2
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
C6
|
3
|
Neznámá 2
|
U2
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
D4
|
1
|
Neznámá 2
|
U2
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
D5
|
2
|
Neznámá 2
|
U2
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
D6
|
3
|
Neznámá 2
|
U2
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
E4
|
1
|
Neznámá 2
|
U2
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E5
|
2
|
Neznámá 2
|
U2
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E6
|
3
|
Neznámá 2
|
U2
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
F4
|
1
|
Neznámá 2
|
U2
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
F5
|
2
|
Neznámá 2
|
U2
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
F6
|
3
|
Neznámá 2
|
U2
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
G4
|
1
|
Neznámá 2
|
U2
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
G5
|
2
|
Neznámá 2
|
U2
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
G6
|
3
|
Neznámá 2
|
U2
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
H4
|
1
|
Neznámá 2
|
U2
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
H5
|
2
|
Neznámá 2
|
U2
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
H6
|
3
|
Neznámá 2
|
U2
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
A7
|
1
|
Neznámá 3
|
U3
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
A8
|
2
|
Neznámá 3
|
U3
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
A9
|
3
|
Neznámá 3
|
U3
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1.0E-10
|
|
P1
|
B7
|
1
|
Neznámá 3
|
U3
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
B8
|
2
|
Neznámá 3
|
U3
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
B9
|
3
|
Neznámá 3
|
U3
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1.0E-09
|
|
P1
|
C7
|
1
|
Neznámá 3
|
U3
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
C8
|
2
|
Neznámá 3
|
U3
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
C9
|
3
|
Neznámá 3
|
U3
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1.0E-08
|
|
P1
|
D7
|
1
|
Neznámá 3
|
U3
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
D8
|
2
|
Neznámá 3
|
U3
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
D9
|
3
|
Neznámá 3
|
U3
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.0E-07
|
|
P1
|
E7
|
1
|
Neznámá 3
|
U3
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E8
|
2
|
Neznámá 3
|
U3
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E9
|
3
|
Neznámá 3
|
U3
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
F7
|
1
|
Neznámá 3
|
U3
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
F8
|
2
|
Neznámá 3
|
U3
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
F9
|
3
|
Neznámá 3
|
U3
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1.0E-05
|
|
P1
|
G7
|
1
|
Neznámá 3
|
U3
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
G8
|
2
|
Neznámá 3
|
U3
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
G9
|
3
|
Neznámá 3
|
U3
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
H7
|
1
|
Neznámá 3
|
U3
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
H8
|
2
|
Neznámá 3
|
U3
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
H9
|
3
|
Neznámá 3
|
U3
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
A10
|
1
|
Kontrolní E2 (max)
|
S
|
E2max1
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.00E-07
|
|
P1
|
A11
|
2
|
Kontrolní E2 (max)
|
S
|
E2max2
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.00E-07
|
|
P1
|
A12
|
3
|
Kontrolní E2 (max)
|
S
|
E2max3
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.00E-07
|
|
P1
|
B10
|
1
|
Kontrolní E2 (IC50)
|
S
|
E2IC501
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B11
|
2
|
Kontrolní E2 (IC50)
|
S
|
E2IC502
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B12
|
3
|
Kontrolní E2 (IC50)
|
S
|
E2IC503
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
C10
|
1
|
Kontrolní NE (max)
|
S
|
Nemax1
|
1.00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1.00E-4,5
|
|
P1
|
C11
|
2
|
Kontrolní NE (max)
|
S
|
Nemax2
|
1.00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1.00E-4,5
|
|
P1
|
C12
|
3
|
Kontrolní NE (max)
|
S
|
Nemax3
|
1.00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1.00E-4,5
|
|
P1
|
D10
|
1
|
Kontrolní NE (IC50)
|
S
|
NEIC501
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D11
|
2
|
Kontrolní NE (IC50)
|
S
|
NEIC502
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D12
|
3
|
Kontrolní NE (IC50)
|
S
|
NEIC503
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
E10
|
1
|
neradioaktivní E2 (vysoká)
|
NSB
|
S1
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E11
|
2
|
neradioaktivní E2 (vysoká)
|
NSB
|
S2
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
E12
|
3
|
neradioaktivní E2 (vysoká)
|
NSB
|
S3
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
F10
|
4
|
neradioaktivní E2 (vysoká)
|
NSB
|
S4
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
F11
|
5
|
neradioaktivní E2 (vysoká)
|
NSB
|
S5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
F12
|
6
|
neradioaktivní E2 (vysoká)
|
NSB
|
S6
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1.0E-06
|
|
P1
|
G10
|
1
|
celková vazba
|
TB
|
TB1
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
|
P1
|
G11
|
2
|
celková vazba
|
TB
|
TB2
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
|
P1
|
G12
|
3
|
celková vazba
|
TB
|
TB3
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
|
P1
|
H10
|
4
|
celková vazba
|
TB
|
TB4
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
|
P1
|
H11
|
5
|
celková vazba
|
TB
|
TB5
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
|
P1
|
H12
|
6
|
celková vazba
|
TB
|
TB6
|
-
|
30
|
60
|
10
|
-
|
100
|
-
|
Dodatek 4
POZNÁMKY PRO ANALÝZU DAT Z KOMPETITIVNÍ VAZEBNÉ ANALÝZY HRER
|
1.
|
Kompetitivní vazebná analýza hrERα měří vazbu jedné koncentrace [3H]17β-estradiolu v přítomnosti zvyšujících se koncentrací zkoušené chemické látky. Křivka kompetitivní vazby se vynáší jako specifická vazba [3H]-17β-estradiolu proti koncentraci (log10 jednotky) kompetitora. Koncentrace zkoušené chemické látky, která inhibuje 50 % maximální specifické vazby [3H]-17β-estradiolu, je hodnota IC50.
|
ANALÝZA DAT PRO REFERENČNÍ ESTROGEN A LÁTKU SE SLABOU VAZBOU (1)
|
2.
|
Data z kontrolních provedení zkoušek se transformují (tj. percentuální specifická vazba [3H]-17β-estradiolu a log koncentrace kontrolní chemické látky) pro další analýzu. Odhady log (IC50) pro pozitivní kontroly (např. referenční estrogen a látku se slabou vazbou) se stanoví pomocí vhodného softwaru pro nelineární vynesení křivky, aby byly vhodné pro čtyřparametrickou Hillovu rovnici (např. BioSoft; GraphPad Prism) (2). Při vynášení těchto křivek může být horní bod, dolní bod, sklon a log(IC50) obvykle ponechán bez omezení. Při určování nejlepších hodnot by měla být použita robustní regrese, pokud není uvedeno zdůvodnění. Metoda zvolená pro robustní regresi by měla být uvedena. U analýz FW nebo CERI hrER nebylo nutné provádět v korekci úbytku ligandu, ale v případě potřeby je možné její provedení zvážit. Po úvodní analýze je třeba každou vazebnou křivku zkontrolovat, aby bylo zajištěno, že je vhodně aplikována na model. Relativní vazebnou afinitu (RBA) pro látku se slabou vazbou lze vypočítat jako procento log (IC50) pro látku se slabou vazbou vzhledem k log (IC50) pro 17β-estradiol. Výsledky z pozitivních kontrol a z kontroly s látkou, která se neváže, se posuzují pomocí měřítek účinnosti analýzy a kritérií přijatelnosti uvedených v této zkušební metodě (odstavec 20), v dodatku 2 (analýza FW, odstavce 41–51) a v dodatku 3 (analýza CERI, odstavce 41–51). Na obrázku 1 jsou uvedeny příklady ze 3 provedení zkoušek na referenční estrogen a látku se slabou vazbou.
|
Obrázek 1
Příklady křivek kompetitivní vazby pro referenční estrogen a kontrolní látku se slabou vazbou.
Analýza dat u zkoušených chemických látek
|
3.
|
Data za všechny zkoušené chemické látky se analyzují postupně, aby bylo zajištěno, že data budou analyzována správně, a aby byla správně klasifikována každá kompetitivní vazebná křivka. Každé provedení zkoušky u zkoušené chemické látky by mělo nejdříve projít standardizovanou analýzou dat, která je stejná jako analýza použitá pro kontroly s referenčním estrogenem a s látkou se slabou vazbou. Po dokončení je třeba provést technickou kontrolu parametrů vynesené křivky a vizuálně zkontrolovat, jak dobře data odpovídají generované kompetitivní vazebné křivce u každého provedení zkoušky. V průběhu této technické kontroly je pozorovaný pokles procenta specificky navázaného [3H]-17β-estradiolu v závislosti na koncentraci, nízká variabilita mezi technickými replikáty při jednotlivých koncentracích chemické látky a konzistentní parametry u tří provedení zkoušky dobrým ukazatelem, že zkouška a analýzy dat byly provedeny správně. Při posuzování výsledků z každého provedení zkoušky za zkoušenou chemickou látku je třeba používat odborný úsudek a data použitá ke klasifikaci jednotlivých zkoušených chemických látek jako látek, které se vážou, nebo látek, které se nevážou, by měla být vědecky obhajitelná.
|
|
4.
|
Občas se mohou vyskytnout příklady dat, která vyžadují větší pozornost, aby byla data o vazbě hrER správně analyzována a interpretována. Předchozí studie ukázaly případy, kdy může být analýza a interpretace dat o kompetitivní vazbě na receptor komplikovaná v důsledku zvýšení percentuální specifické vazby při testování chemických látek při nejvyšších koncentracích (obrázek 2). Je to známý problém, který se vyskytuje při použití protokolů u řady analýz kompetitivní vazby na receptor (3). V těchto případech je pozorována odpověď závislá na koncentraci při nižších koncentracích, ale s tím, jak se koncentrace zkoušené chemické látky blíží mezi rozpustnosti, vytěsňování [3H]17β-estradiolu již neklesá. V těchto případech data u vyšších koncentrací ukazují, že byla dosažena biologická mez analýzy. Tento jev je například často spojován s nerozpustností a srážením chemické látky při vysokých koncentracích, případně může také odrážet překročení kapacity aktivního uhlí potaženého dextranem zachytávat radioizotopově značený ligand v průběhu separace při nejvyšších koncentracích chemické látky. Ponechání těchto datových bodů při nanášení kompetitivních vazebných dat na sigmoidní křivku může někdy vést k chybné klasifikaci vazebného potenciálu zkoušené chemické látky na ER (obrázek 2). Aby k tomu nedocházelo, zahrnuje protokol pro vazebné analýzy FW a CERI hrER možnost vyloučit z analýz datové body, pokud je průměr replikátů pro procento specifické vazby [3H]17β-estradiolu o 10 % nebo více nad průměrem pozorovaným při nižší koncentraci (tj. obvykle se to nazývá pravidlo 10 %). Toto pravidlo lze použít u dané křivky pouze jednou a musí zůstat data pro alespoň 6 koncentrací, aby mohla být křivka správně klasifikována.
|
Obrázek 2
Příklady, kompetitivní vazebné křivky s použitím pravidla 10 % a bez použití tohoto pravidla
|
5.
|
Správné použití pravidla 10 % pro korekci těchto křivek by mělo být pečlivě zváženo a vyhrazeno pouze pro ty případy, kde existuje silná indikace, že jde o látku, která se váže na hrER. Při provádění experimentů pro validační studii analýzy vazby FW hrER bylo pozorováno, že pravidlo 10 % má někdy nezamýšlený a nepředpokládaný důsledek. Chemické látky, které nereagovaly s receptorem (tj. skutečné látky, které se nevážou), často vykazovaly variabilitu kolem 100 % vazby radioligandu, což bylo více než 10 % v celém rozmezí zkoušených koncentrací. Pokud se stalo, že nejnižší hodnota byla při nízké koncentraci, mohla být data při použití pravidla 10 % ze všech vyšších koncentrací z analýzy potenciálně vymazána, ačkoliv tyto koncentrace mohly být užitečné při určení, že tato chemická látka se neváže. Na obrázku 3 jsou uvedeny příklady, kdy použití pravidla 10 % není vhodné.
|
Obrázek 3
Příklady, data o kompetitivní vazbě, kdy použití pravidla 10 % není vhodné
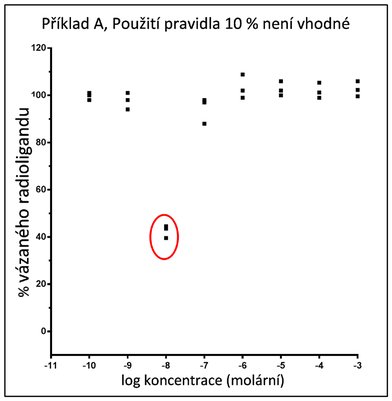
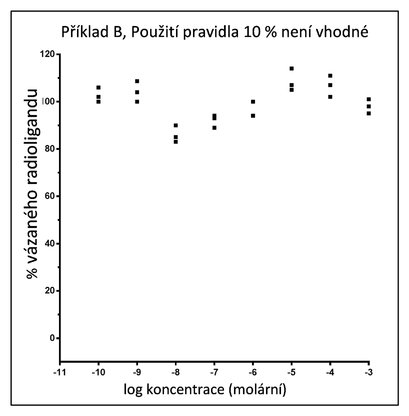
ODKAZY
|
(1)
|
OECD (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 226), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
Motulsky H. and Christopoulos A. (2003). The law of mass action, In Fitting Models to Biological Data Using Linear and Non-linear Regression. GraphPad Software Inc., San Diego, CA, pp 187-191. Www.graphpad.com/manuals/Prism4/RegressionBook.pdf
|
|
(3)
|
Laws SC, Yavanhxay S, Cooper RL, Eldridge JC. (2006). Nature of the Binding Interaction for 50 Structurally Diverse Chemicals with Rat Estrogen Receptors. Toxicological Sci. 94(1):46-56.
|
B.71 ANALÝZY SENZIBILIZACE KŮŽE IN VITRO ZAMĚŘENÉ NA KLÍČOVOU UDÁLOST AKTIVACE DENDRITICKÝCH BUNĚK V DRÁZE NEŽÁDOUCÍCH ÚČINKŮ (AOP) VEDOUCÍCH K SENZIBILIZACI KŮŽE
VŠEOBECNÝ ÚVOD
Zkušební metoda vycházející z klíčové události aktivace dendritických buněk
|
1.
|
Jako látka senzibilizující kůži se označuje látka, která po styku s kůží vyvolává alergickou odpověď, jak ji definuje globálně harmonizovaný systém klasifikace a označování chemických látek Organizace spojených národů (GHS OSN) (1) a nařízení Evropské unie (EU) č. 1272/2008 o klasifikaci, označování a balení látek a směsí (CLP) (99). Existuje všeobecná shoda, pokud jde o klíčové biologické procesy, jež zapříčiňují senzibilizaci kůže. Současné znalosti o chemických a biologických mechanismech souvisejících se senzibilizací kůže byly v rámci programu OECD zkoumajícího dráhy nežádoucích účinků (AOP) (2) shrnuty jako AOP, počínaje molekulární iniciační událostí přes průběžné události až po nepříznivý účinek, a sice alergickou kontaktní dermatitidu. V tomto případě je molekulární iniciační událostí (tj. první klíčová událost) kovalentní vazba elektrofilních látek na nukleofilní centra v kožních proteinech. Druhá klíčová událost v této metodě AOP se odehrává v keratinocytech a zahrnuje zánětlivé odpovědi, jakož i změny v expresi genů spojené se specifickými dráhami buněčné signalizace, např. dráhami závislými na antioxidačním/elektrofilním responzivním elementu (ARE). Třetí klíčovou událostí je aktivace dendritických buněk (DC), typicky hodnocená podle exprese specifických buněčných povrchových markerů, chemokinů a cytokinů. Čtvrtou klíčovou událostí je aktivace a proliferace T buněk, která se nepřímo hodnotí ve zkoušce s vyšetřením lokálních lymfatických uzlin na myších (LLNA) (3).
|
|
2.
|
Tato zkušební metoda je rovnocenná Pokynu OECD pro zkoušení (TG) č. 442E (2017). Popisuje analýzy in vitro, které se zaměřují na mechanismy popsané v klíčové události aktivace dendritických buněk AOP u senzibilizace kůže (2). Tato zkušební metoda zahrnuje zkoušky, které se podpůrně používají k rozlišování mezi látkami senzibilizujícími kůži a látkami nesenzibilizujícími kůži v souladu s GHS OSN a s nařízením CLP.
|
|
Zkoušky popsané v této zkušební metodě jsou:
|
—
|
Zkouška aktivace lidské buněčné linie (h-CLAT)
|
|
—
|
Zkouška aktivace buněčné linie U937 (U-SENS™)
|
|
—
|
Analýza reportérového genu pro interleukin-8 (analýza IL-8 Luc)
|
|
|
|
3.
|
Zkoušky zahrnuté do této zkušební metody a odpovídající pokyn OECD se mohou lišit, pokud jde o postup použitý pro generování dat a naměřené hodnoty, ale pro zkoušení mohou být použity bez rozdílu pro účely požadavků jednotlivých zemí na výsledky zkoušek u klíčové události aktivace dendritických buněk AOP u senzibilizace kůže, přičemž může být využito vzájemné uznávání údajů podle postupu OECD.
|
Obecné informace a principy zkoušek zahrnutých do této zkušební metody vycházející z klíčové události
|
4.
|
Hodnocení senzibilizace kůže bylo obvykle prováděno na pokusných zvířatech. Klasické metody, které používají morčata, Magnussonovu-Klingmannovu maximalizační zkoušku na morčatech (GPMT) a Bühlerovu zkoušku (zkušební metoda B.6) (4), hodnotí indukční i provokační fázi senzibilizace kůže. Zkoušky na myších, LLNA (zkušební metoda TM B.42) (3) a její dvě neradioaktivní modifikace, LLNA: DA (zkušební metoda B.50) (5) a LLNA: BrdU-ELISA (zkušební metoda B.51) (6), posuzují výlučně odpověď na indukci a také se prosadily, neboť ve srovnání se zkouškami na morčatech jsou vhodnější z hlediska dobrého zacházení se zvířaty a objektivního měření indukční fáze senzibilizace kůže.
|
|
5.
|
V poslední době byly přijaty mechanisticky založené zkušební metody in chemico a in vitro zkoumající první klíčovou událost (zkušební metoda B.59; analýza přímé reaktivity peptidů (7)) a druhou klíčovou událost (zkušební metoda B.60; zkušební metoda se stanovením luciferázy ARE-Nrf2 (8)), neboť přispívají k hodnocení potenciální nebezpečnosti chemických látek z hlediska senzibilizace kůže.
|
|
6.
|
Zkoušky popsané v této zkušební metodě kvantifikují změnu exprese buněčného povrchového markeru (markerů) spojenou s procesem aktivace monocytů a dendritických buněk po expozici senzibilizujícím látkám (např. CD54, CD86), nebo změny exprese IL-8, cytokinu souvisejícího s aktivací dendritických buněk. Bylo hlášeno, že látky senzibilizující kůži indukují expresi markerů na buněčné membráně, například CD40, CD54, CD80, CD83 a CD86, vedle indukce prozánětlivých cytokinů, jako jsou IL-1β a TNF-α, a několika chemokinů včetně IL-8 (CXCL8) a CCL3 (9) (10) (11) (12), souvisejících s aktivací dendritických buněk (2).
|
|
7.
|
Protože však aktivace dendritických buněk představuje pouze jednu klíčovou událost AOP (2) (13), informace získané zkouškami měřícími markery aktivace dendritických buněk nemusí samy o sobě stačit k vyvození závěru o existenci či neexistenci potenciálu chemických látek způsobovat senzibilizaci kůže. Proto se navrhuje, aby údaje získané pomocí zkoušek popsaných v této zkušební metodě byly použity jako podpůrné údaje při rozlišení mezi látkami senzibilizujícími kůži (tj. GHS OSN/CLP kategorie 1) a látkami, které kůži nesenzibilizují, při použití v rámci integrovaných přístupů ke zkouškám a posuzování (IATA), společně s dalšími relevantními doplňujícími informacemi, pocházejícími např. ze zkoušek in vitro zkoumajících jiné klíčové události v rámci metody AOP pro stanovení senzibilizace kůže, jakož i z nezkušebních metod včetně údajů odvozených z analogických chemických látek (13). Příklady použití dat získaných z těchto zkoušek v rámci definovaných přístupů, tj. přístupů standardizovaných jak v souvislosti s použitými informačními zdroji, tak i s postupem uplatněným u dat za účelem vypracování předpovědí, byly publikovány (13) a mohou být využity jako užitečné prvky v rámci IATA.
|
|
8.
|
Zkoušky popsané v této zkušební metodě nelze používat samy o sobě, a to ani k zařazení látek senzibilizujících kůži do podkategorií 1A a 1B, jak je definuje systém GHS OSN / CLP, ze strany orgánů zavádějících tyto dvě doplňkové podkategorie, ani k predikci účinnosti těchto látek při rozhodování v rámci posuzování bezpečnosti. V závislosti na regulačním rámci však kladné výsledky získané těmito metodami mohou být použity samy o sobě ke klasifikaci chemické látky v kategorii 1 GHS OSN / CLP.
|
|
9.
|
Termín „zkoušená chemická látka“ se v této zkušební metodě používá k označení toho, co se zkouší (100), a nesouvisí s použitelností zkoušek ke zkoušení jednosložkových látek, vícesložkových látek a/nebo směsí. V současnosti jsou k dispozici jen omezené informace o použitelnosti zkoušek ke zkoušení vícesložkových látek/směsí(14) (15). Tyto zkoušky jsou nicméně technicky použitelné i na zkoušení vícesložkových látek a směsí. Před použitím této zkušební metody ke zkoušení směsi za účelem získání údajů pro zamýšlené použití v právních předpisech by však mělo být zváženo, zda tato metoda může pro tento účel poskytnout přiměřené výsledky, a pokud ano, proč je tomu tak (101). Takové posouzení není nutné, pokud existuje právní požadavek na zkoušení dané směsi. Při zkoušení vícesložkových látek nebo směsí by se mimoto měly uvážit možné rušivé vlivy cytotoxických složek na pozorované odpovědi.
|
LITERATURA
|
(1)
|
United Nations UN (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Sixth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. Dostupný na: https://www.unece.org/trans/danger/publi/ghs/ghs_rev06/06files_e.html.
|
|
(2)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Series on Testing and Assessment No. Series on Testing and Assessment No. 168. Dostupné na: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En.
|
|
(3)
|
Kapitola B.42 této přílohy: Zkouška s vyšetřením lokálních lymfatických uzlin.
|
|
(4)
|
Kapitola B.6 této přílohy: Senzibilizace kůže.
|
|
(5)
|
Kapitola B.50 této přílohy: Senzibilizace kůže: zkouška s vyšetřením lokálních lymfatických uzlin: DA.
|
|
(6)
|
Kapitola B.51 této přílohy: Senzibilizace kůže: zkouška s vyšetřením lokálních lymfatických uzlin: BrdU-ELISA.
|
|
(7)
|
Kapitola B.59 této přílohy: Senzibilizace kůže in chemico: zkouška přímé reaktivity peptidů (DPRA).
|
|
(8)
|
Kapitola B.60 této přílohy: Senzibilizace kůže in vitro: zkušební metoda luciferázou s ARE-Nrf2.
|
|
(9)
|
Steinman RM. (1991). The dendritic cell system and its role in immunogenicity. Annu Rev Immunol 9:271-96.
|
|
(10)
|
Caux C, Vanbervliet B, Massacrier C, Azuma M, Okumura K, Lanier LL, and Banchereau J. (1994). B70/B7-2 is identical to CD86 and is the major functional ligand for CD28 expressed on human dendritic cells. J Exp Med 180:1841-7.
|
|
(11)
|
Aiba S, Terunuma A, Manome H, and Tagami H. (1997). Dendritic cells differently respond to haptens and irritants by their production of cytokines and expression of co-stimulatory molecules. Eur J Immunol 27:3031-8.
|
|
(12)
|
Aiba S, Manome H, Nakagawa S, Mollah ZU, Mizuashi M, Ohtani T, Yoshino Y, and Tagami. H. (2003). p38 mitogen-activated protein kinase and extracellular signal-regulated kinases play distinct roles in the activation of dendritic cells by two representative haptens, NiCl2 and DNCB. J Invest Dermatol 120:390-8.
|
|
(13)
|
OECD (2016). Series on Testing & Assessment No 256: Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, Annex 1 and Annex 2. ENV/JM/HA(2016)29. Organisation for Economic Cooperation and Development, Paris. Dostupné na webových stránkách: https://community.oecd.org/community/iatass.
|
|
(14)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, NishiyamaN, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275-284.
|
|
(15)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. Chem. In Vitro 29, 901-916.
|
Dodatek 1
SENZIBILIZACE KŮŽE IN VITRO: ZKOUŠKA AKTIVACE BUNĚČNÉ LINIE (H-CLAT)
VÝCHOZÍ ÚVAHY A OMEZENÍ
|
1.
|
Zkouška h-CLAT kvantifikuje změny exprese buněčných povrchových markerů spojené s procesem aktivace monocytů a dendritických buněk (DC) (tj. CD86 a CD54) v buněčné linii lidské monocytární leukémie THP-1 po expozici senzibilizujícím látkám (1)(2). Naměřené úrovně exprese buněčných povrchových markerů CD86 a CD54 se poté použijí jako podpůrný údaj pro rozlišení mezi látkami senzibilizujícími kůži a látkami, které kůži nesenzibilizují.
|
|
2.
|
Zkouška h-CLAT byla hodnocena ve validační studii koordinované Referenční laboratoří Evropské unie pro alternativy ke zkouškám na zvířatech (EURL ECVAM) a následně byla podrobena nezávislé odborné revizi, kterou provedl vědecký poradní výbor (ESAC) EURL ECVAM. S ohledem na všechny dostupné důkazy a příspěvky od regulátorů a účastníků doporučil EURL ECVAM (3) zkoušku h-CLAT pro použití v rámci přístupu IATA na podporu rozlišování mezi látkami senzibilizujícími kůži a látkami nesenzibilizujícími kůži za účelem klasifikace nebezpečnosti a označování. Příklady využití údajů h-CLAT v kombinaci s jinými informacemi jsou uvedeny v odborné literatuře (4)(5)(6)(7)(8)(9)(10)(11).
|
|
3.
|
U zkoušky h-CLAT se prokázalo, že ji lze přenést do laboratoří, které mají zkušenosti s technikami používajícími buněčné kultury a analýzou pomocí průtokové cytometrie. Míra reprodukovatelnosti předpovědí, které lze od této zkoušky očekávat, činí řádově 80 % v rámci laboratoří a 80 % mezi laboratořemi (3)(12). Výsledky validační studie (13) a dalších zveřejněných studií (14) celkově ukazují, že v porovnání s výsledky zkušební metody LLNA je přesnost při odlišení kůži senzibilizujících látek (tj. kategorie 1 GHS OSN/CLP) od nesenzibilizujících látek 85 % (N=142) při citlivosti 93 % (94/101) a specifičnosti 66 % (27/41) (na základě opakované analýzy provedené EURL ECVAM (12) při zohlednění všech stávajících dat, přičemž nebyly brány v úvahu negativní výsledky chemických látek s log Kow větším než 3,5, jak je uvedeno v odstavci 4). Falešně negativní předpovědi se u zkoušky h-CLAT pravděpodobněji týkají chemických látek vykazujících nízkou až střední potenci způsobovat senzibilizaci kůže (tj. podkategorie 1B GHS OSN / CLP) než chemických látek vykazujících vysokou potenci způsobovat senzibilizaci kůže (tj. podkategorie 1A GHS OSN / CLP (4)(13)(15). Celkově vzato tyto informace naznačují, že metoda h-CLAT může být užitečná a může přispět k zjištění nebezpečí senzibilizace kůže. Zde uvedené hodnoty přesnosti h-CLAT jako samostatné zkoušky jsou však pouze orientační, neboť tato zkouška by měla být posuzována v kombinaci s jinými zdroji informací v rámci IATA a v souladu s ustanoveními odstavců 7 a 8 ve Všeobecném úvodu. Při hodnocení metod posuzování senzibilizace kůže bez použití zvířat by se dále mělo vést v patrnosti, že ani zkouška LLNA a ani jiné zkoušky na zvířatech nemusí plně odrážet situaci u lidí.
|
|
4.
|
Na základě nyní dostupných údajů bylo prokázáno, že metodu h-CLAT lze použít ke zkoušení chemických látek, které obsahují různé organické funkční skupiny, mají různé reakční mechanismy, různou potenci způsobovat senzibilizaci kůže (jak byla stanovena ve studiích in vivo) a různé fyzikálně-chemické vlastnosti (3)(14) (15). Metodu h-CLAT lze použít ke zkoušení rozpustných chemických látek nebo látek, jež tvoří stabilní disperzi (tj. koloid nebo suspenzi, ve které se zkoušená chemická látka neusazuje ani se neodděluje od rozpouštědla/vehikula do různých fází) ve vhodném rozpouštědle/ vehikulu (viz odstavec 14). Zkoušené chemické látky s log Kow větším než 3,5 mají tendenci dávat falešně negativní výsledky (14). Negativní výsledky zkoušených chemických látek s log Kow větším než 3,5 by proto neměly být brány v úvahu. Pozitivní výsledky získané u zkoušených chemických látek s log Kow větším než 3,5 však přesto mohou být použity na podporu identifikace zkoušené chemické látky jako látky senzibilizující kůži. Z důvodu omezené metabolické schopnosti použité buněčné linie (16) a vzhledem k experimentálním podmínkám mohou prohapteny (tj. látky vyžadující enzymatickou aktivaci, například prostřednictvím enzymů P450) a prehapteny (tj. látky aktivované oxidací) zejména při nízké míře oxidace rovněž vykazovat negativní výsledky ve zkoušce h-CLAT (15). Zkouškou h-CLAT (17) mohou být hodnoceny fluorescenční zkoušené chemické látky, avšak silné fluorescenční zkoušené chemické látky vyzařující na stejné vlnové délce jako fluorescein-isothiokyanát (FITC) nebo jako propidiumiodid (PI) budou narušovat detekci pomocí průtokové cytometrie, a proto nemohou být správně hodnoceny pomocí konjugovaných protilátek FITC nebo PI. V takovém případě mohou být použity jiné protilátky značené fluorochromem, respektive jiné markery cytotoxicity, pokud lze prokázat, že poskytují podobné výsledky jako protilátky značené FITC (viz odstavec 24) nebo PI (viz odstavec 18), např. zkoušením vhodných látek uvedených v dodatku 1-2. Vzhledem k výše uvedenému by se měly negativní výsledky interpretovat v kontextu stanovených omezení a společně s jinými zdroji informací v rámci přístupu IATA. V případech, kdy existují důkazy prokazující nepoužitelnost metody h-CLAT na jiné specifické kategorie zkoušených chemických látek, neměla by se tato metoda ke zkoušení těchto specifických kategorií používat.
|
|
5.
|
Jak bylo popsáno výše, metoda h-CLAT pomáhá rozlišovat mezi látkami senzibilizujícími kůži a látkami nesenzibilujícími kůži. Může však také potenciálně přispět k hodnocení senzibilizační potence (4)(5)(9), je-li používána v rámci integrovaných přístupů, například IATA. Pro určení toho, jak mohou výsledky h-CLAT potenciálně přispět k posuzování potence, nicméně bude nutný další výzkum, přednostně založený na údajích o účincích na člověka.
|
|
6.
|
Definice jsou uvedeny v dodatku 1.1.
|
PODSTATA ZKOUŠKY
|
7.
|
Metoda h-CLAT je analýza in vitro, která kvantifikuje změny exprese buněčných povrchových markerů (tj. CD86 a CD54) v buněčné linii lidské monocytární leukémie, na buňkách THP-1 po 24hodinové expozici zkoušené chemické látce. Tyto povrchové molekuly jsou typickými markery aktivace monocytických THP-1 a mohou napodobovat aktivaci DC, která hraje kritickou roli při primární aktivaci (primingu) T buněk. Změny exprese povrchových markerů se měří pomocí průtokové cytometrie po obarvení buněk protilátkami značenými fluorochromem. Rovněž se provádí paralelní měření cytotoxicity, aby bylo možné posoudit, zda při sub-cytotoxických koncentracích dochází k up-regulaci exprese povrchových markerů. Relativní intenzita fluorescence povrchových markerů v porovnání s kontrolou s rozpouštědlem/vehikulem se vypočítá a použije v predikčním modelu (viz odstavec 26) jako podpůrný údaj pro rozlišení mezi senzibilizujícími a nesenzibilizujícími látkami.
|
PROKÁZÁNÍ ZPŮSOBILOSTI
|
8.
|
Před rutinním používáním zkoušky popsané v tomto dodatku ke zkušební metodě B.71 by měly laboratoře prokázat odbornou způsobilost použitím 10 látek pro prokazování způsobilosti, které jsou uvedeny v dodatku 1.2. Uživatelé zkoušky by si také měli vést historickou databázi údajů pocházejících ze zkoušek reaktivity (viz odstavec 11) a z pozitivních kontrol a kontrol s rozpouštědlem/vehikulem (viz odstavce 20–22) a používat tato data na potvrzení, že v jejich laboratoři je udržována reprodukovatelnost zkoušky v čase.
|
POSTUP
|
9.
|
Tato zkouška je založena na protokolu databázové služby pro alternativní metody k pokusům na zvířatech (DB-ALM) č. 158 (18), což je protokol použitý při validační studii provedené za koordinace EURL ECVAM. Při provádění a používání metody h-CLAT v laboratoři se doporučuje použít tento protokol. Dále je uveden popis hlavních složek a postupů metody h-CLAT, která zahrnuje dva kroky: analýza pro stanovení dávky a měření exprese CD86/CD54.
|
Příprava buněk
|
10.
|
Metoda h-CLAT se používá s buněčnou linií lidské monocytární leukémie THP-1. Doporučuje se, aby byly buňky (TIB-202™) pořízeny z kvalifikované buněčné banky, jako je například Americká sbírka typových kultur.
|
|
11.
|
Buňky THP-1 se kultivují při teplotě 37oC ve zvlhčené atmosféře s 5 % CO2 v médiu RPMI-1640 doplněném 10 % fetálního bovinního séra (FBS), 0,05 mM 2-merkaptoethanolu, 100 jednotkami/ml penicilinu a 100 μg/ml streptomycinu. Použití penicilinu a streptomycinu v kultivačním médiu může být vynecháno. V takovém případě by si však uživatelé měli ověřit, že nepřítomnost antibiotik v kultivačním médiu nemá žádný vliv na výsledky, například otestováním vhodných látek uvedených v dodatku 1.2. Aby se minimalizovalo riziko kontaminace, měly by být v každém případě dodržovány správné postupy při kultivaci buněk nezávisle na tom, zda kultivační médium obsahuje antibiotika. Buňky THP-1 se rutinně nasazují každé 2–3 dny v hustotě 0,1 až 0,2 × 106 buněk/ml. Udržují se při hustotě 0,1 až 1,0 × 106 buněk/ml. Než se buňky použijí pro zkoušení, je třeba zjistit, zda jsou vhodné, provedením kontroly reaktivity. Kontrola reaktivity buněk se provádí pomocí pozitivních kontrol, 2,4-dinitrochlorobenzenu (DNCB) (CAS č. 97-00-7, čistota ≥ 99 %) a síranu nikelnatého (NiSO4) (CAS č. 10101-97-0, čistota ≥ 99 %), a negativní kontroly, kyseliny mléčné (LA) (CAS č. 50-21-5, čistota ≥ 85 %), a to dva týdny po rozmrazení. DNCB i NiSO4 by měly dát pozitivní odpověď buněčných povrchových markerů CD86 i CD54 a LA by měla dát negativní odpověď buněčných povrchových markerů CD86 i CD54. Pro analýzu se používají pouze buňky, které prošly kontrolou reaktivity. Buňky mohou být množeny až dva měsíce po rozmrazení. Počet pasáží by neměl překročit 30. Kontrola reaktivity se provádí podle postupů popsaných v odstavcích 20–24.
|
|
12.
|
Pro zkoušení se buňky THP-1 nasadí v hustotě 0,1 × 106 buněk/ml nebo 0,2 × 106 buněk/ml a předběžně se kultivují v kultivačních baňkách po dobu 72 hodin, respektive 48 hodin. Je důležité, aby byla hustota buněk v kultivační baňce bezprostředně po předběžné kultivaci pokud možno stejná při každém experimentu (použitím jedné ze dvou podmínek předběžné kultivace popsaných výše), protože hustota buněk v kultivační baňce bezprostředně po předběžné kultivaci by mohla ovlivnit expresi CD86/CD54 indukovanou alergeny (19). V den zkoušky se buňky získané z kultivační baňky resuspendují v čerstvém kultivačním médiu při hustotě 2 × 106 buněk/ml. Poté se buňky rozdělí do 24jamkové destičky s plochým dnem (500 μl, 1 × 106 buněk/jamku) nebo do 96jamkové destičky s plochým dnem (80 μl, 1,6 × 105 buněk/jamku).
|
Analýza ke stanovení dávky
|
13.
|
Analýza ke stanovení dávky se provádí za účelem stanovení CV75, což je koncentrace zkoušené chemické látky, která dává 75 % životaschopnost buněk (CV) v porovnání s kontrolou s rozpouštědlem/vehikulem. Hodnota CV75 se používá ke stanovení koncentrace zkoušených chemických látek pro měření exprese CD86/CD54 (viz odstavce 20–24).
|
Příprava zkoušených chemických látek a kontrolních látek
|
14.
|
Zkoušené chemické látky a kontrolní látky se připraví v den zkoušení. Pro metodu h-CLAT se zkoušené chemické látky rozpustí nebo stabilně dispergují (viz také odstavec 4) ve fyziologickém roztoku nebo médiu jako rozpouštědle/vehikulu první volby nebo v dimethylsulfoxidu (DMSO, čistota ≥ 99 %) jako v rozpouštědle/vehikulu druhé volby, jestliže zkoušená chemická látka není rozpustná nebo netvoří stabilní disperzi v předchozích dvou rozpouštědlech/vehikulech, na finální koncentraci 100 mg/ml (ve fyziologickém roztoku nebo médiu) nebo 500 mg/ml (v DMSO). Jestliže je podáno dostatečné vědecké zdůvodnění, mohou být použita jiná rozpouštědla/vehikula než výše uvedená. Je třeba vzít v úvahu stabilitu zkoušené chemické látky ve finálním rozpouštědle/vehikulu.
|
|
15.
|
Z počátečních 100 mg/ml (ve fyziologickém roztoku nebo médiu) nebo 500 mg/ml (v DMSO) zásobních roztoků zkoušených chemických látek se připraví tyto zředěné roztoky:
|
—
|
Pro fyziologický roztok nebo médium jako rozpouštědlo/vehikulum: Připraví se osm zásobních roztoků (osm koncentrací) dvojnásobným postupným ředěním s použitím odpovídajícího rozpouštědla/vehikula. Tyto zásobní roztoky se poté dále naředí padesátinásobně na kultivační médium (pracovní roztoky). Jestliže je nejvyšší konečná koncentrace na destičce 1 000 μg/ml netoxická, je třeba znovu stanovit maximální koncentraci provedením nové zkoušky cytotoxicity. Konečná koncentrace na destičce by neměla přesahovat 5 000 μg/ml zkoušených chemických látek rozpuštěných nebo stabilně dispergovaných ve fyziologickém roztoku nebo médiu.
|
|
—
|
Pro DMSO jako rozpouštědlo/vehikulum: Připraví se osm zásobních roztoků (osm koncentrací) dvojnásobným postupným ředěním s použitím odpovídajícího rozpouštědla/vehikula. Tyto zásobní roztoky se poté dále naředí 250násobně na kultivační médium (pracovní roztoky). Konečná koncentrace na destičce by neměla přesahovat 1 000 μg/ml ani v případě, že je tato koncentrace netoxická.
|
Nakonec se pracovní roztoky použijí pro expozici přidáním stejného objemu pracovního roztoku do objemu buněčné suspenze THP-1 na destičce (viz také odstavec 17), čímž se dosáhne dalšího dvojnásobného ředění) obvykle je konečný rozsah koncentrací na destičce 7,81–1 000 μg/ml).
|
|
16.
|
Jako kontrola s rozpouštědlem/vehikulem se u metody h-CLAT používá kultivační médium (pro zkoušené chemické látky rozpustné nebo stabilně dispergované (viz odstavec 4) v médiu nebo ve fyziologickém roztoku) nebo DMSO (pro zkoušené chemické látky rozpustné nebo stabilně dispergované v DMSO) zkoušené v jedné konečné koncentraci na destičce 0,2 %. Provede se stejné ředění, jak je popsáno u pracovních roztoků v odstavci 15.
|
Aplikace zkoušených chemických látek a kontrolních látek
|
17.
|
Kultivační médium nebo pracovní roztoky popsané v odstavcích 15 a 16 se smíchají 1:1 (obj.) s buněčnými suspenzemi připravenými ve 24jamkové nebo 96jamkové destičce s plochým dnem (viz odstavec 12). Exponované destičky se poté inkubují po dobu 24 ± 0,5 hodin při teplotě 37oC za přítomnosti 5 % CO2. Mělo by se dbát na to, aby nedošlo k vypařování těkavých zkoušených chemických látek a ke vzájemné kontaminaci zkoušenou chemickou látkou mezi jamkami, např. tak, že se destičky před inkubací se zkoušenými chemickými látkami zapečetí (20).
|
Barvení propidiumiodidem (PI)
|
18.
|
Po 24 ± 0,5 hodinách expozice se buňky přenesou do zkumavek a odeberou se odstředěním. Supernatanty se zlikvidují a zbývající buňky se resuspendují s 200 μl (u 96jamkové destičky) nebo 600 μl (u 24jamkové destičky) fosfátového pufru obsahujícího 0,1 % bovinního sérového albuminu (barvicí pufr). 200 μl buněčné suspenze se přenese do 96jamkové destičky s oblým dnem (u 96jamkové destičky) nebo do mikrozkumavky (u 24jamkové destičky) a dvakrát se promyje 200 μl (u 96jamkové destičky) nebo 600 μl (u 24jamkové destičky) barvicího pufru. Nakonec se buňky resuspendují v barvicím pufru (např. 400 μl) a přidá se roztok PI (např. 20 μl) (konečná koncentrace PI činí například 0,625 μg/ml). Mohou být použity jiné markery cytotoxicity, jako je 7-aminoaktinomycin D (7-AAD), trypanová modř či jiné, pokud lze prokázat, například otestováním vhodných látek uvedených v dodatku 1.2, že alternativní barviva dávají podobné výsledky jako PI.
|
Měření cytotoxicity pomocí průtokové cytometrie a odhad hodnoty CV75
|
19.
|
Příjem PI se analyzuje pomocí průtokové cytometrie s akvizičním kanálem FL-3. Připraví se celkem 10 000 živých buněk (PI negativních). Životaschopnost buněk lze vypočítat podle následující rovnice programem cytometrické analýzy. Když je životaschopnost buněk nízká, je třeba připravit až 30 000 buněk včetně odumřelých. Alternativně lze získat údaje za jednu minutu po zahájení analýzy.

Hodnota CV75 (viz odstavec 13), tj. koncentrace vykazující 75 % přežití buněk THP-1 (25 % cytotoxicita), se vypočítá log-lineární interpolací podle této rovnice:
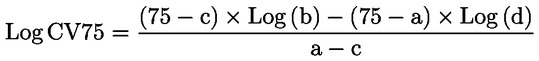
Kde:
a je minimální hodnota životaschopnosti buněk nad 75 %
c je maximální hodnota životaschopnosti buněk pod 75 %
b a d jsou koncentrace ukazující hodnotu životaschopnosti buněk a, respektive c
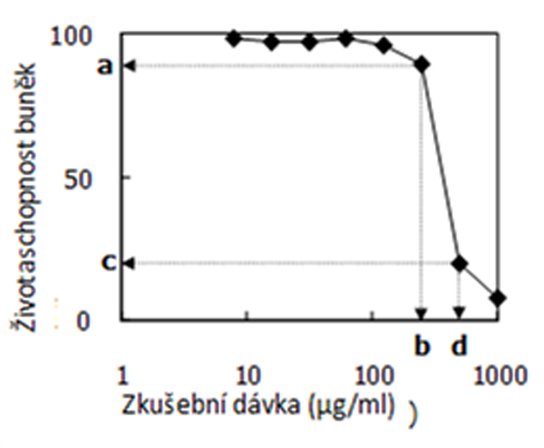
Lze použít i jiné způsoby odvození hodnoty CV75, pokud je prokázáno, že to nemá vliv na výsledky (např. otestováním vhodných látek uvedených).
|
Měření exprese CD86/CD54
Příprava zkoušených chemických látek a kontrolních látek
|
20.
|
Použije se vhodné rozpouštědlo/vehikulum (fyziologický roztok, médium nebo DMSO; viz odstavec 14) na rozpuštění nebo stabilní dispergování zkoušených chemických látek. Zkoušené chemické látky se nejprve naředí na koncentraci odpovídající 100násobku (pro fyziologický roztok nebo médium) nebo 500násobku (pro DMSO) hodnoty 1,2 × CV75 stanovené v analýze pro zjištění dávky (viz odstavec 19). Jestliže nelze stanovit hodnotu CV75 (tj. když v analýze pro zjištění dávky není pozorována dostatečná cytotoxicita), použije se jako výchozí koncentrace nejvyšší rozpustná nebo stabilně dispergovaná koncentrace zkoušené chemické látky připravená s každým rozpouštědlem/vehikulem. Nezapomeňte, že konečná koncentrace na destičce by neměla přesahovat 5 000 μg/ml (v případě fyziologického roztoku nebo média) nebo 1 000 μg/ml (v případě DMSO). Poté se připraví 1,2násobným sériovým ředěním s použitím odpovídajícího rozpouštědla/vehikula zásobní roztoky (osm koncentrací v rozsahu od 100×1,2 × CV75 do 100×0,335 × CV75 (pro fyziologický roztok nebo médium) nebo od 500×1,2 × CV75 do 500×0,335 × CV75 (pro DMSO)), které budou testovány metodou h-CLAT (viz protokol DB-ALM č. 158, kde je uveden příklad dávkování). Zásobní roztoky se poté dále naředí 50násobně (pro fyziologický roztok nebo médium) nebo 250násobně (pro DMSO) na kultivační médium (pracovní roztoky). Tyto pracovní roztoky se nakonec použijí pro expozici s dalším konečným dvojnásobným ředicím faktorem na destičce. Jestliže výsledky nesplňují kritéria přijatelnosti popsaná v odstavcích 29 a 30, pokud jde o životaschopnost buněk, může být zopakována analýza ke stanovení dávky, aby se určila přesnější hodnota CV75. Nezapomeňte, že pro měření exprese CD86/CD54 mohou být použity pouze 24jamkové destičky.
|
|
21.
|
Připraví se kontrola s rozpouštědlem/vehikulem, jak bylo popsáno v odstavci 16. Jako pozitivní kontrola se u metody h-CLAT používá DNCB (viz odstavec 11), přičemž zásobní roztoky se připraví v DMSO a naředí podle popisu ředění zásobních roztoků uvedeného v odstavci 20. Jako pozitivní kontrola pro měření exprese CD86/CD54 se používá DNCB v jedné konečné koncentraci na destičce (obvykle 4,0 μg/ml). Pro získání koncentrace DNCB na destičce 4,0 μg/ml se připraví 2 mg/ml zásobního roztoku DNCB v DMSO, který se dále naředí 250násobně s kultivačním médiem na 8 μg/ml pracovního roztoku. Alternativně může být použita jako koncentrace pozitivní kontroly také hodnota CV75 DNCB, která se stanoví v každém zkušebním zařízení. Lze použít i jiné vhodné pozitivní kontroly, pokud jsou k dispozici dosavadní údaje umožňující odvodit srovnatelná kritéria přijatelnosti daného provedení zkoušky. Jedna konečná koncentrace na destičce u pozitivních kontrol by neměla přesahovat 5 000 μg/ml (v případě fyziologického roztoku nebo média) nebo 1 000 μg/ml (v případě DMSO). Kritéria přijatelnosti provedení zkoušky jsou stejná jako kritéria přijatelnosti popsaná pro zkoušenou chemickou látku (viz odstavec 29), s výjimkou posledního kritéria přijatelnosti, neboť pozitivní kontrola se testuje v jedné koncentraci.
|
Aplikace zkoušených chemických látek a kontrolních látek
|
22.
|
U každé zkoušené chemické látky a kontrolní látky je pro předpověď potřebný jeden experiment. Každý experiment sestává alespoň ze dvou nezávislých provedení zkoušky pro měření exprese CD86/CD54 (viz odstavce 26-28). Každé nezávislé opakování se provádí v různý den nebo ve stejný den, pokud pro každé nezávislé provedení zkoušky: a) jsou připraveny nezávislé čerstvé zásobní roztoky zkoušené chemické látky a roztoky protilátek a b) jsou použity nezávisle sebrané buňky (tj. buňky se sbírají z různých kultivačních baněk); buňky však mohou pocházet ze stejné pasáže. Zkoušené chemické látky a kontrolní látky připravené jako pracovní roztoky (500 μl) se smíchají s 500 μl suspendovaných buněk (1x106 buněk) v poměru 1: 1 a buňky se inkubují po dobu 24 ± 0,5 hodiny, jak je popsáno v odstavcích 20 a 21. Při každém provedení zkoušky stačí jeden replikát pro každou koncentraci zkoušené chemické látky a kontrolní látky, protože predikce se získává alespoň ze dvou nezávislých provedení zkoušky.
|
Barvení a analýza buněk
|
23.
|
Po 24±0,5 hodinách expozice se buňky přenesou z 24jamkové destičky do zkumavek, odeberou se odstředěním a poté se dvakrát promyjí 1 ml barvicího pufru (v případě potřeby může být provedeno další promytí). Po promytí se počet buněk upraví 600 μl tlumivého roztoku (barvicí pufr obsahující 0,01 % (m/v) globulinu (Cohnova frakce II, III, lidský; SIGMA, #G2388-10G nebo ekvivalentní)) a inkubují se při 4oC po dobu 15 min. Po úpravě počtu se buňky rozdělí na tři díly po 180 μl do 96jamkové destičky s oblým dnem nebo mikrozkumavky.
|
|
24.
|
Po odstředění se buňky barví 50 μl protilátky anti-CD86, anti-CD54 nebo myší IgG1 protilátky (izotyp) značené FITC při 4oC po dobu 30 min. Protilátky popsané v protokolu č. 158 pro metodu h-CLAT DB-ALM (18) se používají po naředění 3:25 obj. (pro CD86 (BD-PharMingen, #555657; Klon: Fun-1)) nebo 3:50 obj. (pro CD54 (DAKO, #F7143; Klon: 6.5B5) a IgG1 (DAKO, #X0927)) v barvicím pufru. Vývojáři zkoušky definovali tyto ředicí faktory protilátek jako faktory, které poskytují nejlepší odstup signálu od šumu. Podle zkušeností vývojářů zkoušek je intenzita fluorescence protilátek obvykle u různých šarží konzistentní. Uživatelé však mohou zvážit titraci protilátek v podmínkách své vlastní laboratoře a definovat si tak nejlepší koncentrace pro použití. Mohou být použity jiné protilátky anti-CD86 a/nebo anti-CD54 značené fluorochromem, pokud lze prokázat, například otestováním vhodných látek uvedených v dodatku 1.2, že dávají podobné výsledky jako konjugované protilátky značené FITC. Je třeba poznamenat, že změna klonu nebo dodavatele protilátek, které jsou popsány v protokolu č. 158 pro metodu h-CLAT DB-ALM (18), může ovlivnit výsledky. Po dvojím nebo vícenásobném promytí 150 μl barvicího pufru se buňky resuspendují v barvicím pufru (např. 400 μl) a přidá se roztok PI (např. 20 μl pro dosažení konečné koncentrace 0,625 μg/ml) nebo roztok jiného markeru cytotoxicity (viz odstavec 18). Úrovně exprese CD86 a CD54 a životaschopnosti buněk se analyzují pomocí průtokové cytometrie.
|
ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV
Vyhodnocení údajů
|
25.
|
Exprese CD86 a CD54 se analyzuje pomocí průtokové cytometrie s akvizičním kanálem FL-1. Na základě geometrického průměru intenzity fluorescence (MFI) se vypočítá relativní intenzita fluorescence (RFI) CD86 a CD54 pro buňky pozitivní kontroly (ctrl) a buňky exponované chemické látce podle této rovnice:
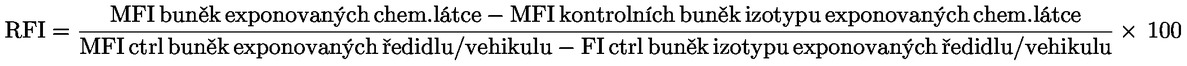
Podle rovnice uvedené v odstavci 19 se vypočítá také životaschopnost buněk z kontrolních (ctrl) buněk izotypu (které jsou obarveny myšími IgG1 (izotyp) protilátkami).
|
Predikční model
|
26.
|
Pro měření exprese CD86/CD54 se každá zkoušená chemická látka otestuje alespoň ve dvou nezávislých provedeních zkoušky, z nichž se odvodí jedna předpověď (POZITIVNÍ nebo NEGATIVNÍ). Předpověď získaná zkouškou h-CLAT se považuje za POZITIVNÍ, je-li splněna alespoň jedna z následujících podmínek ve 2 ze 2 opakování nebo alespoň ve 2 ze 3 nezávislých provedení zkoušky, jinak se předpověď získaná zkouškou h-CLAT považuje za NEGATIVNÍ (obrázek 1):
|
—
|
RFI u CD86 se rovná nebo je větší než 150 % při jakékoliv zkoušené koncentraci (při životaschopnosti buněk ≥ 50 %);
|
|
—
|
RFI u CD54 se rovná nebo je větší než 200 % při jakékoliv zkoušené koncentraci (při životaschopnosti buněk ≥ 50 %).
|
|
|
27.
|
Pokud jsou první dvě provedení zkoušky u CD86 pozitivní anebo jsou pozitivní pro CD54, považuje se předpověď získaná zkouškou h-CLAT za POZITIVNÍ a třetí provedení zkoušky není nutné. Podobně, pokud jsou první dvě provedení zkoušky negativní, považuje se předpověď h-CLAT za NEGATIVNÍ (při zohlednění ustanovení odstavce 30), aniž by bylo nutné opakovat zkoušku potřetí. Pokud však první dvě provedení zkoušky nejsou shodné alespoň u jednoho z markerů (CD54 nebo CD86), je třeba provést zkoušku potřetí, přičemž konečná předpověď bude vycházet z většinového výsledku těchto tří jednotlivých provedení zkoušky (tj. 2 ze 3). V této souvislosti je třeba poznamenat, že pokud se provedou první dvě nezávislé zkoušky a jedna je pozitivní pouze pro CD86 (dále jen P1) a druhá je pozitivní pouze pro CD54 (dále jen P2), je třeba provést zkoušku potřetí. Jestliže je toto třetí provedení zkoušky negativní pro oba markery (dále jen N), považuje se předpověď h-CLAT za NEGATIVNÍ. Jestliže je však toto třetí provedení zkoušky pozitivní pro jeden z markerů (P1 nebo P2) nebo pro oba markery (dále jen P12), považuje se předpověď h-CLAT za POZITIVNÍ.
Obrázek 1: Predikční model používaný při metodě h-CLAT. Předpověď h-CLAT by měla být zvažována v rámci přístupu IATA a v souladu s ustanovením odstavců 7 a 8 Všeobecného úvodu.
P1: provedení zkoušky pozitivní pouze u CD86; P2; provedení zkoušky pozitivní pouze u CD54; P12: provedení zkoušky pozitivní u CD86 i CD54; N: provedení zkoušky není pozitivní u CD86 ani CD54.
*Rámečky ukazují relevantní kombinace výsledků z prvních dvou provedení zkoušky, a to nezávisle na pořadí jejich získání.
#Rámečky ukazují relevantní kombinace výsledků ze tří provedení zkoušky na základě výsledků získaných v prvních dvou provedeních zkoušky znázorněných v rámečku výše, ale nevyjadřují pořadí jejich získání.
|
|
28.
|
Pro zkoušené chemické látky, které jsou na základě metody h-CLAT považovány za POZITIVNÍ, lze doplňkově stanovit dvě hodnoty účinné koncentrace (EC), EC150 pro CD86 a EC200 pro CD54, tj. koncentrace, při níž zkoušené chemické látky indukují RFI 150 nebo 200. Tyto hodnoty EC mohou však také potenciálně přispět k hodnocení senzibilizační potence (9), jsou-li používány v rámci integrovaných přístupů, například IATA (4) (5) (6) (7) (8). Vypočítají se pomocí této rovnice:
EC 150 (for CD86) = Bconc
+ [(150 - BRFI
)/ARFI - BRFI
) × (Aconc - Bconc
)]
EC 200 (for CD86) = Bconc
+ [(200 - BRFI
)/ARFI - BRFI
) × (Aconc - Bconc
)]
kde
Aconc je nejnižší koncentrace v μg/ml s RFI > 150 (CD86) nebo 200 (CD54)
Bconc je nejvyšší koncentrace v μg/ml s RFI < 150 (CD86) nebo 200 (CD54)
ARFI je RFI při nejnižší koncentraci s RFI > 150 (CD86) nebo 200 (CD54)
BRFI je RFI při nejvyšší koncentraci s RFI < 150 (CD86) nebo 200 (CD54)
Pro účely přesnějšího odvození hodnot EC150 a EC200 mohou být potřebná tři nezávislá provedení měření exprese CD86/CD54. Poté se stanoví konečné hodnoty EC150 a EC200 jako medián hodnot EC vypočtených ze tří nezávislých provedení zkoušky. Pokud kritéria pozitivity splňují pouze dvě ze tří nezávislých provedení zkoušky (viz odstavce 26-27), použije se vyšší ze dvou vypočtených hodnot EC150 nebo EC200.
|
Kritéria přijatelnosti
|
29.
|
Při používání metody h-CLAT by měla být splněna tato kritéria přijatelnosti (22) (27).
|
—
|
Životaschopnost buněk u kontroly s médiem a kontroly s rozpouštědlem/vehikulem by měla být vyšší než 90 %.
|
|
—
|
Při kontrole s rozpouštědlem/vehikulem by hodnoty RFI u CD86 i CD54 neměly překročit pozitivní kritéria (CD86 RFI ≥ 150 % a CD54 RFI ≥ 200 %). Hodnoty RFI kontroly s rozpouštědlem/vehikulem se vypočítají podle vzorce uvedeného v odstavci 25 („MFI chemické látky“ se nahradí „MFI rozpouštědla/vehikula“ a „MFI rozpouštědla/vehikula“ se nahradí „MFI kontroly (s médiem)“).
|
|
—
|
U kontroly s médiem i kontroly s rozpouštědlem/vehikulem by mělo být procento MFI u CD86 i CD54 > 105 % kontroly izotypu.
|
|
—
|
Při pozitivní kontrole (DNCB) by hodnoty RFI u CD86 i CD54 měly splňovat pozitivní kritéria (CD86 RFI ≥ 150 a CD54 RFI ≥ 200) a životaschopnost buněk by měla být větší než 50 %.
|
|
—
|
U zkoušené chemické látky by životaschopnost buněk měla být větší než 50 % alespoň u čtyř testovaných koncentrací při každém provedení zkoušky.
|
|
|
30.
|
Negativní výsledky jsou přijatelné pouze u zkoušených chemických látek, které vykazují životaschopnost buněk menší než 90 % při nejvyšší zkoušené koncentraci (tj. 1.2 × CV75 podle schématu sériového ředění popsaného v odstavci 20). Jestliže se životaschopnost buněk při 1,2 × CV75 rovná nebo je vyšší než 90 %, je třeba negativní výsledek vyřadit. V takovém případě se doporučuje pokusit se zpřesnit výběr dávky zopakováním stanovení hodnoty CV75. Je třeba poznamenat, že když se jako maximální experimentální koncentrace zkoušené chemické látky použije 5 000 μg/ml ve fyziologickém roztoku (nebo médiu či jiných rozpouštědlech/vehikulech), 1 000 μg/ml v DMSO nebo nejvyšší rozpustná koncentrace, je negativní výsledek přijatelný i v případě, že je životaschopnost buněk nad 90 %.
|
ZÁVĚREČNÁ ZPRÁVA
|
31.
|
Závěrečná zpráva by měla obsahovat tyto informace.
|
Zkoušená chemická látka
|
Jednosložková látka:
|
chemická identifikace, např. název (názvy) IUPAC nebo CAS, číslo (čísla) CAS, kód SMILES nebo InChI, strukturní vzorec a/nebo jiné identifikátory,
|
|
fyzický vzhled, log Kow, rozpustnost ve vodě, rozpustnost v DMSO, molekulová hmotnost a další důležité fyzikálně-chemické vlastnosti, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
čistota, případně chemická identita nečistot, je-li to prakticky proveditelné atd.,
|
|
případné ošetření před zkoušením (např. zahřívání, mletí),
|
|
podmínky skladování a stabilita, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
zdůvodnění volby rozpouštědla/vehikula pro každou zkoušenou chemickou látku.
|
|
|
Vícesložková látka, UVCB a směs:
|
charakterizovaná v co možná největší míře např. chemickou identitou (viz výše), čistotou, kvantitativním výskytem a důležitými fyzikálně-chemickými vlastnostmi (viz výše) složek, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
fyzický vzhled, rozpustnost ve vodě, rozpustnost v DMSO, a další důležité fyzikálně-chemické vlastnosti, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
molekulová hmotnost nebo zdánlivá molekulová hmotnost v případě směsí/polymerů se známým složením nebo jiné informace důležité pro provedení studie,
|
|
případné ošetření před zkoušením (např. zahřívání, mletí),
|
|
podmínky skladování a stabilita, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
zdůvodnění volby rozpouštědla/vehikula pro každou zkoušenou chemickou látku.
|
|
|
|
Kontroly
|
Pozitivní kontrola:
|
chemická identifikace, např. název (názvy) IUPAC nebo CAS, číslo (čísla) CAS, kód SMILES nebo InChI, strukturní vzorec a/nebo jiné identifikátory,
|
|
případně fyzický vzhled, log Kow, rozpustnost ve vodě, rozpustnost v DMSO, molekulová hmotnost a další důležité fyzikálně-chemické vlastnosti, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
čistota, případně chemická identita nečistot, je-li to prakticky proveditelné atd.,
|
|
případné ošetření před zkoušením (např. zahřívání, mletí),
|
|
podmínky skladování a stabilita, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
odkaz na případné dosavadní výsledky pozitivních kontrol prokazující vhodnost kritérií přijatelnosti provedení.
|
|
|
|
Negativní kontrola a kontrola rozpouštědlem/vehikulem:
|
chemická identifikace, např. název (názvy) IUPAC nebo CAS, číslo (čísla) CAS, kód SMILES nebo InChI, strukturní vzorec a/nebo jiné identifikátory,
|
|
čistota, případně chemická identita nečistot, je-li to prakticky proveditelné atd.,
|
|
fyzický vzhled, molekulová hmotnost a další důležité fyzikálně-chemické vlastnosti v případě použití jiného rozpouštědla/vehikula než rozpouštědel/vehikul uvedených v tomto pokynu, a to v rozsahu, v jakém jsou tyto informace dostupné,
|
|
podmínky skladování a stabilita, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
zdůvodnění volby rozpouštědla/vehikula pro každou zkoušenou chemickou látku.
|
|
|
Zkušební podmínky
|
jméno a adresa zadavatele, zkušebního zařízení a vedoucího studie,
|
|
použité buněčné linie, podmínky skladování a zdroj (např. zařízení, z něhož byly získány),
|
|
použitá průtoková cytometrie (např. model), včetně nastavení přístrojů, použitého globulinu a markeru cytotoxicity,
|
|
postup použitý pro prokázání způsobilosti laboratoře k provádění této zkoušky zkoušením vhodných látek a postup použitý pro prokázání reprodukovatelného provedení zkoušky v čase, např. dosavadní kontrolní údaje anebo dosavadní údaje o kontrolách reaktivity.
|
|
|
Kritéria přijatelnosti zkoušky
|
životaschopnost buněk, hodnoty MFI a RFI získané z kontroly s rozpouštědlem/vehikulem v porovnání s rozsahy přijatelnosti,
|
|
životaschopnost buněk a hodnoty RFI získané z pozitivní kontroly v porovnání s rozsahy přijatelnosti,
|
|
životaschopnost buněk všech testovaných koncentrací zkoušené chemické látky.
|
|
|
Zkušební postup
|
koncentrace zkoušených chemických látek, použité doby aplikace a expozice (pokud se liší od doporučených),
|
|
popis použitých hodnotících kritérií a kritérií rozhodování,
|
|
popis všech změn zkušebního postupu.
|
|
|
Výsledky
|
údaje ve formě tabulky, včetně CV75 (v relevantních případech), jednotlivých geometrických hodnot MFI, RFI, hodnot životaschopnosti buněk, hodnot EC150/EC200 (v relevantních případech) získaných pro zkoušenou chemickou látku a pro pozitivní kontrolu při každém provedení zkoušky a uvedení klasifikace zkoušené chemické látky podle predikčního modelu;
|
|
případně popis jakýchkoli jiných důležitých zjištění z pozorování.
|
|
|
Diskuse o výsledcích
|
diskuse o výsledcích získaných pomocí metody h-CLAT;
|
|
diskuse o výsledcích zkoušky v rámci IATA, jsou-li k dispozici jiné příslušné informace.
|
|
|
LITERATURA
|
(1)
|
Ashikaga T, Yoshida Y, Hirota M, Yoneyama K, Itagaki H, Sakaguchi H, Miyazawa M, Ito Y, Suzuki H, Toyoda H. (2006). Development of an in vitro skin sensitization test using human cell lines: The human Cell Line Activation Test (h-CLAT) I. Optimization of the h-CLAT protocol. Toxicol. Chem. In Vitro 20, 767-773.
|
|
(2)
|
Miyazawa M, Ito Y, Yoshida Y, Sakaguchi H, Suzuki H. (2007). Phenotypic alterations and cytokine production in THP-1 cells in response to allergens. Toxicol. Chem. In Vitro 21, 428-437.
|
|
(3)
|
EC EURL-ECVAM (2013). Recommendation on the human Cell Line Activation Test (h-CLAT) for skin sensitisation testing. Dostupné na: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(4)
|
Takenouchi O, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Hirota M, Ashikaga T, Miyazawa M. (2015). Test battery with the human cell line activation test, direct peptide reactivity assay and DEREK based on a 139 chemical data set for predicting skin sensitizing potential and potency of chemicals. J Appl Toxicol. 35, 1318-1332.
|
|
(5)
|
Hirota M, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Takenouchi O, Ashikaga T, Miyazawa M. (2015). Evaluation of combinations of in vitro sensitization test descriptors for the artificial neural network-based risk assessment model of skin sensitization. J Appl Toxicol. 35, 1333-1347.
|
|
(6)
|
Bauch C, Kolle SN, Ramirez T, Fabian E, Mehling A, Teubner W, van Ravenzwaay B, Landsiedel R. (2012). Putting the parts together: combining in vitro methods to test for skin sensitizing potencials. Regul Toxicol Parmacol. 63, 489-504.
|
|
(7)
|
Van der Veen JW, Rorije E, Emter R, Natch A, van Loveren H, Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol. 69, 371-379.
|
|
(8)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, Natsch A, Emter R, Ashikaga T, Miyazawa M, Sakaguchi H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Parmacol. 71, 337-351.
|
|
(9)
|
Jaworska JS, Natsch A, Ryan C, Strickland J, Ashikaga T, Miyazawa M. (2015). Bayesian integrated testing strategy (ITS) for skin sensitization potency assessment: a decision support system for quantitative weight of evidence and adaptive testing strategy. Arch Toxicol. 89, 2355-2383.
|
|
(10)
|
Strickland J, Zang Q, Kleinstreuer N, Paris M, Lehmann DM, Choksi N, Matheson J, Jacobs A, Lowit A, Allen D, Casey W. (2016). Integrated decision strategies for skin sensitization hazard. J Appl Toxicol. DOI 10.1002/jat.3281.
|
|
(11)
|
Nukada Y, Ashikaga T, Miyazawa M, Hirota M, Sakaguchi H, Sasa H, Nishiyama N. (2012). Prediction of skin sensitization potency of chemicals by human Cell Line Activation Test (h-CLAT) and an attempt at classifying skin sensitization potency. Toxicol. Chem. In Vitro 26, 1150-60.
|
|
(12)
|
EC EURL-ECVAM (2015). Re-analysis of the within and between laboratory reproducibility of the human Cell Line Activation Test (h-CLAT). Dostupné na: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations/eurl-ecvam-recommendation-on-the-human-cell-line-activation-test-h-clat-for-skin-sensitisation-testing
|
|
(13)
|
EC EURL ECVAM (2012). human Cell Line Activation Test (h-CLAT) Validation Study Report Accessible at: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(14)
|
Takenouchi O, Miyazawa M, Saito K, Ashikaga T, Sakaguchi H. (2013). Predictive performance of the human Cell Line Activation Test (h-CLAT) for lipophilic with high octanol-water partition coefficients. A Neuromuscular Screen for Use in Industrial Toxicology. Sci. 38, 599-609.
|
|
(15)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, NishiyamaN, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275-284.
|
|
(16)
|
Fabian E., Vogel D., Blatz V., Ramirez T., Kolle S., Eltze T., van Ravenzwaay B., Oesch F., Landsiedel R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch Toxicol 87, 1683-1969.
|
|
(17)
|
Okamoto K, Kato Y, Kosaka N, Mizuno M, Inaba H, Sono S, Ashikaga T, Nakamura T, Okamoto Y, Sakaguchi H, Kishi M, Kuwahara H, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (6th report): A study for evaluating oxidative hair dye sensitization potential using h-CLAT. AATEX 15, 81-88.
|
|
(18)
|
DB-ALM (INVITTOX) (2014). Protocol 158: human Cell Line Activation Test (h-CLAT), 23pp. Dostupné na: http://ecvam-dbalm.jrc.ec.europa.eu/
|
|
(19)
|
Mizuno M, Yoshida M, Kodama T, Kosaka N, Okamato K, Sono S, Yamada T, Hasegawa S, Ashikaga T, Kuwahara H, Sakaguchi H, Sato J, Ota N, Okamoto Y, Ohno Y. (2008). Effects of pre-culture conditions on the human Cell Line Activation Test (h-CLAT) results; Results of the 4th Japanese inter-laboratory study. AATEX 13, 70-82.
|
|
(20)
|
Sono S, Mizuno M, Kosaka N, Okamoto K, Kato Y, Inaba H, , Nakamura T, Kishi M, Kuwahara H, Sakaguchi H, Okamoto Y, Ashikaga T, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (7th report): Evaluation of volatile, poorly soluble fragrance materials. AATEX 15, 89-96.
|
|
(21)
|
OECD (2005). Guidance Document No 34 on The Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Series on Testing and Assessment. Organization for Economic Cooperation and Development, Paris, France, 2005, 96 pp.
|
|
(22)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Series on Testing and Assessment No. Series on Testing and Assessment No. 168. Dostupné na: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En
|
|
(23)
|
United Nations UN (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Fifth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. Dostupné na: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html
|
|
(24)
|
ECETOC (2003). Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals (Technical Report No 87).
|
|
(25)
|
Ashikaga T, Sakaguchi H, Okamoto K, Mizuno M, Sato J, Yamada T, Yoshida M, Ota N, Hasegawa S, Kodama T, Okamoto Y, Kuwahara H, Kosaka N, Sono S, Ohno Y. (2008). Assessment of the human Cell Line Activation Test (h-CLAT) for Skin Sensitization; Results of the First Japanese Inter-laboratory Study. AATEX 13, 27-35.
|
Dodatek 1.1
DEFINICE
Přesnost
: přesnost shody mezi výsledky zkoušky a přijatými referenčními hodnotami. Je to míra funkčnosti zkoušky a jeden z aspektů její relevantnosti. Tento pojem se často používá místo pojmu soulad a rozumí se jím podíl správných výsledků zkoušky (21).
AOP (mechanismy nežádoucích účinků)
: sled událostí od chemické struktury cílové chemické látky nebo skupiny podobných chemických látek přes molekulární iniciační událost po účinek in vivo, který je předmětem zájmu (22).
Chemická látka
: látka nebo směs.
CV75
: odhad koncentrace ukazující 75 % životaschopnost buněk.
EC150
: koncentrace ukazující hodnoty RFI 150 v expresi CD86.
EC200
: koncentrace ukazující hodnoty RFI 200 v expresi CD54.
Průtoková cytometrie
: cytometrická technika, při níž buňky suspendované v kapalině protékají jedna po druhé přes ohnisko excitačního světla, které je rozptýleno do vzorů charakteristických pro buňky a jejich komponenty; buňky jsou často označeny fluorescenčními markery, aby bylo světlo nejprve absorbováno a poté emitováno v pozměněných frekvencích.
Nebezpečnost
: základní charakteristika látky nebo situace, která má potenciál vyvolat nepříznivé účinky, jsou-li organismus, systém nebo (sub)populace vystaveny této látce.
IATA (jednotný přístup ke zkoušení a posuzování)
: strukturovaný přístup používaný pro zjišťování nebezpečnosti (potenciál), charakterizaci rizik (potence) a/nebo posouzení bezpečnosti (potenciál/potence a expozice) chemické látky nebo skupiny chemických látek, jenž strategicky integruje a váží všechny důležité údaje s cílem poskytnout informace pro přijetí regulačního rozhodnutí, které se týká potenciální nebezpečnosti a/nebo rizika a/nebo potřeby dalšího cíleného, a tudíž minimálního zkoušení.
Kontrola s médiem
: neošetřená replika, která obsahuje všechny složky zkušebního systému. Tento vzorek se zpracovává se vzorky zkoušené chemické látky a jinými kontrolními vzorky, aby se zjistilo, zda rozpouštědlo/vehikulum vzájemně reaguje se zkušebním systémem.
Směs
: směs nebo roztok složený ze dvou nebo více látek.
Jednosložková látka
: látka definovaná svým kvantitativním složením, v níž je přítomna jedna hlavní složka v míře alespoň 80 % hmot.
Vícesložková látka
: látka definovaná svým kvantitativním složením, v níž je přítomna více než jedna hlavní složka v koncentraci ≥ 10 % hmot. a < 80 % hmot. Vícesložková látka je výsledkem výrobního procesu. Rozdíl mezi směsí a vícesložkovou látkou spočívá v tom, že směs se získá smísením dvou nebo více látek bez chemické reakce. Vícesložková látka je výsledkem chemické reakce.
Pozitivní kontrola
: replika obsahující všechny složky zkušebního systému a látku, o níž je známo, že způsobuje pozitivní reakci. Aby se zajistilo, že proměnlivost v reakci na pozitivní kontrolu bude možné časem vyhodnotit, rozsah pozitivní reakce by neměl být nadměrný.
Prehapteny
: chemické látky, z nichž se abiotickou transformací staly senzibilizující látky.
Prohapteny
: chemické látky vyžadující enzymatickou aktivaci, aby vykazovaly potenciál senzibilizace kůže.
Relativní intenzita fluorescence (RFI)
: relativní hodnoty geometrického průměru intenzity fluorescence (MFI) v buňkách exponovaných chemické látce v porovnání s hodnotou MFI v buňkách exponovaných rozpouštědlu/vehikulu.
Relevantnost
: popis vztahu zkoušky k účinku, který je předmětem zájmu, a také toho, zda je zkouška smysluplná a použitelná pro konkrétní účel. Vyjadřuje, do jaké míry zkouška správně měří nebo predikuje biologický účinek, který je předmětem zájmu. Relevantnost zahrnuje také posouzení přesnosti (shody) zkoušky (21).
Spolehlivost
: měření rozsahu, v jakém může být zkouška časem reprodukovatelná v laboratořích a mezi laboratořemi, když se provádí s použitím stejného protokolu. Hodnotí se to výpočtem laboratorní a mezilaboratorní reprodukovatelnosti a laboratorní opakovatelnosti (21).
Provedení zkoušky
: jedna či více zkoušených chemických látek se zkouší současně s kontrolou s rozpouštědlem/vehikulem a s pozitivní kontrolou.
Citlivost
: podíl všech pozitivních/aktivních chemických látek, které jsou na základě zkoušky správně zatříděny. Je to měřítko přesnosti zkoušky, která dává kategorické výsledky, a je důležitým prvkem při hodnocení relevance zkoušky (21).
Barvicí pufr
: fosfátový pufr obsahující 0,1 % bovinního sérového albuminu.
Kontrola s rozpouštědlem/vehikulem
: neexponovaný vzorek obsahující všechny složky zkušebního systému s výjimkou zkoušené chemické látky, ale včetně rozpouštědla/vehikula, které se používá. Používá se ke stanovení účinku na pozadí u vzorků exponovaných zkoušené chemické látce, která je rozpuštěna nebo stabilně dispergována ve stejném rozpouštědle/vehikulu. Pokud se vzorek zkouší souběžnou kontrolou s médiem, ukazuje také, zda rozpouštědlo/vehikulum vzájemně reaguje se zkušebním systémem.
Specifičnost
: podíl všech negativních/neaktivních chemických látek, které jsou na základě zkušební metody správně zatříděny. Je to měřítko přesnosti zkušební metody, která dává kategorické výsledky, a je důležitým prvkem při hodnocení relevance zkušební metody (21).
Látka
: chemický prvek a jeho sloučeniny v přírodním stavu nebo získané výrobním postupem, včetně jakýchkoliv aditiv potřebných pro zachování stability výrobku a všech nečistot pocházejících z použitého postupu, ale s vyloučením všech rozpouštědel, která je možno oddělit bez ovlivnění stability dané látky nebo změny jejího složení.
Zkoušená chemická látka
: jakákoli látka nebo směs zkoušená touto zkušební metodou.
Globálně harmonizovaný systém klasifikace a označování chemických látek Organizace spojených národů (GHS OSN)
: systém navrhující klasifikaci chemických látek (látek a směsí) podle standardizovaných typů a úrovní fyzikálních, zdravotních a environmentálních nebezpečí a řešící příslušné komunikační prvky, jako jsou piktogramy, signální slova, výroky o nebezpečí, výroky o bezpečnostních opatřeních a bezpečnostní datové listy, které obsahují informace o jejich nežádoucích účincích s ohledem na ochranu lidí (včetně zaměstnavatelů, dělníků, přepravců, spotřebitelů a osob reagujících na havárie) a životního prostředí (23).
UVCB
: látky s neznámým nebo proměnlivým složením, komplexní reakční produkty nebo biologický materiál.
Validovaná zkouška
: zkouška, u níž se má za to, že je dostatečně relevantní a spolehlivá pro určitý účel, a která je založena na vědecky spolehlivých zásadách. Zkouška není nikdy validovaná v absolutním smyslu, ale pouze pro určitý konkrétní účel (21).
Dodatek 1.2
LÁTKY PRO PROKÁZÁNÍ ZPŮSOBILOSTI
Dříve než začnou laboratoře běžně používat zkoušku popsanou v tomto dodatku ke zkušební metodě B.71, měly by prokázat odbornou způsobilost správným získáním předpokládané předpovědi h-CLAT u 10 vhodných látek doporučených v tabulce 1 a získáním hodnot CV75, EC150 a EC200, které spadají do příslušného referenčního rozpětí, u nejméně 8 z 10 vhodných látek. Látky pro prokázání způsobilosti byly vybrány tak, aby představovaly rozpětí účinků na nebezpečí senzibilizace kůže. Dalšími kritérii výběru bylo, aby látky byly komerčně dostupné, aby k nim byly k dispozici kvalitní referenční údaje in vivo a také kvalitní údaje in vitro získané metodou h-CLAT. Pro metodu h-CLAT jsou také k dispozici zveřejněné referenční údaje (3) (14).
Tabulka 1
Doporučené látky pro prokázání odborné způsobilosti k používání metody h-CLAT
|
Látky pro prokázání způsobilosti
|
CASRN
|
Skupenství
|
Předpověď in vivo
(102)
|
CV75 Odkaz Rozsah v μg/ml (103)
|
Výsledky h-CLAT pro CD86 (referenční rozmezí EC150 v μg/ml) (103)
|
Výsledky h-CLAT pro CD54 (referenční rozmezí EC200 v μg/ml) (103)
|
|
2,4-dinitrochlorbenzen
|
97-00-7
|
pevná látka
|
senzibilizující (extrémně)
|
2–12
|
pozitivní (0,5–10)
|
pozitivní (0,5–15)
|
|
benzen-1,4-diamin
|
106-50-3
|
pevná látka
|
senzibilizující (silně)
|
5–95
|
pozitivní (< 40)
|
negativní (> 1,5) (104)
|
|
síran nikelnatý
|
10101-97-0
|
pevná látka
|
senzibilizující (středně silně)
|
30–500
|
pozitivní (< 100)
|
pozitivní (10–100)
|
|
2-merkaptbenzothiazol
|
149-30-4
|
pevná látka
|
senzibilizující (středně silně)
|
30–400
|
negativní (> 10) (104)
|
pozitivní (10–140)
|
|
R(+)-limonen
|
5989-27-5
|
kapalina
|
senzibilizující (slabě)
|
> 20
|
negativní (> 5) (104)
|
pozitivní (< 250)
|
|
imidazolidinyl urea
|
39236-46-9
|
pevná látka
|
senzibilizující (slabě)
|
25–100
|
pozitivní (20–90)
|
pozitivní (20–75)
|
|
propan-2-ol
|
67-63-0
|
kapalina
|
není senzibilizující
|
> 5 000
|
negativní (> 5 000 )
|
negativní (> 5 000 )
|
|
glycerol
|
56-81-5
|
kapalina
|
není senzibilizující
|
> 5 000
|
negativní (> 5 000 )
|
negativní (> 5 000 )
|
|
kyselina mléčná
|
50-21-5
|
kapalina
|
není senzibilizující
|
1 500 –5 000
|
negativní (> 5 000 )
|
negativní (> 5 000 )
|
|
kyselina 4-aminobenzoová
|
150-13-0
|
pevná látka
|
není senzibilizující
|
> 1 000
|
negativní (>1 000 )
|
negativní (> 1 000 )
|
|
Zkratky: CAS RN = registrační číslo CAS (Chemical Abstracts Service)
|
Dodatek 2
SENZIBILIZACE KŮŽE IN VITRO: ZKOUŠKA AKTIVACE BUNĚČNÉ LINIE U937 (U-SENS™)
VÝCHOZÍ ÚVAHY A OMEZENÍ
|
1.
|
Zkouška U-SENS™ kvantifikuje změnu exprese buněčného povrchového markeru spojenou s procesem aktivace monocytů a dendritických buněk (DC) (tj. CD86) v buněčné linii lidského histiocytického lymfomu U937 po expozici senzibilizujícím látkám (1). Naměřené úrovně exprese buněčného povrchového markeru CD86 v buněčné linii U937 se poté použijí jako podpůrný údaj pro rozlišení mezi látkami senzibilizujícími kůži a látkami, které kůži nesenzibilizují.
|
|
2.
|
Zkouška U-SENS™ byla hodnocena ve validační studii (2) koordinované společností L’Oreal a poté podrobena odborné revizi poradního výboru (ESAC) referenční laboratoře EU pro alternativy ke zkouškám na zvířatech (EURL ECVAM) (3). S ohledem na všechny dostupné důkazy a příspěvky od regulátorů a účastníků doporučil EURL ECVAM (4) zkoušku U-SENS™ pro použití v rámci přístupu IATA na podporu rozlišování mezi látkami senzibilizujícími kůži a látkami nesenzibilizujícími kůži za účelem klasifikace nebezpečnosti a označování. OECD ve svých pokynech k předkládání zpráv o strukturovaných přístupech k integraci a individuálním zdrojům informací používaným v rámci postupu IATA pro senzibilizaci kůže v současné době rozebírá řadu případových studií popisujících různé strategie zkoušení a predikční modely. Jeden z různých definovaných přístupů vychází z analýzy U-SENS (5). Příklady využití údajů U-SENS™ v kombinaci s jinými informacemi, včetně dosavadních údajů a stávajících platných údajů o humánním použití (6), jsou uvedeny také jinde v odborné literatuře (4) (5) (7).
|
|
3.
|
U zkoušky U-SENS™ se prokázalo, že ji lze přenést do laboratoří, které mají zkušenosti s technikami používajícími buněčné kultury a analýzou pomocí průtokové cytometrie. Míra reprodukovatelnosti předpovědí, které lze od této zkoušky očekávat, činí řádově 90 % v rámci laboratoří a 84 % mezi laboratořemi (8). Výsledky validační studie (8) a dalších zveřejněných studií (1) celkově ukazují, že přesnost při odlišení senzibilizujících látek (tj. kategorie 1 GHS OSN/CLP) od nesenzibilizujících látek činí 86 % (N=166) při citlivosti 91 % (118/129) a specifičnosti 65 % (24/37) ve srovnání s výsledky zkušební metody LLNA. V porovnání s výsledky při humánním použití je přesnost při odlišení senzibilizujících látek (tj. kategorie 1 GHS OSN/CLP) od nesenzibilizujících látek 77 % (N=101) při citlivosti 100 % (58/58) a specifičnosti 47 % (20/43). V porovnání s metodou LLNA se falešně negativní předpovědi u zkoušky U-SENS™ pravděpodobněji týkají chemických látek vykazujících nízkou až střední potenci způsobovat senzibilizaci kůže (tj. podkategorie 1B GHS OSN/CLP) než chemických látek vykazujících vysokou potenci způsobovat senzibilizaci kůže (tj. podkategorie 1A GHS OSN/CLP) (1) (8) (9). Celkově vzato tyto informace naznačují, že zkouška U-SENS™ může být užitečná a může přispět k zjištění nebezpečí senzibilizace kůže. Zde uvedené hodnoty přesnosti U-SENS™ jako samostatné zkoušky jsou však pouze orientační, neboť tato zkouška by měla být posuzována v kombinaci s jinými zdroji informací v rámci IATA a v souladu s ustanoveními odstavců 7 a 8 ve Všeobecném úvodu. Při hodnocení metod posuzování senzibilizace kůže bez použití zvířat by se dále mělo vést v patrnosti, že ani zkouška LLNA a ani jiné zkoušky na zvířatech nemusí plně odrážet situaci u lidí.
|
|
4.
|
Na základě údajů, které jsou v současné době k dispozici, bylo prokázáno, že zkouška U-SENS™ je použitelná u zkoušených chemických látek (včetně složek kosmetických přípravků, například konzervantů, povrchově aktivních látek, účinných látek, barviv) spadajících do nejrůznějších organických funkčních skupin, majících různé fyzikálně chemické vlastnosti, různou schopnost vyvolávat senzibilizaci kůže (podle stanovení ve studiích in vivo) a různé spektrum reakčních mechanismů, o nichž je známo, že souvisí se senzibilizací kůže (tj. Michaelův akceptor, vznik Schiffovy báze, agens acylového přenosu, nukleofilní bimolekulární substituce [SN2] nebo nukleofilní aromatická substituce [SNAr]) (1) (8) (9) (10). Metodu U-SENS™ lze použít ke zkoušení rozpustných chemických látek, které jsou rozpustné nebo které tvoří stabilní disperzi (tj. koloid nebo suspenzi, ve které se zkoušená chemická látka neusazuje ani se neodděluje od rozpouštědla/vehikula do různých fází) ve vhodném rozpouštědle/vehikulu (viz odstavec 13). Chemické látky ve skupině uvedené jako prehapteny (tj. látky aktivované oxidací) nebo prohapteny (tj. látky vyžadující enzymatickou aktivaci, například prostřednictvím enzymu P450) byly ve zkoušce U-SENS™ předpovězeny správně (1) (10). Látky narušující membránu mohou vést k falešně pozitivním výsledkům v důsledku nespecifického zvýšení exprese CD86, neboť 3 ze 7 falešně pozitivních výsledků u referenční klasifikace in vivose týkaly povrchově aktivních látek (1). Proto je třeba pozitivní výsledky u povrchově aktivních látek posuzovat opatrně, zatímco negativní výsledky u povrchových látek přesto mohou být použity na podporu identifikace zkoušené chemické látky jako látky senzibilizující kůži. Zkouškou U-SENS™ (1) mohou být hodnoceny fluorescenční zkoušené chemické látky, avšak silné fluorescenční zkoušené chemické látky vyzařující na stejné vlnové délce jako fluorescein-isothiokyanát (FITC) nebo jako propidiumiodid (PI) budou narušovat detekci pomocí průtokové cytometrie, a proto nemohou být správně hodnoceny pomocí konjugovaných protilátek FITC (potenciálně falešně negativní) nebo PI (životaschopnost neměřitelná). V takovém případě mohou být použity jiné protilátky značené fluorochromem, respektive jiné markery cytotoxicity, pokud lze prokázat, že poskytují podobné výsledky jako protilátky značené FITC nebo PI (viz odstavec 18), např. zkoušením vhodných látek uvedených v dodatku 2.2. Vzhledem k výše uvedenému by se měly pozitivní výsledky u povrchově aktivních látek a negativní výsledky u silně fluorescenčních zkoušených chemických látek interpretovat v kontextu stanovených omezení a společně s jinými zdroji informací v rámci přístupu IATA. V případech, kdy existují důkazy prokazující nepoužitelnost zkoušky U-SENS™ na jiné specifické kategorie zkoušených chemických látek, neměla by se tato metoda ke zkoušení těchto specifických kategorií používat.
|
|
5.
|
Jak bylo popsáno výše, zkouška U-SENS™ pomáhá rozlišovat mezi látkami senzibilizujícími kůži a látkami nesenzibilujícími kůži. Může však také potenciálně přispět k hodnocení senzibilizační potence, je-li používána v rámci integrovaných přístupů, například IATA. Pro určení toho, jak mohou výsledky U-SENS™ potenciálně přispět k posuzování potence, však bude nutný další výzkum, přednostně založený na údajích o účincích na člověka.
|
|
6.
|
Definice jsou uvedeny v dodatku 2.1.
|
PODSTATA ZKOUŠKY
|
7.
|
Zkouška U-SENS™ je analýza in vitro, která kvantifikuje změny exprese buněčného povrchového markeru CD86 na buněčné linii lidského histiocytického lymfomu, buňkách U937, po 45±3 hodinách expozice zkoušené chemické látce. Povrchový marker CD86 je jedním z typických markerů aktivace U937. O markeru CD86 je známo, že je kostimulační molekulou, která může napodobovat aktivaci monocytů, což hraje kritickou roli při primární aktivaci (primingu) T buněk. Změny exprese buněčného povrchového markeru CD86 se měří pomocí průtokové cytometrie po obarvení buněk obvykle protilátkami značenými fluorescein-isothiokyanátem (FITC). Rovněž se provádí paralelní měření cytotoxicity (např. pomocí PI), aby bylo možné posoudit, zda při sub-cytotoxických koncentracích dochází k up regulaci exprese buněčného povrchového markeru CD86. Stimulační index (S.I.) buněčného povrchového markeru CD86 v porovnání s kontrolou s rozpouštědlem/vehikulem se vypočítá a použije v predikčním modelu (viz odstavec 19) jako podpůrný údaj pro rozlišení mezi senzibilizujícími a nesenzibilizujícími látkami.
|
PROKÁZÁNÍ ZPŮSOBILOSTI
|
8.
|
Před rutinním používáním zkoušky popsané v tomto dodatku ke zkušební metodě B.71 by měly laboratoře prokázat odbornou způsobilost použitím 10 látek pro prokazování způsobilosti, které jsou uvedeny v dodatku 2.2, a to v souladu s osvědčenými postupy pro metody in vitro (11). Uživatelé zkoušky by si také měli vést historickou databázi údajů pocházejících ze zkoušek reaktivity (viz odstavec 11) a z pozitivních kontrol a kontrol s rozpouštědlem/vehikulem (viz odstavce 15–16) a používat tato data k potvrzení, že v jejich laboratoři je udržována reprodukovatelnost zkoušky v čase.
|
POSTUP
|
9.
|
Tato zkouška je založena na protokolu databázové služby pro alternativní metody k pokusům na zvířatech (DB-ALM) č. 183 (12) pro U-SENS™. Při zavádění a používání zkoušky U-SENS™ v laboratoři by měly být využívány standardní pracovní postupy. Na provádění zkoušky U-SENS™ může být využit automatizovaný systém, pokud lze prokázat, například otestováním vhodných látek uvedených v dodatku 2.2, že poskytuje podobné výsledky. Dále je uveden popis hlavních složek a postupů zkoušky U-SENS™.
|
Příprava buněk
|
10.
|
Zkouška U-SENS™ se používá s buněčnou linií lidského histiocytického lymfomu U937 (13). Buňky (klon CRL1593.2) by měly být pořízeny z kvalifikované buněčné banky, jako je například Americká sbírka typových kultur.
|
|
11.
|
Buňky U937 se kultivují při teplotě 37 °C ve zvlhčené atmosféře s 5 % CO2 v médiu RPMI-1 640 doplněném 10 % fetálního telecího séra (FCS), 2 mM L-glutaminu, 100 jednotkami/ml penicilinu a 100 μg/ml streptomycinu (kompletní médium). Buňky U937 se rutinně pasážují každé 2–3 dny v hustotě 1,5, respektive 3 × 105 buněk/ml. Hustota buněk by neměla přesahovat 2 × 106 buněk/ml a životaschopnost buněk měřená vyloučením trypanové modři by měla být ≥ 90 % (nepoužije se při první pasáži po rozmrazení). Než se použijí pro zkoušení, je třeba provedením kontroly reaktivity zjistit, zda jsou všechny šarže buněk, FCS nebo protilátek vhodné. Kontrola reaktivity buněk se provádí pomocí pozitivní kontroly, kyseliny picrylsulfonové (kyselina 2,4,6-trinitrobenzensulfonová: TNBS) (CASRN 2 508-19-2, čistota ≥ 99 %) a negativní kontrola pomocí kyseliny mléčné (LA) (CASRN 50-21-5, čistota ≥ 85 %), alespoň jeden týden po rozmrazení. Při kontrole reaktivity je třeba pro každou ze dvou kontrol otestovat šest konečných koncentrací (TNBS: 1, 12,5, 25, 50, 75, 100 μg/ml a LA: 1, 10, 20, 50, 100, 200 μg/ml). TNBS rozpuštěná v kompletním médiu by měla dát pozitivní a na koncentraci závislou odpověď CD86 (např. když po pozitivní koncentraci, stimulační index CD86 ≥ 150, následuje koncentrace s rostoucím stimulačním indexem CD86) a LA rozpuštěná v kompletním médiu by měla dát negativní odpověď CD86 (viz odstavec 21). Pro analýzu se používají pouze šarže buněk, které dvakrát prošly kontrolou reaktivity. Buňky mohou být množeny až sedm týdnů po rozmrazení. Počet pasáží by neměl překročit 21. Kontrola reaktivity se provádí podle postupů popsaných v odstavcích 18–22.
|
|
12.
|
Pro zkoušení se buňky U937 nasadí v hustotě 3 × 105 buněk/ml nebo 6 × 105 buněk/ml a předběžně se kultivují v kultivačních baňkách po dobu 2 dnů, respektive 1 dne. Mohou být použity jiné podmínky předběžné kultivace, než je uvedeno výše, pokud je uvedeno dostatečné vědecké zdůvodnění a lze prokázat, například otestováním vhodných látek uvedených v dodatku 2.2, že poskytují podobné výsledky. V den zkoušky se buňky získané z kultivační baňky resuspendují v čerstvém kultivačním médiu při hustotě 5 × 105 buněk/ml. Poté se buňky rozdělí do 96jamkové destičky s plochým dnem s 100 μl (konečná hustota buněk 0,5 × 105 buněk/jamku).
|
Příprava zkoušených chemických látek a kontrolních látek
|
13.
|
Před zkoušením se provede posouzení rozpustnosti. Za tímto účelem se zkoušené chemické látky rozpustí nebo stabilně dispergují při koncentraci 50 mg/ml v kompletním médiu jako rozpouštědle první volby nebo v dimethylsulfoxidu (DMSO, čistota ≥ 99 %) jako v rozpouštědle druhé volby, jestliže zkoušená chemická látka není rozpustná v kompletním médiu jako rozpouštědle/vehikulu. Pro zkoušení se zkoušená chemická látka rozpustí na konečnou koncentraci 0,4 mg/ml v kompletním médiu, jestliže je chemická látka rozpustná v tomto rozpouštědle/vehikulu. Pokud je chemická látka rozpustná pouze v DMSO, rozpustí se na koncentraci 50 mg/ml. Jestliže je podáno dostatečné vědecké zdůvodnění, mohou být použita jiná rozpouštědla/vehikula než výše uvedená. Je třeba vzít v úvahu stabilitu zkoušené chemické látky ve finálním rozpouštědle/vehikulu.
|
|
14.
|
Zkoušené chemické látky a kontrolní látky se připraví v den zkoušení. Protože se neprovádí analýza ke stanovení dávky, při prvním provedení zkoušky se testuje 6 konečných koncentrací (1, 10, 20, 50, 100 a 200 μg/ml) v odpovídajícím rozpouštědle/vehikulu, a to buď v kompletním médiu, nebo v 0,4 % DMSO v médiu. Pro další provedení zkoušky, od 0,4 mg/ml v kompletním médiu nebo od 50 mg/ml v DMSO, se s použitím odpovídajícího rozpouštědla/vehikula připraví alespoň 4 pracovní roztoky zkoušených chemických látek (tj. alespoň 4 koncentrace). Nakonec se pracovní roztoky použijí pro expozici přidáním stejného objemu buněčné suspenze U937 (viz odstavec 11 výše) do objemu pracovního roztoku na destičce, čímž se dosáhne dalšího dvojnásobného ředění (12). Koncentrace (alespoň 4 koncentrace) pro případné další provedení zkoušky se zvolí na základě jednotlivých výsledků všech předchozích provedení zkoušky (8). Použitelné konečné koncentrace jsou 1, 2, 3, 4, 5, 7.5, 10, 12.5, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 120, 140, 160, 180 a 200 μg/ml. Maximální konečná koncentrace je 200 μg/ml. V případě, že je pozorována pozitivní hodnota CD86 při 1 μg/ml, hodnotí se 0,1 μg/ml, aby byla zjištěna koncentrace zkoušené chemické látky, která nevyvolává CD86 nad pozitivní prahovou hodnotu. Pro každé provedení zkoušky se vypočítá EC150 (koncentrace, při níž chemická látka dosáhne 150 % pozitivní prahové hodnoty CD86, viz odstavec 19), jestliže je pozorována odpověď na pozitivní koncentraci CD86. Pokud zkoušená chemická látka indukuje pozitivní odpověď CD86, která nezávisí na koncentraci, nemusí být výpočet EC150 relevantní, jak je uvedeno v protokolu ke zkušební metodě U-SENS™ DB-ALM č. 183 (12). Pro každé provedení zkoušky se vypočítá CV70 (koncentrace, při níž chemická látka dosáhne 70 % prahové hodnoty cytotoxicity, viz odstavec 19) (12). Pro zkoumání účinku nárůstu koncentrace na odpověď CD86 se zvolí jakékoli koncentrace z použitelných koncentrací rovnoměrně rozdělené mezi EC150 (neboli nejvyšší negativní necytotoxickou koncentraci CD86) a CV70 (neboli nejvyšší povolenou koncentraci tj. 200 μg/ml). Zkouší se minimálně 4 koncentrace v každém provedení zkoušky, přičemž alespoň 2 koncentrace jsou společné s předchozím provedením (provedeními) zkoušky, a to za účelem porovnání.
|
|
15.
|
Jako kontrola s rozpouštědlem/vehikulem se u zkoušky U-SENS™ používá kompletní médium (pro zkoušené chemické látky rozpustné nebo stabilně dispergované v kompletním mediu) (viz odstavec 4) nebo 0,4 % DMSO v kompletním médiu (pro zkoušené chemické látky rozpustné nebo stabilně dispergované v DMSO).
|
|
16.
|
Pozitivní kontrolou používanou u zkoušky U-SENS™ je TNBS (viz odstavec 11) připravený v kompletním médiu. Jako pozitivní kontrola pro měření exprese CD86 se používá TNBS v jedné konečné koncentraci na destičce (50 μg/ml), což dává > 70 % životaschopnost buněk. Pro získání koncentrace TNBS na destičce 50 μg/ml se připraví 1 M (tj. 293 mg/ml) zásobního roztoku TNBS v kompletním médiu, který se dále naředí 2 930násobně s kompletním médiem na 100 μg/ml pracovního roztoku. Jako negativní kontrola se používá kyselina mléčná (LA, CAS 50-21-5) v koncentraci 200 μg/ml rozpuštěná v kompletním médiu (ze zásobního roztoku 0,4 mg/ml). Na každé destičce se při každém provedení zkoušky připraví tři replikáty neexponované kontroly v kompletním médiu, kontroly s rozpouštědlem/vehikulem, negativní kontroly a pozitivní kontroly (12). Lze použít i jiné vhodné pozitivní kontroly, pokud jsou k dispozici dosavadní údaje umožňující odvodit srovnatelná kritéria přijatelnosti daného provedení zkoušky. Kritéria přijatelnosti provedení zkoušky jsou stejná jako u zkoušené chemické látky (odstavec 12).
|
Aplikace zkoušených chemických látek a kontrolních látek
|
17.
|
Kontrola s rozpouštědlem / vehikulem nebo pracovní roztoky popsané v odstavcích 14–16 se smíchají 1: 1 (obj.) s buněčnými suspenzemi připravenými ve 96jamkové destičce s plochým dnem (viz odstavec 12). Exponované destičky se poté inkubují po dobu 45±3 hodin při teplotě 37 °C za přítomnosti 5 % CO2. Před inkubací se destičky zapečetí polopropustnou membránou, aby nedošlo k odpaření těkavých zkoušených chemických látek a křížové kontaminaci mezi buňkami exponovanými zkoušeným chemickým látkám (12).
|
Barvení buněk
|
18.
|
Po 45 ± 3 hodinách expozice se buňky přenesou na mikrotitrační destičku s kónickým dnem a odeberou se odstředěním. Narušení rozpustnosti je definováno jako krystaly nebo kapky pozorované pod mikroskopem 45 ± 3 hodiny po expozici (před obarvením buněk). Supernatanty se zlikvidují a zbývající buňky se jednou promyjí se 100 μl ledově studeného fosfátového pufru (PBS) obsahujícího 5 % fetálního telecího séra (barvicí pufr). Po odstředění se buňky resuspendují se 100 μl barvicího pufru a barví se 5 μl (např. 0,25 μg) protilátek anti-CD86 nebo myšího IgG1 (izotyp) značených FITC při 4 °C po dobu 30 minut chráněny před světlem. Použijí se protilátky uvedené v protokolu ke zkušební metodě U-SENS™ DB-ALM č. 183 (12) (pro CD86: BD-PharMingen #5556 57 klon: Fun-1, nebo Caltag/Invitrogen # MHCD8601 klon: BU63; a pro IgG1: BD-PharMingen #5557 48, nebo Caltag/Invitrogen # GM4992). Podle zkušeností vývojářů zkoušek je intenzita fluorescence protilátek obvykle u různých šarží konzistentní. Pro analýzu mohou být použity jiné klony nebo protilátky od jiných dodavatelů, které prošly kontrolou reaktivity (viz odstavec 11). Uživatelé však mohou zvážit titraci protilátek v podmínkách své vlastní laboratoře, a definovat si tak nejlepší koncentrace pro použití. Může být použit jiný detekční systém, např. protilátky anti-CD86 značené fluorochromem, pokud lze prokázat, například otestováním vhodných látek uvedených v dodatku 2.2, že dávají podobné výsledky jako konjugované protilátky značené FITC. Po dvojím promytí 100 μl barvicího pufru a jednom promytí 100 μl ledově studeného PBS se buňky resuspendují v ledově studeném PBS (např. 125 μl pro vzorky analyzované ručně zkumavku po zkumavce nebo 50 μl s použitím automatické vzorkovací destičky) a přidá se roztok PI (konečná koncentrace 3 μg/ml). Mohou být použity jiné markery cytotoxicity, jako je 7-aminoaktinomycin D (7-AAD) nebo trypanová modř, pokud lze prokázat, například otestováním vhodných látek uvedených v dodatku 2.2, že alternativní barviva dávají podobné výsledky jako PI.
|
Analýza pomocí průtokové cytometrie
|
19.
|
Úroveň exprese CD86 a životaschopnost buněk se analyzuje pomocí průtokové cytometrie. Buňky jsou zobrazeny na dot plot histogramu vyjadřujícím velikost (FSC) a granularitu (SSC) nastaveném na logaritmickou stupnici, aby byla jasně identifikována populace v první oblasti R1 a eliminovány zbytky. Pro každou jamku se připraví cílový počet celkem 10 000 buněk v oblasti R1. Buňky ze stejné oblasti R1 se zobrazí na dot plot diagramu FL3 nebo FL4 / SSC. Životaschopné buňky se vykreslí umístěním druhé oblasti R2 selektující populaci propidiumiodid negativních buněk (kanál FL3 nebo FL4). Životaschopnost buněk lze vypočítat podle následující rovnice programem cytometrické analýzy. Když je životaschopnost buněk nízká, lze připravit až 20 000 buněk včetně odumřelých. Alternativně lze získat údaje za jednu minutu po zahájení analýzy.
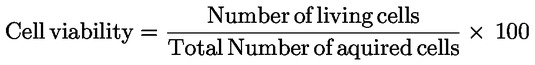
Poté se změří procento FL1 pozitivních buněk z těchto životaschopných buněk gateovaných na R2 (v oblasti R1). Exprese CD86 na buněčném povrchu se analyzuje na dot plot histogramu FL1 / SSC gatovaném na životaschopných buňkách (R2).
U jamek s kompletním médiem / IgG1 se marker analýzy stanoví v blízkosti hlavní populace, aby měly kontroly s kompletním médiem IgG1 v cílové zóně 0,6 až 0,9 %.
Barevná interference se definuje jako posun IgG1 značeného FITC na dot plot diagramu (geometrický průměr stimulačního indexu IgG1 FL1 ≥ 150 %).
Stimulační index (S.I.) CD86 pro kontrolní buňky (neexponované nebo v 0,4 % DMSO) a buňky exponované chemické látce se vypočítá podle této rovnice:.
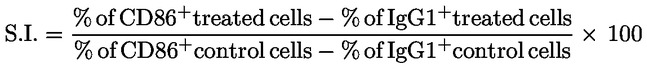
% IgG1+ neexponovaných kontrolních buněk: uvedené jako procento FL1 pozitivních buněk IgG1 definovaných pomocí markeru analýzy (přijatelné rozmezí ≥ 0,6 % a < 1,5 %, viz odstavec 22) z životaschopných neexponovaných buněk.
% IgG1+/CD86+ kontrolních/exponovaných buněk: uvedené jako procento FL1 pozitivních buněk IgG1/CD86 měřeno bez pohybu markeru analýzy z životaschopných kontrolních/exponovaných buněk.
|
ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV
Vyhodnocení údajů
|
20.
|
Ve zkoušce U-SENS™ se vypočítávají tyto parametry: hodnota CV70, tj. koncentrace vykazující 70 % přežití buněk U937 (30 % cytotoxicita) a hodnota EC150, tj. koncentrace, při níž zkoušené chemické látky indukovaly stimulační index (S.I.) CD86 150 %.
CV70 se vypočítá log-lineární interpolací podle této rovnice:
CV70 = C1 + [(V1 - 70) / (V1 – V2) * (C2 – C1)]
kde:
V1 je minimální hodnota životaschopnosti buněk nad 70 %
V2 je maximální hodnota životaschopnosti buněk pod 70 %
C1 a C2 jsou koncentrace ukazující hodnotu životaschopnosti buněk V1, respektive V2.
Lze použít i jiné způsoby odvození hodnoty CV70, pokud je prokázáno, že to nemá vliv na výsledky (např. otestováním vhodných látek uvedených).
EC150 se vypočítá log-lineární interpolací podle této rovnice:
EC150 = C1 + [(150 – S.I.1) / (S.I.2 – S.I.1) * (C2 – C1)]
kde:
C1 je nejvyšší koncentrace v μg/ml se stimulačním indexem CD86 < 150 % (S.I. 1)
C2 je nejnižší koncentrace v μg/ml se stimulačním indexem CD86 > 150 % (S.I. 2).
Hodnoty EC150 a CV70 se vypočítají
|
—
|
pro každé provedení zkoušky: jednotlivé hodnoty EC150 a CV70 se použijí jako nástroj pro zkoumání účinku nárůstu koncentrace na odpověď CD86 (viz odstavec 14),
|
|
—
|
na základě průměrné životaschopnosti se stanoví celková hodnota CV70 (12),
|
|
—
|
na základě průměrných hodnot stimulačního indexu CD86 se stanoví celková hodnota EC150 pro zkoušenou chemickou látku predikovanou při použití metody U-SENS™ jako POZITIVNÍ (viz odstavec 21) (12).
|
|
Predikční model
|
21.
|
Pro měření exprese CD86 se každá zkoušená chemická látka otestuje alespoň ve čtyřech nezávislých provedeních zkoušky (v různé dny), z nichž se odvodí jedna předpověď (NEGATIVNÍ nebo POZITIVNÍ).
|
—
|
Jednotlivé provedení zkoušky U-SENS™ se považuje za negativní (dále jen N), jestliže je stimulační index CD86 menší než 150 % při všech necytotoxických koncentracích (životaschopnost buněk ≥ 70 %) a jestliže není pozorována žádná interference (cytotoxicita, rozpustnost: viz odstavec 18 nebo barva: viz odstavec 19 bez ohledu na necytotoxické koncentrace, při nichž byla detekována interference). Ve všech ostatních případech: stimulační index CD86 vyšší než nebo rovný 150 % a pozorovány interference, je jednotlivé provedení zkoušky U-SENS™ považováno za pozitivní (dále jen P).
|
|
—
|
Predikce na základě zkoušky U-SENS™ je považována za NEGATIVNÍ, jestliže jsou negativní (N) alespoň dvě nezávislá provedení zkoušky (obrázek 1). Pokud jsou první dvě provedení zkoušky negativní (N), považuje se předpověď získaná zkouškou U-SENS™ za NEGATIVNÍ a třetí provedení zkoušky není nutné.
|
|
—
|
Predikce na základě zkoušky U-SENS™ je považována za POZITIVNÍ, jestliže jsou pozitivní (P) alespoň dvě nezávislá provedení zkoušky (obrázek 1). Pokud jsou první dvě provedení zkoušky pozitivní (P), považuje se předpověď získaná zkouškou U-SENS™ za POZITIVNÍ a třetí provedení zkoušky není nutné.
|
|
—
|
Protože se neprovádí analýza ke stanovení dávky, je zde výjimka, pokud je při prvním provedení zkoušky stimulační index CD86 vyšší než nebo rovný 150 % pouze při nejvyšší necytotoxické koncentraci. Toto provedení zkoušky se poté považuje za NEPRŮKAZNÉ (NC) a v dalších provedeních je třeba testovat další koncentrace (mezi nejvyšší necytotoxickou koncentrací a nejnižší cytotoxickou koncentrací – viz odstavec 20). Pokud je provedení zkoušky určeno jako NC, je třeba provést alespoň 2 další zkoušky a v případě, že druhá a třetí zkouška nesouhlasí (N a/nebo P nezávisle), provádí se čtvrtá zkouška (obrázek 1). Další zkoušky budou považovány za pozitivní i v případě, že CD86 vyjde 150 % nebo vyšší pouze u jedné necytotoxické koncentrace, neboť nastavení koncentrace bylo pro tuto konkrétní zkoušenou chemickou látku upraveno. Konečná predikce bude vycházet z většinového výsledku tří nebo čtyř jednotlivých provedení zkoušky (tj. 2 ze 3 nebo 2 ze 4) (obrázek 1).
|
Obrázek 1: Predikční model používaný při zkoušce U-SENS™. Předpověď U-SENS™ by měla být hodnocena v rámci přístupu IATA a v souladu s ustanoveními odstavce 4 a odstavců 7, 8 a 9 Obecného úvodu.
N: Provedení zkoušky bez pozitivního výsledku CD86 a bez pozorované interference;
P: Provedení zkoušky s pozitivním výsledkem CD86 a/nebo s pozorovanou interferencí (interferencemi);
NC: Neprůkazný výsledek. První provedení s neprůkazným výsledkem, když je CD86 pozitivní pouze při nejvyšší necytotoxické koncentraci;
#: Neprůkazný (NC) jednotlivý výsledek zjištěný pouze při prvním provedení zkoušky vede automaticky k nutnosti třetího provedení zkoušky, aby byla dosažena většina pozitivních (P) nebo negativních (N) výsledků při alespoň 2 ze 3 nezávislých provedení zkoušky.
$: Rámečky ukazují relevantní kombinace výsledků ze tří provedení zkoušky na základě výsledků získaných v prvních dvou provedeních zkoušky znázorněných v rámečku výše.
°: Rámečky ukazují relevantní kombinace výsledků ze čtyř provedení zkoušky na základě výsledků získaných v prvních třech provedeních zkoušky znázorněných v rámečku výše.
|
Kritéria přijatelnosti
|
22.
|
Při používání zkoušky U-SENS™ by měla být splněna tato kritéria přijatelnosti (12).
|
—
|
Na konci expozice v délce 45±3 hodin musí být střední životaschopnost trojích neexponovaných buněk U937 > 90 % a nesmí být pozorován žádný posun exprese CD86. Bazální exprese CD86 neexponovaných buněk U937 musí být v rozmezí ≥ 2 % a ≤ 25 %.
|
|
—
|
Když je jako rozpouštědlo použit DMSO, posuzuje se platnost kontroly s vehikulem DMSO pomocí výpočtu stimulačního indexu DMSO v porovnání s neexponovanými buňkami, přičemž střední životaschopnost trojích buněk musí být > 90 %. Kontrola s vehikulem je platná, když je střední hodnota trojího stimulačního indexu CD86 menší než 250 % střední hodnoty trojího stimulačního indexu CD86 neexponovaných buněk U937.
|
|
—
|
Provedení zkoušky se považují za platná, jestliže alespoň dvě ze tří hodnot IgG1 neexponovaných buněk U937 spadají do rozmezí ≥ 0,6 % a < 1,5 %.
|
|
—
|
Souběžně prováděná negativní kontrola (kyselina mléčná) se považuje za platnou, jestliže alespoň dva ze tří replikátů byly negativní (CD86 S.I. < 150 %) a necytotoxické (životaschopnost buněk ≥ 70 %).
|
|
—
|
Pozitivní kontrola (TNBS) se považuje za platnou, jestliže alespoň dva ze tří replikátů byly pozitivní (CD86 S.I. ≥150 %) a necytotoxické (životaschopnost buněk ≥ 70 %).
|
|
ZÁVĚREČNÁ ZPRÁVA
|
23.
|
Závěrečná zpráva by měla obsahovat tyto informace.
|
Zkoušená chemická látka
Jednosložková látka:
|
chemická identifikace, např. název (názvy) IUPAC nebo CAS, číslo (čísla) CAS, kód SMILES nebo InChI, strukturní vzorec a/nebo jiné identifikátory,
|
|
fyzický vzhled, úplná rozpustnost v médiu, rozpustnost v DMSO, molekulová hmotnost a další důležité fyzikálně-chemické vlastnosti, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
čistota, případně chemická identita nečistot, je-li to prakticky proveditelné atd.,
|
|
případné ošetření před zkoušením (např. zahřívání, mletí),
|
|
podmínky skladování a stabilita, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
zdůvodnění volby rozpouštědla/vehikula pro každou zkoušenou chemickou látku.
|
Vícesložková látka, UVCB a směs:
|
charakterizovaná v co možná největší míře např. chemickou identitou (viz výše), čistotou, kvantitativním výskytem a důležitými fyzikálně-chemickými vlastnostmi (viz výše) složek, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
fyzický vzhled, rozpustnost v kompletním médiu, rozpustnost v DMSO, a další důležité fyzikálně-chemické vlastnosti, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
molekulová hmotnost nebo zdánlivá molekulová hmotnost v případě směsí/polymerů se známým složením nebo jiné informace důležité pro provedení studie;,
|
|
případné ošetření před zkoušením (např. zahřívání, mletí),
|
|
podmínky skladování a stabilita, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
zdůvodnění volby rozpouštědla/vehikula pro každou zkoušenou chemickou látku.
|
|
|
Kontroly
Pozitivní kontrola:
|
chemická identifikace, např. název (názvy) IUPAC nebo CAS, číslo (čísla) CAS, kód SMILES nebo InChI, strukturní vzorec a/nebo jiné identifikátory,
|
|
fyzický vzhled, rozpustnost v DMSO, molekulová hmotnost a další důležité fyzikálně-chemické vlastnosti, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
čistota, případně chemická identita nečistot, je-li to prakticky proveditelné atd.,
|
|
případné ošetření před zkoušením (např. zahřívání, mletí),
|
|
podmínky skladování a stabilita, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
odkaz na případné dosavadní výsledky pozitivních kontrol prokazující vhodnost kritérií přijatelnosti provedení.
|
Negativní kontrola a kontrola rozpouštědlem/vehikulem:
|
chemická identifikace, např. název (názvy) IUPAC nebo CAS, číslo (čísla) CAS, kód SMILES nebo InChI, strukturní vzorec a/nebo jiné identifikátory,
|
|
čistota, případně chemická identita nečistot, je-li to prakticky proveditelné atd.,
|
|
fyzický vzhled, molekulová hmotnost a další důležité fyzikálně-chemické vlastnosti v případě použití jiného rozpouštědla/vehikula než rozpouštědel/vehikul uvedených v tomto pokynu, a to v rozsahu, v jakém jsou tyto informace dostupné,
|
|
podmínky skladování a stabilita v rozsahu, v jakém jsou tyto informace dostupné,
|
|
zdůvodnění volby rozpouštědla/vehikula pro každou zkoušenou chemickou látku.
|
|
|
Zkušební podmínky
|
jméno a adresa zadavatele, zkušebního zařízení a vedoucího studie,
|
|
použité buněčné linie, podmínky skladování a zdroj (např. zařízení, z něhož byly získány),
|
|
použitá průtoková cytometrie (např. model), včetně nastavení přístrojů, použitého globulinu a markeru cytotoxicity,
|
|
postup použitý pro prokázání způsobilosti laboratoře k provádění této zkoušky zkoušením vhodných látek a postup použitý pro prokázání reprodukovatelného provedení zkoušky v čase, např. dosavadní kontrolní údaje anebo dosavadní údaje o kontrolách reaktivity.
|
|
|
Kritéria přijatelnosti zkoušky
|
životaschopnost buněk a hodnoty stimulačního indexu CD86 získané z kontroly s rozpouštědlem/vehikulem v porovnání s rozsahy přijatelnosti,
|
|
životaschopnost buněk a hodnoty stimulačního indexu získané z pozitivní kontroly v porovnání s rozsahy přijatelnosti,
|
|
životaschopnost buněk všech testovaných koncentrací zkoušené chemické látky.
|
|
|
Zkušební postup
|
koncentrace zkoušených chemických látek, použité doby aplikace a expozice (pokud se liší od doporučených),
|
|
popis použitých hodnotících kritérií a kritérií rozhodování,
|
|
popis jakýchkoli změn zkušebního postupu.
|
|
|
Výsledky
|
údaje ve formě tabulky, včetně CV70 (v relevantních případech), stimulačního indexu, hodnot životaschopnosti buněk, hodnot EC150 (v relevantních případech) získaných pro zkoušenou chemickou látku a pro pozitivní kontrolu při každém provedení zkoušky a uvedení klasifikace zkoušené chemické látky podle predikčního modelu,
|
|
případně popis jakýchkoli jiných důležitých zjištění z pozorování.
|
|
|
Diskuse o výsledcích
|
diskuse o výsledcích získaných za pomoci zkoušky U-SENS™,
|
|
diskuse o výsledcích zkoušky v rámci IATA, jsou-li k dispozici jiné příslušné informace.
|
|
Závěry
|
LITERATURA
|
(1)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. Chem. In Vitro 29, 901-916.
|
|
(2)
|
EURL ECVAM (2017). The U-SENS™ test method Validation Study Report. Dostupné na: http://ihcp.jrc.ec.europa.eu/our_labs/eurl-ecvam/eurl-ecvam-recommendations
|
|
(3)
|
EC EURL-ECVAM (2016). ESAC Opinion No 2016-03 on the L'Oréal-coordinated study on the transferability and reliability of the U-SENS™ test method for skin sensitisation testing. EUR 28 178 EN; doi 10.2 787/8157 37. Dostupné na: [http://publications.jrc.ec.europa.eu/repository/handle/JRC103705].
|
|
(4)
|
EC EURL-ECVAM (2017). EURL ECVAM Recommendation on the use of non-animal approaches for skin sensitisation testing. EUR 28 553 EN; doi 10.2 760/5889 55. Dostupné na: https://ec.europa.eu/jrc/en/publication/eur-scientific-and-technical-research-reports/eurl-ecvam-recommendation-use-non-animal-approaches-skin-sensitisation-testing.
|
|
(5)
|
Steiling, W. (2016). Safety Evaluation of Cosmetic Ingredients Regarding their Skin Sensitization Potential. doi:10.3390/cosmetics3020014.Cosmetics 3, 14.
|
|
(6)
|
OECD (2016). Guidance Document on The Reporting of Defined Approaches and Individual Information Sources to be Used Within Integrated Approaches to Testing and Assessment (IATA) For Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Organisation for Economic Cooperation and Development, Paris. Dostupné na webových stránkách: [http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm].
|
|
(7)
|
Urbisch, D., Mehling, A., Guth, K., Ramirez, T., Honarvar, N., Kolle, S., Landsiedel, R., Jaworska, J., Kern, P.S., Gerberick, F., Natsch, A., Emter, R., Ashikaga, T., Miyazawa, M., Sakaguchi, H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul. Toxicol. Chem. Acute Exposure of the Neonatal Rat to Triethyltin Results in Persistent Changes in Neurotypic and Gliotypic Proteins. 71, 337-351.
|
|
(8)
|
Alépée, N., Piroird, C., Aujoulat, M., Dreyfuss, S., Hoffmann, S., Hohenstein, A., Meloni, M., Nardelli, L., Gerbeix, C., Cotovio, J. (2015). Prospective multicentre study of the U-SENS test method for skin sensitization testing. Toxicol In Vitro 30, 373-382.
|
|
(9)
|
Reisinger, K., Hoffmann, S., Alépée, N., Ashikaga, T., Barroso, J., Elcombe, C., Gellatly, N., Galbiati, V., Gibbs, S., Groux, H., Hibatallah, J., Keller, D., Kern, P., Klaric, M., Kolle, S., Kuehnl, J., Lambrechts, N., Lindstedt, M., Millet, M., Martinozzi-Teissier, S., Natsch, A., Petersohn, D., Pike, I., Sakaguchi, H., Schepky, A., Tailhardat, M., Templier, M., van Vliet, E., Maxwell, G. (2014). Systematic evaluation of non-animal test methods for skin sensitisation safety assessment. Toxicol. In Vitro 29, 259-270.
|
|
(10)
|
Fabian, E., Vogel, D., Blatz, V., Ramirez, T., Kolle, S., Eltze, T., van Ravenzwaay, B., Oesch, F., Landsiedel, R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch. Toxicol. 87, 1 683-1 696.
|
|
(11)
|
OECD. (2018). Draft Guidance document: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Organisation for Economic Cooperation and Development, Paris. Dostupné na webových stránkách: http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(12)
|
DB-ALM (2016). Protocol no 183: Myeloid U937 Skin Sensitization Test (U-SENS™), 33pp. Dostupné na: [http://ecvam-dbalm.jrc.ec.europa.eu/].
|
|
(13)
|
Sundström, C., Nilsson, K. (1976). Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int. J. Cancer 17, 565-577.
|
|
(14)
|
OECD (2005). Series on Testing and Assessment No. 34: Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Organisation for Economic Cooperation and Development, Paris. Dostupné na webových stránkách: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(15)
|
United Nations UN (2015). Globálně harmonizovaný systém klasifikace a označování chemických látek (GHC). ST/SG/AC.10/30/Rev.6, Sixth Revised Edition, New York & Geneva: United Nations Publications. Dostupné na: http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(16)
|
OECD (2012). Series on Testing and Assessment No 168: The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Series on Testing and Assessment No. Organisation for Economic Cooperation and Development, Paris. Dostupné na webových stránkách: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(17)
|
ECETOC (2003). Technical Report No 87: Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals, Brussels. Dostupné na: https://ftp.cdc.gov/pub/Documents/OEL/06.%20Dotson/References/ECETOC_2003-TR87.pdf.
|
Dodatek 2.1
DEFINICE
Přesnost
: přesnost shody mezi výsledky zkoušky a přijatými referenčními hodnotami. Je to míra funkčnosti zkoušky a jeden z aspektů její relevantnosti. Tento výraz se často používá namísto výrazu soulad a rozumí se jím podíl správných výsledků zkoušky (14).
AOP (mechanismy nežádoucích účinků)
: sled událostí od chemické struktury cílové chemické látky nebo skupiny podobných chemických látek přes molekulární iniciační událost po účinek in vivo, který je předmětem zájmu (15).
Odpověď na koncentraci CD86
: k závislosti na koncentraci (neboli odpovědi na koncentraci) dochází, když po pozitivní koncentraci (CD86 S.I. ≥ 150) následuje koncentrace se zvyšujícím se stimulačním indexem CD86.
Chemická látka
: látka nebo směs.
CV70
: odhad koncentrace ukazující 70 % životaschopnost buněk.
Posun
: posun je definován tak, že i) korigovaná hodnota % CD86+ neexponovaného kontrolního replikátu 3 je menší než 50 % střední hodnoty korigované hodnoty % CD86+ neexponovaných kontrolních replikátů 1 a 2 a ii) korigovaná hodnota % CD86+ negativního kontrolního replikátu 3 je menší než 50 % střední hodnoty korigované hodnoty % CD86+ negativních kontrolních replikátů 1 a 2.
EC150
: odhad koncentrací vykazujících 150 % stimulační index exprese CD86.
Průtoková cytometrie
: cytometrická technika, při níž buňky suspendované v kapalině protékají jedna po druhé přes ohnisko excitačního světla, které je rozptýleno do vzorů charakteristických pro buňky a jejich komponenty; buňky jsou často označeny fluorescenčními markery, aby bylo světlo nejprve absorbováno a poté emitováno v pozměněných frekvencích.
Nebezpečnost
: základní charakteristika látky nebo situace, která má potenciál vyvolat nepříznivé účinky, jsou-li organismus, systém nebo (sub)populace vystaveny této látce.
IATA (jednotný přístup ke zkoušení a posuzování)
: strukturovaný přístup používaný pro zjišťování nebezpečnosti (potenciál), charakterizaci rizik (potence) a/nebo posouzení bezpečnosti (potenciál/potence a expozice) chemické látky nebo skupiny chemických látek, jenž strategicky integruje a váží všechny důležité údaje s cílem poskytnout informace pro přijetí regulačního rozhodnutí, které se týká potenciální nebezpečnosti a/nebo rizika a/nebo potřeby dalšího cíleného, a tudíž minimálního zkoušení.
Směs
: směs nebo roztok složený ze dvou nebo více látek.
Jednosložková látka
: látka definovaná svým kvantitativním složením, v níž je přítomna jedna hlavní složka v míře alespoň 80 % hmot.
Vícesložková látka
: látka definovaná svým kvantitativním složením, v níž je přítomna více než jedna hlavní složka v koncentraci ≥ 10 % hmot. a < 80 % hmot. Vícesložková látka je výsledkem výrobního procesu. Rozdíl mezi směsí a vícesložkovou látkou spočívá v tom, že směs se získá smísením dvou nebo více látek bez chemické reakce. Vícesložková látka je výsledkem chemické reakce.
Pozitivní kontrola
: replika obsahující všechny složky zkušebního systému a látku, o níž je známo, že způsobuje pozitivní reakci. Aby se zajistilo, že proměnlivost v reakci na pozitivní kontrolu bude možné časem vyhodnotit, rozsah pozitivní reakce by neměl být nadměrný.
Prehapteny
: chemické látky, z nichž se abiotickou transformací, např. oxidací, staly senzibilizující látky.
Prohapteny
: chemické látky vyžadující enzymatickou aktivaci, aby vykazovaly potenciál senzibilizace kůže.
Relevantnost
: popis vztahu zkoušky k účinku, který je předmětem zájmu, a také toho, zda je zkouška smysluplná a použitelná pro konkrétní účel. Vyjadřuje, do jaké míry zkouška správně měří nebo predikuje biologický účinek, který je předmětem zájmu. Relevantnost zahrnuje také posouzení přesnosti (shody) zkoušky (14).
Spolehlivost
: měření rozsahu, v jakém může být zkouška časem reprodukovatelná v laboratořích a mezi laboratořemi, když se provádí s použitím stejného protokolu. Hodnotí se to výpočtem laboratorní a mezilaboratorní reprodukovatelnosti a laboratorní opakovatelnosti (14).
Provedení zkoušky
: jedna či více zkoušených chemických látek se zkouší současně s kontrolou s rozpouštědlem/vehikulem a s pozitivní kontrolou.
Citlivost
: podíl všech pozitivních/aktivních chemických látek, které jsou na základě zkoušky správně zatříděny. Je měřítkem přesnosti zkušební metody, která poskytuje kategorické výsledky, a je důležitým prvkem při hodnocení relevantnosti zkušební metody (14).
S.I.
: stimulační index. Relativní hodnoty geometrického průměru intenzity fluorescence v buňkách exponovaných chemické látce v porovnání s buňkami exponovanými rozpouštědlu.
Kontrola s rozpouštědlem/vehikulem
: neexponovaný vzorek obsahující všechny složky zkušebního systému s výjimkou zkoušené chemické látky, ale včetně rozpouštědla/vehikula, které se používá. Používá se ke stanovení účinku na pozadí u vzorků exponovaných zkoušené chemické látce, která je rozpuštěna nebo stabilně dispergována ve stejném rozpouštědle/vehikulu. Pokud se vzorek zkouší souběžnou kontrolou s médiem, ukazuje také, zda rozpouštědlo/vehikulum vzájemně reaguje se zkušebním systémem.
Specifičnost
: podíl všech negativních/neaktivních chemických látek, které jsou na základě zkušební metody správně zatříděny. Je to měřítko přesnosti zkušební metody, která dává kategorické výsledky, a je důležitým prvkem při hodnocení relevance zkušební metody (14).
Barvicí pufr
: fosfátový pufr ve fyziologickém roztoku obsahující 5 % fetálního telecího séra.
Látka
: chemický prvek a jeho sloučeniny v přírodním stavu nebo získané výrobním postupem, včetně jakýchkoliv aditiv potřebných pro zachování stability výrobku a všech nečistot pocházejících z použitého postupu, ale s vyloučením všech rozpouštědel, která je možno oddělit bez ovlivnění stability dané látky nebo změny jejího složení.
Zkoušená chemická látka
: jakákoli látka nebo směs zkoušená touto zkouškou.
Globálně harmonizovaný systém klasifikace a označování chemických látek Organizace spojených národů (GHS OSN)
: systém navrhující klasifikaci látek a směsí podle standardizovaných typů a úrovní fyzikálních, zdravotních a environmentálních nebezpečí a řešící příslušné komunikační prvky, jako jsou piktogramy, signální slova, výroky o nebezpečí, výroky o bezpečnostních opatřeních a bezpečnostní datové listy, které obsahují informace o jejich nežádoucích účincích s ohledem na ochranu lidí (včetně zaměstnavatelů, dělníků, přepravců, spotřebitelů a osob reagujících na havárie) a životního prostředí (16).
UVCB
: látky s neznámým nebo proměnlivým složením, komplexní reakční produkty nebo biologický materiál.
Validovaná zkouška
: zkouška, u níž se má za to, že je dostatečně relevantní a spolehlivá pro určitý účel, a která je založena na vědecky spolehlivých zásadách. Zkouška není nikdy validovaná v absolutním smyslu, ale pouze pro určitý konkrétní účel (14).
Dodatek 2.2
LÁTKY PRO PROKÁZÁNÍ ZPŮSOBILOSTI
Dříve než začnou laboratoře běžně používat zkoušku popsanou v tomto dodatku ke zkušební metodě B.71, měly by prokázat odbornou způsobilost správným získáním předpokládané předpovědi U-SENS™ u 10 vhodných látek doporučených v tabulce 1 a získáním hodnot CV70 a EC150, které spadají do příslušného referenčního rozpětí, u nejméně 8 z 10 vhodných látek. Látky pro prokázání způsobilosti byly vybrány tak, aby představovaly rozpětí účinků na nebezpečí senzibilizace kůže. Dalšími kritérii výběru bylo, aby látky byly komerčně dostupné, aby k nim byly k dispozici kvalitní referenční údaje in vivo a také kvalitní údaje in vitro získané zkouškou U-SENS™. Pro zkoušku U-SENS™ jsou také k dispozici zveřejněné referenční údaje (1) (8).
Tabulka 1
Doporučené látky pro prokázání odborné způsobilosti k provádění zkoušky U-SENS™
|
Látky pro prokázání způsobilosti
|
Fyzikální forma
|
Skupenství
|
Předpověď in vivo
(105)
|
U-SENS Rozpouštědlo/vehikulum
|
U-SENS CV70 Odkaz Rozsah v μg/ml (106)
|
U-SENS (referenční rozmezí EC150 v μg/ml) (106)
|
|
benzen-1,4-diamin
|
106-50-3 Org.
|
pevná látka
|
senzibilizující (silně)
|
kompletní médium (107)
|
< 30
|
pozitivní (≤ 10)
|
|
kyselina picrylsulfonová
|
2508-19-2
|
kapalina
|
senzibilizující (silně)
|
kompletní médium
|
> 50
|
pozitivní (≤ 50)
|
|
diethyl-maleinát
|
141-05-9
|
kapalina
|
senzibilizující (středně silně)
|
DMSO
|
10–100
|
pozitivní (≤ 20)
|
|
resorcin
|
108-46-3
|
pevná látka
|
senzibilizující (středně silně)
|
kompletní médium
|
> 100
|
pozitivní (≤ 50)
|
|
skořicový alkohol
|
104-54-1
|
pevná látka
|
senzibilizující (slabě)
|
DMSO
|
> 100
|
pozitivní (10–100)
|
|
estragol
|
140-67-0
|
kapalina
|
senzibilizující (slabě)
|
DMSO
|
> 100
|
pozitivní (< 200)
|
|
sacharin
|
81-07-2
|
pevná látka
|
není senzibilizující
|
DMSO
|
> 200
|
negativní (> 200)
|
|
glycerol
|
56-81-5
|
kapalina
|
není senzibilizující
|
kompletní médium
|
> 200
|
negativní (> 200)
|
|
kyselina mléčná
|
50-21-5
|
kapalina
|
není senzibilizující
|
kompletní médium
|
> 200
|
negativní (> 200)
|
|
kyselina salicylová
|
69-72-7
|
pevná látka
|
není senzibilizující
|
DMSO
|
> 200
|
negativní (> 200)
|
|
Zkratky: CAS RN = registrační číslo CAS (Chemical Abstracts Service)
|
Dodatek 3
SENZIBILIZACE KŮŽE IN VITRO: ANALÝZA IL-8 LUC
VÝCHOZÍ ÚVAHY A OMEZENÍ
|
1.
|
Na rozdíl od zkoušek analyzujících expresi buněčných povrchových markerů kvantifikuje analýza L8-Luc změny v expresi IL-8, cytokinu spojeného s aktivací dendritických buněk (DC). V reportérové buněčné linii IL-8 odvozené od THP (THP-G8, vytvořené z buněčné linie lidské akutní monocytární leukémie THP-1) se měří exprese IL-8 po expozici senzibilizujícím látkám (1). Poté se použije exprese luciferázy jako pomůcka pro rozlišení mezi látkami senzibilizujícími kůži a látkami nesenzibilujícími kůži.
|
|
2.
|
Analýza IL-8 Luc byla hodnocena ve validační studii (2) provedené Japonským střediskem pro validaci alternativních metod (JaCVAM), ministerstvem hospodářství, obchodu a průmyslu (METI) a Japonskou společností pro alternativy experimentů na zvířatech (JSAAE) a poté byla podrobena nezávislému hodnocení (3) pod záštitou střediska JaCVAM a ministerstva zdravotnictví, práce a sociálních věcí (MHLW) s podporou útvaru pro Mezinárodní spolupráci při alternativních zkušebních metodách (ICATM). S ohledem na všechny dostupné důkazy a příspěvky od regulátorů a účastníků je analýza IL-8 Luc považována za užitečnou v rámci přístupu IATA na rozlišení mezi látkami senzibilizujícími kůži a látkami nesenzibilizujícími kůži za účelem klasifikace nebezpečnosti a označování. Příklady využití údajů z analýzy IL-8 Luc v kombinaci s jinými informacemi jsou uvedeny v odborné literatuře (4) (5) (6).
|
|
3.
|
U analýzy IL-8 Luc se prokázalo, že ji lze přenést do laboratoří, které mají zkušenosti s používáním buněčných kultur a měřením luciferázy. Reprodukovatelnost v rámci laboratoře a mezilaboratorní reprodukovatelnost činily 87,7 %, respektive 87,5 % (2). Údaje získané z validační studie (2) a dalších zveřejněných prací (1) (6) ukazují, že analýza IL-8 Luc určila 118 ze 143 chemických látek jako pozitivních nebo negativních a 25 jako neprůkazných a že přesnost analýzy IL-8 Luc při odlišení senzibilizujících látek (tj. kategorie 1 GHS OSN/CLP) od nesenzibilizujících látek (tj. bez kategorie GHS OSN/CLP) činí 86 % (101/118) při citlivosti 96 % (92/96) a specifičnosti 41 % (9/22). S vyloučením látek mimo oblast použitelnosti, jak je popsáno níže (odstavec 5), analýza IL-8 Luc určila 113 ze 136 chemických látek jako pozitivních nebo negativních a 23 jako neprůkazných, přičemž přesnost analýzy IL-8 Luc činí 89 % (101/113) při citlivosti 96 % (92/96) a specifičnosti 53 % (9/17). Podle údajů o použití u lidí citovaných v Urbisch et al. (7) analýza IL-8 Luc určila 76 z 90 chemických látek jako pozitivních nebo negativních a 14 jako neprůkazných, přičemž přesnost činí 80 % (61/76), citlivosti 93 % (54/58) a specifičnost 39 % (7/18). S vyloučením látek mimo oblast použitelnosti analýza IL-8 Luc určila 71 z 84 chemických látek jako pozitivních nebo negativních a 13 chemických látek jako neprůkazných, přičemž přesnost činí 86 % (61/71) při citlivosti 93 % (54/58) a specifičnosti 54 % (7/13). Falešně negativní předpovědi se u analýzy IL-8 Luc pravděpodobněji týkají chemických látek vykazujících nízkou/střední potenci způsobovat senzibilizaci kůže (podkategorie 1B GHS OSN/CLP) než chemických látek vykazujících vysokou potenci (podkategorie 1A GHS OSN/CLP) (6). Celkově vzato tyto informace podporují roli, kterou může analýza IL-8 Luc sehrát při identifikaci nebezpečí senzibilizace kůže. Uvedená přesnost analýzy IL-8 Luc jako samostatné zkoušky je však pouze orientační, neboť tato zkouška by měla být posuzována v kombinaci s jinými zdroji informací v rámci IATA a v souladu s ustanoveními odstavců 7 a 8 ve Všeobecném úvodu. Při hodnocení zkoušek na senzibilizaci kůže bez použití zvířat by se dále mělo vést v patrnosti, že zkouška LLNA a ani jiné zkoušky na zvířatech nemusí plně odrážet situaci u lidí.
|
|
4.
|
Na základě nyní dostupných údajů bylo prokázáno, že analýzu IL-8 Luc lze použít ke zkoušení chemických látek, které obsahují různé organické funkční skupiny, mají různé reakční mechanismy, různou potenci způsobovat senzibilizaci kůže (jak byla stanovena ve studiích in vivo) a různé fyzikálně-chemické vlastnosti (2) (6).
|
|
5.
|
Ačkoli analýza IL-8 Luc používá jako rozpouštědlo X-VIVOTM 15, správně vyhodnotila chemické látky s log Kow >3,5 a chemické látky s rozpustností ve vodě 100 μg/ml podle výpočtu pomocí EPI SuiteTM a její schopnost detekovat senzibilizující látky špatně rozpustné ve vodě je lepší než v případě analýzy IL-8 Luc používající jako rozpouštědlo dimethylsulfoxid (DMSO) (2). Negativní výsledky u zkoušených chemických látek, které se nerozpouštějí při 20 mg/ml, však mohou dávat falešně negativní výsledky vzhledem k jejich nerozpustnosti v X-VIVOTM 15. Negativní výsledky u těchto zkoušených chemických látek by proto neměly být brány v úvahu. Ve validační studii byla pozorována vysoká míra falešně negativních výsledků u anhydridů. Dále z důvodu omezené metabolické schopnosti buněčné linie (8) a vzhledem k experimentálním podmínkám mohou prohapteny (látky vyžadující metabolickou aktivaci) a prehapteny (látky aktivované oxidací) při analýze vykazovat negativní výsledky. Ačkoliv by však měly být negativní výsledky u pravděpodobných pre/prohaptenů interpretovány opatrně, analýza IL-8 Luc správně posoudila 11 z 11 prehaptenů, 6/6 prohaptenů a 6/8 pre/prohaptenů v souboru dat analýzy IL-8 Luc (2). Na základě nedávno provedené komplexní revize tří zkoušek neprováděných na zvířatech (DPRA, KeratinoSens™ a h-CLAT) na detekci pre- a prohaptenů (9) a na základě skutečnosti, že buňky THP-G8 používané při analýze IL-8 Luc jsou buněčná linie derivovaná z buněk THP-1, které se používají při zkoušce h-CLAT, může analýza IL-8 Luc také přispět ke zvýšení citlivosti zkoušek prováděných na zvířatech, které jsou určeny na detekci pre- a prohaptenů, v kombinaci s jinými zkouškami. Zkoušené povrchově aktivní látky zatím dávaly (falešně) pozitivní výsledky bez ohledu na jejich druh (např. kationtové, aniontové nebo neiontové). Konečně, zkoušené chemické látky, jež ovlivňují luciferázu, mohou zkreslit její aktivitu/měření, neboť způsobují zjevnou inhibici nebo zvýšenou luminiscenci (10). Uvádí se například, že koncentrace fytoestrogenu vyšší než 1 μM ovlivnila luminiscenční signály v jiných zkouškách reportérových genů založených na luciferáze v důsledku nadměrné aktivace reporterového genu luciferázy. V důsledku toho je nutné pečlivě zkoumat exprese luciferázy získané při vysoké koncentraci fytoestrogenů nebo sloučenin, u nichž existuje podezření, že vyvolávají aktivaci reporterového genu luciferázy podobně jako fytoestrogeny (11). Na základě výše uvedeného jsou povrchově aktivní látky, anhydridy a chemické látky interferující s luciferázou mimo oblast použitelnosti této analýzy. V případech, kdy existují důkazy prokazující nepoužitelnost analýzy IL-8 Luc na jiné specifické kategorie zkoušených chemických látek, neměla by se tato zkouška u těchto specifických kategorií používat.
|
|
6.
|
Jak bylo popsáno výše, pomáhá analýza IL-8 Luc rozlišovat mezi látkami senzibilizujícími kůži a látkami nesenzibilujícími kůži. Ke stanovení toho, zda mohou výsledky analýzy IL-8 Luc přispět k hodnocení potence při posuzování v kombinaci s jinými zdroji informací, však bude zapotřebí další výzkum, nejlépe založený na údajích o účincích na člověka.
|
|
7.
|
Definice jsou uvedeny v dodatku 3.1.
|
PODSTATA ZKOUŠKY
|
8.
|
Analýza IL-8 Luc využívá buněčnou linii THP-1 lidské monocytární leukemie, která byla získána z Americké sbírky typových kultur (Manassas, VA, USA). S využitím této buněčné linie vytvořila Katedra dermatologie Lékařské fakulty Univerzity v Tohoku reportérovou buněčnou linii IL-8 derivovanou z buněk THP-1, THP-G8, která obsahuje geny stabilní luciferázové oranže (SLO) a stabilní luciferázové červeně (SLR) pod kontrolou promotérů IL-8, respektive glyceraldehyd-3-fosfátdehydrogenázy (GAPDH) (1). To umožňuje kvantitativní měření indukce genu luciferázy pomocí lumuniscenční detekce za použití známých substrátů pro luciferázu produkujících světlo jako ukazatele aktivity transkripčního faktoru IL-8 a GAPDH v buňkách po expozici senzibilizujícím látkám.
|
|
9.
|
Duální barevný zkušební systém sestává z luciferázy emitující oranžovou (SLO; λmax = 580 nm) (12) pro genovou expresi promotéru IL-8 a z luciferázy emitující červenou (SLR; λmax = 630 nm) (13) pro genovou expresi interního kontrolního promotéru, GAPDH. Tyto dvě luciferázy emitují po reakci s d-luciferinem světlušek různé barvy, přičemž jejich luminiscence se měří současně v jednostupňové reakci rozdělením emise z analyzované směsi pomocí optického filtru (14) (dodatek 3.2).
|
|
10.
|
Buňky THP-G8 se po dobu 16 hodin exponují zkoušené chemické látce a poté se změří aktivita luciferázy SLO (SLO-LA), která vyjadřuje aktivitu promotéru IL-8 a aktivitu luciferázy SLR (SLR-LA), vyjadřující aktivitu promotéru GAPDH. Aby byly zkratky snadno pochopitelné, označují se SLO-LA a SLR-LA jako IL8LA, respektive GAPLA. V tabulce 1 je uveden popis výrazů souvisejících s aktivitou luciferázy při analýze IL-8 Luc. Naměřené hodnoty se používají pro výpočet normalizované hodnoty IL8LA (nIL8LA), což je poměr IL8LA ke GAPLA; indukce nIL8LA (Ind-IL8LA), což je poměr aritmetických průměrů čtyřnásobně změřených hodnot nIL8LA buněk THP-G8 exponovaných zkoušené chemické látce a hodnot nIL8LA neexponovaných buněk THP-G8; a inhibice GAPLA (Inh-GAPLA), což je poměr aritmetických průměrů čtyřnásobně změřených hodnot GAPLA buněk THP-G8 exponovaných zkoušené chemické látce a hodnot GAPLA neexponovaných buněk THP-G8, a použijí se jako ukazatel cytotoxicity.
Tabulka 1
Popis výrazů souvisejících s aktivitou luciferázy při analýze IL-8 Luc
|
Zkratky
|
Definice
|
|
GAPLA
|
Aktivita luciferázy SLR vyjadřující aktivitu promotéru GAPDH
|
|
IL8LA
|
Aktivita luciferázy SLO vyjadřující aktivitu promotéru IL-8
|
|
nIL8LA
|
IL8LA / GAPLA
|
|
Ind-IL8LA
|
nIL8LA buněk THP-G8 exponovaných chemickým látkám / nIL8LA neexponovaných buněk
|
|
Inh-GAPLA
|
GAPLA buněk THP-G8 exponovaných chemickým látkám / GAPLA neexponovaných buněk
|
|
CV05
|
Nejnižší koncentrace chemické látky, při níž je Inh-GAPLA < 0,05.
|
|
|
11.
|
Jsou k dispozici technické parametry (PS) (15), které usnadní validaci pozměněných zkoušek luciferázy IL-8 in vitro obdobných analýze IL-8 Luc a umožní včas provést změnu pokynu OECD 442E, aby do ní mohly být zahrnuty. Vzájemné uznávání údajů podle postupu OECD (MAD) bude zaručeno pouze u zkoušek validovaných podle těchto technických parametrů, pokud tyto zkoušky byly podrobeny revizi a pokud je OECD zahrnula do pokynu ke zkoušení 442E (16).
|
PROKÁZÁNÍ ZPŮSOBILOSTI
|
12.
|
Před rutinním používáním zkoušky popsané v tomto dodatku ke zkušební metodě B.71 by měly laboratoře prokázat odbornou způsobilost použitím 10 látek pro prokazování způsobilosti, které jsou uvedeny v dodatku 3.3, a to v souladu s osvědčenými postupy pro metody in vitro (17). Uživatelé zkoušky by si také měli vést historickou databázi údajů pocházejících ze zkoušek reaktivity (viz odstavec 15) a z pozitivních kontrol a kontrol s rozpouštědlem/vehikulem (viz odstavce 21–24) a používat tato data na potvrzení, že v jejich laboratoři je udržována reprodukovatelnost zkoušky v čase.
|
POSTUP
|
13.
|
Jsou k dispozici standardní pracovní postupy pro analýzu IL-8 Luc a měly by být používány pro provádění této zkoušky (18). Laboratoře, jež chtějí tuto zkoušku provádět, mohou získat rekombinantní buněčnou linii výkonnostních standardů od společnosti GPC Lab. Co. Ltd., Tottori, Japonsko po uzavření smlouvy o prodeji materiálu (MTA) v souladu s podmínkami uvedenými ve vzoru OECD. Dále je uveden popis hlavních složek a postupů analýzy.
|
Příprava buněk
|
14.
|
Při provádění analýzy IL-8 Luc by se měla používat buněčná linie THP-G8 od společnosti GPC Lab. Co. Ltd., Tottori, Japonsko (viz odstavce 8 a 13). Buňky se po obdržení pomnoží (2–4 pasáže) a skladují se zmrazené jako homogenní kmen. Buňky z této zásoby mohou být pasážovány maximálně 12krát nebo maximálně 6 týdnů. K množení se používá kultivační médium RPMI-1640 obsahující 10 % fetálního bovinního séra (FBS), antibiotický/antimykotický roztok (100 jednotek/ml penicilinu G, 100 μg/ml streptomycinu a 0,25 μg/ml amfotericinu B v 0,85 % fyziologickém roztoku) (např. GIBCO kat. č. 15240-062), 0,15 μg/ml Puromycinu (např. CAS:58-58-2) a 300 μg/ml G418 (např. CAS:108321-42-2).
|
|
15.
|
Než se buňky použijí pro zkoušení, je třeba zjistit, zda jsou vhodné, provedením kontroly reaktivity. Tato kontrola se provádí 1–2 týdny nebo 2–4 pasáže po rozmrazení za použití pozitivní kontroly, 4-nitrobenzyl-bromidu (4-NBB) (CAS:100-11-8, čistota ≥ 99 %) a negativní kontroly, kyseliny mléčné (LA) (CAS:50-21-5, čistota ≥ 85 %). 4-NBB by měl dát pozitivní odpověď na Ind-IL8LA (≥ 1,4), zatímco kyselina mléčná by měla dát negativní odpověď na Ind-IL8LA (< 1,4). Pro analýzu se používají pouze buňky, které prošly kontrolou reaktivity. Kontrola se provádí podle postupů popsaných v odstavcích 22–24.
|
|
16.
|
Pro zkoušení se buňky THP-G8 nasadí v hustotě 2 až 5 × 105 buněk/ml a předběžně se kultivují v kultivačních baňkách po dobu 48 až 96 hodin. V den zkoušky se buňky získané z kultivační baňky promyjí RPMI-1640 s obsahem 10 % FBS bez jakýchkoli antibiotik a poté resuspendují s RPMI-1640 s obsahem 10 % FBS bez jakýchkoli antibiotik při 1 × 106 buněk/ml. Poté se buňky rozdělí do 96jamkové destičky s plochým dnem (např. Costar kat. č. 3603) s 50 μl (5 × 104 buněk/jamka).
|
Příprava zkoušené chemické látky a kontrolních látek
|
17.
|
Zkoušená chemická látka a kontrolní látky se připraví v den zkoušení. Pro analýzu IL-8 Luc se zkoušené chemické látky rozpustí v X-VIVOTM 15, komerčně dostupném médiu bez séra (Lonza, 04-418Q), na konečnou koncentraci 20 mg/ml. X-VIVOTM 15 se přidá do 20 mg zkoušené chemické látky (bez ohledu na rozpustnost chemické látky) ve zkumavce do mikroodstředivky a doplní se na objem 1 ml a poté se silně odstředí a promíchává na rotoru při maximální rychlosti 8 ot/min. po dobu 30 minut při okolní teplotě přibližně 20 oC. Pokud jsou pevné chemické látky stále nerozpustné, zkumavka se poté vloží do ultrazvukové lázně, dokud se chemická látka zcela nerozpustí nebo dokud není stabilně dispergována. U zkoušených chemických látek, které jsou rozpustné v X-VIVOTM 15, se roztok naředí přípravkem X-VIVOTM 15 faktorem 5 a použije jako zásobní roztok X-VIVOTM 15 zkoušené chemické látky (4 mg/ml). U zkoušených chemických látek, které nejsou rozpustné v X-VIVOTM 15, se směs opět promíchává po dobu alespoň 30 minut, poté se odstředí při 15000 ot/min (≈ 20000 g) po dobu 5 minut; výsledný supernatant se použije jako zásobní roztok X-VIVOTM 15 zkoušené chemické látky. Pro použití jiných rozpouštědel, jako je DMSO, voda nebo kultivační médium, je třeba uvést vědecké zdůvodnění. Podrobný postup rozpuštění chemických látek je uveden v dodatku 3.5. Roztoky X-VIVOTM 15 popsané v odstavcích 18–23 se smíchají 1: 1 (obj.) s buněčnými suspenzemi připravenými ve 96jamkové destičce s plochým dnem (viz odstavec 16).
|
|
18.
|
Cílem prvního provedení zkoušky je stanovit cytotoxickou koncentraci a zkoumat potenciál chemických látek senzibilizovat kůži. Pomocí X-VIVOTM 15 se připraví sériově naředěné zásobní roztoky X-VIVOTM 15 zkoušených chemických látek při faktoru ředění dva (viz dodatek 3.5) s použitím 96jamkového zkušebního bloku (např. Costar kat. č. EW-01729-03). Poté se 50 μl/jamku naředěného roztoku přidá do 50 μl buněčné suspenze v 96jamkové černé destičce s plochým dnem. Pro zkoušené chemické látky, které jsou rozpustné v X-VIVO TM 15, bude tedy konečná koncentrace zkoušených chemický látek v rozsahu od 0,002 do 2 mg/ml (dodatek 3.5). Pro zkoušené chemické látky, které nejsou rozpustné v X-VIVO TM 15 při 20 mg/ml, se stanoví pouze faktory ředění v rozsahu od 2 do 210, přestože jsou skutečné konečné koncentrace zkoušených chemických látek nejisté a závisí na saturované koncentraci zkoušených chemických látek v zásobním roztoku X-VIVO TM 15.
|
|
19.
|
Při dalších provedeních zkoušky (tj. druhý, třetí a čtvrtý replikát) se zásobní roztoky X-VIVOTM 15 připraví při koncentraci čtyřikrát vyšší, než je koncentrace životaschopnosti buněk 05 (CV05; nejnižší koncentrace, při níž je Inh-GAPLA < 0,05) v prvním experimentu. Jestliže Inh-GAPLA neklesne při prvním provedení zkoušky pod 0,05 při nejvyšší koncentraci, zásobní roztok X-VIVOTM 15 se připraví v nejvyšší koncentraci při prvním provedení zkoušky. Koncentrace CV05 se vypočítá vydělením koncentrace zásobního roztoku v prvním provedení zkoušky faktorem ředění pro CV05 (X) (faktor ředění CV05 (X); faktor ředění potřebný pro naředění zásobního roztoku na CV05) (viz dodatek 3.5). Pro zkoušené látky, které nejsou rozpustné v X-VIVO při 20 mg/ml, se CV05 určí podle koncentrace zásobního roztoku × 1/X. Pro provedení zkoušky 2 až 4 se připraví druhý zásobní roztok jako 4 × CV05 (dodatek 3.5).
|
|
20.
|
Připraví se sériově naředěné druhé zásobní roztoky X-VIVOTM 15 při faktoru ředění 1,5 s použitím 96jamkového zkušebního bloku. Poté se 50 μl/jamku naředěného roztoku přidá do 50 μl buněčné suspenze v jamkách 96jamkové černé destičky s plochým dnem. Každá koncentrace každé zkoušené chemické látky by měla být testována ve 4 jamkách. Vzorky se poté míchají na třepačce a inkubují po dobu 16 hodin při 37oC a 5 % CO2 a poté se změří aktivita luciferázy, jak je popsáno níže.
|
|
21.
|
Kontrola s rozpouštědlem je směs 50 μl/jamku X-VIVOTM 15 a 50 μl/jamku buněčné suspenze v RPMI-1640 s obsahem 10 % FBS.
|
|
22.
|
Doporučená pozitivní kontrola je 4-NBB. 20 mg 4-NBB se připraví ve zkumavce do mikroodstředivky o objemu 1,5 ml a doplní se přípravkem X-VIVOTM 15 na objem 1 ml. Zkumavka se silně odstředí a promíchává na rotoru při maximální rychlosti 8 ot/min. po dobu alespoň 30 minut. Po odstředění při 20000 g po dobu 5 minut se supernatant naředí faktorem 4 přípravkem X-VIVOTM 15 a 500 μl naředěného supernatantu se přenese do jamky 96jamkového zkušebního bloku. Naředěný supernatant se dále naředí přípravkem přípravkem X-VIVOTM 15 při faktoru 2 a 4 a 50 μl roztoku se přidá do 50 μl buněčné suspenze THP-G8 v jamkách 96jamkové černé destičky s plochým dnem (dodatek 3.6). Každá koncentrace pozitivní kontroly by měla být testována ve 4 jamkách. Destička se míchá na třepačce a inkubuje v CO2 po dobu 16 hodin (37 oC, 5 % CO2) a poté se změří aktivita luciferázy, jak je popsáno odstavci 29.
|
|
23.
|
Doporučená negativní kontrola je kyselina mléčná. 20 mg kyseliny mléčné se připraví ve zkumavce do mikroodstředivky o objemu 1,5 ml a doplní se přípravkem X-VIVOTM 15 na objem 1 ml (20 mg/ml). 20 mg/ml roztoku kyseliny mléčné se naředí faktorem 5 přípravkem X-VIVOTM 15 (4 mg/ml); 500 μl tohoto roztoku kyseliny mléčné 4 mg/ml se přenese do jamky 96jamkového zkušebního bloku. Tento roztok se naředí faktorem 2 přípravkem X-VIVOTM 15 a poté se opět naředí faktorem 2, aby vznikly roztoky 2 mg/ml a 1 mg/ml. 50 μl těchto 3 roztoků a kontroly s vehikulem (X-VIVOTM 15) se přidá do 50 μl buněčné suspenze THP-G8 v jamkách 96jamkové černé destičky s plochým dnem. Každá koncentrace negativní kontroly by měla být testována ve 4 jamkách. Destička se míchá na třepačce a inkubuje v CO2 po dobu 16 hodin (37 oC, 5 % CO2) a poté se změří aktivita luciferázy, jak je popsáno odstavci 29.
|
|
24.
|
Lze použít i jiné vhodné pozitivní nebo negativní kontroly, pokud jsou k dispozici dosavadní údaje umožňující odvodit srovnatelná kritéria přijatelnosti daného provedení zkoušky.
|
|
25.
|
Mělo by se dbát na to, aby nedošlo k vypařování těkavých zkoušených chemických látek a ke vzájemné kontaminaci zkoušenou chemickou látkou mezi jamkami, např. tak, že se destičky před inkubací se zkoušenými chemickými látkami zapečetí.
|
|
26.
|
Zkoušené chemické látky a kontrola s rozpouštědlem vyžadují 2 až 4 provedení zkoušky, aby mohla být stanovena pozitivní nebo negativní predikce (viz tabulka 2). Každé opakování se provádí v různý den s čerstvým zásobním roztokem X-VIVOTM 15 zkoušených chemických látek a s nezávisle sklizenými buňkami. Buňky mohou pocházet ze stejné pasáže.
|
Měření aktivity luciferázy
|
27.
|
Luminiscence se měří pomocí luminometru na 96jamkové destičky vybaveného optickými filtry, např. Phelios (ATTO, Tokyo, Japonsko), Tristan 941 (Berthold, Bad Wildbad, Německo) a ARVO series (PerkinElmer, Waltham, MA, USA). Luminometr musí být kalibrován pro každou zkoušku, aby byla zajištěna reprodukovatelnost (19). Pro tuto kalibraci se dodávají rekombinantní luciferázy emitující oranžovou a červenou.
|
|
28.
|
Do každé jamky destičky obsahující buněčnou suspenzi exponovanou nebo neexponovanou chemické látce se přenese 100 μl ohřáté reagencie Tripluc® Luciferase (Tripluc). Destička se protřepává po dobu 10 minut při okolní teplotě přibližně 20oC. Destička se umístí do luminometru a změří se aktivita luciferázy. Bioluminiscence se měří po dobu 3 sekund, a to bez optického filtru (F0) a s optickým filtrem (F1). Při použití alternativního uspořádání, např. v závislosti na modelu použitého luminometru, je třeba uvést zdůvodnění.
|
|
29.
|
Z naměřených hodnot se vypočítají parametry pro každou koncentraci, např. IL8LA, GAPLA, nIL8LA, Ind-IL8LA, Inh-GAPLA, střední hodnota ±SD IL8LA, střední hodnota ±SD GAPLA, střední hodnota ±SD nIL8LA, střední hodnota ±SD Ind-IL8LA, střední hodnota ±SD Inh-GAPLA a 95 % konfidenční interval Ind-IL8LA. Definice parametrů použitých v tomto odstavci jsou uvedeny v dodatcích 3.1 a 3.4.
|
|
30.
|
Před měřením se zajistí barevné rozlišení u vícebarevných reportérových analýz obvykle pomocí detektorů (luminometr a čtečka destiček) vybavených optickými filtry, jako jsou např. sharp-cut (dlouhovlnné nebo krátkovlnné průchodové) filtry nebo pásmové průchodové filtry. Před zkouškou je třeba kalibrovat prostupové koeficienty filtru pro každou barvu bioluminiscenčního signálu podle dodatku 3.2.
|
ÚDAJE A PŘEDKLÁDÁNÍ ZPRÁV
Vyhodnocení údajů
|
31.
|
Kritéria pro pozitivní/negativní rozhodnutí vyžadují, aby při každém provedení zkoušky:
|
—
|
byla predikce při analýze IL-8 Luc určena jako pozitivní, jestliže má zkoušená chemická látka Ind-IL8LA ≥ 1,4 a dolní limit 95 % konfidenčního intervalu Ind-IL8LA ≥ 1,0,
|
|
—
|
byla predikce při analýze IL-8 Luc určena jako negativní, jestliže má zkoušená chemická látka Ind-IL8LA < 1,4 a/nebo dolní limit 95 % konfidenčního intervalu Ind-IL8LA < 1,0.
|
|
Predikční model
|
32.
|
Zkoušené chemické látky, které dávají dva pozitivní výsledky z 1., 2., 3. nebo 4. provedení zkoušky, jsou určeny jako pozitivní, zatímco zkoušené chemické látky, které dávají tři negativní výsledky z 1., 2., 3. nebo 4. provedení zkoušky, jsou určeny jako pravděpodobně negativní (tabulka 2). Z pravděpodobně negativních chemických látek jsou chemické látky, které se rozpouštějí při 20 mg/ml X-VIVOTM 15, určeny jako negativní, zatímco chemické látky, které se nerozpouštějí při 20 mg/ml X-VIVOTM 15, by neměly být brány v úvahu (obrázek 1).
Tabulka 2
Kritéria pro identifikaci pozitivních a pravděpodobně negativních látek
|
1. provedení
|
2. provedení
|
3. provedení
|
4. provedení
|
Konečná predikce
|
|
pozitivní
|
pozitivní
|
–
|
–
|
pozitivní
|
|
negativní
|
pozitivní
|
–
|
pozitivní
|
|
negativní
|
pozitivní
|
pozitivní
|
|
negativní
|
pravděpodobně negativní
|
|
negativní
|
pozitivní
|
pozitivní
|
–
|
pozitivní
|
|
negativní
|
pozitivní
|
pozitivní
|
|
negativní
|
pravděpodobně negativní
|
|
negativní
|
pozitivní
|
pozitivní
|
pozitivní
|
|
negativní
|
pravděpodobně negativní
|
|
negativní
|
–
|
pravděpodobně negativní
|
|
Obrázek 1
Predikční model pro konečné posouzení
Kritéria přijatelnosti
|
33.
|
Při použití zkoušky IL-8 Luc by měla být splněna tato kritéria přijatelnosti:
|
—
|
Ind-IL8LA by mělo být větší než 5,0 alespoň v jedné koncentraci pozitivní kontroly, 4-NBB, při každém provedení zkoušky.
|
|
—
|
Ind-IL8LA by mělo být menší než 1,4 při jakékoli koncentraci negativní kontroly, kyselina mléčná, při každém provedení zkoušky.
|
|
—
|
Údaje z destiček, kde je GAPLA kontrolních jamek s buňkami a přípravkem Tripluc, ale bez chemických látek menší než pětinásobek této hodnoty jamek obsahujících pouze zkoušené médium (50 μl/jamku RPMI-1640 s obsahem 10 % FBS a 50 μl/jamku X-VIVOTM 15), se vyřadí.
|
|
—
|
Údaje z destiček, kde je Inh-GAPLA všech koncentrací zkoušené chemické látky nebo kontrolních chemických látek menší než 0,05, se vyřadí. V takovém případě by měla být první zkouška zopakována, tak aby byla nejvyšší konečná koncentrace opakované zkoušky stejná jako nejnižší konečná koncentrace předchozí zkoušky.
|
|
ZÁVĚREČNÁ ZPRÁVA
|
34.
|
Závěrečná zpráva by měla obsahovat tyto informace:
|
Zkoušené chemické látky
Jednosložková látka:
|
chemická identifikace, např. název (názvy) IUPAC nebo CAS, číslo (čísla) CAS, kód SMILES nebo InChI, strukturní vzorec a/nebo jiné identifikátory,
|
|
fyzický vzhled, rozpustnost ve vodě, molekulová hmotnost a další důležité fyzikálně-chemické vlastnosti, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
čistota, případně chemická identita nečistot, je-li to prakticky proveditelné atd.,
|
|
případné ošetření před zkoušením (např. zahřívání, mletí),
|
|
rozpustnost v X-VIVOTM 15. U chemických látek, které jsou nerozpustné v X-VIVOTM 15, zda je po odstředění pozorována sraženina nebo flotace,
|
|
podmínky skladování a stabilita, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
zdůvodnění volby rozpouštědla/vehikula pro každou zkoušenou chemickou látku, jestliže nebyl použit X-VIVOTM 15.
|
Vícesložková látka, UVCB a směs:
|
charakterizovaná v co možná největší míře např. chemickou identitou (viz výše), čistotou, kvantitativním výskytem a důležitými fyzikálně-chemickými vlastnostmi (viz výše) složek, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
fyzický vzhled, rozpustnost ve vodě a další důležité fyzikálně-chemické vlastnosti, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
molekulová hmotnost nebo zdánlivá molekulová hmotnost v případě směsí/polymerů se známým složením nebo jiné informace důležité pro provedení studie,
|
|
případné ošetření před zkoušením (např. zahřívání, mletí),
|
|
rozpustnost v X-VIVOTM 15. U chemických látek, které jsou nerozpustné v X-VIVOTM 15, zda je po odstředění pozorována sraženina nebo flotace,
|
|
podmínky skladování a stabilita, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
zdůvodnění volby rozpouštědla/vehikula pro každou zkoušenou chemickou látku, jestliže nebyl použit X-VIVOTM 15.
|
|
|
Kontroly
Pozitivní kontrola:
|
chemická identifikace, např. název (názvy) IUPAC nebo CAS, číslo (čísla) CAS, kód SMILES nebo InChI, strukturní vzorec a/nebo jiné identifikátory,
|
|
případně fyzický vzhled, rozpustnost ve vodě, molekulová hmotnost a další důležité fyzikálně-chemické vlastnosti, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
čistota, případně chemická identita nečistot, je-li to prakticky proveditelné atd.,
|
|
případné ošetření před zkoušením (např. zahřívání, mletí),
|
|
podmínky skladování a stabilita, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
odkaz na případné dosavadní výsledky pozitivních kontrol prokazující vhodnost kritérií přijatelnosti.
|
Negativní kontrola:
|
chemická identifikace, např. název (názvy) IUPAC nebo CAS, číslo (čísla) CAS a/nebo jiné identifikátory,
|
|
čistota, případně chemická identita nečistot, je-li to prakticky proveditelné atd.,
|
|
fyzický vzhled, molekulová hmotnost a další důležité fyzikálně-chemické vlastnosti v případě použití jiných negativních kontrol, než které jsou uvedeny v tomto pokynu, a to v rozsahu, v jakém jsou tyto informace dostupné,
|
|
podmínky skladování a stabilita, v rozsahu, v jakém jsou tyto informace dostupné,
|
|
zdůvodnění volby rozpouštědla pro každou zkoušenou chemickou látku.
|
|
|
Zkušební podmínky
|
jméno a adresa zadavatele, zkušebního zařízení a vedoucího studie,
|
|
použitá buněčná linie, podmínky skladování a zdroj (např. zařízení, z něhož byla získána),
|
|
číslo šarže a původ FBS, jméno dodavatele, číslo šarže 96jamkové černé destičky s plochým dnem a číslo šarže reagencie Tripluc,
|
|
číslo pasáže a hustota buněk použitých ke zkoušení,
|
|
metoda počítání buněk použitá při jejich nasazování před zkoušením a opatření přijatá k zajištění homogenní distribuce počtu buněk,
|
|
použitý luminometr (např. model), včetně nastavení přístroje, použitý substrát pro luciferázu a prokázání přiměřeného měření luminiscence na základě kontrolního testu, který je popsán v dodatku 3.2,
|
|
postup použitý pro prokázání způsobilosti laboratoře k provádění této zkoušky (např. zkoušením vhodných látek) nebo pro prokázání reprodukovatelného provedení této zkoušky v průběhu doby.
|
|
|
Zkušební postup
|
počet replikátů a počet provedení zkoušky,
|
|
koncentrace zkoušených chemických látek, postup aplikace a doba expozice (pokud se liší od doporučených),
|
|
popis použitých hodnotících kritérií a kritérií rozhodování,
|
|
popis použitých kritérií přijatelnosti studie,
|
|
popis jakýchkoli změn zkušebního postupu.
|
|
|
Výsledky
|
výpočty pro nIL8LA, Ind-IL8LA a Inh-GAPLA,
|
|
95 % konfidenční interval Ind-IL8LA,
|
|
graf zobrazující křivky dávka-odpověď pro indukci aktivity luciferázy a životaschopnost,
|
|
případně popis jakýchkoli jiných důležitých zjištění z pozorování.
|
|
|
Diskuse o výsledcích
|
diskuse o výsledcích získaných za pomoci analýzy IL-8 Luc,
|
|
diskuse o výsledcích analýzy v rámci IATA, jsou-li k dispozici jiné příslušné informace.
|
|
Závěr
|
LITERATURA
|
(1)
|
Takahashi T, Kimura Y, Saito R, Nakajima Y, Ohmiya Y, Yamasaki K, and Aiba S. (2011). An in vitro test to screen skin sensitizers using a stable THP-1-derived IL-8 reporter cell line, THP-G8. Toxicol Sci 124:359-69.
|
|
(2)
|
OECD (2017). Validation report for the international validation study on the IL-8 Luc assay as a test evaluating the skin sensitizing potential of chemicals conducted by the IL-8 Luc Assay. Series on Testing and Assessment No 267, ENV/JM/MONO(2017)19. Organisation for Economic Cooperation and Development, Paris. Dostupné na webových stránkách: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(3)
|
OECD (2017). Report of the Peer Review Panel for the IL-8 Luciferase (IL-8 Luc) Assay for in vitro skin sensitisation. Series on Testing and Assessment No 258, ENV/JM/MONO(2017)20. Organisation for Economic Cooperation and Development, Paris. Dostupné na webových stránkách: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(4)
|
OECD (2016) Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Organisation for Economic Cooperation and Development, Paris. Dostupné na webových stránkách: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(5)
|
van der Veen JW, Rorije E, Emter R, Natsch A, van Loveren H, and Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol 69:371-9.
|
|
(6)
|
Kimura Y, Fujimura C, Ito Y, Takahashi T, Nakajima Y, Ohmiya Y, and Aiba S. (2015). Optimization of the IL-8 Luc assay as an in vitro test for skin sensitization. Toxicol In Vitro 29:1816-30.
|
|
(7)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, et al. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Pharmacol 71:337-51.
|
|
(8)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, Nishiyama N, and Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Alternatives to laboratory animals: ATLA 38:275-84.
|
|
(9)
|
Patlewicz G, Casati S, Basketter DA, Asturiol D, Roberts DW, Lepoittevin J-P, Worth A and Aschberger K (2016) Can currently available non-animal methods detect pre and pro haptens relevant for skin sensitisation? Regul Toxicol Pharmacol, 82:147-155.
|
|
(10)
|
Thorne N, Inglese J, and Auld DS. (2010). Illuminating insights into firefly luciferase and other bioluminescent reporters used in chemical biology. Chem Biol 17:646-57.
|
|
(11)
|
OECD (2016). Test No 455: Performance-Based Test Guideline for Stably Transfected Transactivation In Vitro Assays to Detect Estrogen Receptor Agonists and Antagonists, OECD Publishing, Paris. http://dx.doi.org/10.1787/9789264265295-en.
|
|
(12)
|
Viviani V, Uchida A, Suenaga N, Ryufuku M, and Ohmiya Y. (2001). Thr226 is a key residue for bioluminescence spectra determination in beetle luciferases. Biochem Biophys Res Commun 280:1286-91.
|
|
(13)
|
Viviani VR, Bechara EJ, and Ohmiya Y. (1999). Cloning, sequence analysis, and expression of active Phrixothrix railroad-worms luciferases: relationship between bioluminescence spectra and primary structures. Biochemistry 38:8271-9.
|
|
(14)
|
Nakajima Y, Kimura T, Sugata K, Enomoto T, Asakawa A, Kubota H, Ikeda M, and Ohmiya Y. (2005). Multicolor luciferase assay system: one-step monitoring of multiple gene expressions with a single substrate. Biotechniques 38:891-4.
|
|
(15)
|
OECD (2017). Bude vydáno - Performance Standards for the assessment of proposed similar or modified in vitro skin sensitisation IL-8 luc test methods. OECD Environment, Health and Safety Publications, Series on Testing and Assessment. OECD, Paris, France
|
|
(16)
|
OECD (2005). Guidance Document the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Environment, Health and Safety publications, OECD Series on Testing and Assessment No 34. OECD, Paris, France.
|
|
(17)
|
OECD (2018). Draft Guidance document: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Organisation for Economic Cooperation and Development, Paris. Dostupné na webových stránkách: http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(18)
|
JaCVAM (2016). IL-8 Luc assay protocol, Available at. http://www.jacvam.jp/en_effort/effort02.html.
|
|
(19)
|
Niwa K, Ichino Y, Kumata S, Nakajima Y, Hiraishi Y, Kato D, Viviani VR, and Ohmiya Y. (2010). Quantum yields and kinetics of the firefly bioluminescence reaction of beetle luciferases. Photochem Photobiol 86:1046-9.
|
|
(20)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins, Part 1: Series on Testing and Assessment No. OECD Environment, Health and Safety Publications, Series on Testing and Assessment No. 168. OECD, Paris, France.
|
|
(21)
|
United Nations (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Sixth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. Dostupné: na webových stránkách: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
Dodatek 3.1
DEFINICE
Přesnost
: přesnost shody mezi výsledky zkoušky a přijatými referenčními hodnotami. Je to míra funkčnosti zkoušky a jeden z aspektů její relevantnosti. Tento výraz se často používá namísto výrazu soulad a rozumí se jím podíl správných výsledků zkoušky (16).
AOP (mechanismy nežádoucích účinků)
: sled událostí od chemické struktury cílové chemické látky nebo skupiny podobných chemických látek přes molekulární iniciační událost po účinek in vivo, který je předmětem zájmu (20).
Chemická látka
: látka nebo směs.
CV05
: životaschopnost buněk 05, tj. minimální koncentrace, při níž vykazují chemické látky méně než 0,05 Inh-GAPLA.
FInSLO-LA
: zkratka, která se používá ve validační zprávě a v předchozích publikacích týkajících se analýzy IL-8 Luc na označení Ind-IL8LA. Viz Ind-IL8LA, kde je uvedena definice.
GAPLA
: luciferázová aktivita stabilní luciferázové červeně (SLR) (λmax = 630 nm), regulované promotérem GAPDH, která prokazuje životaschopnost buněk a počet životaschopných buněk.
Nebezpečnost
: základní charakteristika látky nebo situace, která má potenciál vyvolat nepříznivé účinky, jsou-li organismus, systém nebo (sub)populace vystaveny této látce.
IATA (jednotný přístup ke zkoušení a posuzování)
: strukturovaný přístup používaný pro zjišťování nebezpečnosti (potenciál), charakterizaci rizik (potence) a/nebo posouzení bezpečnosti (potenciál/potence a expozice) chemické látky nebo skupiny chemických látek, jenž strategicky integruje a váží všechny důležité údaje s cílem poskytnout informace pro přijetí regulačního rozhodnutí, které se týká potenciální nebezpečnosti a/nebo rizika a/nebo potřeby dalšího cíleného, a tudíž minimálního zkoušení.
II-SLR-LA
: zkratka, která se používá ve validační zprávě a v předchozích publikacích týkajících se analýzy IL-8 Luc na označení Inh-GAPLA. Viz Inh-GAPLA, kde je uvedena definice.
IL-8 (Interleukin-8)
: cytokin derivovaný z endotelových buněk, fibroblastů, keratinocytů, makrofágů a monocytů, který vyvolává chemotaxi neutrofilů a T-lymfocytů.
IL8LA
: luciferázová aktivita stabilní luciferázové oranžové (SLO) (λmax = 580 nm), regulované promotérem IL-8.
Ind-IL8LA
: násobná indukce nIL8LA. Stanoví se vydělením nIL8LA buněk THP-G8 exponovaných chemickým látkám touto hodnotou nestimulovaných buněk THP-G8 a představuje indukci aktivity promotéru IL-8 chemickými látkami.
Inh-GAPLA
: inhibice GAPLA. Stanoví se vydělením GAPLA buněk THP-G8 exponovaných chemickým látkám touto hodnotou neexponovaných buněk THP-G8 a představuje cytotoxicitu chemických látek.
Minimální prahová hodnota indukce (MIT)
: nejnižší koncentrace, při níž chemická látka splňuje pozitivní kritéria.
Směs
: směs nebo roztok složený ze dvou nebo více látek.
Jednosložková látka
: látka definovaná svým kvantitativním složením, v níž je přítomna jedna hlavní složka v míře alespoň 80 % hmot.
Vícesložková látka
: látka definovaná svým kvantitativním složením, v níž je přítomna více než jedna z hlavních složek v koncentraci ≥ 10 % hmot. a < 80 % hmot. Vícesložková látka je výsledkem výrobního procesu. Rozdíl mezi směsí a vícesložkovou látkou spočívá v tom, že směs se získá smísením dvou nebo více látek bez chemické reakce. Vícesložková látka je výsledkem chemické reakce.
nIL8LA
: aktivita luciferázy SLO vyjadřující aktivitu promotéru IL-8 (IL8LA) normalizovaná aktivitou luciferázy SLR vyjadřující aktivitu promotéru GAPDH (GAPLA). Představuje aktivitu promotéru IL-8 po zohlednění životaschopnosti buněk nebo počtu buněk.
nSLO-LA
: zkratka, která se používá ve validační zprávě a v předchozích publikacích týkajících se analýzy IL-8 Luc na označení nIL8LA. Viz nIL8LA, kde je uvedena definice.
Pozitivní kontrola
: replika obsahující všechny složky zkušebního systému a látku, o níž je známo, že způsobuje pozitivní reakci. Aby se zajistilo, že proměnlivost v reakci na pozitivní kontrolu bude možné časem vyhodnotit, rozsah pozitivní reakce by neměl být nadměrný.
Prehapteny
: chemické látky, z nichž se abiotickou transformací staly senzibilizující látky.
Prohapteny
: chemické látky vyžadující enzymatickou aktivaci, aby vykazovaly potenciál senzibilizace kůže.
Relevantnost
: popis vztahu zkoušky k účinku, který je předmětem zájmu, a také toho, zda je zkouška smysluplná a použitelná pro konkrétní účel. Vyjadřuje, do jaké míry zkouška správně měří nebo predikuje biologický účinek, který je předmětem zájmu. Relevantnost zahrnuje také posouzení přesnosti (shody) zkoušky (16).
Spolehlivost
: měření rozsahu, v jakém může být zkouška časem reprodukovatelná v laboratořích a mezi laboratořemi, když se provádí s použitím stejného protokolu. Hodnotí se to výpočtem laboratorní a mezilaboratorní reprodukovatelnosti a laboratorní opakovatelnosti (16).
Provedení zkoušky
: jedna či více zkoušených chemických látek se zkouší současně s kontrolou s rozpouštědlem/vehikulem a s pozitivní kontrolou.
Citlivost
: podíl všech pozitivních/aktivních chemických látek, které jsou na základě zkoušky správně zatříděny. Je to měřítko přesnosti zkušební metody, která dává kategorické výsledky, a je důležitým prvkem při hodnocení relevance zkušební metody (16).
SLO-LA
: zkratka, která se používá ve validační zprávě a v předchozích publikacích týkajících se analýzy IL-8 Luc na označení IL8LA. Viz IL8LA, kde je uvedena definice.
SLR-LA
: zkratka, která se používá ve validační zprávě a v předchozích publikacích týkajících se analýzy IL-8 Luc na označení GAPLA. Viz GAPLA, kde je uvedena definice.
Kontrola s rozpouštědlem/vehikulem
: neexponovaný vzorek obsahující všechny složky zkušebního systému s výjimkou zkoušené chemické látky, ale včetně rozpouštědla/vehikula, které se používá. Používá se ke stanovení účinku na pozadí u vzorků exponovaných zkoušené chemické látce, která je rozpuštěna nebo stabilně dispergována ve stejném rozpouštědle/vehikulu. Pokud se vzorek zkouší souběžnou kontrolou s médiem, ukazuje také, zda rozpouštědlo/vehikulum vzájemně reaguje se zkušebním systémem.
Specifičnost
: podíl všech negativních/neaktivních chemických látek, které jsou na základě zkušební metody správně zatříděny. Je to měřítko přesnosti zkušební metody, která dává kategorické výsledky, a je důležitým prvkem při hodnocení relevance zkušební metody (16).
Látka
: chemický prvek a jeho sloučeniny v přírodním stavu nebo získané výrobním procesem včetně všech přídatných látek nutných k uchování stability výrobku a všech nečistot vznikajících v použitém procesu, ale s vyloučením všech rozpouštědel, která je možno oddělit bez ovlivnění stability látky nebo změny jejího složení.
Smáčedlo
: nazývané též povrchově aktivní látka je látka jako např. detergent, která je schopna snížit povrchové napětí kapaliny a umožnit jí vytvářet pěnu nebo pronikat do pevných látek; je též známa jako sufraktant. (TG437)
Zkoušená chemická látka
: jakákoli látka nebo směs zkoušená touto zkušební metodou.
THP-G8
: reportérova buněčná linie IL-8, která se používá při analýze IL-8 Luc. Lidská buněčná linie THP-1 podobná makrofágům byla transfekována geny luciferázy SLO a SLR pod kontrolou promotéru IL-8, respektive GAPDH.
Globálně harmonizovaný systém klasifikace a označování chemických látek Organizace spojených národů (GHS OSN)
: systém navrhující klasifikaci chemických látek (látek a směsí) podle standardizovaných typů a úrovní fyzikálních, zdravotních a environmentálních nebezpečí a řešící příslušné komunikační prvky, jako jsou piktogramy, signální slova, výroky o nebezpečí, výroky o bezpečnostních opatřeních a bezpečnostní datové listy, které obsahují informace o jejich nežádoucích účincích s ohledem na ochranu lidí (včetně zaměstnavatelů, dělníků, přepravců, spotřebitelů a osob reagujících na havárie) a životního prostředí (21).
UVCB
: látky s neznámým nebo proměnlivým složením, komplexní reakční produkty nebo biologický materiál.
Validovaná zkušební metoda
: zkouška, u níž se má za to, že je dostatečně relevantní a spolehlivá pro určitý účel, a která je založena na vědecky spolehlivých zásadách. Zkouška není nikdy validovaná v absolutním smyslu, ale pouze pro určitý konkrétní účel.
Dodatek 3.2
PRINCIP MĚŘENÍ AKTIVITY LUCIFERÁZY A STANOVENÍ PROSTUPOVÝCH KOEFICIENTŮ OPTICKÉHO FILTRU PRO SLO A SLR
Multireportérový analytický systém Tripluc se může používat s luminometrem na mikrodestičky s vícebarevným detekčním systémem, který může zahrnovat optický filtr (např. Phelios AB-2350 (ATTO), ARVO (PerkinElmer), Tristar LB941 (Berthold)). Optický filtr použitý při měření je dlouhovlnný nebo krátkovlnný průchodový filtr 600–620 nm nebo pásmový průchodový filtr 600–700 nm.
MĚŘENÍ DVOUBAREVNÝCH LUCIFERÁZ S OPTICKÝM FILTREM.
V tomto příkladu se používá přístroj Phelios AB-2350 (ATTO). Tento luminometr je vybaven dlouhovlnným průchodovým filtrem 600 nm (R60 HOYA Co., 600 nm LP, filtr 1) pro rozdělení luminiscence SLO (λmax = 580 nm) a SLR (λmax = 630 nm).
Pro stanovení prostupových koeficientů filtru 600 nm LP se nejprve pomocí purifikovaných enzymů luciferázy SLO a SLR změří i) SLO a intenzita bioluminiscence SLR bez filtru (F0), ii) intenzita bioluminiscence SLO a SLR, která prošla přes filtr 600 nm LP (filtr 1), a iii) vypočítají se prostupové koeficienty filtru 600 nm LP pro SLO a SLR uvedené níže.
|
Prostupové koeficienty
|
Zkratka
|
Definice
|
|
SLO
|
filtr 1 prostupové koeficienty
|
κOR60
|
prostupový koeficient filtru u SLO
|
|
SLR
|
filtr 1 prostupové koeficienty
|
κRR60
|
prostupový koeficient filtru u SLR
|
Když je ve zkušebním vzorku definována intenzita SLO a SLR jako O, respektive R, i) je intenzita světla bez filtru (celooptický) F0 a ii) intenzita světla, které prochází přes filtr 600 nm LP (filtr 1) F1 popsána, jak je uvedeno níže.
F0 = O + R
F1 = κOR60 x O + κRR60 x R
Tyto vzorce lze vyjádřit takto:
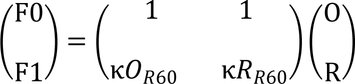
Poté můžete pomocí vypočtených činitelů prostupnosti (κOR60 a κRR60) a změřených hodnot F0 a F1 vypočítat hodnoty O a R takto:
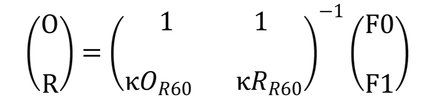
Materiál a metody pro stanovení činitele prostupnosti
|
1)
|
Reagencie
Jednotlivé purifikované luciferázové enzymy:
|
lyofilizovaný purifikovaný enzym SLO
|
|
lyofilizovaný purifikovaný enzym SLR
|
|
(které byly pro validaci získány od společnosti GPC Lab. Co. Ltd., Tottori, Japonsko s buněčnou linií THP-G8)
|
Zkušební reagencie:
|
zkušební reagencie Tripluc® Luciferase (např. od společnosti TOYOBO kat. č. MRA-301)
|
Médium: pro analýzu luciferázy (30 ml, uchováváno při 2–8°C)
|
|
reagencie
|
konc.
|
konečná konc. v médiu
|
potřebné množství
|
|
RPMI-1640
|
–
|
–
|
27 ml
|
|
FBS
|
–
|
10 %
|
3 ml
|
|
2)
|
Příprava enzymatického roztoku
Lyofilizovaný purifikovaný enzym luciferázy rozpusťte ve zkumavce s 200 μl 10 ~ 100 mM Tris/HCl nebo Hepes/HCl (pH 7,5 ~ 8,0) a doplňte 10 % (objemové hmotnosti) glycerolu, enzymatický roztok rozdělte po 10 μl do jednorázových zkumavek o objemu 1,5 ml a uložte je do mrazničky při –80°C. Zmrazený enzymatický roztok můžete používat po dobu 6 měsíců. Při použití přidejte do každé zkumavky obsahující enzymatický roztok (naředěný enzymatický roztok) 1 ml média pro analýzu luciferázy (RPMI-1640 s 10 % FBS) a uložte je na led, abyste zabránili deaktivaci.
|
|
3)
|
Měření bioluminiscence
Rozmrazte zkušební reagencii Tripluc® Luciferase (Tripluc) a nechte ji při pokojové teplotě ve vodní lázni nebo při teplotě okolního vzduchu. Luminometr zapněte 30 minut před zahájením měření, aby se stabilizoval fotonásobič. Přeneste 100 μl naředěného enzymatického roztoku do černé 96jamkové destičky (s plochým dnem) (referenční vzorek SLO do jamek B1, B2, B3, referenční vzorek SLR do jamek D1, D2, D3). Poté napipetujte 100 μl předem ohřátého přípravku Tripluc do každé jamky destičky obsahující naředěný enzymatický vzorek. Pomocí třepačky protřepávejte destičku po dobu 10 minut při pokojové teplotě (přibližně 25°C). Pokud se v roztoku v jamkách objeví bublinky, odstraňte je. Destičku vložte do luminometru a změřte aktivitu luciferázy. Bioluminiscence se měří po dobu 3 sekund, a to bez optického filtru (F0) a s optickým filtrem (F1).
Prostupové koeficienty optického filtru byly vypočteny takto:
Prostupový koeficient (SLO (κOR60)) = (#B1 z F1+ #B2 z F1+ #B3 z F1) / (#B1 z F0+ #B2 z F0+ #B3 z F0)
Prostupový koeficient (SLR (κRR60)) = (#D1 z F1+ #D2 z F1+ #D3 z F1) / (#D1 z F0+ #D2 z F0+ #D3 z F0)
Vypočítané prostupové faktory se použijí pro všechna měření prováděná pomocí stejného luminometru.
|
KONTROLA KVALITY VYBAVENÍ
Použijí se postupy uvedené v protokolu k analýze IL-8 Luc (18).
Dodatek 3.3
LÁTKY PRO PROKÁZÁNÍ ZPŮSOBILOSTI
Dříve než začnou laboratoře běžně používat zkoušku popsanou v tomto dodatku ke zkušební metodě B.71, měly by prokázat odbornou způsobilost získáním předpokládané předpovědi analýzy IL-8 Luc u 9 vhodných látek doporučených v tabulce 1 a získáním hodnot, které spadají do příslušného referenčního rozpětí, u nejméně 8 z 9 vhodných látek (zvolených tak, aby zastupovaly celý rozsah odpovědí pro rizika senzibilizace kůže). Dalšími kritérii výběru bylo, aby látky byly komerčně dostupné, aby k nim byly k dispozici kvalitní referenční údaje in vivo a také kvalitní údaje in vitro získané analýzou IL-8 Luc. Pro analýzu IL-8 Luc jsou také k dispozici zveřejněné referenční údaje (6) (1).
Tabulka 1
Doporučené látky pro prokázání odborné způsobilosti k provádění analýzy IL-8 Luc
|
Látky pro prokázání způsobilosti
|
č. CAS
|
Skupenství
|
Rozpustnost v X-VIVO15 při 20 mg/ml
|
Předpověď in vivo
(108)
|
Predikce podle IL-8 Luc (109)
|
Referenční rozpětí (μg/ml) (110)
|
|
|
CV05
(111)
|
IL-8 Luc MIT (112)
|
|
2,4-dinitrochlorbenzen
|
97-00-7
|
pevná látka
|
nerozpustná
|
senzibilizující (extrémně)
|
pozitivní
|
2,3–3,9
|
0,5–2,3
|
|
formaldehyd
|
50-00-0
|
kapalina
|
rozpustná
|
senzibilizující (silně)
|
pozitivní
|
9–30
|
4–9
|
|
2-sulfanylbenzothiazol
|
149-30-4
|
pevná látka
|
nerozpustná
|
senzibilizující (středně silně)
|
pozitivní
|
250–290
|
60–250
|
|
ethylendiamin
|
107-15-3
|
kapalina
|
rozpustná
|
senzibilizující (středně silně)
|
pozitivní
|
500–700
|
0,1–0,4
|
|
ethylenglykol-dimethakrylát
|
97-90-5
|
kapalina
|
nerozpustná
|
senzibilizující (slabě)
|
pozitivní
|
> 2000
|
0,04–0,1
|
|
4-allylanizol (estragol)
|
140-67-0
|
kapalina
|
nerozpustná
|
senzibilizující (slabě)
|
pozitivní
|
> 2000
|
0,01–0,07
|
|
streptomycin sulfát
|
3810-74-0
|
pevná látka
|
rozpustná
|
není senzibilizující
|
negativní
|
> 2000
|
> 2000
|
|
glycerol
|
56-81-5
|
kapalina
|
rozpustná
|
není senzibilizující
|
negativní
|
> 2000
|
> 2000
|
|
Propan-2-ol
|
67-63-0
|
kapalina
|
rozpustná
|
není senzibilizující
|
negativní
|
> 2000
|
> 2000
|
|
Zkratky: Číslo CAS = registrační číslo Chemical Abstracts Service
|
Dodatek 3.4
INDEXY A KRITÉRIA POSUZOVÁNÍ
nIL8LA (nSLO-LA)
Pro IL8LA (SLO-LA), respektive GAPLA (SLR-LA) se měří j-té opakování (j = 1–4) i-té koncentrace (i = 0–11). Normalizovaná IL8LA, označovaná nIL8LA (nSLO-LA), je definována jako:
nIL8LAij = IL8LAij/GAPLAij
Je to základní jednotka měření v této analýze.
Ind-IL8LA (FInSLO-LA)
Násobné zvýšení zprůměrované nIL8LA (nSLO-LA) pro opakování i-té koncentrace v porovnání s touto hodnotou při koncentraci 0, Ind-IL8LA je primárním měřítkem této analýzy. Tento poměr se zapisuje pomocí následující rovnice:
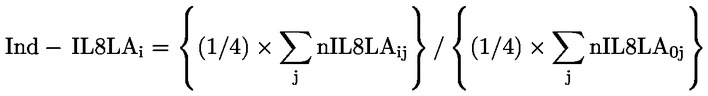
Vedoucí laboratoř navrhla, aby pozitivnímu výsledku pro zkoušenou chemickou látku odpovídala hodnota 1,4. Tato hodnota vychází ze studia dosavadních údajů vedoucí laboratoře. Tým pro správu dat poté používal tuto hodnotu ve všech fázích validační studie. Primární výsledek, Ind-IL8LA, je podíl dvou aritmetických průměrů, jak ukazuje rovnice.
95 % KONFIDENČNÍ INTERVAL (95 % CI)
Může být odhadnut 95 % konfidenční interval (95 % CI) na základě podílu, který ukazuje přesnost tohoto primárního výsledného měřítka. Dolní limit 95 % CI ≥ 1 ukazuje, že nIL8LA v i-té koncentraci je významně větší než tato hodnota při kontrole s rozpouštědlem. 95 % CI lze sestavit několika způsoby. V této studii jsme použili metodu známou jako Fiellerův teorém. Tento teorém 95 % konfidenčního intervalu se získá z následujícího vzorce:
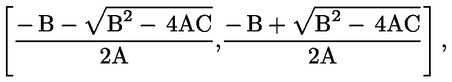
kde .
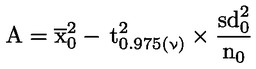
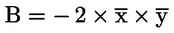
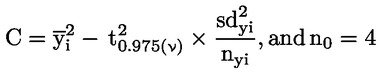
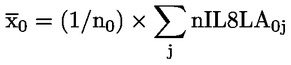
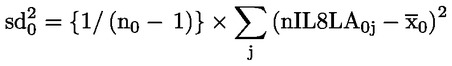
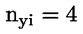
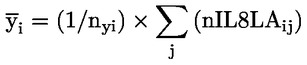
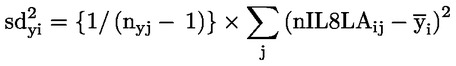
je 97,5 percentil centrálního t rozdělení s ν stupněmi volnosti, kde
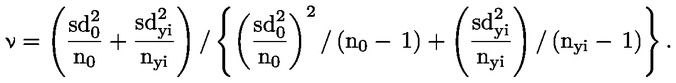
Inh-GAPLA (II-SLR-LA)
Inh-GAPLA je poměr zprůměrované GAPLA (SLR-LA) pro opakování i-té koncentrace v porovnání s koncentrací u kontroly s rozpouštědlem a zapisuje se jako
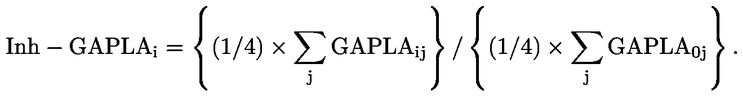
Protože GAPLA je jmenovatelem nIL8LA, působí extrémně malá hodnota velkou odchylku v nIL8LA. Hodnoty Ind-IL8LA s extrémně malou hodnotou Inh-GAPLA (méně než 0,05) mohou být tedy považovány za špatnou přesnost.
Dodatek 3.5
SCHÉMA METOD ROZPOUŠTĚNÍ CHEMICKÝCH LÁTEK PRO ANALÝZU IL-8 LUC.
|
a)
|
Pro chemické látky rozpustné v X-VIVOTM 15 při 20 mg/ml
|
|
b)
|
Pro chemické látky nerozpustné v X-VIVOTM 15 při 20 mg/ml
|
Dodatek 3.6
SCHÉMA METOD ROZPOUŠTĚNÍ 4-NBB PRO POZITIVNÍ KONTROLU ANALÝZY IL-8 LUC.
“ |