|
3.
|
Doplňují se další kapitoly, které znějí:
„B.49. ZKOUŠKA IN VITRO NA PŘÍTOMNOST MIKROJADER V BUŇKÁCH SAVCŮ
ÚVOD
|
1.
|
Zkouška in vitro na přítomnost mikrojader (MNvit) je zkouška genotoxicity pro zjišťování přítomnosti mikrojader v cytoplazmě interfázových buněk. Mikrojádra mohou pocházet z acentrických fragmentů chromozomů (tzn. fragmentů, jimž chybí centromera) nebo z celých chromozomů, které nejsou při buněčném dělení schopny se ve stádiu anafáze rozejít k pólům. Zkouška zjišťuje působení klastogenních a aneugenních chemikálií (látek a směsí) (1) (2) v buňkách, které prodělaly buněčné dělení během expozice nebo po expozici zkoušené látce. Tato zkušební metoda (ZM) umožňuje používat protokoly s inhibitorem polymerace aktinu, cytochalasinem B (cytoB), i bez něho. 5. Přidání cytoB před cílovou mitózou umožňuje identifikaci a selektivní analýzu četnosti výskytu mikrojader v buňkách, které prodělaly jednu mitózu, protože tyto buňky jsou dvoujaderné (3) (4). Tato ZM rovněž umožňuje použití protokolů bez blokování cytokineze, pokud existují důkazy, že analyzovaná populace buněk prodělala mitózu.
|
|
2.
|
Kromě použití zkoušky MNvit pro identifikaci chemikálií (látek a směsí), které vyvolávají vznik mikrojader, lze informace o mechanismech poškození chromozomů a tvorbě mikrojader získat rovněž použitím blokování cytokineze, imunochemického značení kinetochorů nebo hybridizace za použití centromerických/telomerických sond (fluorescenční hybridizace in situ (FISH)) (5) (6) (7) (8) (9) (10) (11) (12) (13) (14) (15) (16). Postupy značení a hybridizace lze použít, pokud dojde ke zvýšené tvorbě mikrojader a zkoušející chce zjistit, zda je toto zvýšení výsledkem klastogenního a/nebo aneugenního působení.
|
|
3.
|
Mikrojádra představují poškození přenesené na dceřiné buňky, zatímco chromozomové aberace zjištěné v metafázových buňkách se přenášet nemusí. Jelikož mikrojádra v interfázových buňkách lze posuzovat poměrně objektivně, laboratorní personál potřebuje pouze zjistit, zda buňky prošly dělením, či nikoli, a kolik buněk obsahuje mikrojádra. Díky tomu lze preparáty vyhodnotit poměrně rychle a analýzu lze automatizovat. Proto je praktické hodnotit při každé zkoušce tisíce namísto stovek buněk, čímž se zvyšuje výkonnost této metody. A konečně, jelikož mikrojádra mohou vznikat z méně vyvinutých chromozomů, je možno zjišťovat látky, které vyvolávají aneuploidii, jež lze obtížně studovat za použití běžných zkoušek na chromozomové aberace, např. podle zkušební metodiky OECD č. 473 (kapitola B.10 této přílohy) (17). Zkouška MNvit však neumožňuje odlišit chemické látky vyvolávající polyploidii od látek vyvolávajících klastogenicitu bez speciálních postupů, jako je například metoda FISH popsaná v odstavci 2.
|
|
4.
|
Zkouška MNvit je metoda in vitro, která obvykle využívá kultivované lidské buňky nebo buňky pocházející z hlodavců. Poskytuje komplexní základ pro vyšetřování potenciálu látek poškozovat chromozomy in vitro, protože jejím prostřednictvím lze detekovat aneugeny i klastogeny.
|
|
5.
|
Zkouška MNvit je robustní a účinná u široké škály různých typů buněk, za přítomnosti nebo nepřítomnosti cytoB. Existuje množství údajů potvrzujících platnost zkoušky MNvit za použití různých buněčných linií pocházejících z hlodavců (CHO, V79, CHL/IU a L5178Y) a také za použití lidských lymfocytů (18) (19) (20) (21) (22) (23) (24) (25) (26) (27) (28) (29) (30) (31). Patří mezi ně zejména mezinárodní validační studie koordinované společností Société Française de Toxicologie Génétique (SFTG) (18) (19) (20) (21) (22) a zprávy z Mezinárodního semináře o zkoušení genotoxicity (4) (16). Dostupné údaje byly rovněž přehodnoceny v retrospektivní validační studii v rámci přístupu založeného na vážení důkazů, provedené Evropským střediskem pro validaci alternativních metod (ECVAM) Evropské komise, a zkušební metoda byla potvrzena jako vědecky platná vědeckým poradním výborem ECVAM (ESAC) (32) (33) (34). Bylo popsáno využití lidské linie lymfoblastoidních buněk TK6 (35), buněk HepG2 (36) (37) a primárních buněk z embryí syrského křečka (38), ačkoli nebyly použity ve validačních studiích.
|
DEFINICE
|
6.
|
Použité definice jsou uvedeny v dodatku 1.
|
VÝCHOZÍ ÚVAHY
|
7.
|
Zkoušky prováděné in vitro obecně vyžadují použití exogenního zdroje metabolické aktivace, pokud nejsou buňky metabolicky kompetentní s ohledem na zkoušené látky. Systém exogenní metabolické aktivace zcela nenapodobuje podmínky in vivo. Je také nutno zabránit vzniku podmínek, které by vedly k uměle pozitivním výsledkům, jež neodrážejí vlastní mutagenitu zkoušené látky, a které mohou nastat působením takových faktorů, jako jsou výrazné změny pH nebo osmolality, nebo v důsledku vysokých úrovní cytotoxicity (39) (40) (41). Pokud zkoušená látka způsobí při přidání změnu pH kultivačního média, je nutno hodnotu pH upravit, nejlépe pufrováním zásobního roztoku tak, aby všechny objemy ve všech zkoušených koncentracích a pro všechny kontroly zůstaly stejné.
|
|
8.
|
Pro analýzu indukce vzniku mikrojader je nezbytné, aby proběhla mitóza v exponovaných i neexponovaných kulturách. Nejinformativnější fází pro hodnocení výskytu mikrojader jsou buňky, které prodělaly jednu mitózu během expozice nebo po expozici zkoušené látce.
|
PODSTATA ZKOUŠKY
|
9.
|
Buněčné kultury lidského a savčího původu jsou vystaveny zkoušené látce za přítomnosti exogenního zdroje metabolické aktivace i bez něho, pokud ovšem nejsou použity buňky s dostatečnou schopností metabolismu. Do všech zkoušek se zařadí souběžné kontroly s rozpouštědlem nebo vehikulem a pozitivní kontrolní látky.
|
|
10.
|
Během nebo po expozici zkoušené látce se buňky kultivují dostatečně dlouho, aby poškození chromozomů nebo dělicího vřeténka vedlo v interfázových buňkách ke vzniku mikrojader. Pro vyvolání aneuploidie by měla být zkoušená látka obvykle přítomna při mitóze. Získané a obarvené interfázové buňky se analyzují na přítomnost mikrojader. V ideálním případě by měla být přítomnost mikrojader zjištěna pouze v těch buňkách, které prodělaly mitózu během expozice zkoušené látce nebo během postexpoziční doby, pokud se používá. V kulturách, které byly ošetřeny blokátorem cytokineze, se toho dosáhne tím, že se zaznamenají pouze dvoujaderné buňky. V případě nepřítomnosti blokátoru cytokineze je nutno prokázat, že analyzované buňky pravděpodobně prodělaly buněčné dělení během expozice nebo po expozici zkoušené látce. U všech protokolů je důležité prokázat, že došlo k proliferaci buněk jak v kontrolních, tak v ošetřených kulturách, a také je třeba v kulturách (nebo v paralelních kulturách), u nichž se hodnotí výskyt mikrojader, posoudit míru cytotoxicity nebo cytostáze.
|
POPIS ZKOUŠKY
Preparáty
|
11.
|
Lze použít kultivované primární lidské lymfocyty z periferní krve (5) (19) (42) (43) a celou řadu buněčných linií pocházejících z hlodavců, například buňky CHO, V79, CHL/IU a L5178Y (18) (19) (20) (21) (22) (25) (26) (27) (28) (30). Použití jiných buněčných linií a druhů by mělo být odůvodněno na základě jejich prokázané funkčnosti ve zkoušce, jak je popsáno v části ‚Kritéria přijatelnosti‘. Jelikož četnost mikrojader v rámci přirozeného pozadí ovlivní citlivost testu, doporučuje se používat druhy buněk s nízkou, stabilní četností tvorby mikrojader v rámci přirozeného pozadí.
|
|
12.
|
Lidské lymfocyty z periferní krve by měly být získány od mladých (přibližně ve věku 18–35 let) zdravých nekuřáků bez jakékoli známé nedávné expozice genotoxickým látkám nebo radiaci. Pokud se společně používají buňky pocházející od více než jednoho dárce, měl by být počet dárců uveden. Četnost mikrojader se zvyšuje s věkem a tato tendence je výraznější u žen než u mužů (44), což je třeba vzít v úvahu při výběru dárcovských buněk pro společné použití.
|
Média a podmínky kultivace
|
13.
|
Pro udržování kultur by se měla používat vhodná kultivační média a inkubační podmínky (kultivační nádoby, koncentrace CO2, teplota a vlhkost). U stabilizovaných buněčných linií a kmenů by se měla pravidelně kontrolovat stabilita modální hodnoty počtu chromozomů a mělo by se zjišťovat, zda nejsou kontaminovány mykoplasmaty. Pokud jsou kultury kontaminované nebo se modální hodnota počtu chromozomů změnila, neměly by se dotyčné kultury používat. Je třeba, aby bylo známo normální trvání buněčného cyklu při kultivačních podmínkách použitých ve zkušební laboratoři. V případě použití metody s blokováním cytokineze by se koncentrace inhibitoru cytokineze měla optimalizovat pro konkrétní typ buněk a mělo by se prokázat, že tato koncentrace poskytuje dostatečné množství dvoujaderných buněk pro vyhodnocení zkoušky.
|
Příprava kultur
|
14.
|
Stabilizované buněčné linie a kmeny: buňky se pomnoží z kmenových kultur, nasadí se do kultivačního média v takové hustotě, aby před odebráním nedosáhly běžné kultury konfluence v monovrstvách a aby suspenzní kultury nedosáhly nadměrné hustoty, a inkubují se při teplotě 37 °C.
|
|
15.
|
Lymfocyty: plná krev ošetřená antikoagulantem (např. heparinem) nebo oddělené lymfocyty se před expozicí zkoušené látce a cytoB kultivují za přítomnosti mitogenu, např. fytohemaglutininu (PHA).
|
Metabolická aktivace
|
16.
|
Při použití buněk s nedostatečnou endogenní metabolickou schopností by se měly použít exogenní metabolizující systémy. Nejčastěji používaným systémem je kofaktorem dotovaná postmitochondriální frakce (S9) připravená z jater hlodavců ošetřených činidlem vyvolávajícím tvorbu enzymů, jako je například Aroclor 1254 (45) (46), nebo směsí fenobarbitonu a β-naftoflavonu (46) (47) (48) (49). Použití uvedené směsi není v rozporu se Stockholmskou úmluvou o perzistentních organických znečišťujících látkách (50) a s nařízením (ES) č. 850/2004 o perzistentních organických znečišťujících látkách (66), přičemž bylo prokázáno, že co se týče vyvolání tvorby oxidáz se smíšenou funkcí, je tato směs stejně účinná jako Aroclor 1254 (46) (47) (48) (49). V konečném zkušebním médiu se frakce S9 obvykle používá v koncentracích od 1 do 10 % (v objemovém vyjádření). Stav systému pro aktivaci metabolismu může záviset na chemické třídě zkoušené látky a v některých případech může být vhodné použít více než jednu koncentraci S9.
|
|
17.
|
Geneticky modifikované buněčné linie vylučující aktivační enzymy specifické pro preparáty získané od lidí a hlodavců mohou eliminovat potřebu exogenního systému pro aktivaci metabolismu a lze je používat jako zkušební buňky. V takových případech je nutno volbu použitých buněčných linií vědecky zdůvodnit, např. důležitostí oxidáz se smíšenou funkcí pro metabolismus zkoušené látky (51) a jejich schopností reagovat na známé klastogeny a aneugeny (viz samostatná část ‚Kritéria přijatelnosti‘). Je třeba počítat s tím, že zkoušené látky nemusí být vyloučenou oxidázou (vyloučenými oxidázami) se smíšenou funkcí metabolizovány; v tom případě by negativní výsledky neprokazovaly, že zkoušená látka nemůže vyvolávat tvorbu mikrojader.
|
Příprava zkoušené látky
|
18.
|
Pevné chemické látky by se měly před použitím k ošetření buněk rozpustit ve vhodných rozpouštědlech nebo vehikulech a popřípadě zředit. Kapalné látky lze přidat přímo do zkušebních systémů a/nebo je lze před použitím k ošetření buněk zředit. Plyny a těkavé látky by se měly zkoušet za použití vhodně upravených standardních protokolů, například prostřednictvím ošetření buněk v neprodyšně uzavřených nádobách (52) (53). Měly by se používat čerstvě připravené zkoušené látky, pokud údaje o stálosti neprokazují možnost skladování.
|
Zkušební podmínky:
Rozpouštědla nebo vehikula
|
19.
|
Rozpouštědlo nebo vehikulum nesmí reagovat se zkoušenou látkou a při použité koncentraci nesmí být neslučitelné s přežitím buněk nebo s udržením aktivity S9. Pokud se používají jiná než zavedená rozpouštědla nebo vehikula (jako jsou například voda, médium pro kultivaci buněk, dimethylsulfoxid), je nutno jejich použití podpořit údaji o jejich kompatibilitě se zkoušenou látkou a o tom, že nejsou genotoxické. Doporučuje se pokud možno nejprve zvážit použití vodného rozpouštědla nebo vehikula.
|
Používání cytoB jako blokátoru cytokineze
|
20.
|
Jedním z nejdůležitějších hledisek při provádění zkoušky MNvit je zajistit, aby hodnocené buňky prodělaly mitózu během expozice zkoušené látce nebo během postexpoziční doby, pokud se používá. CytoB je látka, která se nejčastěji používá k blokování cytokineze, protože inhibuje sestavení aktinu a tím zabraňuje oddělení dceřiných buněk po mitóze, což vede ke vzniku dvoujaderných buněk (5) (54) (55). Proto lze hodnocení výskytu mikrojader omezit pouze na buňky, které prodělaly mitózu během ošetření zkoušenou látkou nebo po něm. Současně lze měřit účinek zkoušené látky na kinetiku buněčné proliferace. CytoB by se měl používat jako blokátor cytokineze při použití lidských lymfocytů, protože délky trvání buněčného cyklu budou u různých kultur a dárců proměnlivé a protože ne všechny lymfocyty budou reagovat na PHA. Při zkoušení buněčných linií za účelem určení, zda se hodnocené buňky rozdělily, se používají i jiné metody, jež jsou uvedeny níže (viz odstavec 26).
|
|
21.
|
V laboratoři by se měla stanovit vhodná koncentrace cytoB pro každý druh buněk, aby se dosáhlo optimální četnosti dvoujaderných buněk v kontrolních kulturách s rozpouštědlem/vehikulem. Vhodná koncentrace cytoB obvykle činí 3 až 6 mg/ml.
|
Měření buněčné proliferace a cytotoxicity a volba expozičních koncentrací
|
22.
|
Při určování nejvyšší koncentrace zkušební látky je nutno se vyhnout koncentracím, které mají schopnost produkovat uměle pozitivní odezvy, například koncentracím, jež vyvolávají velmi vysokou cytotoxicitu, vysrážení v kultivačním médiu a výrazné změny pH nebo osmolality (39) (40) (41).
|
|
23.
|
Měření buněčné proliferace se provádí proto, aby bylo zajištěno, že ošetřené buňky prodělaly během zkoušky mitózu a že se jednotlivá ošetření provádějí při vhodných úrovních cytotoxicity (viz odstavec 29). Cytotoxicita by měla být stanovena s metabolickou aktivací a bez metabolické aktivace u buněk, které vyžadují metabolickou aktivaci pomocí relativního nárůstu počtu buněk (RICC) nebo relativního zdvojnásobení populace (RPD) (příslušné vzorce viz dodatek 2), pokud se ovšem nepoužívá cytoB. Při použití cytoB lze cytotoxicitu stanovit za použití replikačního indexu (RI) (příslušný vzorec viz dodatek 2).
|
|
24.
|
Ošetření kultur cytoB a měření relativních četností jednojaderných, dvoujaderných a vícejaderných buněk v kultuře představuje přesnou metodu kvantifikace účinku na buněčnou proliferaci a cytotoxického nebo cytostatického působení látky použité k ošetření kultury (5) a zajišťuje, že se hodnotí pouze buňky, které prodělaly buněčné dělení během ošetření nebo po něm.
|
|
25.
|
Ve studiích s cytoB lze cytostázi nebo cytotoxicitu kvantifikovat podle proliferačního indexu při blokování cytokineze (CBPI) (5) (26) (56) nebo mohou být odvozeny z RI nejméně 500 buněk v každé kultuře (příslušné vzorce viz dodatek 2). Když se k posouzení buněčné proliferace používá cytoB, měl by se CBPI nebo RI určit nejméně u 500 buněk v každé kultuře. Tato měření lze mimo jiné použít k odhadu cytotoxicity porovnáním hodnot u exponované kultury s hodnotami u kontrolní kultury. Užitečné informace může poskytnout i hodnocení ostatních ukazatelů cytotoxicity (např. konfluence, počtu buněk, apoptózy, nekrózy, počítání metafází).
|
|
26.
|
Ve studiích bez použití cytoB je nutno prokázat, že hodnocené buňky v kultuře prodělaly buněčné dělení během ošetření zkoušenou látkou nebo po něm, jinak mohou vznikat falešně negativní odezvy. Mezi metody, které se doposud používaly k zajištění toho, aby se hodnotily pouze buňky, které prodělaly buněčné dělení, patří vpravení a následná detekce bromdeoxyuridinu (BrdU), který umožňuje identifikovat buňky, které se replikovaly (57), vytváření klonů, když se buňky z permanentních buněčných linií ošetřují a hodnotí in situ na mikroskopickém sklíčku (proliferační index (PI)) (25) (26) (27) (28), nebo měření relativního zdvojnásobení populace (RPD) či relativního nárůstu počtu buněk (RICC) nebo jiné osvědčené metody (16) (56) (58) (59) (příslušné vzorce viz dodatek 2). Užitečné informace může poskytnout i hodnocení ostatních ukazatelů cytotoxicity nebo cytostáze (např. konfluence, počtu buněk, apoptózy, nekrózy, počítání metafází).
|
|
27.
|
Měly by se vyhodnotit nejméně tři analyzovatelné zkušební koncentrace. K tomu může být nezbytné provést experiment za použití většího množství podobných koncentrací a analyzovat tvorbu mikrojader v těch koncentracích, jež poskytují vhodný rozsah hodnot cytotoxicity. Alternativní strategie spočívá v provedení předběžné zkoušky cytotoxicity, aby se zúžil rozsah pro konečnou zkoušku.
|
|
28.
|
Cílem je, aby nejvyšší koncentrace poskytla 55 ± 5 % cytotoxicitu. 32. Vyšší úrovně mohou jako sekundární efekt cytotoxicity vyvolat poškození chromozomů (60). Pokud dochází k výskytu cytotoxicity, měly by zvolené koncentrace pokrývat širokou škálu hodnot, které způsobují různé úrovně cytotoxicity od 55 ± 5 % cytotoxicity až po nízkou nebo žádnou cytotoxicitu.
|
|
29.
|
Pokud není zjištěna žádná cytotoxicita nebo je pozorován vznik sraženiny, měla by nejvyšší zkušební koncentrace odpovídat nejnižší z hodnot 0,01 M, 5 mg/ml nebo 5 μl/ml. Vzájemný rozdíl mezi jednotlivými koncentracemi zvolenými pro analýzu by obecně neměly činit více než 10 %. U zkoušených látek, které vykazují strmé křivky závislosti odezvy na koncentraci, může být nezbytné, zvolit menší kroky mezi jednotlivými koncentracemi zkoušené látky, aby mohly být ohodnoceny i kultury se středními a nízkými rozsahy toxicity.
|
|
30.
|
Když je limitujícím faktorem rozpustnost, měla by se maximální koncentrace, pokud není omezena cytotoxicitou, rovnat nejnižší koncentraci, při které je v kulturách viditelná minimální sraženina, pokud to ovšem neovlivní vyhodnocení. Hodnocení precipitace by se mělo provádět takovými metodami, jako je světelná mikroskopie, přičemž je nutno si všímat sraženiny, která přetrvává nebo se objeví během kultivace (před ukončením expozice).
|
Kontroly
|
31.
|
Součástí každého experimentu by měly být souběžné pozitivní kontroly a kontroly s rozpouštědlem/vehikulem při současné metabolické aktivaci a bez ní.
|
|
32.
|
Pozitivní kontroly jsou nezbytné k prokázání schopnosti použitých buněk a zkušebního protokolu identifikovat klastogeny a aneugeny a potvrdit metabolickou schopnost přípravku S9. Pozitivní kontrola by měla využívat známé látky podněcující tvorbu mikrojader v koncentracích, u nichž se předpokládá malé, avšak reprodukovatelné zvýšení nad hodnoty pozadí, a měla by prokázat citlivost zkušebního systému. Koncentrace pozitivní kontroly by měly být zvoleny tak, aby byl účinek zřetelný, ale aby při odečtu nevyšla ihned najevo totožnost kódovaného preparátu.
|
|
33.
|
Klastogenní látka, která vyžaduje metabolickou aktivaci (např. cyklofosfamid, benzo[a]pyren) by se měla použít k prokázání jak schopnosti metabolismu buněk, tak schopnosti zkušebního systému zjistit přítomnost klastogenů. V odůvodněných případech lze používat i další pozitivní kontroly. Jelikož některé pozitivní kontroly, které vyžadují metabolickou aktivaci, mohou za určitých podmínek ošetření nebo u určitých buněčných linií působit i bez exogenní metabolické aktivace, je nutno u zvolené buněčné linie při zvolených koncentracích otestovat potřebnost metabolické aktivace a rovněž účinnost přípravku S9.
|
|
34.
|
V současné době nejsou známy žádné aneugenní látky, které by ke svému genotoxickému působení vyžadovaly metabolickou aktivaci (16). Mezi v současnosti uznávané pozitivní kontroly aneugenního působení patří například kolchicin a vinblastin. Jiné chemické látky lze použít v případě, že vyvolávají vznik mikrojader výhradně nebo převážně prostřednictvím aneugenního působení. Aby nebyly nutné dvě pozitivní kontroly (na klastogenicitu a aneugenicitu) bez metabolické aktivace, může kontrola aneugenicity sloužit jako pozitivní kontrola bez použití přípravku S9 a kontrolu klastogenicity lze využít k testování vhodnosti použitého systému metabolické aktivace. Pozitivní kontrola na klastogenicitu a aneugenicitu by se měly používat u buněk, které nevyžadují použití přípravku S9. Navrhované pozitivní kontroly jsou uvedeny v dodatku 3.
|
|
35.
|
Lze zvážit použití pozitivní kontroly související s určitou chemickou třídou, pokud jsou k dispozici vhodné chemické látky. Všechny použité pozitivní kontroly by měly být vhodné pro daný druh buněk a podmínky aktivace.
|
|
36.
|
Studie by měla zahrnovat kontroly s rozpouštědlem/vehikulem při každém odběru buněk k vyhodnocení. Kromě toho by se měly používat rovněž neošetřené negativní kontroly (bez rozpouštědla/vehikula), pokud nejsou publikovány nebo pokud nemá laboratoř dřívější kontrolní údaje, jež prokazují, že zvolené rozpouštědlo při použitých koncentracích nevyvolává žádné genotoxické ani jiné škodlivé účinky.
|
ZKUŠEBNÍ POSTUP
Harmonogram aplikace
|
37.
|
Aby se maximalizovala pravděpodobnost detekce aneugenní nebo klastogenní látky působící v určitém stadiu buněčného cyklu, je důležité, aby byl dostatečný počet buněk ošetřen zkoušenou látkou ve všech fázích jejich buněčných cyklů. Harmonogram aplikace pro buněčné linie a primární buněčné kultury se proto může poněkud lišit od harmonogramu aplikace u lymfocytů, které k zahájení svého buněčného cyklu potřebují mitogenní stimulaci, přičemž o jednotlivých harmonogramech pojednávají odstavce 41 až 43 (16).
|
|
38.
|
Teoretické úvahy spolu s publikovanými údaji (18) ukazují, že většina aneugenních a klastogenních látek bude detekována za použití krátkodobé expozice v trvání 3 až 6 hodin za přítomnosti i nepřítomnosti přípravku S9, po níž následuje odstranění zkoušené látky a doba růstu v délce 1,5 až 2,0 buněčných cyklů (6). Vzorky buněk se odebírají v době odpovídající asi 1,5násobku až 2,0násobku trvání běžného buněčného cyklu (tzn. bez aplikace zkoušené látky), buď po začátku nebo na konci aplikace (viz tabulka 1). Intervaly odběru vzorků nebo obnovy kultur lze prodloužit, pokud je známo nebo existuje podezření, že zkoušená látka ovlivňuje trvání buněčného cyklu (například při zkoušení analogů nukleosidů).
|
|
39.
|
Vzhledem k možnému cytotoxickému působení přípravků S9 na kultivované savčí buňky se prodloužená expozice v trvání 1,5násobku až 2,0násobku běžných buněčných cyklů používá jen za nepřítomnosti S9. Při prodloužené expozici existují možnosti, které dovolují ošetřit buňky zkoušenou látkou za nepřítomnosti nebo za přítomnosti cytoB. Tyto možnosti jsou určeny pro situace, kdy mohou být obavy ohledně možných interakcí mezi zkoušenou látkou a cytoB.
|
|
40.
|
Navrhované harmonogramy expozice buněk jsou uvedeny v tabulce 1. Tyto obecné harmonogramy expozice lze upravit v závislosti na stabilitě a reaktivitě zkoušené látky nebo zvláštních růstových charakteristikách použitých buněk. Veškeré expozice by měly být zahájeny a ukončeny v okamžiku, kdy počet buněk exponenciálně narůstá. Tyto harmonogramy jsou podrobněji uvedeny v následujících odstavcích 41 až 47.
Tabulka 1
Doby expozic a odběru buněk k vyhodnocení v rámci zkoušky MNvit
|
Lymfocyty, primární buňky a buněčné linie ošetřené s cytoB
|
+ S9
|
Expozice po dobu 3–6 hod. za přítomnosti S9;
odstranění S9 a expozičního média;
doplnění čerstvého média a cytoB;
odběr k vyhodnocení po uplynutí doby odpovídající 1,5násobku až 2,0násobku trvání běžných buněčných cyklů.
|
|
– S9
Krátká expozice
|
Expozice po dobu 3–6 hod.;
odstranění expozičního média;
doplnění čerstvého média a cytoB;
odběr k vyhodnocení po uplynutí doby odpovídající 1,5násobku až 2,0násobku trvání běžných buněčných cyklů.
|
|
– S9
Prodloužená expozice
|
Možnost A: Expozice po dobu odpovídající 1,5násobku až 2,0násobku trvání běžných buněčných cyklů za přítomnosti cytoB;
odběr k vyhodnocení na konci doby expozice.
Možnost B: Expozice po dobu odpovídající 1,5násobku až 2,0násobku trvání běžných buněčných cyklů;
odstranění zkušební látky;
doplnění čerstvého média a cytoB;
odběr k vyhodnocení po uplynutí doby odpovídající 1,5násobku až 2,0násobku trvání běžných buněčných cyklů.
|
|
Buněčné linie ošetřené bez cytoB
(Harmonogramy jsou shodné s harmonogramy uvedenými výše s výjimkou toho, že se nepřidává žádný cytoB.)
|
|
Lymfocyty, primární buňky a buněčné linie s cytoB
|
41.
|
U lymfocytů je nejúčinnější zahájit expozici zkoušené látce za 44–48 hodin po stimulaci PHA, až zmizí synchronizace cyklů (5). Při úvodní zkoušce se nechá na buňky působit zkoušená látka po dobu 3 až 6 hodin za nepřítomnosti a za přítomnosti S9. Zkušební médium se odstraní a nahradí čerstvým médiem obsahujícím cytoB a buňky se odeberou k vyhodnocení po uplynutí doby odpovídající 1,5násobku až 2,0násobku trvání běžných buněčných cyklů.
|
|
42.
|
Pokud jsou obě úvodní zkoušky s krátkou expozicí (3–6 hod.) negativní nebo nejednoznačné, použije se následná prodloužená expozice bez přítomnosti S9. K dispozici jsou dvě možnosti provedení zkoušky, přičemž obě jsou stejně přijatelné. Může však být vhodnější postupovat podle možnosti A pro stimulované lymfocyty, kde může exponenciální nárůst počtu buněk začít 96 hodin po stimulaci klesat. Při použití možnosti B by také buněčné kultury neměly před konečným odběrem vzorku dosáhnout konfluence.
|
—
|
Možnost A: Buňky jsou vystaveny působení zkoušené látky po dobu odpovídající 1,5násobku až 2,0násobku trvání běžných buněčných cyklů a odeberou se k vyhodnocení na konci doby expozice.
|
|
—
|
Možnost B: Buňky jsou vystaveny působení zkoušené látky po dobu odpovídající 1,5násobku až 2,0násobku trvání běžných buněčných cyklů. Zkušební médium se odstraní a nahradí čerstvým médiem a buňky se odeberou k vyhodnocení po uplynutí doby odpovídající 1,5násobku až 2,0násobku trvání běžných buněčných cyklů.
|
|
|
43.
|
Primární buňky a buněčné linie je nutno ošetřit stejným způsobem jako lymfocyty kromě toho, že není nutno je stimulovat pomocí PHA po dobu 44–48 hodin. Jiné buňky než lymfocyty by měly být vystaveny působení zkoušené látky tak, aby v okamžiku ukončení studie byly buňky stále ve fázi růstu, při které se jejich počet exponenciálně zvyšuje.
|
Buněčné linie bez cytoB
|
44.
|
Buňky je nutno vystavit působení zkoušené látky po dobu 3–6 hodin za přítomnosti a nepřítomnosti S9. Zkušební médium se odstraní a nahradí čerstvým médiem a buňky se odeberou k vyhodnocení po uplynutí doby odpovídající 1,5násobku až 2,0násobku trvání běžných buněčných cyklů.
|
|
45.
|
Pokud jsou obě úvodní zkoušky s krátkou expozicí (3–6 hod.) negativní nebo nejednoznačné, použije se následná prodloužená expozice (bez přítomnosti S9). K dispozici jsou dvě možnosti provedení zkoušky, přičemž obě jsou stejně přijatelné.
|
—
|
Možnost A: Buňky jsou vystaveny působení zkoušené látky po dobu odpovídající 1,5násobku až 2,0násobku trvání běžných buněčných cyklů a odeberou se k vyhodnocení na konci doby expozice.
|
|
—
|
Možnost B: Buňky jsou vystaveny působení zkoušené látky po dobu odpovídající 1,5násobku až 2,0násobku trvání běžných buněčných cyklů. Zkušební médium se odstraní a nahradí čerstvým médiem a buňky se odeberou k vyhodnocení po uplynutí doby odpovídající 1,5násobku až 2,0násobku trvání běžných buněčných cyklů.
|
|
|
46.
|
Na konci doby expozice v trvání 3–6 hodin se v monovrstvách mohou vyskytovat mitotické buňky (které se vyznačují okrouhlým tvarem a odchlipováním od povrchu). Jelikož lze tyto mitotické buňky snadno oddělit, mohou být při odstranění média obsahujícího zkoušenou látku ztraceny. Tyto buňky je třeba při promývání kultur shromáždit a vrátit je zpět, aby při odběru k vyhodnocení nedošlo ke ztrátě buněk, které se nacházejí ve fázi mitózy a jsou ohroženy vznikem mikrojader.
|
Počet kultur
|
47.
|
Pro každou koncentraci zkoušené látky a v případě kultur s vehikulem/rozpouštědlem a kultur negativních kontrol je nutno použít duplicitní kultury. Pokud lze na základě předešlých laboratorních údajů prokázat minimální rozdíl mezi duplicitními kulturami, může být přijatelné použít jen po jedné kultuře. V případě použití vždy jen jedné kultury se doporučuje analyzovat zvýšený počet koncentrací.
|
Odběr buněk a příprava preparátů
|
48.
|
Buňky z každé kultury se odebírají a zpracovávají zvlášť. Příprava buněk může zahrnovat hypotonizaci, ale tento krok není nutný, pokud se dosáhne odpovídajícího rozšíření buněk jiným způsobem. Při přípravě preparátů lze použít různé postupy za předpokladu, že zajistí získání vysoce kvalitních buněčných preparátů pro vyhodnocení. Cytoplazmu buněk je nutno uchovat, aby bylo možné provést detekci mikrojader a (v případě metody s blokováním cytokineze) spolehlivou identifikaci dvoujaderných buněk.
|
|
49.
|
Preparáty lze barvit pomocí různých metod, jako je Giemsa nebo použití fluorescenční barviv se specifickou vazbou na DNA (59). Použitím barviva se specifickou vazbou na DNA (jako je např. akridinová oranž (61) nebo Hoechst 33258 a pyronin-Y (62)) se lze vyhnout některým artefaktům spojeným s použitím barviva bez specifické vazby na DNA. Jsou-li předmětem zájmu informace o mechanismu vzniku mikrojader, lze k identifikaci jejich obsahu (chromozom nebo chromozomální fragment) využít protilátky proti kinetochorům, hybridizační metodu FISH využívající pan-centromerické DNA-sondy, případně značení in situ pomocí primerů se specifickou pan-centromerickou vazbou spolu s vhodným kontrastním barvením DNA (15) (16). K rozlišení mezi klastogeny a aneugeny lze použít i jiné metody, pokud se ukázaly jako účinné.
|
Analýza
|
50.
|
Všechny preparáty včetně preparátů s rozpouštědlem/vehikulem a preparátů kontrol by měly být před analýzou pod mikroskopem nezávisle označeny kódem. Nebo lze kódované vzorky analyzovat za použití validovaného automatického systému pro průtokovou cytometrii nebo systému pro analýzu obrazu.
|
|
51.
|
V případě kultur ošetřených pomocí cytoB by se měla analyzovat četnost výskytu mikrojader nejméně u 2 000 dvoujaderných buněk pro každou koncentraci (nejméně u 1 000 dvoujaderných buněk z každé kultury; po dvou kulturách pro každou koncentraci). V případě použití vždy jen jedné kultury by se mělo z každé kultury vyhodnotit alespoň 2 000 dvoujaderných buněk pro každou koncentraci. Pokud je k dispozici k vyhodnocení pro každou koncentraci podstatně méně než 1 000 dvoujaderných buněk z každé kultury (respektive než 2 000, jestliže se použije jen jedna kultura) a není-li zjištěn výrazný nárůst výskytu mikrojader, musí se zkouška zopakovat za použití většího počtu buněk nebo při méně toxických koncentracích (podle toho, co z těchto dvou možností připadá v úvahu). Je třeba dbát na to, aby se nezapočítávaly dvoujaderné buňky, které mají nepravidelný tvar nebo jejichž dvě jádra se svou velikostí navzájem značně liší; také se nesmějí za dvoujaderné buňky mylně považovat nedostatečně rozšířené jednojaderné buňky. Buňky obsahující více než dvě hlavní jádra by se neměla analyzovat na přítomnost mikrojader, jelikož základní četnost mikrojader může být v těchto buňkách vyšší (63) (64). Započtení jednojaderných buněk je přijatelné, pokud se prokáže, že zkoušená látka narušuje působení cytoB.
|
|
52.
|
U buněčných linií zkoušených bez ošetření cytochalasinem B by se měla vyhodnotit četnost mikrojader alespoň u 2 000 buněk pro každou koncentraci (alespoň u 1 000 buněk z každé kultury; po dvou kulturách pro každou koncentraci). Při použiti pouze jedné kultury pro každou koncentraci by se mělo vyhodnotit nejméně 2 000 buněk z dané kultury.
|
|
53.
|
Při použití cytoB by se měla určit hodnota CBPI nebo RI k posouzení buněčné proliferace (viz dodatek 2) za použití nejméně 500 buněk z každé kultury. Když se expozice provádí bez přítomnosti cytoB, je nezbytné prokázat, že hodnocené buňky prodělaly proliferaci, jak je vysvětleno v odstavcích 24 až 27.
|
Kritéria přijatelnosti
|
54.
|
Laboratoř, která navrhuje používání zkoušky MNvit popsané v této ZM, by měla prokázat svou schopnost spolehlivě a přesně detekovat chemické látky se známým aneugenním a klastogenním působením, při použití metabolické aktivace i bez ní, jakož i známé negativní látky za použití srovnávacích látek uvedených v dodatku 3. Svou schopnost správně provádět tuto ZM by měla laboratoř prokázat předložením důkazů, že buňky, u nichž vyhodnocuje tvorba mikrojader, prodělaly jedno jaderné dělení, pokud se zkouška provádí bez použití cytoB.
|
|
55.
|
Jako srovnávací látky se doporučuje používat chemické látky uvedené v dodatku 3. Náhradní nebo další chemické látky lze rovněž používat, pokud jsou známy jejich účinky, pokud vyvolávají tvorbu mikrojader stejným mechanismem působení a pokud je prokázáno, že mají vztah k chemickým látkám, které se budou testovat za použití postupu MNvit. Součástí zdůvodnění může být validační studie využívající širokou škálu látek nebo zaměřená na užší spektrum na základě chemické třídy zkoušené látky nebo zkoumaného mechanismu vzniku poškození.
|
|
56.
|
Kontrola s rozpouštědlem/vehikulem a neošetřené kultury by měly poskytovat reprodukovatelně nízké a konzistentní četnosti mikrojader (obvykle 5–25 mikrojader na 1 000 buněk v případě druhů buněk uvedených v odstavci 11). Jiné druhy buněk mohou mít odlišné rozsahy odezvy, které by měly být stanoveny při validaci jejich používání v rámci zkoušky MNvit. Údaje z negativních kontrol, kontrol s rozpouštědlem a pozitivních kontrol by měly sloužit ke stanovení dosavadních kontrolních rozmezí. Tyto hodnoty by se měly použít při rozhodování o vhodnosti zařazení souběžných negativních nebo pozitivních kontrol do experimentu.
|
|
57.
|
Jsou-li pro provedení zkoušky navrhovány menší změny protokolu (např. použití automatizovaných namísto ručních postupů vyhodnocování, použití nového druhu buněk), je nutno prokázat účelnost této změny, nežli lze upravený protokol považovat za přijatelný k použití. Součástí prokázání účelnosti je důkaz, že lze zjistit hlavní mechanismy poškození chromozomů a zvýšení nebo snížení jejich počtu a že lze dosáhnout odpovídajících pozitivních i negativních výsledků u třídy jednotlivé látky nebo širokého spektra látek, které se mají testovat.
|
ÚDAJE A VYKAZOVÁNÍ
Zpracování výsledků
|
58.
|
Pokud se používá technika blokování cytokineze, slouží k hodnocení indukce vzniku mikrojader jen četnost dvoujaderných buněk s mikrojádry (nezávisle na počtu mikrojader v každé buňce). Počítání buněk s jedním, dvěma nebo více mikrojádry by mohlo poskytnout užitečné informace, ale není povinné.
|
|
59.
|
Souběžně by se mělo provádět měření cytotoxicity a/nebo cytostáze u všech ošetřených kultur a u kontrolních kultur s rozpouštědlem/vehikulem (58). Při použití metody s blokováním cytokineze, by se měly vypočítat hodnoty CBPI nebo RI u všech ošetřených a kontrolních kultur jako ukazatele zpoždění buněčného cyklu. V případě nepřítomnosti cytoB by se měly použít hodnoty RPD, RICC nebo PI (viz dodatek 2).
|
|
60.
|
Je nutno uvést údaje za jednotlivé kultury. Kromě toho se všechny údaje shrnou do tabulky.
|
|
61.
|
Chemické látky, které vyvolávají vznik mikrojader při zkoušce MNvit, mají tento účinek proto, že vyvolávají poškození chromozomů, ztrátu chromozomů nebo kombinaci obou těchto dějů. Při určování, zda je mechanismus vyvolání tvorby mikrojader důsledkem klastogenního a/nebo aneugenního působení, lze uplatnit další analýzu za použití protilátek proti kinetochorům, sond in situ se specifickou vazbou na centromery nebo jiných metod.
|
Hodnocení a interpretace výsledků
|
62.
|
Žádné ověření jednoznačně pozitivní nebo negativní odezvy pomocí dodatečné zkoušky není nutné. Nejednoznačné výsledky lze objasnit analýzou dalšího 1 000 buněk ze všech kultur, aby se zabránilo ztrátě zaslepení. Pokud tento přístup výsledek neobjasní, mělo by se provést další testování. Při následných experimentech by se mělo zvážit upravení parametrů studie u rozšířeného nebo zúženého rozsahu podmínek podle toho, co přichází v úvahu. Mezi parametry studie, které lze případně upravit, patří velikost kroku mezi jednotlivými zkušebními koncentracemi, načasování expozice a odběru buněk k vyhodnocení a/nebo podmínky metabolické aktivace.
|
|
63.
|
Existuje několik kritérií pro stanovení pozitivního výsledku, například s koncentrací související nebo statisticky významné zvýšení počtu buněk obsahujících mikrojádra. Nejprve je nutno posoudit biologický význam výsledků. Při hodnocení významu biologické odpovědi může být vodítkem posouzení, zda jsou zjištěné hodnoty uvnitř nebo vně rozsahu dosavadních hodnot kontrol. Při hodnocení výsledků zkoušky lze jako pomůcku použít vhodné statistické metody (65). Výsledky statistických testů je však třeba posuzovat s ohledem na závislost odezvy na dávku. V úvahu je nutno vzít také reprodukovatelnost a dosavadní údaje.
|
|
64.
|
Ačkoli většina experimentů poskytne jednoznačně pozitivní nebo negativní výsledky, v některých případech neumožní soubor údajů vyslovit konečný výrok o působení zkoušené látky. Tyto dvojznačné nebo sporné odezvy se mohou vyskytovat bez ohledu na to, kolikrát je experiment opakován.
|
|
65.
|
Pozitivní výsledky zkoušky MNvit znamenají, že zkoušená látka vyvolává v kultivovaných savčích buňkách poškození nebo ztrátu chromozomů. Negativní výsledky znamenají, že zkoušená látka za použitých podmínek zkoušky nevyvolává poškození chromozomů a/nebo nárůst či pokles počtu kultivovaných savčích buněk.
|
Protokol o zkoušce
|
66.
|
Protokol o zkoušce musí obsahovat alespoň tyto informace, pokud jsou pro provedení studie významné:
|
|
Zkoušená látka:
|
—
|
identifikační údaje a registrační číslo CAS a číslo ES,
|
|
—
|
fyzikálně-chemické vlastnosti významné pro provedení studie,
|
|
—
|
reaktivita zkoušené látky s rozpouštědlem/vehikulem nebo médiem pro kultivaci buněk.
|
|
|
|
Rozpouštědlo/vehikulum:
|
—
|
zdůvodnění výběru rozpouštědla/vehikula,
|
|
—
|
rozpustnost a stálost zkoušené látky v rozpouštědle/vehikulu.
|
|
|
|
Buňky:
|
—
|
druh a zdroj použitých buněk,
|
|
—
|
vhodnost použitého druhu buněk,
|
|
—
|
nepřítomnost mykoplasmat, pokud připadá v úvahu,
|
|
—
|
informace o trvání buněčného cyklu, době zdvojnásobení populace nebo proliferačním indexu,
|
|
—
|
při použití lymfocytů je případně nutno uvést pohlaví, věk a počet dárců krve,
|
|
—
|
při použití lymfocytů je nutno uvést, zda se exponuje plná krev nebo odstředěné lymfocyty,
|
|
—
|
počet průchodů, pokud připadá v úvahu,
|
|
—
|
metody udržování buněčných kultur, pokud připadá v úvahu,
|
|
—
|
modální hodnota počtu chromozomů,
|
|
—
|
běžné trvání buněčného cyklu (u negativní kontroly).
|
|
|
|
Zkušební podmínky:
|
—
|
totožnost látky blokující cytokinezi (např. cytoB), pokud je použita, a její koncentrace a trvání expozice buněk,
|
|
—
|
zdůvodnění volby koncentrací a počtu kultur včetně údajů o cytotoxicitě a omezeních rozpustnosti, jsou-li tyto informace k dispozici,
|
|
—
|
složení média, případně koncentrace CO2,
|
|
—
|
hodnoty koncentrace zkoušené látky,
|
|
—
|
koncentrace (a/nebo objem) přidaného vehikula a zkoušené látky,
|
|
—
|
teplota a doba inkubace,
|
|
—
|
doba od expozice do odběru buněk k vyhodnocení,
|
|
—
|
hustota buněk při naočkování média, pokud připadá v úvahu,
|
|
—
|
druh a složení metabolického aktivačního systému včetně kritérií přijatelnosti,
|
|
—
|
látky použité jako pozitivní a negativní kontrola,
|
|
—
|
metody přípravy preparátů a použitý postup barvení,
|
|
—
|
kritéria pro identifikaci mikrojader,
|
|
—
|
počty analyzovaných buněk,
|
|
—
|
metody měření cytotoxicity,
|
|
—
|
jakékoli doplňkové informace týkající se cytotoxicity,
|
|
—
|
kritéria pro označení studií za pozitivní, negativní nebo dvojznačné,
|
|
—
|
použité metody statistické analýzy,
|
|
—
|
tam, kde je to vhodné, je nutno uvést rovněž metody (například využití protilátek proti kinetochorům), které slouží k určení, zda mikrojádra obsahují celé nebo rozlámané chromozomy.
|
|
|
|
Výsledky:
|
—
|
použitý způsob měření cytotoxicity, např. CBPI nebo RI nebo v případě metody blokování cytokineze, respektive RICC nebo RPD nebo PI, pokud se metody blokování cytokineze nepoužívají, popřípadě jiné poznatky, např. konfluence buněk, apoptóza, nekróza, počítání metafází, četnost dvoujaderných buněk,
|
|
—
|
hodnoty pH a osmolality zkušebního média, pokud jsou zjištěny,
|
|
—
|
vymezení přijatelných buněk pro analýzu,
|
|
—
|
rozložení jednojaderných, dvoujaderných a vícejaderných buněk, pokud se používá metoda blokování cytokineze,
|
|
—
|
počet buněk s mikrojádry uvedený zvlášť u každé ošetřené a kontrolní kultury a případně určení, zda se jedná o dvoujaderné nebo jednojaderné buňky,
|
|
—
|
pokud možno závislost odezvy na koncentraci,
|
|
—
|
chemické údaje (koncentrace a rozpouštědla) souběžných negativních kontrol (s rozpouštědlem/vehikulem) a pozitivních kontrol,
|
|
—
|
chemické údaje (koncentrace a rozpouštědla) dřívějších negativních kontrol (s rozpouštědlem/vehikulem) a pozitivních kontrol s uvedením rozsahů, střední hodnoty, směrodatné odchylky a intervalu spolehlivosti (např. 95 %),
|
|
—
|
statistická analýza, případné p hodnoty.
|
|
|
LITERATURA
|
(1)
|
Kirsch-Volders, M. (1997), Towards a validation of the micronucleus test. Mutation Res., 392, 1–4.
|
|
(2)
|
Parry, J.M. and Sors, A. (1993), The detection and assessment of the aneugenic potential of environmental chemicals: the European Community aneuploidy project, Mutation Res., 287, 3–15.
|
|
(3)
|
Fenech, M. and Morley, A.A. (1985), Solutions to the kinetic problem in the micronucleus assay, Cytobios., 43, 233–246.
|
|
(4)
|
Kirsch-Volders, M., Sofuni, T., Aardema, M., Albertini, S., Eastmond, D., Fenech, M., Ishidate, M. Jr, Lorge, E., Norppa, H., Surralles, J., von der Hude, W. and Wakata, A. (2000), Report from the In Vitro Micronucleus Assay Working Group, Environ. Mol. Mutagen., 35, 167–172.
|
|
(5)
|
Fenech, M. (2007), Cytokinesis-block micronucleus cytome assay, Nature Protocols, 2(5), 1084–1104.
|
|
(6)
|
Fenech, M. and Morley, A.A. (1986), Cytokinesis-block micronucleus method in human lymphocytes: effect of in-vivo ageing and low dose X-irradiation, Mutation Res., 161, 193–198.
|
|
(7)
|
Eastmond, D.A. and Tucker, J.D. (1989), Identification of aneuploidy-inducing agents using cytokinesis-blocked human lymphocytes and an antikinetochore antibody, Environ. Mol. Mutagen., 13, 34–43.
|
|
(8)
|
Eastmond, D.A. and Pinkel, D. (1990), Detection of aneuploidy and aneuploidy-inducing agents in human lymphocytes using fluorescence in-situ hybridisation with chromosome-specific DNA probes, Mutation Res., 234, 9–20.
|
|
(9)
|
Miller, B.M., Zitzelsberger, H.F., Weier, H.U. and Adler, I.D. (1991), Classification of micronuclei in murine erythrocytes: immunofluorescent staining using CREST antibodies compared to in situ hybridization with biotinylated gamma satellite DNA, Mutagenesis, 6, 297–302.
|
|
(10)
|
Farooqi, Z., Darroudi, F. and Natarajan, A.T. (1993), The use of fluorescence in-situ hybridisation for the detection of aneugens in cytokinesis-blocked mouse splenocytes, Mutagenesis, 8, 329–334.
|
|
(11)
|
Migliore, L., Bocciardi, R., Macri, C. and Lo Jacono, F. (1993), Cytogenetic damage induced in human lymphocytes by four vanadium compounds and micronucleus analysis by fluorescence in situ hybridization with a centromeric probe, Mutation Res., 319, 205–213.
|
|
(12)
|
Norppa, H., Renzi, L. and Lindholm, C. (1993), Detection of whole chromosomes in micronuclei of cytokinesis-blocked human lymphocytes by antikinetochore staining and in situ hybridization, Mutagenesis, 8, 519–525.
|
|
(13)
|
Eastmond, D.A, Rupa, D.S. and Hasegawa, L.S. (1994), Detection of hyperdiploidy and chromosome breakage in interphase human lymphocytes following exposure to the benzene metabolite hydroquinone using multicolor fluorescence in situ hybridization with DNA probes, Mutation Res., 322, 9–20.
|
|
(14)
|
Marshall, R.R., Murphy, M., Kirkland, D.J. and Bentley, K.S. (1996), Fluorescence in situ hybridisation (FISH) with chromosome-specific centromeric probes: a sensitive method to detect aneuploidy, Mutation Res., 372, 233–245.
|
|
(15)
|
Zijno, P., Leopardi, F., Marcon, R. and Crebelli, R. (1996), Analysis of chromosome segregation by means of fluorescence in situ hybridization: application to cytokinesis-blocked human lymphocytes, Mutation Res., 372, 211–219.
|
|
(16)
|
Kirsch-Volders, M., Sofuni, T., Aardema, M., Albertini, S., Eastmond, D., Fenech, M., Ishidate Jr., M., Lorge, E., Norppa, H., Surrallés, J., von der Hude, W. and Wakata, A. (2003), Report from the in vitro micronucleus assay working group. Mutation Res., 540, 153–163.
|
|
(17)
|
OECD (1997), In Vitro Mammalian Chromosome Aberration Test, Test Guideline No. 473, OECD Guidelines for Testing of Chemicals, OECD, Paris. K dispozici na adrese: [www.oecd.org/env/testguidelines].
|
|
(18)
|
Lorge, E., Thybaud, V., Aardema, M.J., Oliver, J., Wakata, A., Lorenzon G. and Marzin, D. (2006), SFTG International collaborative Study on in vitro micronucleus test. I. General conditions and overall conclusions of the study, Mutation Res., 607, 13–36.
|
|
(19)
|
Clare, G., Lorenzon, G., Akhurst, L.C., Marzin, D., van Delft, J., Montero, R., Botta, A., Bertens, A., Cinelli, S., Thybaud, V. and Lorge, E. (2006), SFTG International collaborative study on the in vitro micronucleus test. II. Using human lymphocytes, Mutation Res., 607, 37–60.
|
|
(20)
|
Aardema, M.J., Snyder, R.D., Spicer, C., Divi, K., Morita, T., Mauthe, R.J., Gibson, D.P., Soelter, S., Curry, P.T., Thybaud, V., Lorenzon, G., Marzin, D. and Lorge, E. (2006), SFTG International collaborative study on the in vitro micronucleus test, III. Using CHO cells, Mutation Res., 607, 61–87.
|
|
(21)
|
Wakata, A., Matsuoka, A., Yamakage, K., Yoshida, J., Kubo, K., Kobayashi, K., Senjyu, N., Itoh, S., Miyajima, H., Hamada, S., Nishida, S., Araki, H., Yamamura, E., Matsui, A., Thybaud, V., Lorenzon, G., Marzin, D. and Lorge, E. (2006), SFTG International collaborative study on the in vitro micronucleus test, IV. Using CHO/IU cells, Mutation Res., 607, 88–124.
|
|
(22)
|
Oliver, J., Meunier, J.-R., Awogi, T., Elhajouji, A., Ouldelhkim, M.-C., Bichet, N., Thybaud, V., Lorenzon, G., Marzin, D. and Lorge, E. (2006), SFTG International collaborative study on the in vitro micronucleus test, V. Using L5178Y cells, Mutation Res., 607, 125–152.
|
|
(23)
|
Albertini, S., Miller, B., Chetelat, A.A. and Locher, F. (1997), Detailed data on in vitro MNT and in vitro CA: industrial experience, Mutation Res., 392, 187–208.
|
|
(24)
|
Miller, B., Albertini, S., Locher, F., Thybaud, V. and Lorge, E. (1997), Comparative evaluation of the in vitro micronucleus test and the in vitro chromosome aberration test: industrial experience, Mutation Res., 392, 45–59.
|
|
(25)
|
Miller, B., Potter-Locher, F., Seelbach, A., Stopper, H., Utesch, D. and Madle, S. (1998), Evaluation of the in vitro micronucleus test as an alternative to the in vitro chromosomal aberration assay: position of the GUM Working Group on the in vitro micronucleus test. Gesellschaft fur Umwelt-Mutations-forschung, Mutation Res., 410, 81–116.
|
|
(26)
|
Kalweit, S., Utesch, U., von der Hude, W. and Madle, S. (1999), Chemically induced micronucleus formation in V79 cells – comparison of three different test approaches, Mutation Res. 439, 183–190.
|
|
(27)
|
Kersten, B., Zhang, J., Brendler Schwaab, S.Y., Kasper, P. and Müller, L. (1999), The application of the micronucleus test in Chinese hamster V79 cells to detect drug-induced photogenotoxicity, Mutation Res. 445, 55–71.
|
|
(28)
|
von der Hude, W., Kalweit, S., Engelhardt, G., McKiernan, S., Kasper, P., Slacik-Erben, R., Miltenburger, H.G., Honarvar, N., Fahrig, R., Gorlitz, B., Albertini, S., Kirchner, S., Utesch, D., Potter-Locher, F., Stopper, H. and Madle, S. (2000), In vitro micronucleus assay with Chinese hamster V79 cells - results of a collaborative study with in situ exposure to 26 chemical substances, Mutation Res., 468, 137–163.
|
|
(29)
|
Garriott, M.L., Phelps, J.B. and Hoffman, W.P. (2002), A protocol for the in vitro micronucleus test, I. Contributions to the development of a protocol suitable for regulatory submissions from an examination of 16 chemicals with different mechanisms of action and different levels of activity, Mutation Res., 517, 123–134.
|
|
(30)
|
Matsushima, T., Hayashi, M., Matsuoka, A., Ishidate, M. Jr., Miura, K.F., Shimizu, H., Suzuki, Y., Morimoto, K., Ogura, H., Mure, K., Koshi, K. and Sofuni, T. (1999), Validation study of the in vitro micronucleus test in a Chinese hamster lung cell line (CHL/IU), Mutagenesis, 14, 569–580.
|
|
(31)
|
Elhajouji, A., and Lorge, E. (2006), Special Issue: SFTG International collaborative study on in vitro micronucleus test, Mutation Res., 607, 1–152.
|
|
(32)
|
ECVAM (2006), Statement by the European Centre for the Validation of Alternative Methods (ECVAM) Scientific Advisory Committee (ESAC) on the scientific validity of the in vitro micronucleus test as an alternative to the in vitro chromosome aberration assay for genotoxicity testing. ESAC 25th meeting, 16–17 November, 2006, K dispozici na adrese: [http://ecvam.jrc.it/index.htm].
|
|
(33)
|
ESAC (2006), ECVAM Scientific Advisory Committee (ESAC) Peer Review, Retrospective Validation of the In Vitro Micronucleus Test, Summary and Conclusions of the Peer Review Panel, K dispozici na adrese: [http://ecvam.jrc.it/index.htm].
|
|
(34)
|
Corvi, R., Albertini, S., Hartung, T., Hoffmann, S., Maurici, D., Pfuhler, S, van Benthem, J., Vanparys P. (2008), ECVAM Retrospective Validation of in vitro Micronucleus Test (MNT), Mutagenesis, 23, 271–283.
|
|
(35)
|
Zhang, L.S., Honma, M., Hayashi, M., Suzuki, T., Matsuoka, A. and Sofuni, T. (1995), A comparative study of TK6 human lymphoblastoid and L5178Y mouse lymphoma cell lines in the in vitro micronucleus test, Mutation Res., 347, 105–115.
|
|
(36)
|
Ehrlich, V., Darroudi, F., Uhl, M., Steinkellner, S., Zsivkovits, M. and Knasmeuller, S. (2002), Fumonisin B1 is genotoxic in human derived hepatoma (HepG2) cells, Mutagenesis, 17, 257–260.
|
|
(37)
|
Knasmüller, S., Mersch-Sundermann, V., Kevekordes, S., Darroudi, F., Huber, W.W., Hoelzl, C., Bichler, J. and Majer, B.J. (2004), Use of human-derived liver cell lines for the detection of environmental and dietary genotoxicants; current state of knowledge, Toxicol., 198, 315–328.
|
|
(38)
|
Gibson, D.P., Brauninger, R., Shaffi, H.S., Kerckaert, G.A., LeBoeuf, R.A., Isfort, R.J. and Aardema, M.J. (1997), Induction of micronuclei in Syrian hamster embryo cells: comparison to results in the SHE cell transformation assay for National Toxicology Program test chemicals, Mutation Res., 392, 61–70.
|
|
(39)
|
Scott, D., Galloway, S.M., Marshall, R.R., Ishidate, M. Jr., Brusick, D., Ashby, J. and Myhr, B.C. (1991), International Commission for Protection Against Environmental Mutagens and Carcinogens, Genotoxicity under extreme culture conditions. A report from ICPEMC Task Group 9, Mutation Res., 257, 147–205.
|
|
(40)
|
Morita, T., Nagaki, T., Fukuda, I. and Okumura, K. (1992), Clastogenicity of low pH to various cultured mammalian cells, Mutation Res., 268, 297–305.
|
|
(41)
|
Brusick, D. (1986), Genotoxic effects in cultured mammalian cells produced by low pH treatment conditions and increased ion concentrations, Environ. Mutagen., 8, 789–886.
|
|
(42)
|
Fenech, M. and Morley, A.A. (1985), Measurement of micronuclei in lymphocytes, Mutation Res., 147, 29–36.
|
|
(43)
|
Fenech, M. (1997), The advantages and disadvantages of cytokinesis-blood micronucleus method, Mutation Res., 392, 11–18.
|
|
(44)
|
Bonassi, S., Fenech, M., Lando, C., Lin, Y.P., Ceppi, M., Chang, W.P., Holland, N., Kirsch-Volders, M., Zeiger, E., Ban, S., Barale, R., Bigatti, M.P., Bolognesi, C., Jia, C., Di Giorgio, M., Ferguson, L.R., Fucic, A., Lima, O.G., Hrelia, P., Krishnaja, A.P., Lee, T.K., Migliore, L., Mikhalevich, L., Mirkova, E., Mosesso, P., Muller, W.U., Odagiri, Y., Scarffi, M.R., Szabova, E., Vorobtsova, I., Vral, A. and Zijno, A. (2001), HUman MicroNucleus Project: international database comparison for results with the cytokinesis-block micronucleus assay in human lymphocytes, I. Effect of laboratory protocol, scoring criteria and host factors on the frequency of micronuclei, Environ. Mol. Mutagen. 37, 31–45.
|
|
(45)
|
Maron, D.M. and Ames, B.N. (1983), Revised methods for the Salmonella mutagenicity test, Mutation Res., 113, 173–215.
|
|
(46)
|
Ong, T.-m., Mukhtar, M., Wolf, C.R. and Zeiger, E. (1980), Differential effects of cytochrome P450-inducers on promutagen activation capabilities and enzymatic activities of S-9 from rat liver, J. Environ. Pathol. Toxicol., 4, 55–65.
|
|
(47)
|
Elliott, B.M., Combes, R.D., Elcombe, C.R., Gatehouse, D.G., Gibson, G.G., Mackay, J.M. and Wolf, R.C. (1992), Alternatives to Aroclor 1254-induced S9 in in-vitro genotoxicity assays. Mutagenesis, 7, 175–177.
|
|
(48)
|
Matsushima, T., Sawamura, M., Hara, K. and Sugimura, T. (1976), A safe substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems, In: de Serres, F.J., Fouts, J. R., Bend, J.R. and Philpot, R.M. (eds), In Vitro Metabolic Activation in Mutagenesis Testing, Elsevier, North-Holland, pp. 85–88.
|
|
(49)
|
Johnson, T.E., Umbenhauer, D.R. and Galloway, S.M. (1996), Human liver S-9 metabolic activation: proficiency in cytogenetic assays and comparison with phenobarbital/beta-naphthoflavone or Aroclor 1254 induced rat S-9, Environ. Mol. Mutagen., 28, 51–59.
|
|
(50)
|
UNEP (2001), Stockholm Convention on Persistent Organic Pollutants, United Nations Environment Programme (UNEP). K dispozici na adrese: [http://www.pops.int/].
|
|
(51)
|
Doherty, A.T., Ellard, S., Parry, E.M. and Parry, J.M. (1996), An investigation into the activation and deactivation of chlorinated hydrocarbons to genotoxins in metabolically competent human cells, Mutagenesis, 11, 247–274.
|
|
(52)
|
Krahn, D.F., Barsky, F.C. and McCooey, K.T. (1982), CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids, In: Tice, R.R., Costa, D.L. and Schaich, K.M. (eds), Genotoxic Effects of Airborne Agents. New York, Plenum, pp. 91–103.
|
|
(53)
|
Zamora, P.O., Benson, J.M., Li, A.P. and Brooks, A.L. (1983), Evaluation of an exposure system using cells grown on collagen gels for detecting highly volatile mutagens in the CHO/HGPRT mutation assay, Environ. Mutagenesis 5, 795–801.
|
|
(54)
|
Fenech, M. (1993), The cytokinesis-block micronucleus technique: a detailed description of the method and its application to genotoxicity studies in human populations, Mutation Res., 285, 35–44.
|
|
(55)
|
Phelps, J.B., Garriott, M.L., and Hoffman, W.P. (2002), A protocol for the in vitro micronucleus test. II. Contributions to the validation of a protocol suitable for regulatory submissions from an examination of 10 chemicals with different mechanisms of action and different levels of activity, Mutation Res., 521, 103–112.
|
|
(56)
|
Kirsch-Volders, M., Sofuni, T., Aardema, M., Albertini, S., Eastmond, D., Fenech, M., Ishidate, M. Jr., Kirchner, S., Lorge, E., Morita, T., Norppa, H., Surralles, J., Vanhauwaert, A. and Wakata, A. (2004), Corrigendum to ‚Report from the in vitro micronucleus assay working group‘, Mutation Res., 564, 97–100.
|
|
(57)
|
Pincu, M., Bass, D. and Norman, A. (1984), An improved micronuclear assay in lymphocytes, Mutation Res., 139, 61–65.
|
|
(58)
|
Lorge, E., Hayashi, M., Albertini, S. and Kirkland, D. (2008), Comparison of different methods for an accurate assessment of cytotoxicity in the in vitro micronucleus test. I. Theoretical aspects, Mutation Res., 655, 1–3.
|
|
(59)
|
Surralles, J., Xamena, N., Creus, A., Catalan, J., Norppa, H. and Marcos, R. (1995), Induction of micronuclei by five pyrethroid insecticides in whole-blood and isolated human lymphocyte cultures, Mutation Res., 341, 169–184.
|
|
(60)
|
Galloway, S. (2000), Cytotoxicity and chromosome aberrations in vitro: Experience in industry and the case for an upper limit on toxicity in the aberration assay, Environ. Molec. Mutagenesis 35, 191–201.
|
|
(61)
|
Hayashi, M., Sofuni, T., and Ishidate, M. Jr. (1983), An Application of Acridine Orange Fluorescent Staining to the Micronucleus Test, Mutation Res., 120, 241–247.
|
|
(62)
|
MacGregor, J. T., Wehr, C. M., and Langlois, R. G. (1983), A Simple Fluorescent Staining Procedure for Micronuclei and RNA in Erythrocytes Using Hoechst 33258 and Pyronin Y, Mutation Res., 120, 269–275.
|
|
(63)
|
Hayashi, M., Sofuni, T. and Ishidate, M. Jr. (1983), An application of acridine orange fluorescent staining to the micronucleus test, Mutation Res., 120, 241–247.
|
|
(64)
|
Fenech, M., Chang, W.P., Kirsch-Volders, M., Holland, N., Bonassi, S. and Zeiger, E. (2003), HUMN project: detailed description of the scoring criteria for the cytokinesis-block micronucleus assay using isolated human lymphocyte cultures, Mutation Res., 534, 65–75.
|
|
(65)
|
Hoffman, W.P., Garriott, M.L. and Lee, C. (2003), In vitro micronucleus test, In: Encyclopedia of Biopharmaceutical Statistics, Second edition. S. Chow (ed.), Marcel Dekker, Inc. New York, NY, pp. 463–467.
|
|
(66)
|
Nařízení Evropského parlamentu a Rady (ES) č. 850/2004 ze dne 29. dubna 2004 o perzistentních organických znečišťujících látkách a o změně směrnice 79/117/EHS (Úř. věst. L 229, 30.4.2004, s. 5).
|
Dodatek 1
Definice
Aneugen: jakákoli látka nebo proces, který v interakci se složkami mitotického a meiotického cyklu buněčného dělení vede k aneuploidii v buňkách nebo organismech.
Aneuploidie: jakákoli odchylka od normálního diploidního (nebo haploidního) počtu chromozomů o jeden nebo více chromozomů, avšak nikoli o celou sadu (nebo více sad) chromozomů (polyploidie).
Apoptóza: programovaná buněčná smrt, která se vyznačuje řadou kroků vedoucích k rozpadu buněk na membránově vázané části, které jsou poté odstraněny fagocytózou nebo rozkladem.
Buněčná proliferace: zvyšování počtu buněk v důsledku jejich mitotického dělení.
Centromera: oblast DNA v chromozomu, kde se stýkají obě chromatidy a k níž jsou bok po boku připojeny oba kinetochory.
Klastogen: jakákoli látka nebo proces, který způsobuje strukturální chromozomální aberace v populacích buněk nebo organismů.
Cytokineze: proces rozdělení buňky ihned po mitóze, čímž vzniknou dvě dceřiné buňky, z nichž každá obsahuje jedno jádro.
Proliferační index při blokování cytokineze (CBPI): podíl počtu buněk z druhého dělení v ošetřené populaci k jejich počtu v neošetřené kontrole (příslušný vzorec viz dodatek 2).
Cytostáze: inhibice růstu buněk (příslušný vzorec viz dodatek 2).
Cytotoxicita: škodlivé účinky na strukturu nebo funkci buňky, které nakonec způsobí její smrt.
Genotoxický: obecný termín zahrnující všechny typy poškození DNA nebo chromozomů včetně jejich rozlámání, přestavby aduktů, mutací, chromozomálních aberací a aneuploidie. Ne všechny druhy genotoxických účinků způsobují mutace nebo stálé poškození chromozomů.
Interfázové buňky: buňky, které se nenacházejí ve stádiu mitózy.
Kinetochor: struktura obsahující proteiny, jež se vytváří u centromery chromozomu, na kterou se při dělení buněk napojují vlákna dělicího vřeténka, což umožňuje správný pohyb dceřiných chromozomů k pólům dceřiných buněk.
Mikrojádra: malá jádra existující odděleně od hlavních jader a vedle nich, vytvářená během telofáze mitózy (meiózy) nereplikujícími se (lagging) chromozomovými fragmenty nebo celými chromozomy.
Mitóza: proces dělení buněčného jádra, který se obvykle člení na profázi, prometafázi, metafázi, anafázi a telofázi.
Mitotický index: poměr buněk v metafázi vydělený celkovým počtem buněk zjištěných v populaci buněk; udává stupeň buněčné proliferace této populace.
Mutagenní: produkující dědičné změny sekvence (sekvencí) párů bází DNA v genech nebo struktury chromozomů (chromozomální aberace).
Nondisjunkce: děj, při kterém se párové chromatidy nerozloučí a správně nerozdělí do vznikajících dceřiných buněk, což vede ke vzniku dceřiných buněk s abnormálními počty chromozomů.
Polyploidie: numerické chromozomální aberace v buňkách nebo organismech, které se týkají celé sady (celých sad) chromozomů, na rozdíl od jednoho chromozomu nebo jednotlivých chromozomů (aneuploidie).
Proliferační index (PI): metoda pro měření cytotoxicity bez použití cytoB (příslušný vzorec viz dodatek 2).
Relativní nárůst počtu buněk (RICC): metoda pro měření cytotoxicity bez použití cytoB (příslušný vzorec viz dodatek 2).
Relativní zdvojnásobení populace (RPD): metoda pro měření cytotoxicity bez použití cytoB (příslušný vzorec viz dodatek 2).
Replikační index (RI): podíl počtu dokončených cyklů buněčného dělení v ošetřené kultuře k jejich počtu v neošetřené kontrole během expoziční doby a obnovy kultury (příslušný vzorec viz dodatek 2).
Zkoušená látka: Jakákoli látka nebo směs, která se zkouší za použití této zkušební metody.
Dodatek 2
Vzorce pro hodnocení cytotoxicity
|
1.
|
Při použití cytoB by mělo být hodnocení cytotoxicity založeno na proliferačním indexu při blokování cytokineze (CBPI) nebo na replikačním indexu (RI) (16) (58). Hodnota CBPI udává průměrný počet buněčných cyklů na buňku v době jejího vystavení cytochalasinu B a lze ji použít k výpočtu proliferace buněk. Hodnota RI udává relativní počet jader v ošetřených kulturách ve srovnání s kontrolními kulturami a lze ji použít k výpočtu procenta cytostáze:
% cytostáze = 100 – 100{(CBPIT – 1) ÷ (CBPIC – 1)}
přičemž:
|
T
|
=
|
kultura ošetřená zkoušenou látkou
|
|
C
|
=
|
kontrolní kultura s vehikulem
|
kde:
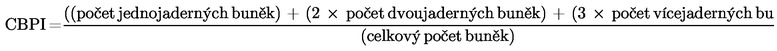
Hodnota CBPI = 1 (všechny buňky jsou jednojaderné) tedy odpovídá 100 % cytostázi.
Cytostáze = 100 – RI
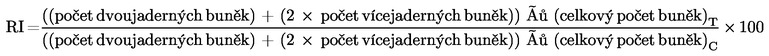
|
T
|
=
|
ošetřené kultury
|
|
C
|
=
|
kontrolní kultury
|
|
|
2.
|
Hodnota RI = 53 % tedy znamená, že v porovnání s počtem buněk, z nichž v kontrolní kultuře vznikly rozdělením dvoujaderné a vícejaderné buňky, se v ošetření kultuře rozdělilo pouze 53 % tohoto počtu, což znamená 47 % cytostázi.
|
|
3.
|
Když se cytoB nepoužije, doporučuje se hodnotit cytotoxicitu na základě relativního nárůstu počtu buněk (RICC) nebo relativního zdvojnásobení populace (RPD) (58), jelikož oba tyto ukazatele zohledňují poměr buněčné populace, která se rozdělila.

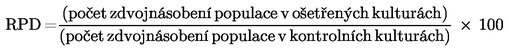
kde:
Zdvojnásobení populace = [log (počet buněk po ošetření ÷ počáteční počet buněk)] ÷ log 2
|
|
4.
|
Hodnota RICC nebo RPD ve výši 53 % tedy znamená 47 % cytotoxicitu/cytostázi.
|
|
5.
|
Za použití indexu proliferace (PI) lze posoudit cytotoxicitu spočítáním klonů tvořených 1 buňkou (cl1), 2 buňkami (cl2), 3 až 4 buňkami (cl4) a 5 až 8 buňkami (cl8)
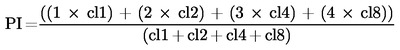
|
|
6.
|
Hodnota PI se používá jako cenný a spolehlivý parametr cytotoxicity i pro buněčné linie kultivované in situ bez přítomnosti cytoB (25) (26) (27) (28).
|
Dodatek 3
Srovnávací látky doporučené pro hodnocení funkčnosti
(16)
|
Kategorie
|
Chemická látka
|
Číslo CAS
|
Číslo ES
|
| 1. Klastogeny působící bez metabolické aktivace
|
|
|
cytarabin
|
147-94-4
|
205-705-9
|
|
|
mitomycin C
|
50-07-7
|
200-008-6
|
| 2. Klastogeny vyžadující metabolickou aktivaci
|
|
|
benzo[a]pyren
|
50-32-8
|
200-028-5
|
|
|
cyklofosfamid
|
50-18-0
|
200-015-4
|
| 3. Aneugeny
|
|
|
kolchicin
|
64-86-8
|
200-598-5
|
|
|
vinblastin
|
143-67-9
|
205-606-0
|
| 4. Negativní látky
|
|
|
bis(2-ethylhexyl)ftalát
|
117-81-7
|
204-211-0
|
|
|
kyselina nalidixová
|
389-08-2
|
206-864-7
|
|
|
pyren
|
129-00-0
|
204-927-3
|
|
|
chlorid sodný
|
7647-14-5
|
231-598-3
|
B.50. SENZIBILIZACE KŮŽE: ZKOUŠKA S VYŠETŘENÍM LOKÁLNÍCH LYMFATICKÝCH UZLIN: DA
ÚVOD
|
1.
|
Metodiky OECD pro testování chemických látek a zkušební metody EU jsou pravidelně přezkoumávány s ohledem na vědecký pokrok, změny regulačních potřeb a dobré životní podmínky zvířat. První zkušební metoda (ZM) (kapitola B.42 této přílohy) pro stanovení senzibilizace kůže u myší, tzv. zkouška s vyšetřením lokálních lymfatických uzlin (LLNA; zkušební metodika OECD č. 429) byla revidována (1). Podrobnosti o validaci metody LLNA a přehled souvisejících prací byly publikovány (2)(3) (4) (5) (6) (7) (8) (9). V rámci metody LLNA se k měření proliferace lymfocytů využívají radioizotopy thymidinu nebo jódu, a proto má tato zkouška omezené použití, pokud získání, používání nebo likvidace radioaktivních látek představují problém. Metoda LLNA: DA (kterou vyvinula firma Daicel Chemical Industries, Ltd.) je neradioaktivní modifikací metody LLNA, která kvantifikuje obsah adenosintrifosfátu (ATP) prostřednictvím bioluminiscence, jež slouží jako indikátor proliferace lymfocytů. Zkušební metoda LLNA: DA byla ověřena, přezkoumána a doporučena mezinárodní komisí pro vzájemné hodnocení jakožto metoda, která je s určitými omezeními považována za užitečnou pro identifikaci chemických látek způsobujících nebo naopak nezpůsobujících senzibilizaci kůže (10) (11) (12) (13). Tato ZM je určena k hodnocení potenciálu chemikálií (látek a směsí) způsobovat senzibilizaci kůže u zvířat. Zkušební metoda podle kapitoly B.6 této přílohy a zkušební metodiky OECD č. 406 využívá zkoušek na morčatech, a to zvláště maximalizační zkoušku na morčatech a Bühlerův test (14). Zkušební metoda LLNA (podle kapitoly B.42 této přílohy a zkušební metodiky OECD č. 429 a její dvě neradioaktivní modifikace, metoda LLNA DA (podle kapitoly B.50 této přílohy a zkušební metodiky OECD č. 442 A) a metoda LLNA: BrdU-ELISA (podle kapitoly B.51 této přílohy a zkušební metodiky OECD č. 442 B) poskytují výhody ve srovnání se zkouškami na morčatech podle kapitoly B.6 a zkušební metodiky OECD č. 406 (14), pokud jde o snížení počtu využívaných zvířat a zdokonalení způsobu jejich využívání.
|
|
2.
|
Podobně jako metoda LLNA i metoda LLNA: DA zkoumá indukční fázi senzibilizace kůže a poskytuje kvantitativní údaje, které jsou vhodné pro hodnocení závislosti odezvy na dávce. Schopnost detekovat látky senzibilizující kůži bez nutnosti použití radioizotopů pro značení DNA navíc odstraňuje možnost expozice radioaktivitě při práci a problémy s likvidací odpadů. To zase může umožnit zvýšené využívání myší k detekování látek senzibilizujících kůži, což by mohlo dále snížit využívání morčat k testování potenciálu chemických látek způsobovat senzibilizaci kůže (tzn. metodou podle kapitoly B.6 a zkušební metodiky OECD č. 406 (14).
|
DEFINICE
|
3.
|
Použité definice jsou uvedeny v dodatku 1.
|
VÝCHOZÍ ÚVAHY A OMEZENÍ
|
4.
|
Zkušební metoda LLNA: DA je modifikovanou metodou LLNA, která s určitými omezeními slouží k identifikaci chemických látek, jež mohou způsobovat senzibilizaci kůže. To však nutně neznamená, že by se metoda LLNA: DA měla používat ve všech případech namísto metody LLNA nebo namísto zkoušek na morčatech (tzn. podle kapitoly B.6 této přílohy a zkušební metodiky OECD č. 406 (14); znamená to spíše, že tato metoda má stejnou hodnotu a že ji lze použít jako alternativní metodu, jejíž pozitivní ani negativní výsledky obvykle již nevyžadují žádné další potvrzování (10) (11). Zkušební laboratoř by měla před provedením studie zohlednit všechny dostupné informace o zkoušené látce. Mezi tyto informace patří totožnost a chemická struktura zkoušené látky, její fyzikálně-chemické vlastnosti, výsledky jakýchkoli jiných zkoušek toxicity in vitro a in vivo za použití zkoušené látky a toxikologické údaje o strukturně příbuzných látkách. Tyto informace je nutno posoudit, aby se zjistilo, zda je metoda LLNA: DA vhodná pro danou látku (s ohledem na neslučitelnost některých druhů chemických látek s metodou LLNA: DA (viz odstavec 5)), a také aby se usnadnila volba dávkování.
|
|
5.
|
Zkušební metoda LLNA: DA je metodou in vivo, a tudíž neeliminuje používání zvířat při hodnocení alergického kontaktního senzibilizujícího působení. Umožňuje však omezit využívání zvířat pro tento účel ve srovnání se zkouškami na morčatech (podle kapitoly B.6 a zkušební metodiky OECD č. 406 (14). Kromě toho metoda LLNA: DA nabízí podstatné zlepšení způsobu zacházení se zvířaty (méně bolesti a utrpení), která se využívají ke zkoušení kontaktní alergické senzibilizace, jelikož na rozdíl od metody podle kapitoly B.6 a zkušební metodiky OECD č. 406 metoda LLNA: DA nevyžaduje umělé vyvolání kožní přecitlivělosti. Ale i přes výhody, které má metoda LLNA: DA ve srovnání s metodou popsanou v kapitole B.6 a zkušební metodice OECD č. 406 (14), existují určitá omezení, která si mohou vyžádat použití metody popsané v kapitole B.6 nebo zkušební metodice OECD č. 406 (např. falešně negativní výsledky při použití metody LLNA pro určité kovy, falešně pozitivní výsledky u určitých látek způsobujících podráždění kůže (jako jsou například některé chemikálie typu povrchově aktivních látek) (6) (1 a kapitola B.42 této přílohy) nebo rozpustnost zkoušené látky). Kromě toho si použití zkoušek na morčatech (tzn. podle kapitoly B.6 této přílohy nebo zkušební metodiky OECD č. 406 (14) mohou vyžádat také některé třídy chemických látek nebo látky obsahující funkční skupiny, u nichž je prokázáno, že mohou způsobovat zkreslení výsledků (16). Doporučuje se, aby se omezení zjištěná u metody LLNA (1 a kapitola B.42 této přílohy) vztahovala rovněž na metodu LLNA: DA (10). Použití metody LLNA: DA navíc nemusí být vhodné pro zkoušení látek, které ovlivňují hladiny ATP (např. látek, jež působí jako inhibitory ATP), nebo látek, které ovlivňují přesnost měření intracelulárního ATP (např. přítomnost enzymů snižujících hladinu ATP nebo přítomnost extracelulárního ATP v lymfatických uzlinách). S výjimkou těchto známých omezení by měla být metoda LLNA: DA použitelná k testování jakýchkoli látek, nejsou-li s těmito látkami spojeny vlastnosti, které mohou nepříznivě ovlivnit přesnost metody LLNA: DA. Kromě toho je třeba zvážit možnost hraničních pozitivních výsledků při získání hodnot indexu stimulace (SI) v rozmezí od 1,8 do 2,5 (viz odstavce 31 až 32). To je založeno na validační databázi 44 látek za použití hodnot SI ≥ 1,8 (viz odstavec 6), u kterých metoda LLNA: DA správně identifikovala všech 32 látek identifikovaných jako senzibilizující látky metodou LLNA, ale nesprávně identifikovala tři z 12 látek identifikovaných metodou LLNA jako nesenzibilizující látky s hodnotami SI v rozmezí od 1,8 do 2,5 (tzn. hraniční pozitivní) (10). Jelikož však byl stejný soubor údajů použit k stanovení hodnot SI a k výpočtu prediktivních vlastností zkoušky, mohou uvedené výsledky představovat nadhodnocení skutečných prediktivních vlastností této metody.
|
PODSTATA ZKUŠEBNÍ METODY
|
6.
|
Základní podstata metody LLNA: DA spočívá v tom, že senzibilizující látky vyvolávají proliferaci lymfocytů v lymfatických uzlinách drenujících místo aplikace zkoušené látky. Tato proliferace je úměrná dávce a potenciálu aplikovaného alergenu a představuje jednoduchý prostředek k získání kvantitativního údaje o míře senzibilizace. Proliferace se měří porovnáním průměrné proliferace u každé zkušební skupiny s průměrnou proliferací u kontrolní skupiny s vehikulem. Určí se poměr průměrné proliferace u každé z exponovaných skupin k průměrné proliferaci u souběžně testované kontrolní skupiny s vehikulem, nazývaný index stimulace (SI), přičemž jeho hodnota by měla být ≥ 1,8, aby bylo oprávněné další hodnocení zkoušené látky jako potenciálního senzibilizátoru kůže. Zde popsané postupy jsou založeny na použití měření obsahu ATP za použití bioluminescence (o níž je známo, že její intenzita je úměrná počtu živých buněk) 17) k indikaci zvýšeného počtu proliferujících buněk lymfatických uzlin drenujících aurikulární oblast (18) (19). Bioluminescenční metoda využívá enzym luciferázu ke katalyzaci tvorby světla z ATP a luciferin v rámci této reakce:
ATP + Luciferin + O
2
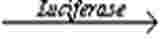 Oxyluciferin + AMP + PPi
+ CO
2 + Light
Oxyluciferin + AMP + PPi
+ CO
2 + Light
Intenzita vyzařovaného světla je přímo úměrná koncentraci ATP a měří se pomocí luminometru. Zkouška založená na použití luciferinu a luciferázy je citlivou metodou pro určení množství ATP, která se používá k široké škále účelů (20).
|
POPIS ZKOUŠKY
Volba druhu zvířat
|
7.
|
Vhodným druhem pro tuto zkoušku je myš. Validační studie metody LLNA: DA byly prováděny výhradně s kmenem CBA/J, který je proto považován za upřednostňovaný kmen (12) (13). Používají se mladé dospělé samice myší, které musí být nullipary a nesmějí být březí. Při zahájení studie by zvířata měla být přibližně 8 až 12 týdnů stará, přičemž odchylky v hmotnosti zvířat by měly být pouze minimální a neměly by překročit 20 % průměrné hmotnosti. Případně lze používat i jiné kmeny a samce, pokud je k dispozici dostatek údajů, které dokazují, že v rámci reakcí neexistují u metody LLNA: DA významné specifické odlišnosti mezi kmeny a/nebo pohlavími.
|
Podmínky chovu a krmení
|
8.
|
Myši by měly být chovány ve skupinách (21), není-li předloženo dostatečné vědecké zdůvodnění, proč by měly být chovány jednotlivě. Teplota v místnosti pro pokusná zvířata by měla být 22 ± 3 °C. Ačkoli by relativní vlhkost vzduchu měla být minimálně 30 % a pokud možno by kromě doby úklidu místnosti neměla přesáhnout 70 %, cílem by měla být hodnota 50–60 %. Osvětlení by mělo být umělé se střídáním 12 hodin světla a 12 hodin tmy. Ke krmení lze používat běžnou laboratorní stravu a k pití by měly mít myši k dispozici neomezené množství vody.
|
Příprava zvířat
|
9.
|
Zvířata se náhodně vyberou, pro usnadnění individuální identifikace se označí (nesmí se však použít žádná forma značení uší) a chovají se v klecích minimálně pět dnů před začátkem aplikování zkoušené látky, aby se mohla přizpůsobit laboratorním podmínkám. Před začátkem aplikace látky se všechna zvířata vyšetří, aby se ověřilo, že nemají žádné pozorovatelné kožní léze.
|
Příprava dávkovacích roztoků
|
10.
|
Pevné chemické látky by se měly před nanesením na ucho myši rozpustit nebo suspendovat ve vhodných rozpouštědlech nebo vehikulech a popřípadě zředit. Kapalné látky lze aplikovat bez přísad nebo je lze před dávkováním rozpustit. Nerozpustné látky, jaké se nejčastěji používají k výrobě zdravotnických prostředků, by měly být vystaveny intenzívní extrakci ve vhodném rozpouštědle, aby se před nanesením na ucho myši odhalily všechny extrahovatelné složky, které je nutno testovat. Zkoušené látky by se měly připravovat každý den čerstvé, pokud údaje o stálosti neprokazují přijatelnost jejich skladování.
|
Kontrola spolehlivosti
|
11.
|
K prokázání řádné funkčnosti zkoušky slouží pozitivní kontrolní chemikálie, a to tak, že reagují s dostatečnou a reprodukovatelnou citlivostí na zkoušenou senzibilizující látku, u níž je dobře popsána síla odezvy. Zařazení souběžné pozitivní kontroly se doporučuje proto, že prokazuje způsobilost laboratoře úspěšně provést každou zkoušku a umožňuje posoudit vnitrolaboratorní a mezilaboratorní reprodukovatelnost a srovnatelnost. Některé regulační orgány také vyžadují při každé studii pozitivní kontrolu, a proto se uživatelům doporučuje před provedením zkoušky LLNA: DA příslušné orgány konzultovat. Doporučuje se tudíž běžně používat souběžnou pozitivní kontrolu, aby nebylo nutné další zkoušení na zvířatech ke splnění požadavků, které by mohly vzniknout při používání periodické pozitivní kontroly (viz odstavec 12). Pozitivní kontrola by měla v rámci metody LLNA: DA vyvolat pozitivní odezvu při úrovni expozice, u které se očekává zvýšení indexu stimulace (SI) > 1,8 ve srovnání s negativní kontrolní skupinou. Dávka pozitivní kontroly by měla být zvolena tak, aby nevyvolala nadměrné podráždění kůže nebo systémovou toxicitu a přitom aby indukce byla reprodukovatelná, avšak nikoli nadměrná (tzn. že hodnota SI > 10 by byla považována za příliš velkou). K upřednostňovaným látkám pro pozitivní kontrolu patří 25 % roztok 2-benzylidenoktanalu (reg. č. Chemical Abstracts Service (CAS) 101-86-0) nebo eugenolu (číslo CAS 97-53-0) v acetonu s olivovým olejem (v objemovém poměru 4:1). Mohou existovat určité okolnosti, za nichž lze při řádném zdůvodnění použít i jiné pozitivní kontrolní látky, které splňují výše uvedená kritéria.
|
|
12.
|
I když se zařazení skupiny souběžných pozitivních kontrol doporučuje, mohou existovat situace, kdy může být periodické testování (tzn. v intervalech ≤ 6 měsíců) pozitivních kontrol vhodné u laboratoří, které provádějí zkoušky LLNA: DA pravidelně (tzn. nejméně jednou měsíčně) a mají k dispozici databázi dřívějších údajů pozitivních kontrol, která prokazuje schopnost laboratoře získat při použití pozitivních kontrol reprodukovatelné a přesné výsledky. Odpovídající zběhlost v používání metody LLNA: DA lze úspěšně prokázat dosažením shodných pozitivních výsledků při použití pozitivní kontroly alespoň v 10 nezávislých zkouškách provedených během přiměřené doby (tzn. během méně než jednoho roku).
|
|
13.
|
Skupina souběžných pozitivních kontrol by se měla zařadit vždy, když dojde k procedurální změně metody LLNA: DA (např. při změně složení vyškoleného personálu, při změně materiálů a/nebo činidel či vybavení, jež se používají v rámci zkušební metody, nebo při změně zdroje pokusných zvířat), přičemž takové změny by se měly vždy zdokumentovat v laboratorních zprávách. Při určování nezbytnosti vytvořit novou databázi ke zdokumentování shodnosti výsledků pozitivních kontrol by se měl zvážit dopad těchto změn na přiměřenost dosavadní databáze.
|
|
14.
|
Zkoušející by si měli uvědomit, že rozhodnutí používat pozitivní kontroly periodicky namísto souběžně má závažné dopady na přiměřenost a přijatelnost negativních výsledků studie získaných bez souběžných pozitivních kontrol během intervalu mezi jednotlivými periodickými použitími pozitivních kontrol. Například obdržení falešně negativního výsledku při periodickém používání pozitivních kontrol může zpochybnit negativní výsledky zkoušené látky získané v období mezi poslední přijatelnou periodickou studií pozitivních kontrol a nepřijatelnou periodickou studií pozitivních kontrol. Důsledky, jež z toho vyplývají, je třeba pečlivě zvážit při rozhodování, zda do zkoumání zařadit souběžné pozitivní kontroly, nebo používat pouze periodické pozitivní kontroly. Je také nutno vzít v úvahu možnost použít v rámci skupiny souběžných pozitivních kontrol méně zvířat, jestliže je to vědecky opodstatněné a jestliže laboratoř na základě svých vlastních dosavadních údajů prokáže, že lze používat méně myší (22).
|
|
15.
|
Ačkoli by se pozitivní kontroly měly zkoušet ve vehikulu, o němž je známo, že vždy vyvolává shodnou reakci (např. aceton s olivovým olejem v objemovém poměru 4:1), mohou nastat určité situace, kdy je z důvodů požadovaných v právních předpisech nezbytné provést zkoušku s použitím nestandardního vehikula (klinicky/chemicky relevantní složení) (23). Pokud se souběžná pozitivní kontrola zkouší v jiném vehikulu než zkoušená látka, měla by být součástí zkoušky se souběžnou pozitivní kontrolou také samostatná kontrola s vehikulem.
|
|
16.
|
V případech, kdy se hodnotí látky, které patří do určité třídy chemických látek nebo vykazují určitou škálu odezev, mohou být užitečné také srovnávací látky, jelikož mohou prokázat, že zkušební metoda je schopna správně zjistit potenciál těchto druhů látek vyvolávat senzibilizaci kůže. Vhodné srovnávací látky by měly mít tyto vlastnosti:
|
—
|
strukturální a funkční podobnost s třídou, do níž patří zkoušená látka,
|
|
—
|
známé fyzikálně-chemické vlastnosti,
|
|
—
|
podklady ze zkoušky LLNA: DA,
|
|
—
|
podklady získané při použití jiných zvířecích modelů a/nebo lidí.
|
|
ZKUŠEBNÍ POSTUP
Počet zvířat a dávkování
|
17.
|
Na každou dávkovou skupinu se použijí alespoň čtyři zvířata, přičemž se použijí minimálně tři koncentrace zkoušené látky, dále se použije souběžná negativní kontrolní skupina, k jejíž expozici se použije pouze vehikulum zkoušené látky, a pozitivní kontrola (souběžná nebo z nedávné jiné zkoušky – v závislosti na postupech dané laboratoře při zohlednění odstavců 11–15). Mělo by se zvážit zkoušení většího počtu dávek pozitivní kontroly, a to zejména v případě, že se pozitivní kontrola nepoužívá soustavně. Se zvířaty v kontrolních skupinách by se s výjimkou aplikace zkoušené látky mělo zacházet stejně jako se zvířaty v exponovaných skupinách.
|
|
18.
|
Volba dávkování a vehikula by měla být založena na doporučeních uvedených v literatuře – (2) a (24). Po sobě jdoucí dávky se běžně vybírají z vhodné řady koncentrací, například 100 %, 50 %, 25 %, 10 %, 5 %, 2,5 %, 1 %, 0,5 % atd. Volba této řady koncentrací by měla být dostatečně vědecky zdůvodněna. Při výběru tří po sobě jdoucích koncentrací je třeba zvážit veškeré existující toxikologické informace (např. o akutní toxicitě a kožní dráždivosti) a informace o struktuře a fyzikálně-chemických vlastnostech zkoušené látky (a/nebo strukturně příbuzných látek), jsou-li k dispozici, aby nejvyšší koncentrace maximalizovala expozici, aniž by současně vyvolala systémovou toxicitu a/nebo nadměrné lokální podráždění kůže (24) (25). Nejsou-li takové informace k dispozici, může být nezbytné provést úvodní předběžnou screeningovou zkoušku (viz odstavce 21 až 24).
|
|
19.
|
Vehikulum by nemělo nepříznivě ovlivňovat ani jinak zkreslovat výsledky zkoušky a mělo by být vybráno tak, aby se maximalizovala rozpustnost umožňující dosáhnout nejvyšší možné koncentrace a zároveň připravit roztok nebo suspenzi, jež jsou vhodné pro aplikaci zkoušené látky. K doporučeným vehikulům patří aceton s olivovým olejem (v objemovém poměru 4:1), N,N-dimethylformamid, ethyl(methyl)keton, propan-1,2-diol a dimethylsulfoxid (6), avšak při dostatečném vědeckém zdůvodnění lze použít i jiná vehikula. V určitých situacích může být nezbytné použít jako doplňující kontrolu klinicky relevantní rozpouštědlo nebo běžně prodejný přípravek obsahující zkoušenou látku. Zvláště je třeba dbát na to, aby byly hydrofilní látky zapracovány do takového vehikula, které zvlhčí kůži a nesteče ihned z pokožky. Toho lze dosáhnout použitím vhodných solubilizérů (jako je např. 1 % Pluronic® L92). Není tudíž vhodné používat zcela vodná vehikula.
|
|
20.
|
Zpracování lymfatických uzlin z jednotlivých myší umožňuje posoudit variabilitu mezi jednotlivými zvířaty a provést statistické porovnání rozdílu mezi hodnotami zkoušené látky a hodnotami naměřenými u kontrolní skupiny se samotným vehikulem (viz odstavec 33). Posoudit možnost snížit počet myší v pozitivní kontrolní skupině je kromě toho vhodné jen tehdy, když se zaznamenávají údaje od jednotlivých zvířat (22). Sběr údajů od jednotlivých zvířat navíc vyžadují i některé regulační orgány. Pravidelný sběr údajů od jednotlivých zvířat je výhodnější z hlediska dobrých životních podmínek zvířat, jelikož nevyžaduje duplicitní zkoušení, které by bylo nezbytné, pokud by výsledky pro určitou zkoušenou látku, jež byly původně shromážděny jedním způsobem (např. za použití souhrnných údajů od všech zvířat), měly být později posouzeny regulačními orgány s jinými požadavky (např. vyžadujícími použití údajů od jednotlivých zvířat).
|
Předběžná screeningová zkouška
|
21.
|
Nejsou-li k dispozici informace pro stanovení nejvyšší dávky, která má být předmětem zkoušky (viz odstavec 18), měla by se provést předběžná screeningová zkouška umožňující vymezit vhodné dávkování, které se bude metodou LLNA: DA zkoušet. Účelem předběžné screeningové zkoušky je poskytnout vodítko pro volbu maximálního dávkování, které se má použít v hlavní studii LLNA: DA, pokud nejsou k dispozici informace o koncentraci, která vyvolává systémovou toxicitu (viz odstavec 24) a/nebo nadměrné lokální podráždění kůže (viz odstavec 23). Maximální zkoušenou dávkou by mělo být 100 % zkoušené látky v případě kapalin, respektive maximální možná koncentrace v případě pevných látek nebo suspenzí.
|
|
22.
|
Předběžná screeningová zkouška se provádí za stejných podmínek jako hlavní studie LLNA: DA až na to, že se nehodnotí proliferace buněk z lymfatických uzlin a lze použít méně zvířat na dávkovou skupinu. Navrhuje se počet jednoho až dvou zvířat na dávkovou skupinu. Všechny myši se denně pozorují, zda nevykazují klinické příznaky systémové toxicity nebo lokálního podráždění v místě aplikace. Před zahájením zkoušky a před jejím ukončením (den 8) se zaznamenají hodnoty tělesné hmotnosti zvířat. Na obou uších každé myši se sleduje výskyt erytému, který se vyhodnotí podle tabulky 1 (25). Tloušťka ucha se změří tloušťkoměrem (např. digitálním mikrometrem nebo tloušťkoměrem s kruhovou stupnicí) v den 1 (před aplikací první dávky), v den 3 (přibl. 48 hodin po první dávce) a v den 7 (24 hodin před ukončením) a v den 8. Kromě toho lze v den 8 určit tloušťku ucha na základě stanovení hmotnosti části ucha vyseknuté speciálními kleštěmi, což by se mělo provádět až poté, co se zvířata humánně usmrtí. Známkou nadměrného lokálního podráždění je zarudnutí ohodnocené 3 nebo více body a/nebo zvýšení tloušťky ucha o 25 % nebo více v kterýkoli den měření (26) (27). Jako nejvyšší dávka se pro hlavní studii LLNA: DA zvolí nejbližší nižší dávka v rámci řady koncentrací zjištěných při předběžné screeningové zkoušce (viz odstavec 18), která nevyvolává systémovou toxicitu a/nebo nadměrné lokální podráždění kůže.
Tabulka 1
Bodové hodnocení erytému
|
Pozorované příznaky
|
Bodové ohodnocení
|
|
Žádný erytém
|
0
|
|
Velmi slabý erytém (sotva patrný)
|
1
|
|
Zřetelně viditelný erytém
|
2
|
|
Mírný až výrazný erytém
|
3
|
|
Těžký erytém (silné zarudnutí) nebo tvorba příškvaru znemožňující posouzení erytému
|
4
|
|
|
23.
|
Kromě 25 % zvýšení tloušťky ucha (26) (27) se k identifikaci dráždivých látek při zkoušce LLNA používá také statisticky významné zvýšení tloušťky ucha u myší ze zkušební skupiny ve srovnání s kontrolními zvířaty (28) (29) (30) (31) (32) (33) (34). Ačkoli mohou nastat případy statisticky významného zvýšení, i když se tloušťka ucha zvýší o méně než 25 %, nebyly tyto případy spojeny výhradně s nadměrným podrážděním (30) (31) (32) (33) (34).
|
|
24.
|
O systémové toxicitě (35) mohou při použití v rámci integrovaného hodnocení svědčit tyto poznatky z klinického pozorování, a tudíž mohou indikovat maximální dávku, která by se měla použít v hlavní studii LLNA: DA: změny fungování nervového systému (např. piloerekce, ataxie, třes a křeče), změny chování (např. agresivita, změna činností týkajících se péče o srst, výrazná změna úrovně aktivity), změny způsobů dýchání (tj. změny frekvence a intenzity dýchání, např. dušnost, lapání po dechu a chropy) a změny v přijímání potravy a vody. Při hodnocení je navíc nutno brát v úvahu známky letargie a/nebo absence reagování, veškeré klinické příznaky více než mírné nebo momentální bolesti či utrpení nebo snížení tělesné hmotnosti ode dne 1 do dne 8 o více než 5 % a také úmrtnost. Umírající zvířata nebo zvířata, která jeví známky silné bolesti a utrpení, by se měla humánně usmrtit (36).
|
Harmonogram zkoušek v rámci hlavní studie
|
25.
|
Harmonogram zkoušky je následující:
— Den 1: Každé zvíře se zváží a zaznamená se jeho hmotnost a jakékoli poznatky z klinického pozorování. Na hřbet každého ucha se čtyřmi až pěti tahy štětečku namočeného do 1 % vodného roztoku natrium-dodecyl-sulfátu (SLS) nanese tento roztok tak, aby pokryl celou plochu hřbetu ucha. Jednu hodinu po nanesení roztoku SLS se na hřbet každého ucha aplikuje 25 μl vhodně zředěného roztoku zkoušené látky, samotné vehikulum nebo pozitivní kontrola (tu lze provádět buď souběžně, nebo lze použít pozitivní kontrolu z nedávné jiné zkoušky – v závislosti na postupech dané laboratoře při zohlednění odstavců 11–15).
— Den 2, den 3 a den 7: Zopakuje se postup předběžného ošetření 1 % vodným roztokem SLS a nanesení zkoušené látky provedený v den 1.
— Den 4, den 5 a den 6: Žádná aplikace.
— Den 8: Zaznamená se hmotnost každého zvířete a jakékoli poznatky z klinického pozorování. Přibližně 24 až 30 hodin po zahájení aplikace v den 7 se zvířata humánně usmrtí. U každé myši se vyjmou lymfatické uzliny drenující aurikulární oblast obou uší a zpracují se ve fosfátovém pufrovém fyziologickém roztoku (PBS) od každého zvířete zvlášť. Podrobnosti a grafické znázornění vyhledání uzlin a jejich disekce lze nalézt v literatuře (22). Pro účely dalšího sledování lokální kožní reakce v rámci hlavní studie lze do protokolu studie zařadit i další parametry, jako je bodové ohodnocení erytému ucha nebo údaje o tloušťce ucha (získané buď pomocí tloušťkoměru, nebo na základě stanovení hmotnosti části ucha vyseknuté speciálními kleštěmi při pitvě).
|
Příprava buněčných suspenzí
|
26.
|
Od každé myši se připraví jednobuněčná suspenze obsahující buňky z vyňatých lymfatických uzlin drenujících oblast obou uší; suspenze se připraví tak, že se lymfatické uzliny vloží mezi dvě podložná sklíčka a lehkým tlakem se rozdrtí. Po potvrzení, že je tkáň rozprostřena v tenké vrstvě, se sklíčka odtáhnou od sebe. Tkáně z obou sklíček se suspendují v roztoku PBS tak, že se každé sklíčko podrží nakloněné nad Petriho miskou a oplachuje se roztokem PBS při současném seškrabování tkáně ze sklíčka stěrkou na buňky. Kromě toho jsou lymfatické uzliny u zvířat z negativní kontrolní skupiny malé, takže je třeba postupovat opatrně, aby se zabránilo jakémukoli umělému ovlivnění hodnot SI. K opláchnutí obou sklíček by se měl použít celkový objem 1 ml PBS. Suspenzi buněk lymfatických uzlin je nutno v Petriho misce zlehka homogenizovat stěrkou na buňky. Mikropipetou se pak nabere 20μL poměrný díl suspenze buněk z lymfatických uzlin – přičemž je třeba nenabrat membránu, která je viditelná pouhým okem – a následně se smísí s 1,98 ml PBS, abychom dostali 2 ml vzorku. Druhý vzorek o objemu 2 ml se pak připraví stejným způsobem, aby vzniky od každého zvířete dva vzorky.
|
Stanovení buněčné proliferace (měření obsahu ATP v lymfocytech)
|
27.
|
Zvýšení obsahu ATP v lymfatických uzlinách se zjišťuje metodou, která je založena na použití luciferinu a luciferázy, za pomoci soupravy pro měření obsahu ATP, která měří bioluminescenci v relativních jednotkách luminiscence (RLU). U všech jednotlivých zvířat je nutno dodržet stejnou dobu od usmrcení do změření obsahu ATP, která by měla činit přibližně 30 minut, protože se má za to, že obsah ATP postupně klesá v závislosti na době uplynulé od usmrcení zvířete 12). Proto je nutno celou řadu úkonů od vynětí aurikulárních lymfatických uzlin až do změření obsahu ATP dokončit do 20 minut podle předem stanoveného harmonogramu, který je u každého zvířete stejný. Luminescenci ATP je nutno změřit u každého 2 ml vzorku, takže se získají dvě hodnoty ATP za každé zvíře. Pak se určí průměrná luminescence ATP, která se použije v následných výpočtech (viz odstavec 30).
|
ZÍSKANÉ POZNATKY
Poznatky z klinického pozorování
|
28.
|
Každá myš se jednou denně pečlivě vyšetří, zda nevykazuje jakékoli klinické příznaky lokálního podráždění v místě aplikace nebo příznaky systémové toxicity. Všechny získané poznatky se systematicky zaznamenají, přičemž záznamy se vedou zvlášť za každou myš. Plány sledování by měly obsahovat kritéria umožňující rychle identifikovat myši, jež vykazují známky systémové toxicity, nadměrného lokálního podráždění pokožky nebo poleptání kůže, aby mohly být usmrceny (36).
|
Hodnoty tělesné hmotnosti
|
29.
|
Jak je uvedeno v odstavci 25, tělesnou hmotnost jednotlivých zvířat je nutno zjistit na začátku zkoušky a v době plánovaného humánního usmrcení zvířete.
|
VÝPOČET VÝSLEDKŮ
|
30.
|
Výsledky za každou exponovanou skupinu se vyjádří jako průměrný index stimulace (SI). Hodnota SI se stanoví jako podíl průměrné hodnoty RLU na zvíře jak u každé exponované skupiny, tak u pozitivní kontrolní skupiny, a průměrné hodnoty RLU na zvíře u kontrolní skupiny s rozpouštědlem/vehikulem. Průměrná hodnota SI u kontrolních skupin s vehikulem je pak rovna 1.
|
|
31.
|
Při rozhodování o výsledku zkoušky se za pozitivní výsledek považuje, pokud hodnota SI ≥ 1,8 10). Při rozhodování, zda hraniční výsledek (tzn. hodnotu SI v rozmezí od 1,8 do 2,5) prohlásit za pozitivní, lze také použít míru závislosti odezvy na dávce, statistickou významnost a jednotnost výsledků kontrol s rozpouštědlem/vehikulem a pozitivních kontrol (2) (3) (37).
|
|
32.
|
U hraniční pozitivní odezvy v rozmezí hodnot SI od 1,8 do 2,5 mohou uživatelé zvážit další informace, například závislost odezvy na dávce, známky systémové toxicity nebo nadměrného podráždění, a tam, kde je to vhodné, také statistickou významnost spolu s hodnotami SI, aby se potvrdilo, že jsou takové výsledky pozitivní (10). Měla by se rovněž věnovat pozornost různým vlastnostem zkoušené látky včetně toho, zda se její struktura podobá struktuře známých senzibilizátorů kůže, zda u myší způsobuje nadměrné lokální podráždění kůže, a také povaze zjištěné závislosti odezvy na dávce. Těmito faktory a dalšími aspekty, které je třeba zvážit, se podrobně zabývá literatura (4).
|
|
33.
|
Shromažďování údajů úrovni jednotlivých myší umožní provést statistickou analýzu, kterou se na základě těchto údajů zjistí existence a míra závislosti odezvy na dávce. Případné statistické hodnocení by mohlo obsahovat posouzení závislosti odezvy na dávce, jakož i vhodně upravená srovnání zkušebních skupin (např. skupiny s párově srovnávaným dávkováním oproti souběžné kontrolní skupině s rozpouštědlem/vehikulem). Statistické analýzy mohou zahrnovat např. lineární regresi nebo Williamovu zkoušku k posouzení trendů závislosti odezvy na dávce a Dunnettovu zkoušku pro párová srovnání. Při výběru vhodné metody pro statistickou analýzu by si měl být zkoušející vědom možných nestejných odchylek a jiných souvisejících problémů, které si mohou vyžádat transformaci údajů nebo neparametrickou statistickou analýzu. V každém případě může být nezbytné, aby zkoušející provedl výpočty SI a statistické analýzy jednak za použití určitých krajních hodnot (někdy označovaných výrazem ‚odlehlé hodnoty‘) a jednak bez nich.
|
ÚDAJE A VYKAZOVÁNÍ
Údaje
|
34.
|
Údaje je nutno shrnout do tabulky s hodnotami RLU od jednotlivých zvířat, průměrnou hodnotou RLU na zvíře za celou skupinu, příslušným chybovým výrazem (např. směrodatnou odchylkou – SD, směrodatnou odchylkou průměrných hodnot – SEM) a průměrnou hodnotou SI za každou dávkovou skupinu oproti souběžné kontrolní skupině s rozpouštědlem nebo vehikulem.
|
Protokol o zkoušce
|
35.
|
Protokol o zkoušce musí obsahovat tyto informace:
|
|
Zkoušená látka a kontrolní látka:
|
—
|
identifikační údaje (např. číslo CAS a číslo ES, jsou-li k dispozici, zdroj, čistota, známé nečistoty, číslo šarže),
|
|
—
|
skupenství a fyzikálně-chemické vlastnosti (např. prchavost, stálost, rozpustnost),
|
|
—
|
jedná-li se o směs, je nutno uvést její složení a poměrné procentuální zastoupení složek.
|
|
|
|
Rozpouštědlo/vehikulum:
|
—
|
identifikační údaje (čistota, popřípadě koncentrace, použitý objem),
|
|
—
|
zdůvodnění volby vehikula.
|
|
|
|
Pokusná zvířata:
|
—
|
zdroj myší z kmene CBA,
|
|
—
|
mikrobiologický stav zvířat, je-li znám,
|
|
—
|
původ zvířat, podmínky chovu, strava atd.
|
|
|
|
Zkušební podmínky:
|
—
|
zdroj, číslo šarže a údaje od výrobce soupravy pro měření obsahu ATP, které se týkají prokazování a kontroly kvality,
|
|
—
|
podrobnosti o přípravě a aplikaci zkoušené látky,
|
|
—
|
zdůvodnění volby dávkování (včetně výsledků předběžné screeningové zkoušky, pokud byla provedena),
|
|
—
|
použité koncentrace vehikula a zkoušené látky a celkové aplikované množství zkoušené látky,
|
|
—
|
podrobnosti o jakosti vody a potravy (včetně druhu/zdroje stravy, zdroje vody),
|
|
—
|
podrobnosti o harmonogramech expozice a odběru vzorků,
|
|
—
|
metody měření toxicity,
|
|
—
|
kritéria pro označení studie za pozitivní nebo negativní,
|
|
—
|
údaje o případných odchylkách od protokolu a vysvětlení, jakým způsobem taková odchylka ovlivňuje koncepci studie a její výsledky.
|
|
|
|
Kontrola spolehlivosti:
|
—
|
souhrn výsledků nejnovější kontroly spolehlivosti včetně informací o použité zkoušené látce, koncentraci a vehikulu,
|
|
—
|
údaje ze souběžných a/nebo dřívějších pozitivních nebo negativních kontrolních skupin (s rozpouštědlem/vehikulem) zkoušených v dotyčné laboratoři,
|
|
—
|
pokud souběžná pozitivní kontrola nebyla součástí studie, je nutno uvést datum a laboratorní zprávu nejnovější periodické pozitivní kontroly a podrobnou zprávu s údaji z dřívějších pozitivních kontrol zkoušených v dotyčné laboratoři, které odůvodňují rozhodnutí neprovádět souběžné pozitivní kontroly.
|
|
|
|
Výsledky:
|
—
|
hmotnost jednotlivých myší na počátku aplikace a v době plánovaného usmrcení, jakož i průměrná hodnota a příslušný chybový výraz (např. SD, SEM) za každou exponovanou skupinu,
|
|
—
|
u každého zvířete časový průběh nástupu příznaků toxicity, včetně podráždění kůže v místě aplikace, pokud k němu dojde,
|
|
—
|
doba usmrcení a doba změření množství ATP u každého zvířete,
|
|
—
|
tabulka hodnot RLU a hodnot SI od jednotlivých myší za každou exponovanou skupinu,
|
|
—
|
průměrná hodnota RLU na zvíře a příslušný chybový výraz (např. SD, SEM) za každou exponovanou skupinu a výsledky analýzy odlehlých hodnot za každou exponovanou skupinu,
|
|
—
|
vypočtená hodnota SI a příslušná míra variability, která zohledňuje variabilitu mezi jednotlivými zvířaty jak z testování zkoušené látky, tak z kontrolních skupin,
|
|
—
|
závislost odezvy na dávce,
|
|
—
|
případné statistické analýzy.
|
|
|
|
Rozbor výsledků:
|
—
|
stručný komentář k výsledkům, k analýze závislosti odezvy na dávce a k případným statistickým analýzám a dále závěr, zda se má zkoušená látka považovat za senzibilizátor kůže.
|
|
|
LITERATURA
|
(1)
|
OECD (2010), Skin Sensitisation: Local Lymph Node Assay, Test Guideline No. 429, Guidelines for the Testing of Chemicals, OECD, Paris. K dispozici na adrese: [http://www.oecd.org/env/testguidelines].
|
|
(2)
|
Chamberlain, M. and Basketter, D.A. (1996), The local lymph node assay: status of validation. Food Chem, Toxicol., 34, 999–1002.
|
|
(3)
|
Basketter, D.A., Gerberick, G.F., Kimber, I. and Loveless, S.E. (1996), The local lymph node assay: A viable alternative to currently accepted skin sensitisation tests. Food Chem, Toxicol., 34, 985–997.
|
|
(4)
|
Basketter, D.A., Gerberick, G.F. and Kimber, I. (1998), Strategies for identifying false positive responses in predictive sensitisation tests. Food Chem. Toxicol., 36, 327–333.
|
|
(5)
|
Van Och, F.M.M., Slob, W., De Jong, W.H., Vandebriel, R.J. and Van Loveren, H. (2000), A quantitative method for assessing the sensitising potency of low molecular weight chemicals using a local lymph node assay: employment of a regression method that includes determination of uncertainty margins. Toxicol., 146, 49–59.
|
|
(6)
|
ICCVAM (1999), The murine local lymph node Assay: A test method for assessing the allergic contact dermatitis potential of chemicals/compounds: The results of an independent peer review evaluation coordinated by the Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program Center for the Evaluation of Alternative Toxicological Methods (NICETAM). NIH Publication No: 99-4494. Research Triangle Park, N.C. K dispozici na adrese: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/llna/llnarep.pdf].
|
|
(7)
|
Dean, J.H., Twerdok, L.E., Tice, R.R., Sailstad, D.M., Hattan, D.G., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: II. Conclusions and recommendations of an independent scientific peer review panel. Reg. Toxicol. Pharmacol. 34, 258–273.
|
|
(8)
|
Haneke, K.E., Tice, R.R., Carson, B.L., Margolin, B.H., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: III. Data analyses completed by the national toxicology program interagency center for the evaluation of alternative toxicological methods. Reg. Toxicol. Pharmacol. 34, 274–286.
|
|
(9)
|
Sailstad, D.M., Hattan, D., Hill, R.N., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: I. The ICCVAM review process. Reg. Toxicol. Pharmacol. 34, 249–257.
|
|
(10)
|
ICCVAM (2010), ICCVAM Test Method Evaluation Report. Nonradioactive local lymph node assay: modified by Daicel Chemical Industries, Ltd., based on ATP content test method protocol (LLNA: DA). NIH Publication No. 10-7551 A/B. Research Triangle Park, NC: National Institute of Environmental Health Sciences. K dispozici na adrese: [http://iccvam.niehs.nih.gov/methods/immunotox/llna-DA/TMER.htm].
|
|
(11)
|
ICCVAM (2009), Independent Scientific Peer Review Panel Report: Updated validation status of new versions and applications of the murine local lymph node assay: a test method for assessing the allergic contact dermatitis potential of chemicals and products. Research Triangle Park, NC: National Institute of Environmental Health Sciences. K dispozici na adrese: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/LLNAPRPRept2009.pdf].
|
|
(12)
|
Idehara, K., Yamagishi, G., Yamashita, K. and Ito, M. (2008), Characterization and evaluation of a modified local lymph node assay using ATP content as a non-radio isotopic endpoint. J. Pharmacol. Toxicol. Meth., 58, 1–10.
|
|
(13)
|
Omori, T., Idehara, K., Kojima, H., Sozu, T., Arima, K., Goto, H., Hanada, T., Ikarashi, Y., Inoda, T., Kanazawa, Y., Kosaka, T., Maki, E., Morimoto, T., Shinoda, S., Shinoda, N., Takeyoshi, M., Tanaka, M., Uratani, M., Usami, M., Yamanaka, A., Yoneda, T., Yoshimura, I. and Yuasa, A. (2008), Interlaboratory validation of the modified murine local lymph node assay based on adenosine triphosphate measurement. J. Pharmacol. Toxicol. Meth., 58, 11–26.
|
|
(14)
|
OECD (1992), Skin Sensitisation, Test Guideline No. 406, Guidelines for Testing of Chemicals, OECD, Paris. K dispozici na adrese: [http://www.oecd.org/env/testguidelines].
|
|
(15)
|
Kreiling, R., Hollnagel, H.M., Hareng, L., Eigler, L., Lee, M.S., Griem, P., Dreessen, B., Kleber, M., Albrecht, A., Garcia, C. and Wendel, A. (2008), Comparison of the skin sensitising potential of unsaturated compounds as assessed by the murine local lymph node assay (LLNA) and the guinea pig maximization test (GPMT). Food Chem. Toxicol., 46, 1896–1904.
|
|
(16)
|
Basketter, D., Ball, N., Cagen, S., Carrillo, J.C., Certa, H., Eigler, D., Garcia, C., Esch, H., Graham, C., Haux, C., Kreiling, R. and Mehling, A. (2009), Application of a weight of evidence approach to assessing discordant sensitisation datasets: implications for REACH. Reg. Toxicol. Pharmacol., 55, 90–96.
|
|
(17)
|
Crouch, S.P., Kozlowski, R., Slater, K.J. and Fletcher J. (1993), The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J. Immunol. Meth., 160, 81–88.
|
|
(18)
|
Ishizaka, A., Tono-oka, T. and Matsumoto, S. (1984), Evaluation of the proliferative response of lymphocytes by measurement of intracellular ATP. J. Immunol. Meth., 72, 127–132.
|
|
(19)
|
Dexter, S.J., Cámara, M., Davies, M. and Shakesheff, K.M. (2003), Development of a bioluminescent ATP assay to quantify mammalian and bacterial cell number from a mixed population. Biomat., 24, 27–34.
|
|
(20)
|
Lundin A. (2000), Use of firefly luciferase in ATP-related assays of biomass, enzymes, and metabolites. Meth. Enzymol., 305, 346–370.
|
|
(21)
|
ILAR (1996), Institute of Laboratory Animal Research (ILAR) Guide for the Care and Use of Laboratory Animals. 7th ed. Washington, DC: National Academies Press.
|
|
(22)
|
ICCVAM (2009), Recommended Performance Standards: Murine Local Lymph Node Assay, NIH Publication Number 09-7357, Research Triangle Park, NC: National Institute of Environmental Health Science. K dispozici na adrese: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/llna-ps/LLNAPerfStds.pdf]
|
|
(23)
|
McGarry, H.F. (2007), The murine local lymph node assay: regulatory and potency considerations under REACH. Toxicol., 238, 71–89.
|
|
(24)
|
Kimber, I., Dearman, R.J., Scholes E.W. and Basketter, D.A. (1994), The local lymph node assay: developments and applications. Toxicol., 93, 13–31.
|
|
(25)
|
OECD (2002), Acute Dermal Irritation/Corrosion, Test Guideline No. 404, Guidelines for Testing of Chemicals, OECD, Paris. K dispozici na adrese: [http://www.oecd.org/env/testguidelines].
|
|
(26)
|
Reeder, M.K., Broomhead, Y.L., DiDonato, L. and DeGeorge, G.L. (2007), Use of an enhanced local lymph node assay to correctly classify irritants and false positive substances. Toxicologist, 96, 235.
|
|
(27)
|
ICCVAM (2009), Nonradioactive Murine Local Lymph Node Assay: Flow Cytometry Test Method Protocol (LLNA: BrdU-FC) Revised Draft Background Review Document. Research Triangle Park, NC: National Institute of Environmental Health Sciences. K dispozici na adrese: [http://iccvam.niehs.nih.gov/methods/immunotox/fcLLNA/BRDcomplete.pdf].
|
|
(28)
|
Hayes, B.B., Gerber, P.C., Griffey, S.S. and Meade, B.J. (1998), Contact hypersensitivity to dicyclohexylcarbodiimide and diisopropylcarbodiimide in female B6C3F1 mice. Drug. Chem. Toxicol., 21, 195–206.
|
|
(29)
|
Homey, B., von Schilling, C., Blumel, J., Schuppe, H.C., Ruzicka, T., Ahr, H.J., Lehmann, P. and Vohr, V.W. (1998), An integrated model for the differentiation of chemical-induced allergic and irritant skin reactions. Toxicol. Appl. Pharmacol., 153, 83–94.
|
|
(30)
|
Woolhiser, M.R., Hayes, B.B. and Meade, B.J. (1998), A combined murine local lymph node and irritancy assay to predict sensitisation and irritancy potential of chemicals. Toxicol. Meth., 8, 245–256.
|
|
(31)
|
Hayes, B.B. and Meade, B.J. (1999), Contact sensitivity to selected acrylate compounds in B6C3F1 mice: relative potency, cross reactivity, and comparison of test methods. Drug Chem. Toxicol., 22, 491–506.
|
|
(32)
|
Ehling, G., Hecht, M., Heusener, A., Huesler, J., Gamer, A.O., van Loveren, H., Maurer, T., Riecke, K., Ullmann, L., Ulrich, P., Vandebriel, R. and Vohr, H.W. (2005), A European inter-laboratory validation of alternative endpoints of the murine local lymph node assay: first round. Toxicol., 212, 60–68.
|
|
(33)
|
Vohr, H.W. and Ahr, H.J. (2005), The local lymph node assay being too sensitive? Arch. Toxicol., 79, 721–728.
|
|
(34)
|
Patterson, R.M., Noga, E. and Germolec, D. (2007), Lack of evidence for contact sensitisation by Pfiesteria extract. Environ. Health Perspect., 115, 1023–1028.
|
|
(35)
|
ICCVAM (2009), Report on the ICCVAM-NICEATM/ECVAM/JaCVAM Scientific Workshop on Acute Chemical Safety Testing: Advancing In Vitro Approaches and Humane Endpoints for Systemic Toxicity Evaluations. Research Triangle Park, NC: National Institute of Environmental Health Sciences. K dispozici na adrese: [http://iccvam.niehs.nih.gov/methods/acutetox/Tox_workshop.htm].
|
|
(36)
|
OECD (2000), Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation, Environmental Health and Safety Monograph Series on Testing and Assessment No. 19, ENV/JM/MONO(2000)7, OECD, Paris. K dispozici na adrese: [http://www.oecd.org/env/testguidelines].
|
|
(37)
|
Kimber, I., Hilton, J., Dearman, R.J., Gerberick, G.F., Ryan, C.A., Basketter, D.A., Lea, L., House, R.V., Ladies, G.S., Loveless, S.E. and Hastings, K.L. (1998), Assessment of the skin sensitisation potential of topical medicaments using the local lymph node assay: An interlaboratory exercise. J. Toxicol. Environ. Health, 53 563–79.
|
|
(38)
|
OECD (2005), Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment, Environment, Health and Safety Monograph Series on Testing and Assessment No. 34, ENV/JM/MONO(2005)14, OECD, Paris. K dispozici na adrese: [http://www.oecd.org/env/testguidelines].
|
Dodatek 1
DEFINICE
Přesnost: Blízkot shody mezi výsledky zkušební metody a uznávanými referenčními hodnotami. Je to míra funkčnosti zkušební metody a jeden z aspektů její relevantnosti. Tento výraz se často používá namísto výrazu „soulad“ a rozumí se jím podíl správných výsledků zkušební metody (38).
Srovnávací látka: Senzibilizující nebo nesenzibilizující látka použitá jako standard pro srovnání se zkoušenou látkou. Srovnávací látka by měla mít tyto vlastnosti: i) konzistentní a spolehlivý zdroj (konzistentní a spolehlivé zdroje); ii) strukturální a funkční podobnost s třídou zkoušených látek; iii) známé fyzikálně-chemické vlastnosti; iv) podklady o známých účincích a v) známá účinnost v rozsahu žádoucí odezvy.
Falešně negativní: Látka je zkušební metodou nesprávně označena za negativní nebo neaktivní, ačkoli ve skutečnosti je pozitivní nebo aktivní.
Falešně pozitivní: Látka je zkušební metodou nesprávně označena za pozitivní nebo aktivní, ačkoli ve skutečnosti je negativní nebo neaktivní.
Nebezpečnost: Potenciál látky působit nepříznivě na zdraví nebo životní prostředí. Nepříznivý účinek se projevuje pouze při dostatečné úrovni expozice.
Mezilaboratorní reprodukovatelnost: Míra, v jaké mohou různé kvalifikované laboratoře za použití stejného protokolu a při testování stejných zkoušených látek získat kvalitativně a kvantitativně podobné výsledky. Mezilaboratorní reprodukovatelnost se určuje během předvalidačních a validačních postupů a ukazuje, do jaké míry lze zkoušku úspěšně přenášet mezi různými laboratořemi (38).
Vnitrolaboratorní reprodukovatelnost: Určení míry, v jaké může kvalifikovaný personál ve stejné laboratoři jindy úspěšně dosáhnout stejných výsledků za použití určitého protokolu (38).
Odlehlá hodnota: Odlehlá hodnota je poznatek, který se výrazně liší od ostatních hodnot v náhodném vzorku od celé populace.
Prokazování kvality: Řídicí proces, v jehož rámci se hodnotí dodržování zkušebních norem a standardů laboratoře, příslušných požadavků a postupů vedení záznamů, jakož i přesnost přenosu údajů, přičemž tyto prvky jsou hodnoceny osobami, které jsou nezávislé na pracovnících, kteří zkoušky provádějí.
Spolehlivost: Míra, v jaké lze zkušební metodu provádět reprodukovatelně v rámci téže laboratoře i v rámci různých laboratoří v průběhu doby, jestliže se provádí za použití stejného protokolu. Hodnotí se na základě výpočtu vnitrolaboratorní a mezilaboratorní reprodukovatelnosti (38).
Senzibilizace kůže: Imunologický proces, který nastává, když je vnímavá osoba vystavena místně aplikovanému chemickému alergenu, který vyvolává kožní imunitní odezvu, jež může vést ke vzniku kontaktní senzibilizace.
Index stimulace (SI): Hodnota vypočtená za účelem posouzení potenciálu zkoušené látky vyvolat senzibilizaci kůže. Vyjadřuje poměr hodnoty proliferace u exponovaných skupin k hodnotě proliferace u souběžné kontrolní skupiny s vehikulem.
Zkoušená látka: Jakákoli látka nebo směs, která se zkouší za použití této zkušební metody.
B.51. SENZIBILIZACE KŮŽE: ZKOUŠKA S VYŠETŘENÍM LOKÁLNÍCH LYMFATICKÝCH UZLIN: BRDU-ELISA
ÚVOD
|
1.
|
Metodiky OECD pro testování chemických látek a zkušební metody EU jsou pravidelně přezkoumávány s ohledem na vědecký pokrok, změny regulačních potřeb a dobré životní podmínky zvířat. První zkušební metoda (ZM) (kapitola B.42 této přílohy) pro stanovení senzibilizace kůže u myší, tzv. zkouška s vyšetřením lokálních lymfatických uzlin (LLNA; zkušební metodika OECD č. 429) byla revidována (1 a kapitola B.42 této přílohy). Podrobné údaje o validaci metody LLNA a přehled prací spojených s touto metodou byly publikovány (2) (3) (4) (5) (6) (7) (8) (9). V rámci metody LLNA se k měření proliferace lymfocytů využívají radioizotopy thymidinu nebo jódu, a proto má tato zkouška omezené použití, pokud získání, používání nebo likvidace radioaktivních látek představují problém. Zkušební metoda LLNA: BrdU-ELISA [Enzyme-Linked Immunosorbent Assay] je neradioaktivní modifikace zkušební metody LLNA, která k měření proliferace lymfocytů využívá neradioaktivně značený 5-brom-2-deoxyuridin (BrdU) (Reg. č. Chemical Abstracts Service [CAS] 59-14-3) v rámci systému založeném na zkoušce ELISA. Metoda LLNA: BrdU-ELISA byla ověřena, přezkoumána a doporučena mezinárodní vědeckou komisí pro vzájemné hodnocení jakožto metoda, která je s určitými omezeními považována za užitečnou pro identifikaci chemických látek způsobujících nebo naopak nezpůsobujících senzibilizaci kůže (10) (11) (12). Tato ZM je určena k hodnocení potenciálu chemikálií (látek a směsí) způsobovat senzibilizaci kůže u zvířat. Zkušební metoda podle kapitoly B.6 této přílohy a zkušební metodiky OECD č. 406 využívá zkoušek na morčatech, a to zvláště maximalizační zkoušku na morčatech a Bühlerův test (13). Metoda LLNA (kapitola B.42 této přílohy; zkušební metodika OECD č. 429) a její dvě neradioaktivní modifikace, metoda LLNA: BrdU-ELISA (kapitola B.51 této přílohy; zkušební metodika OECD č. 442 B) a metoda LLNA: DA (kapitola B.50 této přílohy; zkušební metodika OECD č. 442 A) poskytují výhody ve srovnání se zkouškami na morčatech podle kapitoly B.6 a zkušební metodiky OECD č. 406 (13), pokud jde o snížení počtu využívaných zvířat a zdokonalení způsobu jejich využívání.
|
|
2.
|
Podobně jako metoda LLNA i metoda LLNA: BrdU-ELISA zkoumá indukční fázi senzibilizace kůže a poskytuje kvantitativní údaje, které jsou vhodné pro hodnocení závislosti odezvy na dávce. Schopnost detekovat látky senzibilizující kůži bez nutnosti použití radioizotopů pro značení DNA navíc odstraňuje možnost expozice radioaktivitě při práci a problémy s likvidací odpadů. To zase může umožnit zvýšené využívání myší k detekování látek senzibilizujících kůži, což by mohlo dále snížit využívání morčat k testování potenciálu chemických látek způsobovat senzibilizaci kůže (tzn. metodou podle kapitoly B.6 a zkušební metodiky OECD č. 406) (13).
|
DEFINICE
|
3.
|
Použité definice jsou uvedeny v dodatku 1.
|
VÝCHOZÍ ÚVAHY A OMEZENÍ
|
4.
|
Zkušební metoda LLNA: BrdU-ELISA je modifikovanou metodou LLNA, která s určitými omezeními slouží k identifikaci chemických látek, jež mohou způsobovat senzibilizaci kůže. To však nutně neznamená, že by se metoda LLNA: BrdU-ELISA měla používat ve všech případech namísto metody LLNA nebo namísto zkoušek na morčatech (tzn. podle kapitoly B.6 této přílohy a zkušební metodiky OECD č. 406) (13); znamená to spíše, že tato metoda má stejnou hodnotu a že ji lze použít jako alternativní metodu, jejíž pozitivní ani negativní výsledky obvykle již nevyžadují žádné další potvrzování (10) (11). Zkušební laboratoř by měla před provedením studie zohlednit všechny dostupné informace o zkoušené látce. Mezi tyto informace patří totožnost a chemická struktura zkoušené látky, její fyzikálně-chemické vlastnosti, výsledky jakýchkoli jiných zkoušek toxicity in vitro a in vivo za použití zkoušené látky a toxikologické údaje o strukturně příbuzných látkách. Tyto informace je nutno posoudit, aby se zjistilo, zda je metoda LLNA: BrdU-ELISA vhodná pro danou látku (s ohledem na neslučitelnost některých druhů chemických látek s metodou LLNA: BrdU-ELISA [viz odstavec 5]), a také aby se usnadnila volba dávkování.
|
|
5.
|
Zkušební metoda LLNA: BrdU-ELISA je metodou in vivo, a tudíž neeliminuje používání zvířat při hodnocení alergického kontaktního senzibilizujícího působení. Umožňuje však omezit využívání zvířat pro tento účel ve srovnání se zkouškami na morčatech (podle kapitoly B.6 a zkušební metodiky OECD č. 406) (13). Kromě toho metoda LLNA: BrdU-ELISA nabízí podstatné zlepšení způsobu zacházení se zvířaty, která se využívají ke zkoušení kontaktní alergické senzibilizace, jelikož na rozdíl od metody podle kapitoly B.6 a zkušební metodiky OECD č. 406 metoda LLNA: BrdU-ELISA nevyžaduje umělé vyvolání kožní přecitlivělosti. Kromě toho metoda LLNA: BrdU-ELISA nevyžaduje ani používání adjuvantu, jak je tomu v případě maximalizační zkoušky na morčatech (podle kapitoly B.6 této přílohy) (13). Díky tomu metoda LLNA: BrdU-ELISA snižuje utrpení zvířat. Ale i přes výhody, které má metoda LLNA: BrdU-ELISA ve srovnání s metodou popsanou v kapitole B.6 a zkušební metodice OECD č. 406 (13), existují určitá omezení, která si mohou vyžádat použití metody popsané v kapitole B.6 nebo zkušební metodice OECD č. 406 (např. zkoušení některých kovů, falešně pozitivní výsledky u určitých látek způsobujících podráždění kůže (jako jsou například některé chemikálie typu povrchově aktivních látek) (6) (1 a kapitola B.42 této přílohy) nebo rozpustnost zkoušené látky). Kromě toho si použití zkoušek na morčatech (tzn. podle kapitoly B.6 nebo zkušební metodiky OECD č. 406) (13) mohou vyžádat také některé třídy chemických látek nebo látky obsahující funkční skupiny, u nichž je prokázáno, že mohou způsobovat zkreslení výsledků (15). Doporučuje se, aby se omezení zjištěná u metody LLNA (1 a kapitola B.42 této přílohy) vztahovala rovněž na metodu LLNA: BrdU-ELISA (10). S výjimkou těchto známých omezení by měla být metoda LLNA: BrdU-ELISA použitelná k testování jakýchkoli látek, nejsou-li s těmito látkami spojeny vlastnosti, které mohou nepříznivě ovlivnit přesnost metody LLNA: BrdU-ELISA. Kromě toho je třeba zvážit možnost hraničních pozitivních výsledků při získání hodnot indexu stimulace (SI) v rozmezí od 1,6 do 1,9 (viz odstavce 31 až 32). To je založeno na validační databázi 43 látek za použití hodnot SI ≥ 1,6 (viz odstavec 6), u kterých metoda LLNA: BrdU-ELISA správně identifikovala všech 32 látek identifikovaných jako senzibilizující látky metodou LLNA, ale nesprávně identifikovala dvě z 11 látek identifikovaných metodou LLNA jako nesenzibilizující látky s hodnotami SI v rozmezí od 1,6 do 1,9 (tzn. hraniční pozitivní) (10). Jelikož však byl stejný soubor údajů použit k stanovení hodnot SI a k výpočtu prediktivních vlastností zkoušky, mohou uvedené výsledky představovat nadhodnocení skutečných prediktivních vlastností této metody.
|
PODSTATA ZKUŠEBNÍ METODY
|
6.
|
Základní podstata metody LLNA: BrdU-ELISA spočívá v tom, že senzibilizující látky vyvolávají proliferaci lymfocytů v lymfatických uzlinách drenujících místo aplikace zkoušené látky. Tato proliferace je úměrná dávce a potenciálu aplikovaného alergenu a představuje jednoduchý prostředek k získání kvantitativního údaje o míře senzibilizace. Proliferace se měří porovnáním průměrné proliferace u každé zkušební skupiny s průměrnou proliferací u kontrolní skupiny s vehikulem. Určí se poměr průměrné proliferace u každé z exponovaných skupin k průměrné proliferaci u souběžně testované kontrolní skupiny s vehikulem, nazývaný index stimulace (SI), přičemž jeho hodnota by měla být ≥ 1,6, aby bylo oprávněné další hodnocení zkoušené látky jako potenciálního senzibilizátoru kůže. Zde popsané postupy jsou založeny na použití měření obsahu BrdU k indikaci zvýšeného počtu proliferujících buněk v lymfatických uzlinách drenujících aurikulární oblast. Látka BrdU je analogem thymidinu a začleňuje se podobným způsobem do DNA proliferujících buněk. Začlenění BrdU se měří metodou ELISA, která využívá protilátku specifickou pro BrdU, která je rovně značí peroxidázou. Po přidání substrátu peroxidáza reaguje se substrátem při vzniku barevného produktu, jehož množství se určuje při určité absorbanci za použití čtečky mikrotitračních destiček.
|
POPIS ZKOUŠKY
Volba druhu zvířat
|
7.
|
Vhodným druhem pro tuto zkoušku je myš. Validační studie metody LLNA: BrdU-ELISA byly prováděny výhradně s kmenem CBA/JN, který je proto považován za upřednostňovaný kmen (10) (12). Používají se mladé dospělé samice myší, které musí být nullipary a nesmějí být březí. Při zahájení studie by zvířata měla být přibližně 8 až 12 týdnů stará, přičemž odchylky v hmotnosti zvířat by měly být pouze minimální a neměly by překročit 20 % průměrné hmotnosti. Případně lze používat i jiné kmeny a samce, pokud je k dispozici dostatek údajů, které dokazují, že v rámci reakcí neexistují u metody LLNA: BrdU-ELISA významné specifické odlišnosti mezi kmeny a/nebo pohlavími.
|
Podmínky chovu a krmení
|
8.
|
Myši by měly být chovány ve skupinách (16), není-li předloženo dostatečné vědecké zdůvodnění, proč by měly být chovány jednotlivě. Teplota v místnosti pro pokusná zvířata by měla být 22 ± 3 °C. Ačkoli by relativní vlhkost vzduchu měla být minimálně 30 % a pokud možno by kromě doby úklidu místnosti neměla přesáhnout 70 %, cílem by měla být hodnota 50–60 %. Osvětlení by mělo být umělé se střídáním 12 hodin světla a 12 hodin tmy. Ke krmení lze používat běžnou laboratorní stravu a k pití by měly mít myši k dispozici neomezené množství vody.
|
Příprava zvířat
|
9.
|
Zvířata se náhodně vyberou, pro usnadnění individuální identifikace se označí (nesmí se však použít žádná forma značení uší) a chovají se v klecích minimálně pět dnů před začátkem aplikování zkoušené látky, aby se mohla přizpůsobit laboratorním podmínkám. Před začátkem aplikace látky se všechna zvířata vyšetří, aby se ověřilo, že nemají žádné pozorovatelné kožní léze.
|
Příprava dávkovacích roztoků
|
10.
|
Pevné chemické látky by se měly před nanesením na ucho myši rozpustit nebo suspendovat ve vhodných rozpouštědlech nebo vehikulech a popřípadě zředit. Kapalné látky lze aplikovat bez přísad nebo je lze před dávkováním rozpustit. Nerozpustné látky, jaké se nejčastěji používají k výrobě zdravotnických prostředků, by měly být vystaveny intenzívní extrakci ve vhodném rozpouštědle, aby se před nanesením na ucho myši odhalily všechny extrahovatelné složky, které je nutno testovat. Zkoušené látky by se měly připravovat každý den čerstvé, pokud údaje o stálosti neprokazují přijatelnost jejich skladování.
|
Kontrola spolehlivosti
|
11.
|
K prokázání řádné funkčnosti zkoušky slouží pozitivní kontrolní chemikálie, a to tak, že reagují s dostatečnou a reprodukovatelnou citlivostí na zkoušenou senzibilizující látku, u níž je dobře popsána síla odezvy. Zařazení souběžné pozitivní kontroly se doporučuje proto, že prokazuje způsobilost laboratoře úspěšně provést každou zkoušku a umožňuje posoudit vnitrolaboratorní a mezilaboratorní reprodukovatelnost a srovnatelnost. Některé regulační orgány také vyžadují při každé studii pozitivní kontrolu, a proto se uživatelům doporučuje před provedením zkoušky LLNA: BrdU-ELISA příslušné orgány konzultovat. Doporučuje se tudíž běžně používat souběžnou pozitivní kontrolu, aby nebylo nutné další zkoušení na zvířatech ke splnění požadavků, které by mohly vzniknout při používání periodické pozitivní kontroly (viz odstavec 12). Pozitivní kontrola by měla v rámci metody LLNA: BrdU-ELISA vyvolat pozitivní odezvu při úrovni expozice, u které se očekává zvýšení indexu stimulace (SI) > 1,6 ve srovnání s negativní kontrolní skupinou. Dávka pozitivní kontroly by měla být zvolena tak, aby nevyvolala nadměrné podráždění kůže nebo systémovou toxicitu a přitom aby indukce byla reprodukovatelná, avšak nikoli nadměrná (tzn. že hodnota SI > 14 by byla považována za příliš velkou). K upřednostňovaným látkám pro pozitivní kontrolu patří 25 % roztok 2-benzylidenoktanalu (č. CAS 101-86-0) nebo eugenolu (číslo CAS 97-53-0) v acetonu s olivovým olejem (v objemovém poměru 4:1). Mohou existovat určité okolnosti, za nichž lze při řádném zdůvodnění použít i jiné pozitivní kontrolní látky, které splňují výše uvedená kritéria.
|
|
12.
|
I když se zařazení skupiny souběžných pozitivních kontrol doporučuje, mohou existovat situace, kdy může být periodické testování (tzn. v intervalech ≤ 6 měsíců) pozitivních kontrol vhodné u laboratoří, které provádějí zkoušky LLNA: BrdU-ELISA pravidelně (tzn. nejméně jednou měsíčně) a mají k dispozici databázi dřívějších údajů pozitivních kontrol, která prokazuje schopnost laboratoře získat při použití pozitivních kontrol reprodukovatelné a přesné výsledky. Odpovídající zběhlost v používání metody LLNA: BrdU-ELISA lze úspěšně prokázat dosažením shodných pozitivních výsledků při použití pozitivní kontroly alespoň v 10 nezávislých zkouškách provedených během přiměřené doby (tzn. během méně než jednoho roku).
|
|
13.
|
Skupina souběžných pozitivních kontrol by se měla zařadit vždy, když dojde k procedurální změně metody LLNA: BrdU-ELISA (např. při změně složení vyškoleného personálu, při změně materiálů a/nebo činidel či vybavení, jež se používají v rámci zkušební metody, nebo při změně zdroje pokusných zvířat), přičemž takové změny by se měly vždy zdokumentovat v laboratorních zprávách. Při určování nezbytnosti vytvořit novou databázi ke zdokumentování shodnosti výsledků pozitivních kontrol by se měl zvážit dopad těchto změn na přiměřenost dosavadní databáze.
|
|
14.
|
Zkoušející by si měli uvědomit, že rozhodnutí používat pozitivní kontroly periodicky namísto souběžně má závažné dopady na přiměřenost a přijatelnost negativních výsledků studie získaných bez souběžných pozitivních kontrol během intervalu mezi jednotlivými periodickými použitími pozitivních kontrol. Například obdržení falešně negativního výsledku při periodickém používání pozitivních kontrol může zpochybnit negativní výsledky zkoušené látky získané v období mezi poslední přijatelnou periodickou studií pozitivních kontrol a nepřijatelnou periodickou studií pozitivních kontrol. Důsledky, jež z toho vyplývají, je třeba pečlivě zvážit při rozhodování, zda do zkoumání zařadit souběžné pozitivní kontroly, nebo používat pouze periodické pozitivní kontroly. Je také nutno vzít v úvahu možnost použít v rámci skupiny souběžných pozitivních kontrol méně zvířat, jestliže je to vědecky opodstatněné a jestliže laboratoř na základě svých vlastních dosavadních údajů prokáže, že lze používat méně myší (17).
|
|
15.
|
Ačkoli by se pozitivní kontroly měly zkoušet ve vehikulu, o němž je známo, že vždy vyvolává shodnou reakci (např. aceton s olivovým olejem v objemovém poměru 4:1), mohou nastat určité situace, kdy je z důvodů požadovaných v právních předpisech nezbytné provést zkoušku s použitím nestandardního vehikula (klinicky/chemicky relevantní složení) (18). Pokud se souběžná pozitivní kontrola zkouší v jiném vehikulu než zkoušená látka, měla by být součástí zkoušky se souběžnou pozitivní kontrolou také samostatná kontrola s vehikulem.
|
|
16.
|
V případech, kdy se hodnotí zkoušené látky, které patří do určité třídy chemických látek nebo vykazují určitou škálu odezev, mohou být užitečné také srovnávací látky, jelikož mohou prokázat, že zkušební metoda je schopna správně zjistit potenciál těchto druhů zkoušených látek vyvolávat senzibilizaci kůže. Vhodné srovnávací látky by měly mít tyto vlastnosti:
|
—
|
strukturální a funkční podobnost s třídou, do níž patří zkoušená látka,
|
|
—
|
známé fyzikálně-chemické vlastnosti,
|
|
—
|
podklady ze zkoušky LLNA: BrdU-ELISA,
|
|
—
|
podklady získané při použití jiných zvířecích modelů a/nebo lidí.
|
|
ZKUŠEBNÍ POSTUP
Počet zvířat a dávkování
|
17.
|
Na každou dávkovou skupinu se použijí alespoň čtyři zvířata, přičemž se použijí minimálně tři koncentrace zkoušené látky, dále se použije souběžná negativní kontrolní skupina, k jejíž expozici se použije pouze vehikulum zkoušené látky, a pozitivní kontrolní skupina (souběžná nebo z nedávné jiné zkoušky – v závislosti na postupech dané laboratoře při zohlednění odstavců 11–15). Mělo by se zvážit zkoušení většího počtu dávek pozitivní kontroly, a to zejména v případě, že se pozitivní kontrola nepoužívá soustavně. Se zvířaty v kontrolních skupinách by se s výjimkou aplikace zkoušené látky mělo zacházet stejně jako se zvířaty v exponovaných skupinách.
|
|
18.
|
Volba dávkování a vehikula by měla být založena na doporučeních uvedených v literatuře – (2) a (19). Po sobě jdoucí dávky se běžně vybírají z vhodné řady koncentrací, například 100 %, 50 %, 25 %, 10 %, 5 %, 2,5 %, 1 %, 0,5 % atd. Volba této řady koncentrací by měla být dostatečně vědecky zdůvodněna. Při výběru tří po sobě jdoucích koncentrací je třeba zvážit veškeré existující toxikologické informace (např. o akutní toxicitě a kožní dráždivosti) a informace o struktuře a fyzikálně-chemických vlastnostech zkoušené látky (a/nebo strukturně příbuzných látek), jsou-li k dispozici, aby nejvyšší koncentrace maximalizovala expozici, aniž by současně vyvolala systémovou toxicitu a/nebo nadměrné lokální podráždění kůže (19) (20 a kapitola B.4 této přílohy). Nejsou-li takové informace k dispozici, může být nezbytné provést úvodní předběžnou screeningovou zkoušku (viz odstavce 21 až 24).
|
|
19.
|
Vehikulum by nemělo nepříznivě ovlivňovat ani jinak zkreslovat výsledky zkoušky a mělo by být vybráno tak, aby se maximalizovala rozpustnost umožňující dosáhnout nejvyšší možné koncentrace a zároveň připravit roztok nebo suspenzi, jež jsou vhodné pro aplikaci zkoušené látky. K doporučeným vehikulům patří aceton s olivovým olejem (v objemovém poměru 4:1), N,N-dimethylformamid, ethyl(methyl)keton, propan-1,2-diol a dimethylsulfoxid (6), avšak při dostatečném vědeckém zdůvodnění lze použít i jiná vehikula. V určitých situacích může být nezbytné použít jako doplňující kontrolu klinicky relevantní rozpouštědlo nebo běžně prodejný přípravek obsahující zkoušenou látku. Zvláště je třeba dbát na to, aby byly hydrofilní zkoušené látky zapracovány do takového vehikula, které zvlhčí kůži a nesteče ihned z pokožky. Toho lze dosáhnout použitím vhodných solubilizérů (jako je např. 1 % Pluronic® L92). Není tudíž vhodné používat zcela vodná vehikula.
|
|
20.
|
Zpracování lymfatických uzlin z jednotlivých myší umožňuje posoudit variabilitu mezi jednotlivými zvířaty a provést statistické porovnání rozdílu mezi zkoušenou látkou a hodnotami naměřenými u kontrolní skupiny se samotným vehikulem (viz odstavec 33). Posoudit možnost snížit počet myší v pozitivní kontrolní skupině je kromě toho vhodné jen tehdy, když se zaznamenávají údaje od jednotlivých zvířat (17). Sběr údajů od jednotlivých zvířat navíc vyžadují i některé regulační orgány. Pravidelný sběr údajů od jednotlivých zvířat je výhodnější z hlediska dobrých životních podmínek zvířat, jelikož nevyžaduje duplicitní zkoušení, které by bylo nezbytné, pokud by výsledky pro určitou zkoušenou látku, jež byly původně shromážděny jedním způsobem (např. za použití souhrnných údajů od všech zvířat), měly být později posouzeny regulačními orgány s jinými požadavky (např. vyžadujícími použití údajů od jednotlivých zvířat).
|
Předběžná screeningová zkouška
|
21.
|
Nejsou-li k dispozici informace pro stanovení nejvyšší dávky, která má být předmětem zkoušky (viz odstavec 18), měla by se provést předběžná screeningová zkouška umožňující vymezit vhodné dávkování, které se bude metodou LLNA: BrdU-ELISA zkoušet. Účelem předběžné screeningové zkoušky je poskytnout vodítko pro volbu maximálního dávkování, které se má použít v hlavní studii LLNA: BrdU-ELISA, pokud nejsou k dispozici informace o koncentraci, která vyvolává systémovou toxicitu (viz odstavec 24) a/nebo nadměrné lokální podráždění kůže (viz odstavec 23). Maximální zkoušenou dávkou by měla být 100 % koncentrace zkoušené látky v případě kapalin, respektive maximální možná koncentrace v případě pevných látek nebo suspenzí.
|
|
22.
|
Předběžná screeningová zkouška se provádí za stejných podmínek jako hlavní studie LLNA: BrdU-ELISA až na to, že se nehodnotí proliferace buněk z lymfatických uzlin a lze použít méně zvířat na dávkovou skupinu. Navrhuje se počet jednoho až dvou zvířat na dávkovou skupinu. Všechny myši se denně pozorují, zda nevykazují klinické příznaky systémové toxicity nebo lokálního podráždění v místě aplikace. Před zahájením zkoušky a před jejím ukončením (den 6) se zaznamenají hodnoty tělesné hmotnosti zvířat. Na obou uších každé myši se sleduje výskyt erytému, který se vyhodnotí podle tabulky 1 (20 a kapitola B.4 této přílohy). Tloušťka ucha se změří tloušťkoměrem (např. digitálním mikrometrem nebo tloušťkoměrem s kruhovou stupnicí) v den 1 (před aplikací první dávky), v den 3 (přibl. 48 hodin po první dávce) a v den 6. Kromě toho lze v den 6 určit tloušťku ucha na základě stanovení hmotnosti části ucha vyseknuté speciálními kleštěmi, což by se mělo provádět až poté, co se zvířata humánně usmrtí. Známkou nadměrného lokálního podráždění je zarudnutí ohodnocené 3 nebo více body a/nebo zvýšení tloušťky ucha o 25 % nebo více v kterýkoli den měření (21) (22). Jako nejvyšší dávka se pro hlavní studii LLNA: BrdU-ELISA v rámci řady koncentrací zjištěných při předběžné screeningové zkoušce (viz odstavec 18) zvolí nejbližší nižší dávka, která nevyvolává systémovou toxicitu a/nebo nadměrné lokální podráždění kůže.
Tabulka 1
Bodové hodnocení erytému
|
Pozorované příznaky
|
Bodové ohodnocení
|
|
Žádný erytém
|
0
|
|
Velmi slabý erytém (sotva patrný)
|
1
|
|
Zřetelně viditelný erytém
|
2
|
|
Mírný až výrazný erytém
|
3
|
|
Těžký erytém (silné zarudnutí) nebo tvorba příškvaru znemožňující posouzení erytému
|
4
|
|
|
23.
|
Kromě 25 % zvýšení tloušťky ucha (21) (22) se k identifikaci dráždivých látek při zkoušce LLNA používá také statisticky významné zvýšení tloušťky ucha u myší ze zkušební skupiny ve srovnání s kontrolními zvířaty (22) (23) (24) (25) (26) (27) (28). Ačkoli mohou nastat případy statisticky významného zvýšení, i když se tloušťka ucha zvýší o méně než 25 %, nebyly tyto případy spojeny výhradně s nadměrným podrážděním (25) (26) (27) (28) (29).
|
|
24.
|
O systémové toxicitě (30) mohou při použití v rámci integrovaného hodnocení svědčit tyto poznatky z klinického pozorování, a tudíž mohou indikovat maximální dávku pro hlavní studii LLNA: BrdU-ELISA: změny fungování nervového systému (např. piloerekce, ataxie, třes a křeče), změny chování (např. agresivita, změna činností týkajících se péče o srst, výrazná změna úrovně aktivity), změny způsobů dýchání (tj. změny frekvence a intenzity dýchání, např. dušnost, lapání po dechu a chropy) a změny v přijímání potravy a vody. Při hodnocení je navíc nutno brát v úvahu známky letargie a/nebo absence reagování, veškeré klinické příznaky více než mírné nebo momentální bolesti či utrpení nebo snížení tělesné hmotnosti ode dne 1 do dne 6 o více než 5 % a také úmrtnost. Umírající zvířata nebo zvířata, která jeví známky silné bolesti a utrpení, by se měla humánně usmrtit (31).
|
Harmonogram zkoušek v rámci hlavní studie
|
25.
|
Harmonogram zkoušky je následující:
— Den 1: Každé zvíře se zváží a zaznamená se jeho hmotnost a jakékoli poznatky z klinického pozorování. Na hřbet každého ucha se aplikuje 25 μl vhodně zředěného roztoku zkoušené látky, samotné vehikulum nebo pozitivní kontrola (tu lze provádět buď souběžně, nebo lze použít pozitivní kontrolu z nedávné jiné zkoušky – v závislosti na postupech dané laboratoře při zohlednění odstavců 11 až 15).
— Den 2 a den 3: Zopakuje se postup aplikace provedený v den 1.
— Den 4: Žádná aplikace.
— Den 5: Injekčně se intraperitoneálně aplikuje 0,5 ml (5 mg/myš) roztoku BrdU (10 mg/ml).
— Den 6: Zaznamená se hmotnost každého zvířete a jakékoli poznatky z klinického pozorování. Přibližně 24 hodin (24 hod.) po injekčním podání BrdU se zvířata humánně usmrtí. U každé myši se vyjmou lymfatické uzliny drenující aurikulární oblast obou uší a zpracují se ve fosfátovém pufrovém fyziologickém roztoku (PBS) od každého zvířete zvlášť. Podrobnosti a grafické znázornění vyhledání uzlin a jejich disekce lze nalézt v literatuře (17). Pro účely dalšího sledování lokální kožní reakce v rámci hlavní studie lze do protokolu studie zařadit i další parametry, jako je bodové ohodnocení erytému ucha nebo údaje o tloušťce ucha (získané buď pomocí tloušťkoměru, nebo na základě stanovení hmotnosti části ucha vyseknuté speciálními kleštěmi při pitvě).
|
Příprava buněčných suspenzí
|
26.
|
Od každé myši se připraví jednobuněčná suspenze obsahující buňky z vyňatých lymfatických uzlin drenujících oblast obou uší; suspenze se připraví jemnou mechanickou deagregací přes 200 μm sítko z korozivzdorné oceli nebo jiným přijatelným postupem vytvoření jednobuněčné suspenze (např. rozdrcením lymfatických uzlin za použití jednorázové plastové paličky a následným protlačením tkáně nylonovým sítkem # 70). Postup přípravy suspenze buněk z lymfatických uzlin je v této zkoušce rozhodující, a proto by si měl každý pracovník osvojit tuto dovednost předem. Kromě toho jsou lymfatické uzliny u zvířat z negativní kontrolní skupiny malé, takže je třeba postupovat opatrně, aby se zabránilo jakémukoli umělému ovlivnění hodnot SI. V každém případě je nutno cílový objem suspenze buněk z lymfatických uzlin upravit na stanovený optimální objem (přibližně 15 ml). Optimální objem je založen na dosažení průměrné absorbance negativní kontrolní skupiny v rozmezí 0,1–0,2.
|
Stanovení buněčné proliferace (měření obsahu BrdU v DNA lymfocytů)
|
27.
|
Obsah BrdU se měří metodou ELISA za použit komerční soupravy (např. Roche Applied Science, Mannheim, Německo, katalogové číslo 11 647 229 001). Postup lze stručně popsat tak, že 100 ml suspenze buněk z lymfatických uzlin se nakape do jamek tří mikrotitračních destiček s plochým dnem. Po fixaci a denaturaci buněk z lymfatických uzlin se do každé jamky přidá protilátka proti BrdU a obsah jamek se nechá zreagovat. Následně se protilátka proti BrdU odstraní vymytím, přidá se roztok substrátu a nechá se vzniknout chromogen. Pak se změří absorbance při vlnové délce 370 nm s referenční vlnovou délkou 492 nm. Ve všech případech je nutno optimalizovat zkušební podmínky (viz odstavec 26).
|
ZÍSKANÉ POZNATKY
Poznatky z klinického pozorování
|
28.
|
Každá myš se jednou denně pečlivě vyšetří, zda nevykazuje jakékoli klinické příznaky lokálního podráždění v místě aplikace nebo příznaky systémové toxicity. Všechny získané poznatky se systematicky zaznamenají, přičemž záznamy se vedou zvlášť za každou myš. Plány sledování by měly obsahovat kritéria umožňující rychle identifikovat myši, jež vykazují známky systémové toxicity, nadměrného lokálního podráždění pokožky nebo poleptání kůže, aby mohly být usmrceny (31).
|
Hodnoty tělesné hmotnosti
|
29.
|
Jak je uvedeno v odstavci 25, tělesnou hmotnost jednotlivých zvířat je nutno zjistit na začátku zkoušky a v době plánovaného humánního usmrcení zvířete.
|
VÝPOČET VÝSLEDKŮ
|
30.
|
Výsledky za každou exponovanou skupinu se vyjádří jako průměrný index stimulace (SI). Hodnota SI se stanoví jako podíl průměrné hodnoty indexu značení BrdU na zvíře jak u každé exponované skupiny, tak u pozitivní kontrolní skupiny, a průměrné hodnoty indexu značení BrdU na zvíře u kontrolní skupiny s rozpouštědlem/vehikulem. Průměrná hodnota SI u kontrolních skupin s vehikulem je pak rovna 1.
Index značení BrdU je definován jako:
Index značení BrdU = (ABSem – ABS blankem) – (ABSref – ABS blankref)
kde: em = vlnová délka záření; ref = referenční vlnová délka.
|
|
31.
|
Při rozhodování o výsledku zkoušky se za pozitivní výsledek považuje, pokud hodnota SI ≥ 1,6 (10). Při rozhodování, zda hraniční výsledek (tzn. hodnotu SI v rozmezí od 1,6 do 1,9) prohlásit za pozitivní, lze také použít míru závislosti odezvy na dávce, statistickou významnost a jednotnost výsledků kontrol s rozpouštědlem/vehikulem a pozitivních kontrol (3) (6) (32).
|
|
32.
|
U hraniční pozitivní odezvy v rozmezí hodnot SI od 1,6 do 1,9 mohou uživatelé zvážit další informace, například závislost odezvy na dávce, známky systémové toxicity nebo nadměrného podráždění, a tam, kde je to vhodné, také statistickou významnost spolu s hodnotami SI, aby se potvrdilo, že jsou takové výsledky pozitivní (10). Měla by se rovněž věnovat pozornost různým vlastnostem zkoušené látky včetně toho, zda se její struktura podobá struktuře známých senzibilizátorů kůže, zda u myší způsobuje nadměrné lokální podráždění kůže, a také povaze zjištěné závislosti odezvy na dávce. Těmito body a dalšími aspekty, které je třeba zvážit, se podrobně zabývá literatura (4).
|
|
33.
|
Shromažďování údajů úrovni jednotlivých myší umožní provést statistickou analýzu, kterou se na základě těchto údajů zjistí existence a míra závislosti odezvy na dávce. Případné statistické hodnocení by mohlo obsahovat posouzení závislosti odezvy na dávce, jakož i vhodně upravená srovnání zkušebních skupin (např. skupiny s párově srovnávaným dávkováním oproti souběžné kontrolní skupině s rozpouštědlem/vehikulem). Statistické analýzy mohou zahrnovat např. lineární regresi nebo Williamsovu zkoušku k posouzení trendů závislosti odezvy na dávce a Dunnettovu zkoušku pro párová srovnání. Při výběru vhodné metody pro statistickou analýzu by si měl být zkoušející vědom možných nestejných odchylek a jiných souvisejících problémů, které si mohou vyžádat transformaci údajů nebo neparametrickou statistickou analýzu. V každém případě může být nezbytné, aby zkoušející provedl výpočty SI a statistické analýzy jednak za použití určitých krajních hodnot (někdy označovaných výrazem ‚odlehlé hodnoty‘) a jednak bez nich.
|
ÚDAJE A VYKAZOVÁNÍ
Údaje
|
34.
|
Údaje je nutno shrnout do tabulky s hodnotami indexu značení BrdU od jednotlivých zvířat, průměrnou hodnotou indexu značení BrdU na zvíře za celou skupinu, příslušným chybovým výrazem (např. směrodatnou odchylkou – SD, směrodatnou odchylkou průměrných hodnot – SEM) a průměrnou hodnotou SI za každou dávkovou skupinu oproti souběžné kontrolní skupině s rozpouštědlem nebo vehikulem.
|
Protokol o zkoušce
|
35.
|
Protokol o zkoušce musí obsahovat tyto informace:
|
|
Zkoušená látka a kontrolní látka:
|
—
|
identifikační údaje (např. číslo CAS a číslo ES, jsou-li k dispozici, zdroj, čistota, známé nečistoty, číslo šarže),
|
|
—
|
skupenství a fyzikálně-chemické vlastnosti (např. prchavost, stálost, rozpustnost),
|
|
—
|
jedná-li se o směs, je nutno uvést její složení a poměrné procentuální zastoupení složek.
|
|
|
|
Rozpouštědlo/vehikulum:
|
—
|
identifikační údaje (čistota, popřípadě koncentrace, použitý objem),
|
|
—
|
zdůvodnění volby vehikula.
|
|
|
|
Pokusná zvířata:
|
—
|
zdroj myší z kmene CBA,
|
|
—
|
mikrobiologický stav zvířat, je-li znám,
|
|
—
|
původ zvířat, podmínky chovu, strava atd.
|
|
|
|
Zkušební podmínky:
|
—
|
zdroj, číslo šarže a údaje od výrobce soupravy ELISA, které se týkají prokazování a kontroly kvality (citlivost a specifičnost detekce protilátek a mez detekce),
|
|
—
|
podrobnosti o přípravě a aplikaci zkoušené látky,
|
|
—
|
zdůvodnění volby dávkování (včetně výsledků předběžné screeningové zkoušky, pokud byla provedena),
|
|
—
|
použité koncentrace vehikula a zkoušené látky a celkové aplikované množství zkoušené látky,
|
|
—
|
podrobnosti o jakosti vody a potravy (včetně druhu/zdroje stravy, zdroje vody),
|
|
—
|
podrobnosti o harmonogramech expozice a odběru vzorků,
|
|
—
|
metody měření toxicity,
|
|
—
|
kritéria pro označení studie za pozitivní nebo negativní,
|
|
—
|
údaje o případných odchylkách od protokolu a vysvětlení, jakým způsobem taková odchylka ovlivňuje koncepci studie a její výsledky.
|
|
|
|
Kontrola spolehlivosti:
|
—
|
souhrn výsledků nejnovější kontroly spolehlivosti včetně informací o použité zkoušené látce, koncentraci a vehikulu,
|
|
—
|
údaje ze souběžných a/nebo dřívějších pozitivních nebo negativních kontrolních skupin (s rozpouštědlem/vehikulem) zkoušených v dotyčné laboratoři,
|
|
—
|
pokud souběžná pozitivní kontrola nebyla součástí studie, je nutno uvést datum a laboratorní zprávu nejnovější periodické pozitivní kontroly a podrobnou zprávu s údaji z dřívějších pozitivních kontrol zkoušených v dotyčné laboratoři, které odůvodňují rozhodnutí neprovádět souběžné pozitivní kontroly.
|
|
|
|
Výsledky:
|
—
|
hmotnost jednotlivých myší na počátku aplikace a v době plánovaného humánního usmrcení, jakož i průměrná hodnota a příslušný chybový výraz (např. SD, SEM) za každou exponovanou skupinu,
|
|
—
|
u každého zvířete časový průběh nástupu příznaků toxicity, včetně podráždění kůže v místě aplikace, pokud k němu dojde,
|
|
—
|
tabulka hodnot indexu značení BrdU a hodnot SI od jednotlivých myší za každou exponovanou skupinu;
|
|
—
|
průměrná hodnota indexu značení BrdU na zvíře a příslušný chybový výraz (např. SD, SEM) za každou exponovanou skupinu a výsledky analýzy odlehlých hodnot za každou exponovanou skupinu,
|
|
—
|
vypočtená hodnota SI a příslušná míra variability, která zohledňuje variabilitu mezi jednotlivými zvířaty jak z testování zkoušené látky, tak z kontrolních skupin,
|
|
—
|
závislost odezvy na dávce,
|
|
—
|
případné statistické analýzy.
|
|
|
|
Rozbor výsledků:
|
—
|
stručný komentář k výsledkům, k analýze závislosti odezvy na dávce a k případným statistickým analýzám a dále závěr, zda se má zkoušená látka považovat za senzibilizátor kůže.
|
|
|
LITERATURA
|
(1)
|
OECD (2010), Skin Sensitisation: Local Lymph Node Assay, Test Guideline No. 429, Guidelines for the Testing of Chemicals, OECD, Paris. K dispozici na adrese: [http://www.oecd.org/env/testguidelines].
|
|
(2)
|
Chamberlain, M. and Basketter, D.A. (1996), The local lymph node assay: status of validation. Food Chem. Toxicol., 34, 999–1002.
|
|
(3)
|
Basketter, D.A., Gerberick, G.F., Kimber, I. and Loveless, S.E. (1996), The local lymph node assay: A viable alternative to currently accepted skin sensitisation tests. Food Chem. Toxicol., 34, 985–997.
|
|
(4)
|
Basketter, D.A., Gerberick, G.F. and Kimber, I. (1998), Strategies for identifying false positive responses in predictive sensitisation tests. Food Chem. Toxicol., 36, 327–33.
|
|
(5)
|
Van Och, F.M.M., Slob, W., De Jong, W.H., Vandebriel, R.J. and Van Loveren, H. (2000), A quantitative method for assessing the sensitising potency of low molecular weight chemicals using a local lymph node assay: employment of a regression method that includes determination of uncertainty margins. Toxicol., 146, 49–59.
|
|
(6)
|
ICCVAM (1999), The murine local lymph node Assay: A test method for assessing the allergic contact dermatitis potential of chemicals/compounds: The results of an independent peer review evaluation coordinated by the Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program Center for the Evaluation of Alternative Toxicological Methods (NICETAM). NIH Publication No: 99-4494. Research Triangle Park, N.C. K dispozici na adrese: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/llna/llnarep.pdf].
|
|
(7)
|
Dean, J.H., Twerdok, L.E., Tice, R.R., Sailstad, D.M., Hattan, D.G., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: II. Conclusions and recommendations of an independent scientific peer review panel. Reg. Toxicol. Pharmacol., 34(3), 258–273.
|
|
(8)
|
Haneke, K.E., Tice, R.R., Carson, B.L., Margolin, B.H., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: III. Data analyses completed by the national toxicology program interagency center for the evaluation of alternative toxicological methods. Reg. Toxicol. Pharmacol., 34(3), 274–286.
|
|
(9)
|
Sailstad, D.M., Hattan, D., Hill, R.N., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: I. The ICCVAM review process. Reg. Toxicol. Pharmacol., 34(3), 249–257.
|
|
(10)
|
ICCVAM (2010), ICCVAM Test Method Evaluation Report. Nonradioactive local lymph node assay: BrdU-ELISA Test Method Protocol (LLNA: BrdU-ELISA). NIH Publication No. 10-7552 A/B. Research Triangle Park, NC: National Institute of Environmental Health Sciences. K dispozici na adrese: [http://iccvam.niehs.nih.gov/methods/immunotox/llna-ELISA/TMER.htm].
|
|
(11)
|
ICCVAM (2009), Independent Scientific Peer Review Panel Report: Updated validation status of new versions and applications of the murine local lymph node assay: a test method for assessing the allergic contact dermatitis potential of chemicals and products. Research Triangle Park, NC: National Institute of Environmental Health Sciences. K dispozici na adrese: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/LLNAPRPRept2009.pdf].
|
|
(12)
|
Takeyoshi, M., Iida, K., Shiraishi, K. and Hoshuyama, S. (2005), Novel approach for classifying chemicals according to skin sensitising potency by non-radioisotopic modification of the local lymph node assay. J. Appl. Toxicol., 25, 129–134.
|
|
(13)
|
OECD (1992), Skin Sensitisation, Test Guideline No. 406, Guidelines for Testing of Chemicals, OECD, Paris. K dispozici na adrese: [http://www.oecd.org/env/testguidelines]
|
|
(14)
|
Kreiling, R., Hollnagel, H.M., Hareng, L., Eigler, L., Lee, M.S., Griem, P., Dreessen, B., Kleber, M., Albrecht, A., Garcia, C. and Wendel, A. (2008), Comparison of the skin sensitising potential of unsaturated compounds as assessed by the murine local lymph node assay (LLNA) and the guinea pig maximization test (GPMT). Food Chem. Toxicol., 46, 1896–1904.
|
|
(15)
|
Basketter, D., Ball, N., Cagen, S., Carrilo, J.C., Certa, H., Eigler, D., Garcia, C., Esch, H., Graham, C., Haux, C., Kreiling, R. and Mehling, A. (2009), Application of a weight of evidence approach to assessing discordant sensitisation datasets: implications for REACH. Reg. Toxicol. Pharmacol., 55, 90–96.
|
|
(16)
|
ILAR (1996), Institute of Laboratory Animal Research (ILAR) Guide for the Care and Use of Laboratory Animals. 7th ed. Washington, DC: National Academies Press.
|
|
(17)
|
ICCVAM (2009), Recommended Performance Standards: Murine Local Lymph Node Assay. NIH Publication Number 09-7357. Research Triangle Park, NC: National Institute of Environmental Health Sciences. K dispozici na adrese: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/llna-ps/LLNAPerfStds.pdf].
|
|
(18)
|
McGarry, H.F. (2007), The murine local lymph node assay: regulatory and potency considerations under REACH. Toxicol., 238, 71–89.
|
|
(19)
|
Kimber, I., Dearman, R.J., Scholes E.W. and Basketter, D.A. (1994), The local lymph node assay: developments and applications. Toxicol., 93, 13–31.
|
|
(20)
|
OECD (2002), Acute Dermal Irritation/Corrosion, Test Guideline No. 404, Guidelines for Testing of Chemicals, OECD, Paris. K dispozici na adrese: [http://www.oecd.org/env/testguidelines].
|
|
(21)
|
Reeder, M.K., Broomhead, Y.L., DiDonato, L. and DeGeorge, G.L. (2007), Use of an enhanced local lymph node assay to correctly classify irritants and false positive substances. Toxicologist, 96, 235.
|
|
(22)
|
ICCVAM (2009), Nonradioactive Murine Local Lymph Node Assay: Flow Cytometry Test Method Protocol (LLNA: BrdU-FC) Revised Draft Background Review Document. Research Triangle Park, NC: National Institute of Environmental Health Sciences. K dispozici na adrese: [http://iccvam.niehs.nih.gov/methods/immunotox/fcLLNA/BRDcomplete.pdf].
|
|
(23)
|
Hayes, B.B., Gerber, P.C., Griffey, S.S. and Meade, B.J. (1998), Contact hypersensitivity to dicyclohexylcarbodiimide and diisopropylcarbodiimide in female B6C3F1 mice. Drug Chem. Toxicol., 21, 195–206.
|
|
(24)
|
Homey, B., von Schilling, C., Blumel, J., Schuppe, H.C., Ruzicka, T., Ahr, H.J., Lehmann, P. and Vohr, V.W. (1998), An integrated model for the differentiation of chemical-induced allergic and irritant skin reactions. Toxicol. Appl. Pharmacol., 153, 83–94.
|
|
(25)
|
Woolhiser, M.R., Hayes, B.B. and Meade, B.J. (1998), A combined murine local lymph node and irritancy assay to predict sensitisation and irritancy potential of chemicals. Toxicol. Meth., 8, 245–256.
|
|
(26)
|
Hayes, B.B. and Meade, B.J. (1999), Contact sensitivity to selected acrylate compounds in B6C3F1 mice: relative potency, cross reactivity, and comparison of test methods. Drug. Chem. Toxicol., 22, 491–506.
|
|
(27)
|
Ehling, G., Hecht, M., Heusener, A., Huesler, J., Gamer, A.O., van Loveren, H., Maurer, T., Riecke, K., Ullmann, L., Ulrich, P., Vandebriel, R. and Vohr, H.W. (2005), A European inter- laboratory validation of alternative endpoints of the murine local lymph node assay: first round. Toxicol., 212, 60–68.
|
|
(28)
|
Vohr, H.W. and Ahr, H.J. (2005), The local lymph node assay being too sensitive? Arch. Toxicol., 79, 721–728.
|
|
(29)
|
Patterson, R.M., Noga, E. and Germolec D. (2007), Lack of evidence for contact sensitisation by Pfiesteria extract. Environ. Health Perspect., 115, 1023–1028.
|
|
(30)
|
ICCVAM (2009), Report on the ICCVAM-NICEATM/ECVAM/JaCVAM Scientific Workshop on Acute Chemical Safety Testing: Advancing In Vitro Approaches and Humane Endpoints for Systemic Toxicity Evaluations. Research Triangle Park, NC: National Institute of Environmental Health Sciences. K dispozici na adrese: [http://iccvam.niehs.nih.gov/methods/acutetox/Tox_workshop.htm].
|
|
(31)
|
OECD (2000), Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation, Environmental Health and Safety Monograph Series on Testing and Assessment No. 19, ENV/JM/MONO(2000)7, OECD, Paris. K dispozici na adrese: [http://www.oecd.org/env/testguidelines].
|
|
(32)
|
Kimber, I., Hilton, J., Dearman, R.J., Gerberick, G.F., Ryan, C.A., Basketter, D.A., Lea, L., House, R.V., Ladies, G.S., Loveless, S.E. and Hastings, K.L. (1998), Assessment of the skin sensitisation potential of topical medicaments using the local lymph node assay: An interlaboratory exercise. J. Toxicol. Environ.l Health, 53, 563–79.
|
|
(33)
|
OECD (2005), Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment, Environment, Health and Safety Monograph Series on Testing and Assessment No. 34, ENV/JM/MONO(2005)14, OECD, Paris. Available at: [http://www.oecd.org/env/testguidelines].
|
Dodatek 1
DEFINICE
Přesnost: Blízkost shody mezi výsledky zkušební metody a uznávanými referenčními hodnotami. Je to míra funkčnosti zkušební metody a jeden z aspektů její relevantnosti. Tento výraz se často používá namísto výrazu „soulad“ a rozumí se jím podíl správných výsledků zkušební metody (33).
Srovnávací látka: Senzibilizující nebo nesenzibilizující látka použitá jako standard pro srovnání se zkoušenou látkou. Srovnávací látka by měla mít tyto vlastnosti: i) konzistentní a spolehlivý zdroj (konzistentní a spolehlivé zdroje); ii) strukturální a funkční podobnost s třídou zkoušených látek; iii) známé fyzikálně-chemické vlastnosti; iv) podklady o známých účincích a v) známá účinnost v rozsahu žádoucí odezvy.
Falešně negativní: Zkoušená látka je zkušební metodou nesprávně označena za negativní nebo neaktivní, ačkoli ve skutečnosti je pozitivní nebo aktivní (33).
Falešně pozitivní: Zkoušená látka je zkušební metodou nesprávně označena za pozitivní nebo aktivní, ačkoli ve skutečnosti je negativní nebo neaktivní (33).
Nebezpečnost: Potenciál látky působit nepříznivě na zdraví nebo životní prostředí. Nepříznivý účinek se projevuje pouze při dostatečné úrovni expozice.
Mezilaboratorní reprodukovatelnost: Míra, v jaké mohou různé kvalifikované laboratoře za použití stejného protokolu a při testování stejné zkoušené látky získat kvalitativně a kvantitativně podobné výsledky. Mezilaboratorní reprodukovatelnost se určuje během předvalidačních a validačních postupů a ukazuje, do jaké míry lze zkoušku úspěšně přenášet mezi různými laboratořemi (33).
Vnitrolaboratorní reprodukovatelnost: Určení míry, v jaké může kvalifikovaný personál ve stejné laboratoři jindy úspěšně dosáhnout stejných výsledků za použití určitého protokolu (33).
Odlehlá hodnota: Odlehlá hodnota je poznatek, který se výrazně liší od ostatních hodnot v náhodném vzorku od celé populace.
Prokazování kvality: Řídicí proces, v jehož rámci se hodnotí dodržování zkušebních norem a standardů laboratoře, příslušných požadavků a postupů vedení záznamů, jakož i přesnost přenosu údajů, přičemž tyto prvky jsou hodnoceny osobami, které jsou nezávislé na pracovnících, kteří zkoušky provádějí.
Spolehlivost: Míra, v jaké lze zkušební metodu provádět reprodukovatelně v rámci téže laboratoře i v rámci různých laboratoří v průběhu doby, jestliže se provádí za použití stejného protokolu. Hodnotí se na základě výpočtu vnitrolaboratorní a mezilaboratorní reprodukovatelnosti (33).
Senzibilizace kůže: Imunologický proces, který nastává, když je vnímavá osoba vystavena místně aplikovanému chemickému alergenu, který vyvolává kožní imunitní odezvu, jež může vést ke vzniku kontaktní senzibilizace.
Index stimulace (SI): Hodnota vypočtená za účelem posouzení potenciálu zkoušené látky vyvolat senzibilizaci kůže. Vyjadřuje poměr hodnoty proliferace u exponovaných skupin k hodnotě proliferace u souběžné kontrolní skupiny s vehikulem.
Zkoušená látka: Jakákoli látka nebo směs, která se zkouší za použití této zkušební metody.
“ |