(ES) č. 761/2009Nařízení Komise (ES) č. 761/2009 ze dne 23. července 2009, kterým se přizpůsobuje technickému pokroku nařízení (ES) č. 440/2008, kterým se stanoví zkušební metody podle nařízení Evropského parlamentu a Rady (ES) č. 1907/2006 o registraci, hodnocení, povolování a omezování chemických látek (Text s významem pro EHP)
| Publikováno: | Úř. věst. L 220, 24.8.2009, s. 1-94 | Druh předpisu: | Nařízení |
| Přijato: | 23. července 2009 | Autor předpisu: | Evropská komise |
| Platnost od: | 27. srpna 2009 | Nabývá účinnosti: | 27. srpna 2009 |
| Platnost předpisu: | Ano | Pozbývá platnosti: | |
Text předpisu s celou hlavičkou je dostupný pouze pro registrované uživatele.
NAŘÍZENÍ KOMISE (ES) č. 761/2009
ze dne 23. července 2009,
kterým se přizpůsobuje technickému pokroku nařízení (ES) č. 440/2008, kterým se stanoví zkušební metody podle nařízení Evropského parlamentu a Rady (ES) č. 1907/2006 o registraci, hodnocení, povolování a omezování chemických látek
(Text s významem pro EHP)
KOMISE EVROPSKÝCH SPOLEČENSTVÍ,
s ohledem na Smlouvu o založení Evropského společenství,
s ohledem na nařízení Evropského parlamentu a Rady (ES) č. 1907/2006 ze dne 18. prosince 2006 o registraci, hodnocení, povolování a omezování chemických látek, o zřízení Evropské agentury pro chemické látky, o změně směrnice 1999/45/ES a o zrušení nařízení Rady (EHS) č. 793/93, nařízení Komise (ES) č. 1488/94, směrnice Rady 76/769/EHS a směrnic Komise 91/155/EHS, 93/67/EHS, 93/105/ES a 2000/21/ES (1), a zejména na čl. 13 odst. 3 uvedeného nařízení,
vzhledem k těmto důvodům:
|
(1) |
Nařízení Komise (ES) č. 440/2008 (2) obsahuje zkušební metody pro určování fyzikálně-chemických vlastností látek, jejich toxicity a ekotoxicity, které se mají používat pro účely nařízení (ES) č. 1907/2006. |
|
(2) |
Je třeba aktualizovat nařízení (ES) č. 440/2008 tak, aby se zohlednily změny některých zkušebních metod a nově zahrnuly některé nové zkušební metody přijaté OECD. Zúčastněné strany byly ohledně tohoto návrhu konzultovány. Změny slouží přizpůsobení předmětných metod vědecko-technickému pokroku. |
|
(3) |
Je třeba zrevidovat ustanovení o tlaku par tak, aby se zahrnula nová efuzní metoda. |
|
(4) |
Je třeba vložit novou metodu měření délkově váženého geometrického průměru vláken. |
|
(5) |
Je třeba aktualizovat nařízení (ES) č. 440/2008 tak, aby se přednostně zahrnula nová zkušební metoda in vitro u dráždění kůže, s cílem snížit počet zvířat používaných k pokusným účelům v souladu se směrnicí Rady 86/609/EHS ze dne 24. listopadu 1986 o sbližování právních a správních předpisů členských států týkajících se ochrany zvířat používaných pro pokusné a jiné vědecké účely (3). Přestože předloha zkušební metody in vitro u dráždění kůže je ještě předmětem rozpravy v OECD, je třeba v tomto výjimečném případě zahrnout metodu B 46 do tohoto nařízení. Metoda B 46 by se měla co možná nejdříve aktualizovat, tj. jakmile dojde v rámci OECD k dohodě nebo jakmile budou k dispozici další informace svědčící ve prospěch této revize. |
|
(6) |
Je třeba zrevidovat ustanovení o zkušební metodě inhibice růstu řas tak, aby se zahrnuly další druhy a dodržely požadavky na posouzení rizika a klasifikaci chemických látek. |
|
(7) |
Je třeba vložit novou metodu simulační zkoušky biologické rozložitelnosti k měření aerobní mineralizace v povrchových vodách a novou zkušební metodu inhibice růstu k posuzování toxicity pro druh Lemna. |
|
(8) |
Nařízení (ES) č. 440/2008 by proto mělo být odpovídajícím způsobem změněno. |
|
(9) |
Opatření stanovená tímto nařízením jsou v souladu se stanoviskem výboru zřízeného článkem 133 nařízení (ES) č. 1907/2006, |
PŘIJALA TOTO NAŘÍZENÍ:
Článek 1
Příloha nařízení (ES) č. 440/2008 se mění takto:
|
1) |
Část A se mění takto:
|
|
2) |
Část B se mění takto: Doplňuje se nová kapitola B.46 uvedená v příloze III tohoto nařízení. |
|
3) |
Část C se mění takto:
|
Článek 2
Toto nařízení vstupuje v platnost třetím dnem po vyhlášení v Úředním věstníku Evropské unie.
Toto nařízení je závazné v celém rozsahu a přímo použitelné ve všech členských státech.
V Bruselu dne 23. července 2009.
Za Komisi
Stavros DIMAS
člen Komise
(1) Úř. věst. L 396, 30.12.2006, s. 1.
(2) Úř. věst. L 142, 31.5.2008, s. 1.
(3) Úř. věst. L 358, 18.12.1986, s. 1.
PŘÍLOHA I
|
A.4 |
TLAK PAR |
1. METODA
Tato metoda je rovnocenná Pokynu OECD pro zkoušení 104 (2004).
1.1 ÚVOD
Tato přepracovaná verze metody A.4 (1) zahrnuje jednu doplňkovou metodu, a to metodu efusní: isotermální termogravimetrii určenou pro látky s velmi nízkými tlaky (až 10–10 Pa). Vzhledem k tomu, že je zapotřebí postupů, zejména v souvislosti se stanovením tlaku par u látek s nízkou tenzí par, další postupy se přehodnocující v rámci této metody s ohledem na další rozmezí použitelnosti.
Při termodynamické rovnováze je tlak par čisté látky pouze funkcí teploty. Základní principy jsou popsány v literatuře (2, 3).
Neexistuje žádný postup vhodný pro celý rozsah tlaku par od méně než 10–10 do 105 Pa. Do této metody spadá osm metod měření tlaku par, které lze používat v různých rozmezích tlaku par. Jednotlivé metody jsou srovnány z hlediska použití a rozsahu měření v tabulce 1. Tyto metody lze použít pouze u sloučenin, které se za podmínek zkoušky nerozkládají. V případě, kdy nelze experimentální metody z technických důvodů použít, lze tlak par rovněž odhadnout, přičemž doporučená metoda odhadu je uvedena v dodatku.
1.2 DEFINICE A JEDNOTKY
Tlak par látky je definován jako tlak nasycené páry nad pevnou nebo kapalnou látkou.
Měla by být používána jednotka tlaku v soustavě SI, tj. pascal (Pa). Dále jsou uvedeny některé dříve používané jednotky s odpovídajícími přepočítávacími faktory:
|
1 torr |
= |
1 mm Hg |
= |
1,333 × 102 Pa |
|
1 fyzikální atmosféra (atm) |
= |
1,013 × 105 Pa |
|
|
|
1 bar |
= |
105 Pa |
|
|
Jednotkou termodynamické teploty v soustavě SI je kelvin (K). Převod stupňů Celsia na kelviny se provádí podle vzorce:
T = t+ 273,15
kde T je teplota v kelvinech, neboli termodynamická teplota, a t je teplota ve stupních Celsia.
Tabulka 1
|
Metoda měření |
Látky |
Odhad opakovatelnosti |
Odhad reprodukovatelnosti |
Doporučená oblast |
|
|
pevné |
kapalné |
||||
|
Dynamická metoda |
s nízkým bodem tání |
ano |
do 25 % 1 až 5 % |
do 25 % 1 až 5 % |
103 Pa až 2 × 103 Pa 2 × 103 Pa až 105 Pa |
|
Statická metoda |
ano |
ano |
5 až 10 % |
5 až 10 % |
10 Pa až 105 Pa 10–2 Pa až 105 Pa (1) |
|
Metoda za použití isoteniskopu |
ano |
ano |
5 až 10 % |
5 až 10 % |
102 Pa až 105 Pa |
|
Efusní metoda: váhy pro měření tlaku par |
ano |
ano |
5 až 20 % |
do 50 % |
10–3 Pa až 1 Pa |
|
Efusní metoda: Knudsenova komůrka |
ano |
ano |
10 až 30 % |
— |
10–10 Pa až 1 Pa |
|
Efusní metoda: isotermální termogravimetrie |
ano |
ano |
5 až 30 % |
do 50 % |
10–10 Pa až 1 Pa |
|
Metoda sycení plynu |
ano |
ano |
10 až 30 % |
do 50 % |
10–10 Pa až 103 Pa |
|
Metoda rotujícího tělíska |
ano |
ano |
10 až 20 % |
— |
10–4 Pa až 0,5 Pa |
1.3 PODSTATA ZKOUŠKY
Obecně se tlak par stanovuje při různých teplotách. V omezeném rozsahu teplot je logaritmus tlaku par čisté látky nepřímo úměrný termodynamické teplotě podle zjednodušené Clapeyronovy-Clausiovy rovnice:
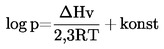
kde:
|
p |
= |
tlak par v pascalech, |
|
ΔHv |
= |
výparné teplo v J mol–1, |
|
R |
= |
univerzální molární plynová konstanta (8,314 J mol–1 K–1), |
|
T |
= |
termodynamická teplota v K. |
1.4 REFERENČNÍ LÁTKY
Referenční látky není nutné vždy používat. Slouží v první řadě k občasné kontrole provedení metody a ke vzájemnému porovnávání výsledků získaných jinými metodami.
1.5 POPIS METODY
1.5.1 Dynamická metoda (Cottrellova metoda)
1.5.1.1 Podstata
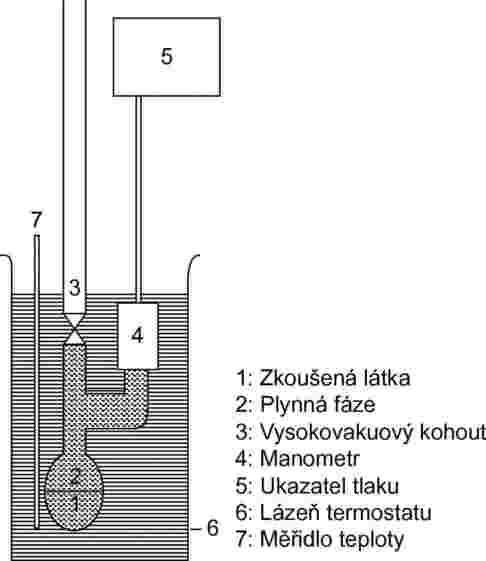
Pro stanovení tlaku par látky se měří její teplota varu při různých specifikovaných tlacích mezi asi 103 a 105 Pa. Tato metoda se rovněž doporučuje pro stanovení bodu varu. K tomuto účelu je metoda vhodná až do teploty 600 K. Teploty varu kapalin jsou v hloubce 3 až 4 cm přibližně o 0,1 °C vyšší než na povrchu, a to kvůli hydrostatickému tlaku sloupce kapaliny. Při Cottrellově metodě (4) je teploměr umístěn do páry nad povrch kapaliny a vroucí kapalina se nepřetržitě čerpá okolo baňky teploměru. Baňku teploměru pokrývá tenká vrstva kapaliny, která je v rovnováze s párou při atmosférickém tlaku. Teploměr tak odečítá skutečnou teplotu varu bez chyb způsobených přehřátím nebo hydrostatickým tlakem. Vývěva původně používaná Cottrellem je zachycena na obrázku 1. Vroucí kapalina se nachází ve zkumavce A. Platinový drátek B zatavený do dna usnadňuje jednotný průběh vření. Boční trubička C vede do kondenzátoru a pouzdro D brání styku studeného kondenzátu a teploměru E. Když kapalina v A vře, bubliny a kapalina zachycená nálevkou se přelévá přes baňku teploměru prostřednictvím dvou ramen vývěvy F.
|
Obrázek 1
|
Obrázek 2
|
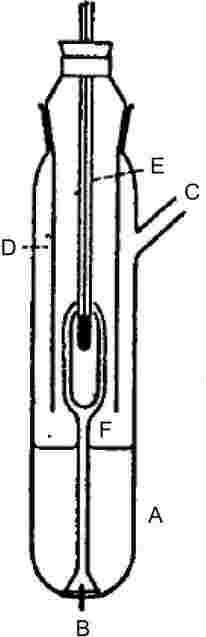
Cottrellova vývěva (4)
|
A: |
Termočlánek |
|
B: |
Vyrovnávací prostor vakua |
|
C: |
Manometr |
|
D: |
Vakuum |
|
E: |
Měřicí bod |
|
F: |
Topný článek asi 150 W |
1.5.1.2 Aparatura
Velmi přesná aparatura využívající Cottrellův princip je zachycena na obrázku 2. Sestává se z trubice s varnou částí ve spodní části, chladiče ve střední části a výpusti a příruby v horní části. Cottrellova vývěva je umístěna ve varné části, která je ohřívána elektrickou topnou vložkou. Teplota se měří plášťovým termočlánkem nebo odporovým teploměrem zasunutým přes přírubu v horní části. Výpusť je připojena na systém regulace tlaku. Systém pro regulaci tlaku se skládá z vývěvy, vyrovnávacího objemu vakua, manostatu pro připouštění dusíku k ovládaní tlaku a manometru.
1.5.1.3 Postup měření
Látka se umístí do varné části. Při plnění pevných látek, které nejsou ve formě prášku, může dojít k problémům, kterým se však lze někdy vyhnout zahřátím chladicího pláště. Aparatura se uzavře přírubou a látka se odplyní. Touto metodou nelze měřit látky, které pění.
Poté se nastaví nejnižší požadovaný tlak a zapne se ohřev. Současně se teplotní čidlo připojí k zapisovači.
Rovnováhy je dosaženo, je-li při konstantním tlaku zaznamenána konstantní teplota varu. Je třeba věnovat zvláštní pozornost tomu, aby se zabránilo prudkému uvolňování par během varu. Navíc musí dojít k úplné kondenzaci v chladiči. Při stanovování tlaku par u nízkotajících pevných látek je třeba dbát na to, aby nedošlo k ucpání chladiče.
Po zaznamenání naměřeného rovnovážného teplotního bodu se nastaví vyšší tlak. Takto se pokračuje, dokud se nedosáhne tlaku 105 Pa (celkem asi 5 až 10 bodů měření). Pro kontrolu se musí stanovení rovnovážných bodů opakovat při klesajících hodnotách tlaku.
1.5.2 Statická metoda
1.5.2.1 Podstata
Statickou metodou (5) se měří tlak par, který se ustaví při termodynamické rovnováze při dané teplotě. Tato metoda je vhodná pro látky a vícesložkové kapaliny a pevné látky v rozsahu od 10–1 do 105 Pa a při potřebné pečlivosti lze tuto metodu použít také pro oblast od 1 do 10 Pa.
1.5.2.2 Aparatura
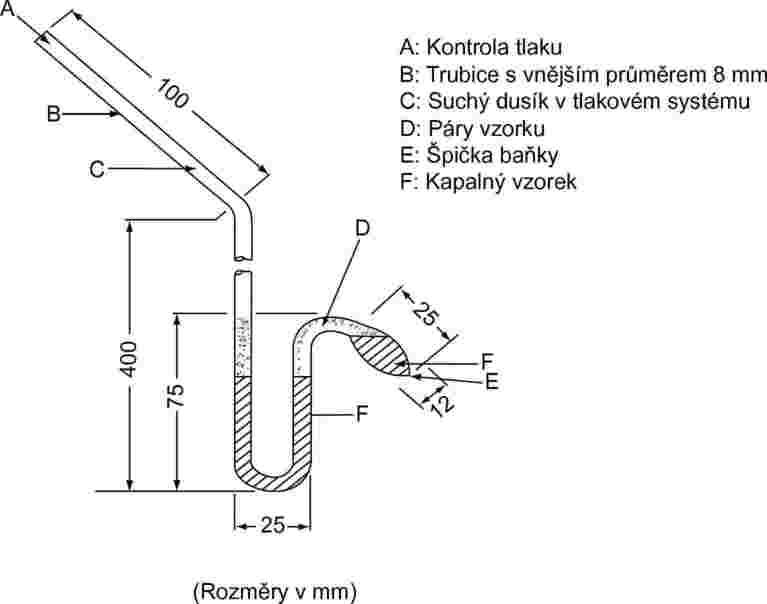
Zařízení se skládá z lázně o konstantní teplotě (preciznost ±0,2 K), zásobníku vzorku připojenému k podtlakovému potrubí, manometru a soustavy k regulaci tlaku. Baňka na vzorek (obrázek 3a) je připojena k podtlakovému potrubí přes ventil a diferenciální manometr (U-trubice obsahující vhodnou manometrickou kapalinu), který slouží jako ukazatel nuly. V závislosti na rozsahu tlaků a v závislosti na chemickém chování zkoušené látky jsou k použití v diferenciálním manometru vhodné rtuť, silikonové oleje a ftaláty. Avšak s ohledem na životní prostředí je třeba se vyvarovat použití rtuti, bude-li to možné. Zkoušená látka se nesmí znatelně rozpouštět ani nesmí reagovat s kapalinou v U-trubici. Namísto U-trubice lze použít manometr (obrázek 3b). V rozsahu od normálního tlaku do 102 Pa lze v manometru používat rtuť, zatímco silikonové oleje a ftaláty je vhodné používat pro tlaky pod 102 Pa až do 10 Pa. Existují též jiná měřidla tlaku, která lze použít pro tlak nižší než 102 Pa, a ohřívatelné membránové kapacitní manometry lze dokonce používat pro tlak nižší než 10–1 Pa. Teplota se měří na vnější stěně baňky se vzorkem nebo v baňce samotné.
1.5.2.3 Postup měření
Při použití aparatury popsané na obrázku 3a se U-trubice naplní zvolenou kapalinou, která musí být před použitím odplyněna za zvýšené teploty. Zkoušená látka se vloží do aparatury a odplyní se za snížené teploty. V případě vícesložkového vzorku musí být teplota dostatečně nízká, aby se zajistilo, že se složení materiálu nezmění. Rovnováhy lze rychleji dosáhnout mícháním. Vzorek může být podchlazen kapalným dusíkem nebo suchým ledem, je ale nutno zabránit kondenzaci vzduchu nebo kapaliny z vývěvy. Při otevřeném ventilu nádoby se vzorkem se připojí na několik minut odsávání, aby se odstranil vzduch. Je-li to nutné, odplyňovací postup se několikrát opakuje.
|
Obrázek 3a
|
Obrázek 3b
|
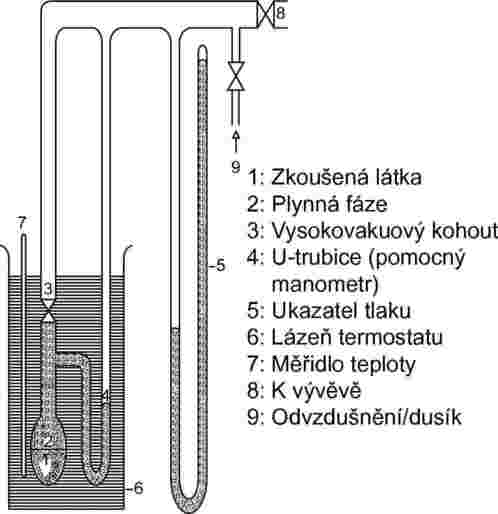
Při zahřívání vzorku za uzavřeného ventilu roste tlak par. To mění rovnováhu kapaliny v U-trubici. Aby se změna kompenzovala, připouští se ventilem dusík nebo vzduch do té doby, dokud není indikátor rozdílu tlaku opět na nule. Tlak potřebný k ustavení nulové hodnoty může být odečten přesným manometrem nebo přístrojem o vyšší preciznosti. Tento tlak odpovídá tenzi par látky při teplotě měření. Při použití aparatury popsané na obrázku 3b se tlak páry odečítá přímo.
Tlak par se stanoví ve vhodných krátkých intervalech teplot (celkem asi 5 až 10 bodů měření) až do požadovaného teplotního maxima.
Odečty při nízkých teplotách se musí pro kontrolu opakovat. Neleží-li hodnoty zjištěné z opakovaných odečtů na křivce zjištěné pro zvyšující se teplotu, může to být způsobeno jednou z těchto příčin:
|
i) |
vzorek stále obsahuje vzduch (např. u vysoce viskózních materiálů) nebo obsahuje látky s nízkým bodem varu, které jsou při zahřátí uvolňovány, |
|
ii) |
v látce probíhá ve vyšetřovaném teplotní rozsahu chemická reakce (např. rozklad, polymerace). |
1.5.3 Metoda za použití isoteniskopu
1.5.3.1 Podstata
Isoteniskop (6) vychází z podstaty statické metody. Metoda spočívá v uložení vzorku do baňky udržované při konstantní teplotě a připojené k manometru a vývěvě. Odplyněním za sníženého tlaku se odstraní nečistoty těkavější než vyšetřovaná látka. Tlak par vzorku při zvolených teplotách je vyrovnáván známým tlakem interního plynu. Isoteniskop byl vyvinut k měření tlaku par určitých kapalných uhlovodíků, ale hodí se rovněž k vyšetřování pevných látek. Metoda obvykle není vhodná pro vícesložkové systémy. U vzorků obsahujících netěkavé nečistoty jsou výsledky zatíženy pouze mírnými chybami. Doporučený rozsah je od 102 do 105 Pa.
1.5.3.2 Aparatura
Příklad měřicího zařízení je zachycen na obrázku 4. Úplný popis lze nalézt v ASTM D 2879–86 (6).
1.5.3.3 Postup měření
V případě kapalin slouží látka samotná jako indikační sloupec v diferenciálním manometru. Do isoteniskopu se odměří množství kapaliny postačující k naplnění baňky a krátkého ramene manometru. Isoteniskop se připojí k vakuovému systému, evakuuje se a poté se naplní dusíkem. Evakuace a výplach systému se opakuje dvakrát, aby se odstranil veškerý zbytkový kyslík. Naplněný isoteniskop se umístí do horizontální polohy, aby se vzorek rozprostřel v tenké vrstvě v baňce se vzorkem a v manometru. Tlak v systému se sníží na 133 Pa a vzorek se opatrně zahřeje právě k varu (odstranění rozpuštěných plynů). Poté se isoteniskop vrátí do původní polohy tak, aby se vzorek vrátil do baňky a krátkého ramene manometru. Tlak se udržuje na 133 Pa. Špička baňky se vzorkem se ohřívá malým plamenem, dokud uvolněná pára vzorku neexpanduje natolik, že přetlačí část vzorku z horní části baňky a ramene manometru do manometru, přičemž se vytvoří prostor bez dusíku naplněný výhradně parami. Isoteniskop se poté vloží do lázně se stálou teplotou a tlak dusíku se upraví tak, aby se rovnal tlaku vzorku. V rovnováze se rovná tlak dusíku tenzi par látky.
Obrázek 4
U pevných látek se v závislosti na oblasti tlaku a teploty používají manometrické kapaliny, jako jsou například silikonové kapaliny nebo ftaláty. Odplyněná manometrická kapalina se naplní do rozšířené části dlouhého ramene isoteniskopu. Poté se vyšetřovaná látka naplní do baňky a odplyní se při zvýšené teplotě. Isoteniskop se poté nakloní, aby manometrická kapalina natekla do U-trubice.
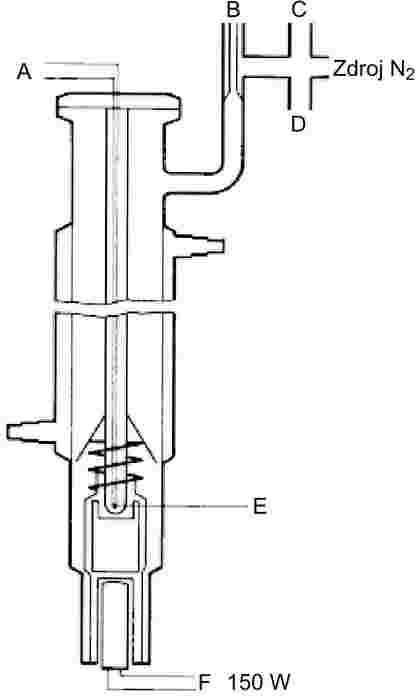
1.5.4 Efusní metoda: váhy pro měření tlaku par (7)
1.5.4.1 Podstata
Vzorek zkoušené látky se ohřeje v malé pícce a umístí se pod evakuovaný skleněný zvon. Pícka se zakryje víkem, které má malé otvory o známém průměru. Pára látky unikající jedním z otvorů je vedena na misku vysoce citlivých vah, která je rovněž umístěna pod evakuovaným skleněným zvonem. U některých konstrukcí je miska vah uzavřena v chladicím bloku, který zajišťuje rozptyl tepla do vnějšího okolí vedením tepla, a je ochlazována vyzařováním tak, aby unikající pára na ní kondenzovala. Hybnost proudu par působí jako síla proti misce vah. Tlak par lze odvodit dvěma způsoby: přímo ze síly působící na misku vah a rovněž z rychlosti vypařování s využitím Hertzovy-Knudsenovy rovnice (2):
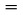
kde:
|
G |
= |
rychlost vypařování (kg s–1 m–2), |
|
M |
= |
molární hmotnost (g mol–1), |
|
T |
= |
termodynamická teplota (K), |
|
R |
= |
univerzální molární plynová konstanta, (J mol–1 K–1), |
|
p |
= |
tlak páry (Pa). |
Doporučený rozsah je od 10–3 do 1 Pa.
1.5.4.2 Aparatura
Obecný princip aparatury je znázorněn na obrázku 5.
Obrázek 5
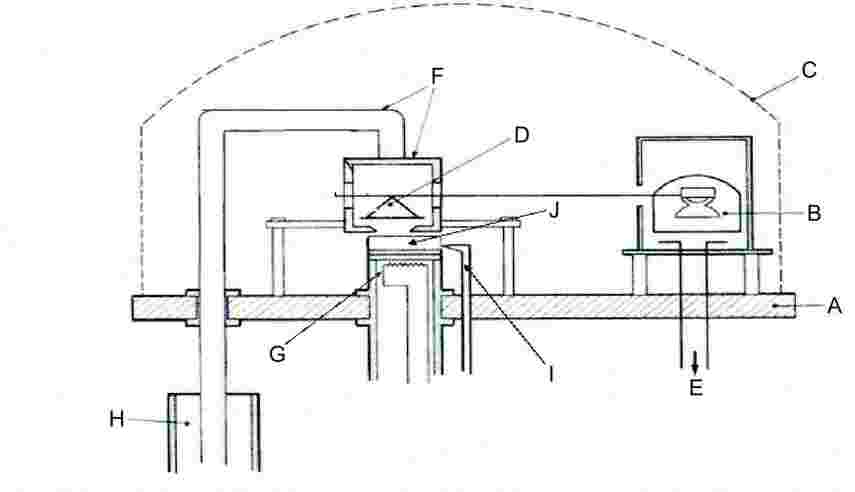
|
A: |
Základní deska |
F: |
Chladicí blok a chladicí tyč |
|
B: |
Přístroj s pohyblivou cívkou |
G: |
Odpařovací pícka |
|
C: |
Skleněný zvon |
H: |
Dewarova nádoba s kapalným dusíkem |
|
D: |
Miska vah se stupnicí |
I: |
Měření teploty vzorku |
|
E: |
Zařízení pro měření vakua |
J: |
Zkoušená látka |
1.5.5 Efusní metoda: Knudsenova komůrka
1.5.5.1 Podstata
Metoda je založena na odhadu hmotnosti par zkoušené látky unikající za jednotku času z Knudsenovy komůrky (8) mikrodýzou za podmínek vysokého vakua. Hmotnost difundujících par může být zjištěna buď stanovením úbytku hmotnosti komůrky nebo kondenzací par při nízké teplotě a stanovením jejich množství chromatografickou analýzou. Tlak par se vypočte za použití Hertzovy-Knudsenovy rovnice (viz bod 1.5.4.1) pomocí korekčních faktorů, které závisí na parametrech přístroje (9). Doporučený rozsah je od 10–10 do 1 Pa (10, 11, 12, 13, 14).
1.5.5.2 Aparatura
Obecný princip aparatury je znázorněn na obrázku 6.
Obrázek 6
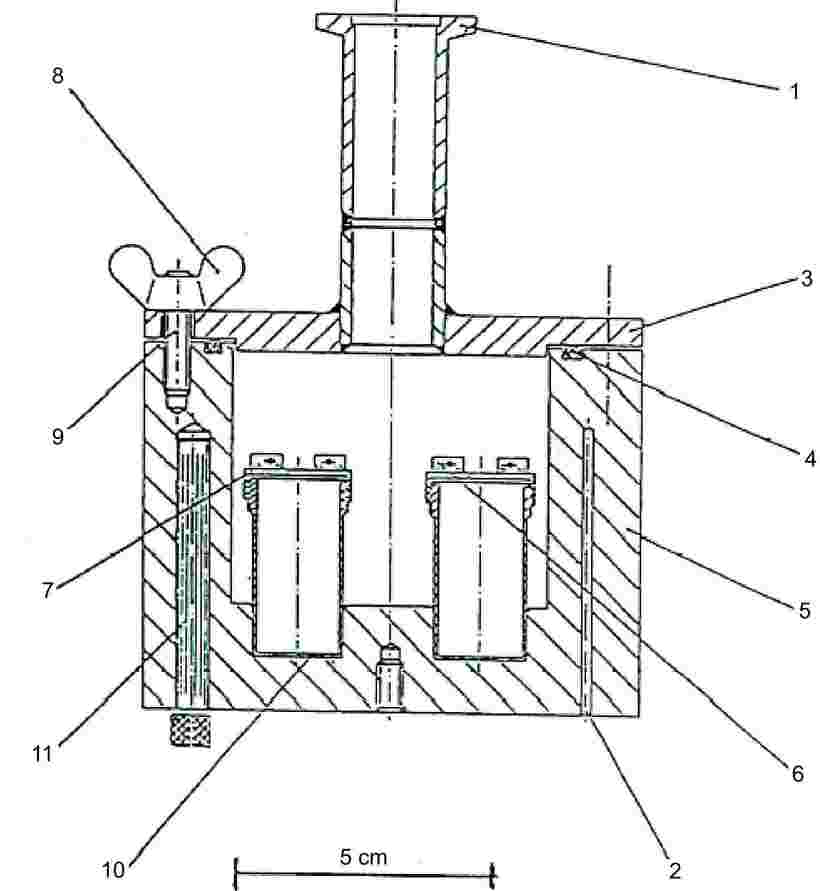
|
1: |
Přípojka k vakuu |
7: |
Šroubovací víko |
|
2: |
Otvory pro platinový odporový teploměr nebo pro systém měření a kontroly teploty |
8: |
Křídlové matice |
|
3: |
Víko vakuového bloku |
9: |
Šrouby |
|
4: |
O-kroužek |
10: |
Efusní komůrky z korozivzdorné oceli |
|
5: |
Hliníkový vakuový blok |
11: |
Topné těleso |
|
6: |
Zařízení pro zasouvání a vytahování efusních komůrek |
|
|
1.5.6 Efusní metoda: isotermální termogravimetrie
1.5.6.1 Podstata
Metoda je založena na stanovení zvýšených rychlostí vypařování zkoušené látky při zvýšených teplotách a tlaku okolí s využitím termogravimetrie (10, 15, 16, 17, 18, 19, 20). Rychlosti vypařování vT jsou výsledkem vystavení zvolené sloučeniny působení pomalu protékajícího inertního plynu, přičemž se sleduje úbytek hmotnosti při definovaných isotermálních teplotách T v kelvinech během příslušných časových období. Tlaky par pT se vypočítají z hodnot vT s využitím lineární závislosti mezi logaritmem tlaku par a logaritmem rychlosti vypařování. V případě potřeby lze provést extrapolaci na teploty 20 °C a 25 °C regresní analýzou log pT versus 1/T. Tato metoda je vhodná pro látky s tlakem páry na úrovni 10–10 Pa (10–12 mbar) při čistotě co nejvíce se blížící 100 %, aby se zabránilo chybné interpretaci naměřených hmotnostních úbytků.
1.5.6.2 Aparatura
Obecný princip měřicí soustavy je zachycen na obrázku 7.
Obrázek 7
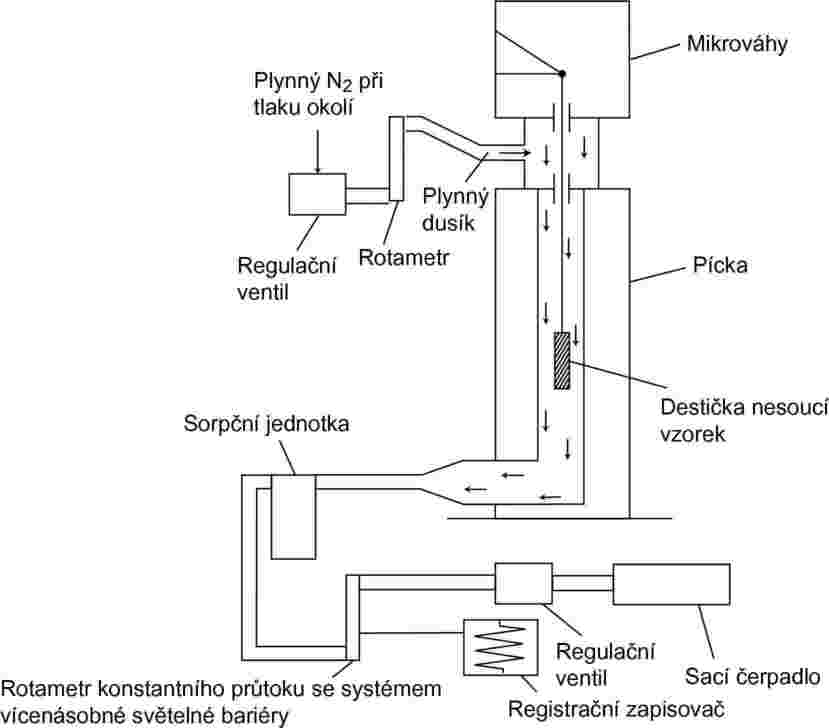
Destička nesoucí vzorek, zavěšená na mikrováhách v komůrce s kontrolovanou teplotou, je vlečena proudem suchého plynného dusíku, který unáší molekuly par zkoušené látky. Po průchodu komůrkou se proud plynu čistí v sorpční jednotce.
1.5.6.3 Postup měření
Zkoušená látka se nanese na povrch zdrsněné skleněné destičky jako homogenní vrstva. U pevných látek se destička rovnoměrně navlhčí roztokem látky ve vhodném rozpouštědle a vysuší se v inertní atmosféře. Pro měření se pokrytá destička zavěsí na termogravimetrický analyzátor a následně se průběžně měří úbytek hmotnosti jako funkce času.
Rychlost vypařování vT při určité teplotě se vypočítá ze ztráty hmotnosti Δm destičky se vzorkem pomocí rovnice
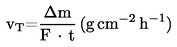
kde F je plocha povrchu pokrytého zkoušenou látkou, obvykle je to povrchová plocha destičky se vzorkem, a t je čas, během něhož dojde k úbytku hmotnosti Δm.
Tlak par pT se vypočítá jako funkce rychlosti vypařování vT :
Log pT = C + D log vT
kde C a D jsou konstanty specifické pro použité experimentální uspořádání v závislosti na průměru měřicí komůrky a rychlosti toku plynu. Tyto konstanty se musí jednorázově stanovit měřením množiny sloučenin se známým tlakem par a určit pomocí lineární regrese ze závislosti log pT na log vT (11, 21, 22).
Vztah mezi tlakem par pT a teplotou T v kelvinech popisuje rovnice
Log pT = A + B 1/T
kde A a B jsou konstanty získané lineární regresí ze závislosti log pT na 1/T. Pomocí této rovnice lze vypočítat tlak par extrapolací pro jakoukoliv teplotu.
1.5.7 Metoda nasycení plynu (23)
1.5.7.1 Podstata
Inertní plyn se vede při pokojové teplotě a známém průtoku přes vzorek zkoušené látky nebo nad ním, a to dostatečně pomalu, aby bylo zajištěno nasycení. Dosažení nasycení v plynné fázi je zásadně důležité. Transportovaná látka se zachytí, obvykle za použití sorbentu, a stanoví se její množství. Jako alternativní metody zachycení páry a následné analýzy mohou být ke stanovení množství hmoty transportovaného materiálu použity průtokové analytické techniky, jako je plynová chromatografie. Tlak par se vypočítá s tím, že se předpokládá platnost zákona o ideálním plynu a že celkový tlak směsi plynů je roven součtu tlaků plynných složek. Dílčí tlak zkoušené látky, tj. tlak par, se vypočítá ze známého celkového objemu plynu z hmotnosti transportovaného materiálu.
Metoda nasycení plynu je použitelná u pevných nebo kapalných látek. Lze ji použít pro tlaky par do 10–10 Pa (10, 11, 12, 13, 14). Metoda je nejspolehlivější pro tlaky par nižší než 103 Pa. Při tlaku nad 103 Pa jsou tlaky par všeobecně nadsazené, pravděpodobně kvůli tvorbě aerosolu. Protože se měření tlaku par provádí při pokojové teplotě, není nezbytné extrapolovat údaje získané za vysokých teplot a extrapolace vysokých teplot, která často způsobuje závažné chyby, se neprovádí.
1.5.7.2 Aparatura
Proces vyžaduje použití bloku s konstantní teplotou. Nákres na obrázku 8 ukazuje blok obsahující tři držáky pevných vzorků a tři držáky kapalných vzorků, které umožňují trojitou analýzu buď pevného, nebo kapalného vzorku. Teplota je regulována s precizností ±0,5 °C nebo lepší.
Obrázek 8
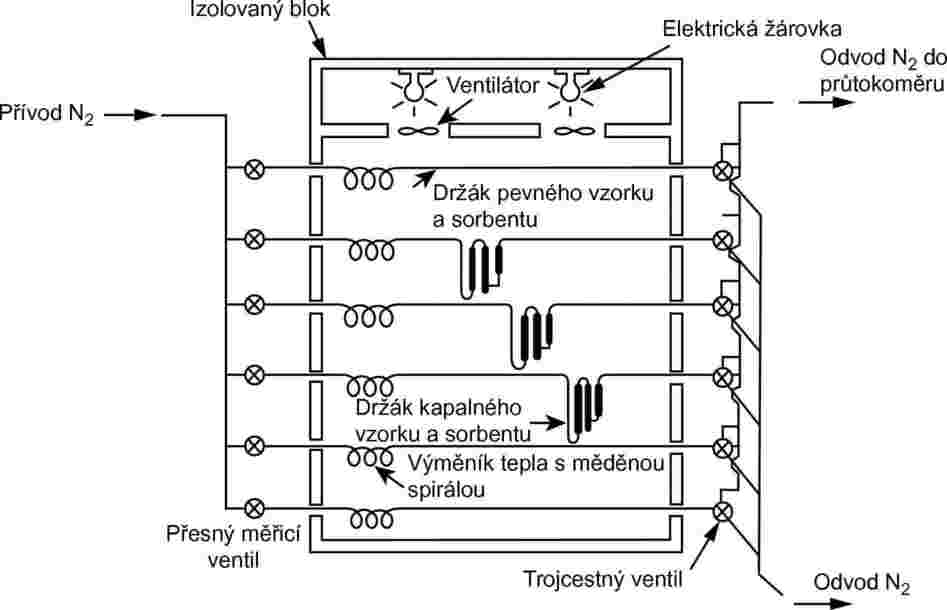
Obecně se jako inertní nosný plyn používá dusík, ale někdy může být nutno použít jiný plyn (24). Nosný plyn musí být suchý. Proud plynu se rozdělí do 6 proudů ovládaných jehlovými ventily (otvor přibližně 0,79 mm) a protéká do bloku měděnou trubkou o vnitřním průměru 3,8 mm. Po vyrovnání teplot plyn protéká okolo vzorku a sorbentovým lapačem a vychází z bloku.
Pevné vzorky se umístí do trubice o vnitřním průměru 5 mm mezi zátky ze skelné vaty (viz obrázek 9). Obrázek 10 ukazuje držák kapalného vzorku a systém se sorbentem. Metoda pro měření tlaku par kapalin s nejvyšší reprodukovatelností spočívá v nanesení kapaliny na skleněné kuličky nebo na inertní sorbent, například na oxid křemičitý, a obalení držáku těmito kuličkami. Druhou možností je nechat procházet nosný plyn hrubou fritou a probublávat sloupcem s kapalnou zkoušenou látkou.
|
Obrázek 9
|
Obrázek 10
|
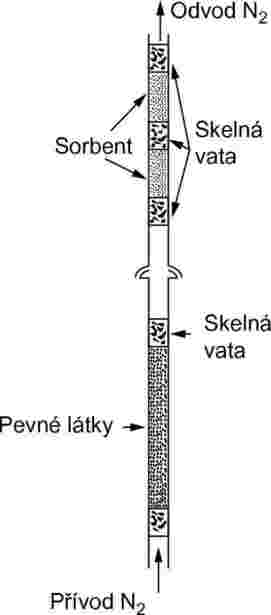
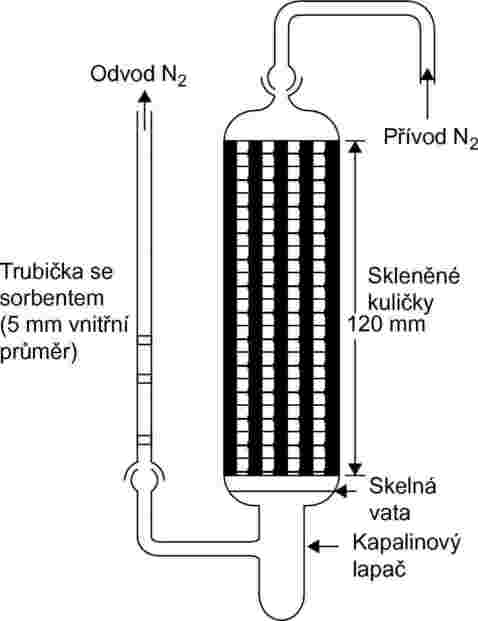
Systém se sorbentem obsahuje přední a zadní část se sorbentem. Při velmi nízkých tlacích páry se na sorbentu zachytí pouze malá množství a adsorpce na skelné vatě a ve skleněném potrubí mezi vzorkem a sorbentem může představovat závažný problém.
Lapače chlazené pevným CO2 jsou dalším účinným způsobem pro zachytávání materiálu v podobě par. Nevyvolávají žádný protitlak v syticí koloně a rovněž lze snadno kvantitativně odstranit zachycený materiál.
1.5.7.3 Postup měření
Průtok vytékajícího nosného plynu se měří při pokojové teplotě. Průtok se během experimentu často kontroluje, aby se zajistilo, že je přesně zjištěna hodnota celkového objemu nosného plynu. Dává se přednost nepřetržitému sledování hmotnostním průtokoměrem. Nasycení plynné fáze může vyžadovat značnou kontaktní dobu, a tím i dosti nízké průtoky plynu (25).
Po skončení experimentu se samostatně analyzují jak přední, tak zadní část se sorbentem. Sloučenina v každé části se desorbuje přidáním rozpouštědla. Výsledné roztoky se analyzují kvantitativně, aby se stanovila hmotnost desorbovaná z každé části. Volba analytické metody (rovněž volba sorbentu a desorbčního rozpouštědla) je určována povahou zkoušeného materiálu. Účinnost desorpce je stanovena vstříknutím známého množství vzorku do sorbentu, jeho desorpcí a analýzou zpětně získaného množství. Je důležité kontrolovat účinnost desorpce při koncentraci vzorku za podmínek zkoušení nebo v její blízkosti.
Aby bylo zajištěno nasycení nosného plynu zkoušenou látkou, používají se tři odlišné průtoky plynu. Jestliže vypočítaný tlak par nevykazuje závislost na průtoku, předpokládá se nasycení plynu.
Tlak par se vypočítá pomocí rovnice:
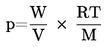
kde:
|
p |
= |
tlak páry (Pa), |
|
W |
= |
hmotnost odpařené zkoušené látky (g), |
|
V |
= |
objem nasyceného plynu (m3), |
|
R |
= |
univerzální molární plynová konstanta 8,314 (J mol–1 K–1), |
|
T |
= |
termodynamická teplota (K), |
|
M |
= |
molární hmotnost zkoušené látky (g mol–1). |
Naměřené objemy musí být korigovány v důsledku rozdílů tlaků a teplot mezi průtokoměrem a syticí kolonou.
1.5.8 Metoda rotujícího tělíska
1.5.8.1 Podstata
Tato metoda využívá měřiče viskozity s rotujícím tělískem, kde je měřicím prvkem malá ocelová kulička zavěšená v magnetickém poli, která je uváděna do rotačního pohybu rotujícími magnetickými poli (26, 27, 28). Zvedací cívky umožňují měření rotační rychlosti. Když kulička dosáhne stanovené rotační rychlosti, obvykle přibližně 400 otáček za sekundu, zastaví se další napájení a zahájí se zpomalování vyvolané brzdicím účinkem plynu. Pokles rotační rychlosti je měřen jako funkce času. Tlak par je odvozen ze zpomalení rotující ocelové kuličky v závislosti na tlaku. Doporučený rozsah je od 10–4 do 0,5 Pa.
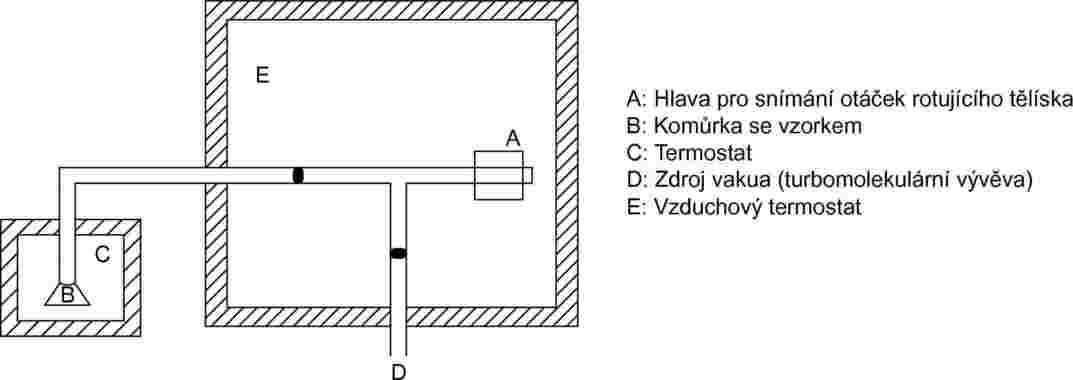
1.5.8.2 Aparatura
Schematický nákres měřicí soustavy je zachycen na obrázku 11. Měřicí hlava je umístěna v prostoru s konstantní teplotou, v němž je teplota regulována s přesností na 0,1 °C. Nádobka se vzorkem je umístěna do samostatného prostoru, v němž je teplota rovněž regulována s přesností na 0,1 °C. Všechny ostatní části soustavy jsou udržovány při vyšší teplotě, aby nedocházelo ke kondenzaci. K zařízení se připojí vysokovakuová vývěva.
Obrázek 11
2. DATA A ZPRÁVY
2.1 DATA
Stanovení tlaku par kteroukoli z výše popsaných metod by se mělo provést nejméně při dvou teplotách. Aby se ověřil lineární průběh křivky tlaku par v oblasti teplot od 0 °C do 50 °C, doporučuje se měření při třech nebo více teplotách. V případě efusní metody (Knudsenova komůrka a isotermální termogravimetrie) a metody nasycení plynu se jako rozsah měřicích teplot doporučuje 120 °C až 150 °C namísto 0 °C až 50 °C.
2.2 PROTOKOL O ZKOUŠCE
Protokol o zkoušce musí obsahovat tyto údaje:
|
— |
použitá metoda, |
|
— |
přesná specifikace látky (identifikace a nečistoty) a popř. informace o provedeném předběžném čištění, |
|
— |
nejméně dvě hodnoty tlaku par a teploty – pokud možno tři či více – v oblasti 0 °C až 50 °C (nebo 120 °C až 150 °C), |
|
— |
nejméně jedna z teplot by měla činit 25 °C nebo méně, bude-li to technicky proveditelné při zvolené metodě, |
|
— |
všechny původní údaje, |
|
— |
křivka závislosti log p na 1/T, |
|
— |
odhadnutá hodnota tlaku par při 20 °C nebo 25 °C. |
Zjistí-li se změna stavu (fázový přechod, rozklad), měly by být uvedeny tyto skutečnosti:
|
— |
druh změny, |
|
— |
teplota při atmosférickém tlaku, při k které ke změně dochází, |
|
— |
hodnoty tlaku par při 10 °C a 20 °C pod teplotou přechodu a nad teplotou přechodu (s výjimkou přechodů z pevného do plynného skupenství). |
Musí být uvedeny všechny informace a poznámky, které jsou důležité pro interpretaci výsledků, zejména pokud jde o nečistoty a fyzikální stav látky.
3. LITERATURA
|
(1) |
Úřední věstník Evropských společenství L 383 A, 26–47 (1992). |
|
(2) |
Ambrose, D. (1975). Experimental Thermodynamics, Vol. II, Le Neindre, B., Vodar, B., (eds.), Butterworths, London. |
|
(3) |
Weissberger R., (ed.) (1959). Technique of Organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed., Vol. I, Part I. Chapter IX, Interscience Publ., New York. |
|
(4) |
Glasstone, S. (1946). Textbook of Physical Chemistry, 2nd ed., Van Nostrand Company, New York. |
|
(5) |
NF T 20–048 AFNOR (September 1985). Chemical products for industrial use – Determination of vapour pressure of solids and liquids within a range from 10–1 to 105 Pa – Static method. |
|
(6) |
ASTM D 2879–86, Standard test method for vapour pressure – temperature relationship and initial decomposition temperature of liquids by isoteniscope. |
|
(7) |
NF T 20–047 AFNOR (September 1985). Chemical products for industrial use – Determination of vapour pressure of solids and liquids within range from 10–3 to 1 Pa – Vapour pressure balance method. |
|
(8) |
Knudsen, M. (1909). Ann. Phys. Lpz., 29, 1979; (1911), 34, 593. |
|
(9) |
Ambrose, D., Lawrenson, I.J., Sprake, C.H.S. (1975). J. Chem. Thermodynamics 7, 1173. |
|
(10) |
Schmuckler, M.E., Barefoot, A.C., Kleier, D.A., Cobranchi, D.P. (2000), Vapor pressures of sulfonylurea herbicides; Pest Management Science 56, 521–532. |
|
(11) |
Tomlin, C.D.S. (ed.), The Pesticide Manual, Twelfth Edition (2000). |
|
(12) |
Friedrich, K., Stammbach, K., Gas chromatographic determination of small vapour pressures determination of the vapour pressures of some triazine herbicides. J. Chromatog. 16 (1964), 22–28. |
|
(13) |
Grayson, B.T., Fosbraey, L.A., Pesticide Science 16 (1982), 269–278. |
|
(14) |
Rordorf, B.F., Prediction of vapor pressures, boiling points and enthalpies of fusion for twenty-nine halogenated dibenzo-p-dioxins, Thermochimia Acta 112 Issue 1 (1987), 117–122. |
|
(15) |
Gückel, W., Synnatschke, G., Ritttig, R., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection; Pesticide Science 4 (1973) 137–147. |
|
(16) |
Gückel, W., Synnatschke, G., Ritttig, R., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection II. Application to Formulated Products; Pesticide Science 5 (1974) 393–400. |
|
(17) |
Gückel, W., Kaestel, R., Lewerenz, J., Synnatschke, G., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection. Part III: The Temperature Relationship between Vapour Pressure and Evaporation Rate; Pesticide Science 13 (1982) 161–168. |
|
(18) |
Gückel, W., Kaestel, R., Kroehl, T., Parg, A., Methods for Determining the Vapour Pressure of Active Ingredients Used in Crop Protection. Part IV: An Improved Thermogravimetric Determination Based on Evaporation Rate; Pesticide Science 45 (1995) 27–31. |
|
(19) |
Kroehl, T., Kaestel, R., Koenig, W., Ziegler, H., Koehle, H., Parg, A., Methods for Determining the Vapour Pressure of Active Ingredients Used in Crop Protection. Part V: Thermogravimetry Combined with Solid Phase MicroExtraction (SPME); Pesticide Science, 53 (1998) 300–310. |
|
(20) |
Tesconi, M., Yalkowsky, S.H., A Novel Thermogravimetric Method for Estimating the Saturated Vapor Pressure of Low-Volatility Compounds; Journal of Pharmaceutical Science 87(12) (1998) 1512–20. |
|
(21) |
Lide, D.R. (ed.), CRC Handbook of Chemistry and Physics, 81th ed.(2000), Vapour Pressure in the Range – 25 °C to 150 °C. |
|
(22) |
Meister, R.T. (ed.), Farm Chemicals Handbook, Vol. 88 (2002). |
|
(23) |
40 CFR, 796. (1993). s. 148–153, Office of the Federal Register, Washington DC. |
|
(24) |
Rordorf B.F. (1985). Thermochimica Acta 85, 435. |
|
(25) |
Westcott et al. (1981). Environ. Sci. Technol. 15, 1375. |
|
(26) |
Messer G., Röhl, P., Grosse G., Jitschin W. (1987). J. Vac. Sci. Technol. (A), 5(4), 2440. |
|
(27) |
Comsa G., Fremerey J.K., Lindenau, B. (1980). J. Vac. Sci. Technol. 17(2), 642. |
|
(28) |
Fremerey, J.K. (1985). J. Vac. Sci. Technol. (A), 3(3), 1715. |
(1) Při použití kapacitního manometru.
Dodatek
Metoda odhadu
ÚVOD
Odhadnuté hodnoty tlaku par lze využít:
|
— |
k rozhodnutí, která z experimentálních metod je vhodná, |
|
— |
k provedení odhadu nebo ke stanovení mezní hodnoty v případech, kdy nelze z technických důvodů použít experimentální metodu. |
METODA ODHADU
Tlak par kapalin a pevných látek může být odhadnut pomocí modifikovaného Watsonova korelačního vztahu (a). Požaduje se pouze jeden experimentální údaj, a to teplota varu za normálního tlaku. Metoda je použitelná pro tlaky od 105 do 10–5 Pa.
Podrobné informace o metodě jsou uvedeny v příručce Handbook of Chemical Property Estimation Methods (b). Viz rovněž OECD Environmental Monograph No. 67 (c).
POSTUP VÝPOČTU
Tlak par se vypočte podle vzorce:
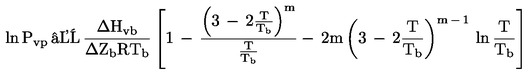
kde:
|
T |
= |
teplota, pro kterou je tlak par vypočítáván, |
|
T b |
= |
teplota varu při normálním tlaku, |
|
P vp |
= |
tlak par při teplotě T, |
|
ΔH vb |
= |
výparné teplo, |
|
ΔZ b |
= |
faktor stlačitelnosti (použije se hodnota 0,97), |
|
m |
= |
empirický faktor, jehož hodnota závisí na fyzikálním stavu při teplotě, pro níž je prováděn výpočet. |
Dále
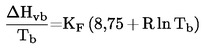
kde K F je empirický součinitel zohledňující polaritu látky. V příručce (b) jsou uvedeny hodnoty faktoru K F pro několik typů sloučenin.
Velmi často jsou k dispozici data o teplotě varu při sníženém tlaku. V takovém případě se tlak par vypočte podle tohoto vztahu:
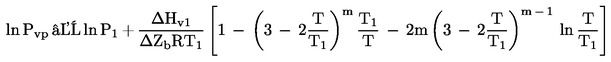
kde T 1 je teplota varu při sníženém tlaku P 1.
ZPRÁVA
Je-li použita metoda odhadu, musí zpráva obsahovat úplný postup výpočtu.
LITERATURA
|
(a) |
Watson, K.M. (1943). Ind. Eng. Chem., 35, 398. |
|
(b) |
Lyman, W.J., Reehl, W.F., Rosenblatt, D.H. (1982). Handbook of Chemical Property Estimation Methods, McGraw-Hill. |
|
(c) |
OECD Environmental Monograph No. 67. Application of Structure-Activity Relationships to the Estimation of Properties Important in Exposure Assessment (1993). |
PŘÍLOHA II
|
A.22 |
DÉLKOVĚ VÁŽENÝ STŘEDNÍ GEOMETRICKÝ PRŮMĚR VLÁKEN |
1. METODA
1.1 ÚVOD
Tato metoda popisuje postup měření délkově váženého středního geometrického průměru (DVGSP) umělých minerálních vláken (UMV). Jelikož DVGSP statistického souboru bude s pravděpodobností 95 % mezi hladinami významnosti 95 % (DVGSP ± dvě střední směrodatné chyby) vzorku, ohlášená hodnota (testová hodnota) bude nižší hladina významnosti 95 % vzorku (tj. DVGSP – 2 dvě střední směrodatné chyby). Metoda je založena na aktualizaci (červen 1994) návrhu průmyslového postupu HSE (Výkonný výbor pro zdraví a bezpečnost) dohodnutého na schůzce HSE a ECFIA (Evropská asociace odvětví keramických vláken) v Chesteru dne 26. září 1993 a vyvinutého druhou mezilaboratorní zkouškou (1, 2). Tato měřicí metoda může být použita pro charakterizaci průměru vláken sypkých látek či výrobků obsahujících UMV, včetně vysokotavných keramických vláken (VKV), umělých skelných vláken (USV), krystalických a polykrystalických vláken.
Délkové vážení je způsobem kompenzace vlivu rozlomení dlouhých vláken při odběru vzorků či při manipulaci s vlákny na rozložení jejich průměru. Pro měření rozložení velikosti průměrů UMV jsou použita geometrická statistická data (geometrický průměr), jelikož tyto průměry obvykle mají rozložení velikosti blížící se normálnímu.
Měření délky a průměru je jak únavné, tak dlouhé, ovšem pokud se měří pouze ta vlákna, která se dotýkají nekonečně tenké čáry na zorném poli rastrovacího elektronového mikroskopu, pak je pravděpodobnost výběru daného vlákna přímo úměrná jeho délce. Jelikož tím se zohledňuje délka při délkově vážených výpočtech, jediné vyžadované měření je měření průměru a DVGSP-2SE může být vypočten uvedeným způsobem.
1.2 DEFINICE
Částice: objekt s poměrem délky k šířce menším než 3:1.
Vlákno: objekt s poměrem délky k šířce (charakteristický poměr) nejméně 3:1.
1.3 PLATNOST A OMEZENÍ
Metoda je určena pro pohled na rozdělení průměrů o středním průměru mezi 0,5 μm a 6 μm. Větší průměry mohou být měřeny pomocí menších zvětšení rastrového elektronového mikroskopu, ale tato metoda bude stále více limitována vzhledem k rozdělení jemných vláken a je-li střední průměr menší než 0,5 μm, doporučujeme měření transmisním elektronovým mikroskopem (TEM).
1.4 Princip měřicí metody
Z vláknové pokrývky či z volných sypkých vláken se odebere několik reprezentativních jádrových vzorků. Sypká vlákna jsou délkově omezena pomocí procesu drcení a reprezentativní podíl vzorku se rozpustí ve vodě. Poměrné podíly se vyloučí a přefiltrují přes polykarbonátový filtr o rozměrech pórů 0,2 μm a připraví se na pozorování pomocí technik rastrovacího elektronového mikroskopu (SEM). Průměry vláken se měří při zvětšení obrazovky × 10 000 nebo vyšším (1) pomocí čárové zadržovací metody, aby se získal nezaujatý odhad středního průměru. Vypočte se nižší hladina významnosti 95 % (na základě jednostranného testu), a tím se získá odhad nejnižších hodnot geometrického středního průměru vláken materiálu.
1.5 Popis měřicí metody
1.5.1 Bezpečnostní opatření
Je nutno minimalizovat vystavení osob zvířeným vláknům a při manipulaci se suchými vlákny je nutno používat digestoř či suchou skříň. Je nutno periodicky provádět monitoring osobní expozice pro určení efektivity kontrolních metod. Při manipulaci s UMV je nutno nosit jednorázové rukavice pro omezení podráždění kůže a pro zabránění křížové kontaminaci.
1.5.2 Přístroje/zařízení
|
— |
lis a lisovadla (schopná vyvinout tlak 10 MPa), |
|
— |
polykarbonátové filtry s kapilárními póry o velikosti 0,2 μm (průměr 25 mm), |
|
— |
membránový filtr z esteru buničiny o rozměru pórů 5 μm jako filtr záložní, |
|
— |
skleněný filtrační přístroj (či použitelné filtrační systémy) pro filtry o průměru 25 cm (např. skleněná mikroanalytická sada Millipore typ XX10 025 00), |
|
— |
čerstvá voda destilovaná přes filtr o rozměru pórů 0,2 μm pro odstranění mikroorganismů, |
|
— |
katodový rozprašovač s anodou ze zlata či ze zlata/palladia, |
|
— |
rastrovací elektronový mikroskop schopný rozlišení až 10 nm a pracující při zvětšení × 10 000, |
|
— |
různé: stěrky, skalpel typu 24, pinzeta, tubusy SEM, uhlíkové lepidlo či uhlíková lepící páska, stříbrná tyčinka, |
|
— |
ultrazvuková sonda či ultrazvuková lázeň na desce laboratorního stolu, |
|
— |
odběrač vzorků sedimentu či korkovrt pro odebírání jádrových vzorků z pokrývky UMV. |
1.5.3 Postup měření
1.5.3.1 Odběr vzorků
U pokrývek a roun se používá odběrač vzorků velikosti 25 mm či korkovrt pro odebírání vzorků z řezu. Vzorky by měly být rovnoměrně rozprostřeny po šířce a z malé délky pokrývky, nebo by měly být odebrány z náhodných ploch, je-li k dispozici pokrývka o velké délce. Stejné zařízení může být použito pro odběr náhodných vzorků z volných vláken. Je-li to možné, mělo by být odebráno šest vzorků tak, aby byly zohledněny prostorové odchylky v sypkém materiálu.
Těchto šest jádrových vzorků je nutno rozdrtit v lisovadle o průměru 50 mm tlakem 10 MPa. Materiál se rozmíchá stěrkou a znovu se stlačí tlakem 10 MPa. Poté se materiál vyjme z lisovadla a umístí v utěsněné skleněné láhvi.
1.5.3.2 Příprava vzorku
Je-li to nutné, organické pojivo může být odstraněno umístěním vláken do pece o teplotě 450 °C po dobu jedné hodiny.
Vytvarujte vzorek do kužele a rozčtvrťte jej (to by mělo být provedeno uvnitř prachové skříně).
Přidejte stěrkou malé množství vzorku (< 0,5 g) do 100 ml čerstvé destilované vody, která byla přefiltrována přes membránový filtr 0,2 μm (je možno použít jiný zdroj velmi čisté vody, ukáže-li se být uspokojivý). Řádně rozmíchejte vzorek pomocí ultrazvukové sondy provozované s výkonem 100 W a vyladěné tak, aby se objevila kavitace (není-li sonda k dispozici, postupujte takto: opakovaně vzorek protřepte a otočte jej na dobu 30 sekund; po dobu pěti minut vystavte vzorek ultrazvuku v ultrazvukové lázni; poté opět několikrát protřepte a otočte vzorek po dobu dalších 30 sekund).
Ihned po promíchání vláken odeberte několik poměrných dílů (např. tři poměrné části o 3, 6 a 10 ml) pomocí široké pipety (o objemu 2–5 ml).
Vakuově přefiltrujte každý poměrný díl přes polykarbonátový filtr 0,2 μm s pomocí podpůrného filtru MEC o velikosti pórů 5 µm pomocí skleněného filtračního tunelu 25 mm s válcovou nádrží. Přibližně 5 ml filtrované destilované vody by mělo být umístěno do tunelu a poměrný díl by měl být pomalu pipetován do vody, okraj pipety se drží pod prohnutou čočkou. Po pipetování musí být pipeta a nádrž řádně propláchnuta, jelikož tenká vlákna mají snahu ulpívat dále na povrchu.
Opatrně odstraňte filtr a oddělte jej od podpůrného filtru před jeho umístěním do obalu za účelem jeho vysušení.
Uřízněte čtvrtinu či polovinu filtrované části filtrovaného depozitu pomocí skalpelu typu 24 trhnutím. Opatrně připojte odříznutou část k bloku SEM pomocí lepící uhlíkové pásky či uhlíkového lepidla. Je nutno použít alespoň na třech místech stříbrnou tyčinku pro zlepšení elektrického kontaktu na okrajích filtru a bloku. Je-li lepidlo/stříbrná tyčinka suchá, katodově rozprašte cca 50 nm zlata či zlata/palladia na povrch depozitu.
1.5.3.3 Kalibrace a provoz SEM
1.5.3.3.1 Kalibrace
Kalibrace SEM by měla být zkontrolována minimálně jednou za týden (ideálně jednou denně) pomocí certifikované kalibrační mřížky. Kalibrace musí být kontrolována vůči certifikovanému standardu a není-li naměřená hodnota (SEM) v rozsahu ±2 % hodnoty certifikované, potom musí být kalibrace SEM seřízena a znovu zkontrolována.
SEM by měl být schopen rozlišit alespoň minimální viditelný průměr 0,2 µm pomocí reálné vzorkovací matice při zvětšení × 2 000.
1.5.3.3.2 Provoz
SEM by měl být provozován při zvětšení 10 000 (2) za podmínek, které dávají dobré rozlišení při přijatelném snímku při pomalých snímacích rychlostech např. 5 sekund na rámec. I když se provozní požadavky různých SEM mohou lišit, obecně by mělo být použito urychlovací napětí 5–10 keV s nastavením malé velikosti bodu a krátkou pracovní vzdáleností; tím se získá nejlepší viditelnost a rozlišení s materiály o relativně nízkých atomových hmotnostech. Při provádění lineárního příčného pohybu musí být použit sklon 0° pro minimalizaci přeostření, nebo má-li SEM eucentrickou fázi, musí být použita eucentrická pracovní vzdálenost. Menší zvětšení může být použito, neobsahuje-li materiál vlákna o malých průměrech a průměry vláken jsou velké (> 5 μm)
1.5.3.4 Třídění dle rozměrů
1.5.3.4.1 Pozorování při malém zvětšení pro vyhodnocení vzorku
Zpočátku by měl být vzorek pozorován při malém zvětšení pro ujištění se o rozdrcení velkých vláken a pro vyhodnocení hustoty vláken. V případě velkého rozdrcení je doporučeno připravit nový vzorek.
Kvůli statistické přesnosti je nezbytné změřit minimální počet vláken a může se zdát být výhodná vysoká hustota vláken, jelikož pozorováním prázdných polí ztrácíme čas a nijak k analýze nepřispíváme. Ovšem při přetížení filtru je obtížné změřit všechna měřitelná vlákna a jelikož malá vlákna mohou být zakryta vlákny většími, mohou být vynechána.
Je-li hustota vláken větší než 150 vláken na milimetr lineárního příčného posuvu, může dojít k nežádoucímu vlivu nadhodnocení DVGSP. Na druhé straně malá koncentrace vláken zvyšuje dobu analýzy a často je nákladově efektivní připravit vzorek v hustotou vlákna blíže hodnotě optimální, než zůstat u počítání vláken o menších koncentracích. optimální hustota vláken by měla dávat průměrně jedno či dvě počítatelná vlákna na pole z záběru o zvětšení 5 000. Nicméně optimální hustota bude záviset na velikosti (průměru) vláken, takže je nezbytné, aby obsluha používala odborný úsudek za účelem rozhodnutí, zda je hustota vláken blízko hodnotě optimální či nikoliv.
1.5.3.4.2 Délkové vážení průměrů vláken
Pouze ta vlákna, která se dotýkají (nebo kříží) (nekonečně) tenkou čáru nakreslenou na obrazovce SEM, se započítávají. Z tohoto důvodu je přes střed obrazovky nakreslena horizontální (či vertikální) čára.
Alternativně může být ve středu obrazovky umístěn jediný bod a zahájí se spojité snímání v jednom směru přes filtr. Každé vlákno o charakteristickém poměru větším než 3:1 dotýkající se při procházející tímto bodem má změřený a zaznamenaný průměr.
1.5.3.4.3 Třídění vláken
Doporučuje se měření minimálně 300 vláken. Každé vlákno je měřeno pouze jednou v bodě průsečíku s čárou či bodem nakresleným na snímku (nebo blízko průsečíku, jsou-li okraje vlákna zakrytá). Jestliže se objeví vlákna s nerovnoměrným průřezem, je nutno použít měření reprezentující střední průměr vlákna. Je nutno dbát na definování okraje a měřit nejkratší vzdálenost mezi okraji vlákna. Třídění může být prováděno on-line, nebo off-line na uložených snímcích či fotografiích. Doporučeny jsou poloautomatické snímkovací měřicí systémy, které stahují data přímo do tabulky, jelikož šetří čas, eliminují chyby v přepisech a mohou být zautomatizovány výpočty.
Konce dlouhých vláken by měla být zkontrolovány při malém zvětšení, aby bylo zajištěno, že se nepřesunou zpět do měřicího zorného pole a že budou měřeny pouze jednou.
2. DATA
2.1 NAKLÁDÁNÍ S VÝSLEDKY
Průměry vláken obvykle nemají normální rozdělení. Ovšem provedením logaritmické transformace je možno získat rozdělení, které se blíží normálnímu.
Vypočtěte aritmetický průměr (střední (mean) lnD) a standardní odchylku (SDlnD) logaritmem o základu e hodnoty (lnD) průměrů vláken (D).
|
|
(1) |
|
|
(2) |
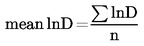
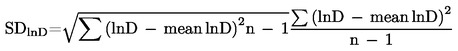
Standardní odchylka je dělena druhou odmocninou počtu měření (n), čímž získáme střední směrodatnou chybu (SElnD).
|
|
(3) |
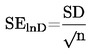
Odečtěte od průměrné hodnoty dvojnásobek střední směrodatné chyby a vypočtěte exponenciál této hodnoty (průměr mínus dvě střední směrodatné chyby), což dává střední geometrickou hodnotu mínus dvě geometrické směrodatné chyby.
|
|
(4) |
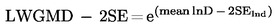
3. ZPRÁVY
ZPRÁVA O MĚŘENÍ
Zpráva o měření by měla obsahovat minimálně tyto informace:
|
— |
hodnotu DVGSP-2SE, |
|
— |
veškeré odchylky a zejména ty, které mohou mít vliv na přesnost výsledků, s patřičným odůvodněním. |
4. LITERATURA
|
(1) |
B. Tylee SOP MF 240. Health and Safety Executive. Únor 1999. |
|
(2) |
G. Burdett and G. Revell. Development of a standard method to measure the length-weigthed geometric mean fibre diameter: Results of the Second inter-laboratory exchange. IR/L/MF/94/07. Projekt R42.75 HPD. Health and Safety Executive. Research and Laboratory Services Division. 1994. |
(1) Tato hodnota zvětšení je uvedena pro vlákna o průměru 3 µm, pro vlákna o průměru 6 µm je vhodnější zvětšení × 5 000.
(2) U vláken 3 μm viz předchozí poznámka.
PŘÍLOHA III
|
B.46 |
DRÁŽDĚNÍ KŮŽE IN VITRO: ZKOUŠKA POMOCÍ MODELU REKONSTRUOVANÉ LIDSKÉ EPIDERMIS |
1. METODA
1.1 ÚVOD
Dráždění kůže znamená tvorbu reverzibilního poškození kůže po použití zkoušené látky v délce 4 hodin (jak ji definuje globálně harmonizovaný systém klasifikace a označování chemických látek Organizace spojených národů (OSN) (GHS)) (1). Tato zkušební metoda poskytuje postup in vitro, který v závislosti na informačních požadavcích může povolovat stanovení dráždivých účinků látek na kůži jakožto samostatná náhradní zkouška v rámci strategie zkoušení v přístupu založeném na vážení důkazů (2).
Hodnocení dráždění kůže typicky zahrnovalo používání laboratorních zvířat (viz metoda B.4) (3). V souvislosti s ohledy na dobré zacházení se zvířaty metoda B.4 umožňuje stanovit poleptání/dráždění kůže uplatněním strategie postupného zkoušení, použitím validovaných metod in vitro a ex vivo, což vylučuje bolest a utrpení zvířat. Tři validované zkušební metody in vitro nebo zkušební pokyny B.40, B.40a a TG 435 (4, 5, 6) jsou užitečné pro tu část strategie postupného zkoušení B.4, jež se týká poleptání.
Tato zkušební metoda je založena na modelech rekonstruované lidské epidermis, které svým celkovým uspořádáním (použitím keratinocytů získaných z lidské epidermis jakožto zdroje buněk, reprezentativní tkáně a cytoarchitektury) věrně napodobují biochemické a fyziologické vlastnosti svrchních částí lidské kůže, tj. epidermis. Postup popsaný podle této zkušební metody dovoluje rozpoznat nebezpečí dráždivých látek v souladu s kategorií 2 GHS OSN. Tato zkušební metoda rovněž zahrnuje množinu standardů chování pro hodnocení podobných a modifikovaných zkušebních metod založených na rekonstruované lidské epidermis (7).
Pro dvě zkušební metody in vitro (8, 9, 10, 11, 12, 13, 14, 15, 16, 17), komerčně dostupné jako EpiSkin™ a EpiDerm™, které používají modely rekonstruované lidské epidermis, byly dokončeny prevalidační, optimalizační a validační studie. Tyto odkazy byly založeny na větě označující rizikovost R38. Některými aspekty přepočtu pro účely GHS se zabývá v rámci použité literatury odkaz 25. Metody, u nichž chování odpovídá EpiSkin™ (validovaná referenční metoda 1), jsou doporučeny jakožto samostatná náhradní zkušební metoda pro zkoušku na králících in vivo, která slouží ke klasifikaci dráždivých látek kategorie 2 GHS. Metody, u nichž chování odpovídá EpiDerm™ (validovaná referenční metoda 2), jsou doporučeny pouze jako rozřaďovací zkušební metoda nebo jako součást strategie postupného zkoušení u přístupu založeného na vážení důkazů pro klasifikaci dráždivých látek kategorie 2 GHS. Předtím, než lze pro regulační účely použít navrhovanou zkoušku s modelem rekonstruované lidské epidermis in vitro k podráždění kůže, je zapotřebí stanovit její spolehlivost, relevanci (přesnost) a omezení vztahující se na její navrhované použití, aby se zajistila její srovnatelnost s uvedenými parametry validované referenční metody 1 v souladu se standardy chování stanovenými v této zkušební metodě (dodatek).
V souladu s požadavky podle této zkušební metody byly validovány dvě další zkušební metody s rekonstruovanou lidskou epidermis in vitro a vykazují podobné výsledky jako validovaná referenční metoda 1 (18). Jsou to modifikovaná zkušební metoda EpiDerm™ (upravená referenční metoda 2) a zkušební metoda SkinEthic RHE™ (metoda 1 „me-too“).
1.2 DEFINICE
V rámci této zkušební metody se používají následující definice:
Přesnost: Blízkost shody mezi výsledky zkušební metody a přijatými referenčními hodnotami. Je to míra chování zkušební metody a jeden aspekt relevance. Termín se často používá zaměnitelně za „shodu“, čímž se má na mysli podíl správných výsledků zkušební metody.
Kontrolní látka šarže: Srovnávací látka vyvolávající u tkáně odezvu životaschopnosti buněk středního rozsahu.
Životaschopnost buněk: Parametr měřící celkovou aktivitu buněčné populace, např. jako schopnost buněčných mitochondriálních dehydrogenáz redukovat vitální barvivo MTT (3–(4,5-dimethylthiazol-2-yl)-2,5-difenyl-2H-tetrazolium-bromid, thiazolylová modř), které v závislosti na naměřeném cílovém bodu a použitém designu zkoušky koreluje s celkovým počtem nebo vitalitou živých buněk.
ET50 : Doba expozice požadovaná pro snížení životaschopnosti buněk o 50 % po nasazení látky markeru ve specifikované pevné koncentraci, viz rovněž IC50.
Míra falešné negativity: Podíl všech pozitivních látek falešně identifikovaných zkušební metodou jako negativní. Je to jeden ukazatel chování zkušební metody.
Míra falešné pozitivity: Podíl všech negativních (neúčinných) látek, které jsou falešně identifikovány jako pozitivní. Je to jeden ukazatel chování zkušební metody.
Nekonečná dávka: Množství zkušební látky nanesené na kůži, které přesahuje množství požadované pro úplné a rovnoměrné pokrytí povrchu kůže.
GHS (globálně harmonizovaný systém klasifikace a označování chemických látek): Systém navrhující klasifikaci látek a směsí podle standardizovaných typů a úrovní fyzikálních, zdravotních a environmentálních nebezpečí a řešící příslušné komunikační prvky, jako jsou piktogramy, signální slova, výroky o nebezpečí, výroky o bezpečnostních opatřeních a bezpečnostní datové listy, které obsahují informace o jejich nežádoucích účincích s ohledem na ochranu lidí (včetně zaměstnavatelů, dělníků, přepravců, spotřebitelů a osob reagujících na havárie) a životního prostředí (1); tento systém je v EU proveden nařízením (ES) č. 1272/2008.
IC50 : Koncentrace, při níž látka markeru snižuje životaschopnost tkání o 50 % (IC50) po pevně stanovené době expozice, viz rovněž ET50.
Standardy chování: Standardy založené na validované referenční metodě, které slouží jako základna pro vyhodnocení srovnatelnosti navrhované zkušební metody, jež je mechanicky a funkčně podobná. Mezi ně patří I) zásadně důležité složky zkušební metody, II) minimální seznam referenčních látek vybraných z látek používaných k prokázání přijatelného chování validované referenční metody a III) srovnatelné hladiny přesnosti a spolehlivosti založené na tom, co bylo získáno pro validovanou referenční metodu, které by navrhovaná zkušební metoda měla prokázat při vyhodnocení za použití minimálního seznamu referenčních látek.
Spolehlivost: Měří rozsah, v němž lze provádět zkušební metodu reprodukovatelně v rámci laboratoří a mezi nimi navzájem po danou dobu, přičemž se metoda provádí s využitím stejného protokolu. Vyhodnocuje se na základě výpočtu vnitrolaboratorní a mezilaboratorní reprodukovatelnosti.
Citlivost: Podíl všech pozitivních/účinných látek, které jsou správně klasifikovány zkouškou. Je to měřítko přesnosti zkušební metody, která dává kategorické výsledky, a je důležitým prvkem při hodnocení relevance zkušební metody.
Specifičnost: Podíl všech negativních/neúčinných látek, které jsou správně zkouškou klasifikovány. Je to měřítko přesnosti zkušební metody, která dává kategorické výsledky, a je důležitým prvkem při hodnocení relevance zkušební metody.
Dráždění kůže: Tvorba reverzibilního poškození kůže po použití zkoušené látky až po dobu 4 hodin. Dráždění kůže je lokálně vznikající neimunogenní reakce, která se objevuje krátce po stimulaci (24). Její hlavní charakteristikou je reverzibilní proces zahrnující zánětlivé reakce a většinu klinických charakteristických příznaků dráždění (erytém, edém, svědění a bolest) v souvislosti se zánětlivým procesem.
1.3 ROZSAH A OMEZENÍ
Omezení zkoušek rekonstruované lidské epidermis spadajících do této zkušební metody spočívá v tom, že pouze klasifikují látky jako dráždivé pro kůži podle kategorie 2 GHS OSN. Protože nedovolují klasifikaci látek podle volitelné kategorie 3, jak ji definuje GHS OSN, všechny zbývající látky klasifikovány nebudou (bez kategorie). V závislosti na potřebách regulace a na možném budoucím zahrnutí nových cílových bodů, zlepšení nebo vývoji nových zkoušek typu „me-too“ bude možná nutné tuto zkušební metodu revidovat.
Tato zkušební metoda dovoluje rozpoznat nebezpečí dráždivých jednosložkových látek (19), ale neposkytuje adekvátní informace o poleptání kůže. Plyny a aerosoly zkoušet nelze, zatímco směsi dosud nebyly ve validační studii hodnoceny.
1.4 PODSTATA ZKUŠEBNÍ METODY
Zkoušená látka se topicky nanese na trojrozměrný model rekonstruované lidské epidermis obsahující normální lidské epidermální keratinocyty, které byly kultivovány s cílem vytvořit vícevrstevnatý, vysoce diferenciovaný model lidské epidermis. Skládá se z organizovaných bazálních, výběžkovitých a granulárních vrstev a vícevrstevnatého stratum corneum, jež obsahuje mezibuněčné lamelární lipidové vrstvy uspořádané do vzorců analogických k těm, které se nachází in vivo.
Princip zkoušky na modelu rekonstruované lidské epidermis je založen na předpokladu, že dráždivé látky jsou schopny proniknout přes stratum corneum difúzí a že jsou cytotoxické pro buňky v podkladových vrstvách. Životaschopnost buněk se měří přeměnou vitálního barviva MTT [3–(4,5-dimethylthiazol-2-yl)-2,5-difenyl-2H-tetrazolium-bromid, thiazolylová modř, číslo EINECS 206–069–5, číslo CAS 298–93–1] účinkem dehydrogenázy na sůl modrého formazanu, který se kvantitativně měří po extrakci z tkání (20). Dráždivé látky se identifikují na základě jejich schopnosti snižovat životaschopnost buněk pod definované prahové úrovně (tj. ≤ 50 % pro dráždivé látky kategorie 2 GHS OSN). Látky, které produkují životaschopnosti buněk nad definovanou prahovou hladinu, klasifikovány nebudou (tj. > 50 %, žádná kategorie).
Modelové systémy rekonstruované lidské epidermis lze použít k testování pevných látek, kapalin, polotuhých látek a vosků. Kapaliny mohou být vodné či nevodné, pevné látky mohou být ve vodě rozpustné či nerozpustné. Pevné látky je zapotřebí zkoušet ve formě jemného prášku, bude-li to možné. Protože do validace zkušebních systémů založených na modelu rekonstruované lidské epidermis bylo zahrnuto 58 pečlivě vybraných látek reprezentujících široké spektrum chemických tříd, očekává se, že metody budou obecně použitelné napříč chemickými třídami (16). Validace zahrnuje 13 dráždivých látek kategorie 2 GHS. Je třeba poznamenat, že do validace nebyly zařazeny nežíravé kyseliny, zásady, soli a jiné anorganické látky a nebyly do ní zahrnuty ani některé známé třídy organických dráždivých látek, například hydroperoxidy, fenoly a povrchově aktivní látky, či byly zařazeny jen v omezené míře.
1.5 PROKÁZÁNÍ ODBORNOSTI
Před rutinním používáním validované metody, která se řídí touto zkušební metodou, mohou laboratoře projevit přání prokázat technickou odbornost při použití deseti látek doporučených v tabulce 1. Podle této zkušební metody se volitelná kategorie 3 GHS OSN nepovažuje za kategorii. U originálních podobných zkušebních metod (typu „me-too“) vyvinutých podle této zkušební metody, které jsou strukturálně a funkčně podobné validovaným referenčním metodám, nebo u úprav validovaných metod je zapotřebí použít standardy chování popsané v dodatku k této zkušební metodě pro prokázání srovnatelné spolehlivosti a přesnosti nové zkušební metody před jejím použitím pro regulační zkoušení.
Tabulka 1
Látky k prokázání odbornosti, které jsou podmnožinou referenčních látek uvedených na seznamu v dodatku
|
Látka |
Číslo CAS |
Skóre in vivo |
Skupenství |
Kategorie GHS |
|
kyselina naftalen-1-octová |
86–87–3 |
0 |
pevné |
žádná kategorie |
|
isopropylalkohol |
67–63–0 |
0,3 |
kapalné |
žádná kategorie |
|
methyl-stearát |
112–61–8 |
1 |
pevné |
žádná kategorie |
|
heptyl-butyrát |
5870–93–9 |
1,7 |
kapalné |
volitelná kategorie 3 |
|
hexyl-salicylát |
6259–76–3 |
2 |
kapalné |
volitelná kategorie 3 |
|
cyklamenaldehyd |
103–95–7 |
2,3 |
kapalné |
kategorie 2 |
|
1-bromhexan |
111–25–1 |
2,7 |
kapalné |
kategorie 2 |
|
butyl-methakrylát |
97–88–1 |
3 |
kapalné |
kategorie 2 |
|
3-fenyl-1-methylpiperazin |
5271–27–2 |
3,3 |
pevné |
kategorie 2 |
|
heptanal |
111–71–7 |
4 |
kapalné |
kategorie 2 |
1.6 POPIS METODY
Následuje popis složek a postupů zkoušky založené na modelu rekonstruované lidské epidermis při hodnocení dráždění kůže. Model rekonstruované lidské epidermis lze sestavit, připravit nebo získat komerčně (např. EpiSkin™, EpiDerm™ a SkinEthic RHE™). Protokoly standardní zkušební metody pro EpiSkin™, EpiDerm™ a SkinEthic RHE™ lze získat na adrese [http://ecvam.jrc.ec.europa.eu] (21, 22, 23). Zkoušení lze provádět následujícím způsobem:
1.6.1 Složky modelu rekonstruované lidské epidermis
1.6.1.1 Všeobecné podmínky modelu
K sestavení epitelu je zapotřebí použít normální lidské keratinocyty. Pod funkčním stratum corneum by měly být přítomny vícenásobné vrstvy životaschopných epitelových buněk (bazální vrstva, stratum spinosum, stratum granulosum). Stratum corneum by mělo být vícevrstevnaté a obsahovat profil esenciálních lipidů k vytvoření funkční bariéry robustní natolik, aby vzdorovala rychlému průniku cytotoxických látek markeru, např. dodecylsulfátu sodného (SDS) nebo přípravku Triton X-100. Funkci bariéry lze hodnotit buď stanovením koncentrace, při níž látka markeru snižuje životaschopnost tkání o 50 % (IC50) po pevně stanovené expoziční době, nebo stanovením expoziční doby požadované ke snížení životaschopnosti buněk o 50 % (ET50) po nasazení látky markeru ve specifikované pevně stanovené koncentraci. Zadržovací vlastnosti modelu by měly zabránit průchodu materiálu v oblasti stratum corneum do životaschopné tkáně, což by způsobilo špatné modelování expozice kůže. Model kůže by neměl být kontaminován bakteriemi, viry, mykoplazmou ani mykózami.
1.6.1.2 Funkční podmínky modelu
1.6.1.2.1 Životaschopnost
MTT (20) je upřednostňovanou kvantitativní analýzou pro stanovení míry životaschopnosti. Optická hustota (OD) extrahovaného (rozpustitelného) barviva z tkáně ošetřené negativní kontrolou (NC) by měla být nejméně dvacetinásobně větší než OD extrakčního rozpouštědla samotného. Je nutné prokázat, že tkáň ošetřená NC je stabilní v kultuře (poskytnout podobná měření životaschopnosti) po dobu trvání lhůty zkušební expozice.
1.6.1.2.2 Bariérová funkce
Stratum corneum a jeho lipidové složení by mělo dostačovat na to, aby odolávalo rychlému průniku cytotoxických látek markeru, např. SDS nebo Triton X-100, podle odhadu na základě IC50 nebo ET50.
1.6.1.2.3 Morfologie
Histologické vyšetření rekonstruované kůže/epidermis by měl provádět vhodně kvalifikovaný personál a mělo by prokázat strukturu podobnou lidské kůži/epidermis (včetně vícevrstevnatého stratum corneum).
1.6.1.2.4 Reprodukovatelnost
Výsledky metody používající specifický model by měly prokázat reprodukovatelnost v průběhu času, nejlépe pomocí vhodné kontrolní (srovnávací) látky šarže (viz dodatek).
1.6.1.2.5 Kontroly kvality (QC) modelu
Každá šarže použitého epidermálního modelu by měla splňovat definovaná produkční kritéria uvolnění do oběhu, kdy mezi nejvýznamnější patří kritérium životaschopnosti (bod 1.6.1.2.1) a bariérové funkce (bod 1.6.1.2.2). Rozsah přijatelnosti (horní a dolní mez) pro IC50 nebo ET50 je zapotřebí zjistit u dodavatele modelu kůže (nebo experimentátora, pokud se používá vlastní model). Bariérové vlastnosti tkání je nutno ověřit laboratorně po dodání tkání. Pro spolehlivou předpověď dráždivých účinků mohou být přijatelné pouze výsledky vyprodukované pomocí náležitých tkání. Jako příklad jsou dále uvedeny rozsahy přijatelnosti pro validované referenční metody.
Tabulka 2
Příklady kritérií kontroly kvality pro uvolnění šarže do oběhu
|
|
Dolní mez přijatelnosti |
Průměr rozsahu přijatelnosti |
Horní mez přijatelnosti |
|
Validovaná referenční metoda 1 (18 hodin ošetření SDS) |
IC50 = 1,0 mg/ml |
IC50 = 2,32 mg/ml |
IC50 = 3,0 mg/ml |
|
Validovaná referenční metoda 2 (1 % Triton X100) |
ET50 = 4,8 h |
ET50 = 6,7 h |
ET50 = 8,7 h |
1.6.1.3 Nasazení zkoušené a kontrolní látky
Pro každou léčbu a pro kontroly (minimálně tři replikáty na jednu zkoušku) je zapotřebí použít dostatečný počet tkáňových replikátů. U kapalných i u pevných látek je zapotřebí nanášet dostatečné množství zkušební látky, aby rovnoměrně pokryla povrch kůže, a současně se vyhnout nekonečné dávce (viz bod 1.2 Definice), tj. minimálně by se mělo použít 25 μl/cm2 nebo 25 mg/cm2. U pevných látek by měl být povrch epidermis před nanesením zvlhčen deionizovanou nebo destilovanou vodou, aby se zajistil dobrý kontakt s kůží. Pevné látky je zapotřebí zkoušet ve formě jemného prášku, bude-li to možné. Na konci expoziční lhůty je nutné zkušební látku pečlivě spláchnout z povrchu kůže vodným pufrem nebo roztokem 0,9 % NaCl. V závislosti na použitém modelu rekonstruované lidské epidermis se může doba expozice měnit od 15 do 60 minut a teplota inkubace v rozmezí 20 až 37 °C. Podrobnosti naleznete ve standardních prováděcích postupech pro tyto tři metody (21, 22, 23).
Souběžné NC a pozitivní kontroly (PC) je nutné používat pro každou studii, aby se prokázalo, že životaschopnost (NC), bariérová funkce a výsledná tkáňová citlivost (PC) tkání je v rámci definovaného rozpětí historické přijatelnosti. Navrhovanou látkou PC je 5 % vodný SDS. Navrhovanými látkami NC jsou voda a fosfátový pufrový fyziologický roztok (PBS).
1.6.1.4 Měření životaschopnosti buněk
Nejdůležitějším prvkem zkušebního postupu je to, že se měření životaschopnosti neprovádí okamžitě po expozici působení zkoušených látek, ale po dostatečně dlouhé inkubační době po ošetření opláchnutých tkání v čerstvém médiu. Toto období umožňuje jak regeneraci po slabých dráždivých účincích, tak i po vzniku jasných cytotoxických účinků. Během optimalizační fáze zkoušky (9, 10, 11, 12, 13) se jako optimální ukázala 42hodinová inkubační doba po ošetření, a byla proto použita při validaci referenčních zkušebních metod.
Kvantitativní analýza konverze MTT je kvantitativní validovaná metoda, kterou je nutné používat k měření životaschopnosti buněk. Je kompatibilní s použitím soustavy trojrozměrné tkáně. Vzorek kůže se umístí do roztoku MTT o vhodné koncentraci (např. 0,3 až 1 mg/ml) na 3 hodiny. Vysrážený modrý formazan se poté extrahuje z tkáně pomocí rozpouštědla (např. isopropylalkohol, kyselý isopropylalkohol) a koncentrace formazanu se měří stanovením OD při 570 nm pomocí pásmové propustnosti maximálně ±30 nm.
Optické vlastnosti zkoušené látky nebo její chemické působení na MTT může interferovat s kvantitativní analýzou, což vede k nepravdivému odhadu životaschopnosti (protože zkoušená látka může bránit tvorbě barvy nebo ji zpětně převracet a stejně tak ji i způsobovat). K tomu může docházet v případě, že se specifická zkoušená látka zcela neodstraní z kůže oplachováním, nebo v případě, že pronikne přes epidermis. Jestliže zkoušená látka působí přímo na MTT, má přírodní zabarvení nebo se zabarví během ošetřování tkáně, pak jsou zapotřebí ke zjištění interference zkušební látky s technikou měření životaschopnosti a k její korekci dodatečné kontroly. Podrobný popis postupu testování přímé redukce MTT naleznete v protokolu zkušební metody pro validované referenční metody (21, 22, 23). Nespecifická barva (NSC) způsobená těmito interferencemi by neměla překročit 30 % NC (pro korekce). Jestliže je NSC > 30 %, zkoušená látka bude považována za látku se zkouškou nekompatibilní.
1.6.1.5 Kritéria přijatelnosti kvantitativní analýzy
U každé kvantitativní analýzy využívající platných šarží (viz bod 1.6.1.2.5) je zapotřebí, aby tkáně ošetřené NC vykazovaly OD odrážející kvalitu tkání, která vznikla po všech přepravních a přejímacích krocích a celém procesu dráždění dle protokolu. Hodnoty OD kontrol by neměly být pod historicky zjištěnými dolními hranicemi. Podobně by tkáně ošetřené PC, tj. 5 % vodným SDS, měly reflektovat citlivost, kterou si tkáně uchovaly, a jejich schopnost reagovat na dráždivou látku v podmínkách každé individuální kvantitativní analýzy (např. životaschopnost ≤ 40 % pro validovanou referenční metodu 1 a ≤ 20 % pro validovanou referenční metodu 2). Je nutno definovat související a vhodná měření variability mezi tkáňovými replikáty (např. pokud se používají standardní odchylky, měly by být ≤ 18 %).
2. ÚDAJE
2.1 ÚDAJE
Pro každou léčbu by údaje z individuálních vzorků zkoušek replikátu (např. hodnoty OD a údaje vypočítané procentuální životaschopnosti pro každou zkoušenou látku včetně klasifikace) měly být uváděny ve formě tabulky včetně údajů z opakovaných experimentů v případě potřeby. Navíc je pro každý pokus zapotřebí udávat průměry ± standardní odchylky. Pozorované interakce s činidlem MTT a barevnými zkoušenými látkami je nutno uvádět pro každou zkoušenou látku.
2.2 VÝKLAD VÝSLEDKŮ
Hodnoty OD získané s každým zkušebním vzorkem lze použít pro výpočet procentuálního podílu životaschopnosti v porovnání s NC, která je stanovena na 100 %. Je zapotřebí jasně definovat, dokumentovat a v případě potřeby prokázat hodnotu hraniční meze („cut-off“) procentuálního podílu životaschopnosti buněk odlišujícího dráždivou látku od neklasifikovaných zkoušených látek a statistický postup (statistické postupy) použité k hodnocení výsledků a identifikaci dráždivých látek. Mezní hodnoty pro předpověď dráždění spojené s validovanými referenčními hodnotami jsou uvedeny níže:
Zkoušená látka je považována za dráždivou pro kůži v souladu s kategorií 2 GHS OSN:
|
i) |
jestliže je životaschopnost tkáně po expozici a inkubaci po ošetření menší nebo rovna (≤) 50 %. |
Zkoušená látka se považuje za nezařazenou do žádné kategorie:
|
ii) |
jestliže je životaschopnost tkáně po expozici a inkubaci po ošetření větší (>) 50 %. |
3. ZPRÁVY
3.1 PROTOKOL O ZKOUŠCE
Protokol o zkoušce má pokud možno obsahovat následující informace:
Zkoušená a kontrolní látka:
|
— |
chemický název (názvy), např. dle IUPAC nebo název CAS a číslo CAS, jsou-li známy, |
|
— |
čistota a složení látky (v hmotnostních procentech), |
|
— |
fyzikálně-chemické vlastnosti významné pro provádění studie (např. skupenství, stabilita a těkavost, pH, rozpustnost ve vodě, pokud je známa), |
|
— |
případné ošetření zkoušených/kontrolních látek před zkouškou (např. zahřívání, mletí), |
|
— |
podmínky uchovávání. |
Odůvodnění modelu kůže a použitého protokolu.
Zkušební podmínky:
|
— |
použitý buněčný systém, |
|
— |
informace o kalibraci měřicího zařízení a pásma propustnosti použitého pro měření životaschopnosti buněk (např. spektrofotometr), |
|
— |
úplné podpůrné informace pro specifický použitý model kůže včetně jeho chování. Ten by měl zejména zahrnovat:
|
|
— |
podrobnosti použitého zkušebního postupu, |
|
— |
použité zkušební dávky, doba expozice a doba inkubace po ošetření, |
|
— |
popis jakýchkoli úprav zkušebního postupu, |
|
— |
odkaz na historické údaje modelu. Ten by měl zejména zahrnovat:
|
|
— |
popis použitých hodnotících kritérií včetně odůvodnění výběru bodu (bodů) hraniční meze pro model předpovědí. |
Výsledky:
|
— |
údaje z individuálních zkušebních vzorků ve formě tabulky, |
|
— |
popis ostatních pozorovaných účinků. |
Diskuse o výsledcích.
Závěry.
4. LITERATURA
|
(1) |
Organizace spojených národů (OSN) (2007). Globálně harmonizovaný systém klasifikace a označování chemických látek Organizace spojených národů (OSN) (GHS), OSN New York a Ženeva, druhé revidované vydání, 2007. K dispozici na adrese: http://www.unece.org/trans/danger/publi/ghs/ghs_rev02/02files_e.html. |
|
(2) |
REACH: Pokyn pro požadavky na informace a hodnocení chemické bezpečnosti. K dispozici na adrese: http://guidance.echa.europa.eu/docs/guidance_document/information_requirements_en.htm?time=1232447649. |
|
(3) |
Zkušební metoda B.4. AKUTNÍ TOXICITA, DRÁŽDIVÉ A LEPTAVÉ ÚČINKY NA KŮŽI. |
|
(4) |
Zkušební metoda B.40. LEPTAVÉ ÚČINKY NA KŮŽI IN VITRO: ZKOUŠKA TRANSKUTÁNNÍHO ELEKTRICKÉHO ODPORU (TER). |
|
(5) |
Zkušební metoda B.40a. LEPTAVÉ ÚČINKY NA KŮŽI IN VITRO: ZKOUŠKA POMOCÍ MODELU LIDSKÉ KŮŽE. |
|
(6) |
OECD (2006). Zkušební pokyn 435. Pokyn OECD ke zkoušení chemikálií. Metoda membránového bariérového testu in vitro. Schváleno 19. července 2006. K dispozici na adrese: http://www.oecd.org/document/22/0,2340,en_2649_34377_1916054_1_1_1_1,00.html. |
|
(7) |
ECVAM (2009) Performance Standards for applying human skin models to in vitro skin irritation. K dispozici jako studijní dokumenty ke stažení (Download Study Documents) na adrese: http://ecvam.jrc.ec.europa.eu. |
|
(8) |
Fentem, J.H., Briggs, D., Chesné, C., Elliot, G.R., Harbell, J.W., Heylings, J.R., Portes, P., Roguet, R., van de Sandt, J.J.M. & Botham, P. (2001). A prevalidation study on in vitro tests for acute skin irritation. Results and evaluation by the Management Team. Toxicology in Vitro 15, 57–93. |
|
(9) |
Portes, P., Grandidier, M.H., Cohen, C. & Roguet, R.(2002). Refinement of the EPISKIN protocol for the assessment of acute skin irritation of chemicals: follow-up to the ECVAM prevalidation study. Toxicology in Vitro 16, 765–770. |
|
(10) |
Kandárová, H., Liebsch, M., Genschow, E., Gerner, I., Traue, D., Slawik, B. & Spielmann, H. (2004). Optimisation of the EpiDerm test protocol for the upcoming ECVAM validation study on in vitro skin irritation tests. ALTEX 21, 107–114. |
|
(11) |
Kandárová, H., Liebsch, M., Gerner, I., Schmidt, E., Genschow, E., Traue, D. & Spielmann H. (2005) The EpiDerm Test Protocol fort the Upcoming ECVAM Validation Study on In Vitro Skin Irritation Tests – An Assessment of the Performance of the Optimised Test. ATLA 33, 351–367. |
|
(12) |
Cotovio, J., Grandidier, M.–H., Portes, P., Roguet, R. & G. Rubinsteen. (2005). The In Vitro Acute Skin Irritation of Chemicals: Optimisation of the EPISKIN Prediction Model within the Framework of the ECVAM Validation Process. ATLA 33, 329–249. |
|
(13) |
Zuang, V., Balls, M., Botham, P.A., Coquette, A., Corsini, E., Curren, R.D., Elliot, G.R., Fentem, J.H., Heylings, J.R., Liebsch, M., Medina, J., Roguet, R., van De Sandt, J.J.M., Wiemann, C. & Worth, A.(2002). Follow-up to the ECVAM prevalidation study on in vitro tests for acute skin irritation. ECVAM Skin Irritation Task Force Report 2. ATLA 30,109–129. |
|
(14) |
Spielmann, H., Hoffmann, S., Liebsch, M., Botham, P., Fentem, J., Eskes, C., Roguet, R., Cotovió, J., Cole, T., Worth, A., Heylings, J., Jones, P., Robles, C., Kandárová, H., Gamer, A., Remmele, M., Curren, R., Raabe, H., Cockshott, A., Gerner, I. and Zuang, V. (2007) The ECVAM International Validation Study on In Vitro Tests for Acute Skin Irritation: Report on the Validity of the EPISKIN and EpiDerm Assays and on the Skin Integrity Function Test. ATLA 35, 559–601. |
|
(15) |
Hoffmann, S. (2006). ECVAM Skin Irritation Validation Study Phase II: Analysis of the Primary Endpoint MTT and the Secondary Endpoint IL1-α. 135 pp. + annexes. K dispozici jako studijní dokumenty ke stažení (Download Study Documents) na adrese: http://ecvam.jrc.ec.europa.eu. |
|
(16) |
Eskes, C., Cole, T., Hoffmann, S., Worth, A., Cockshott, A., Gerner, I. & Zuang. V (2007) ECVAM International Validation Study on In Vitro Tests for Acute Skin Irritation: Selection of Test Chemicals. ATLA 35, 603–619. |
|
(17) |
J. Cotovio, M.–H. Grandidier, D. Lelièvre, R. Roguet, E. Tinois-Tessonneaud, J. Leclaire (2007). In vitro acute skin irritancy of chemicals using the validated EPISKIN model in a tiered strategy – Results and performances with 184 cosmetic ingredients, AATEX, Special Issue-proceedings from WC6. Vol. 14, 351–358. |
|
(18) |
Prohlášení ESAC o aktualizovaných analýzách EpiDerm a podobných analýzách SkinEthic. 5. listopad 2008. |
|
(19) |
ES (2006). Nařízení Evropského parlamentu a Rady (ES) č. 1907/2006 ze dne 18. prosince 2006 o registraci, hodnocení, povolování a omezování chemických látek, o zřízení Evropské agentury pro chemické látky, o změně směrnice 1999/45/ES a o zrušení nařízení Rady (EHS) č. 793/93, nařízení Komise (ES) č. 1488/94, směrnice Rady 76/769/EHS a směrnic Komise 91/155/EHS, 93/67/EHS, 93/105/ES a 2000/21/ES. Úřední věstník Evropské unie, L 396/1, 30.12.2006. OPOCE, Lucemburk. |
|
(20) |
Mosman, T. (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Journal of Immunological Methods 65, 55–63. |
|
(21) |
EpiSkin™ SOP, Version 1.6 (January 2005). Validation of the EpiSkin Skin Irritation Test – 42 Hours Assay For The Prediction of Acute Skin Irritation of Chemicals. K dispozici jako studijní dokumenty ke stažení (Download Study Documents) na adrese: http://ecvam.jrc.ec.europa.eu. |
|
(22) |
EpiDerm™ SOP, Version 5.0 (October 2004). Draft Standard Operating Procedure. In Vitro Skin Irritation Test: Human Skin Model. Model: EpiDerm™- 200. K dispozici jako studijní dokumenty ke stažení (Download Study Documents) na adrese: http://ecvam.jrc.ec.europa.eu. |
|
(23) |
SkinEthic RHE™ SOP. K dispozici jako studijní dokumenty ke stažení (Download Study Documents) na adrese: http://ecvam.jrc.ec.europa.eu. |
|
(24) |
Harvell, J.D., Lammintausta, K., Maibach H.I. (1995) Irritant contact dermatitis IN: Guin J.D. Practical Contact Dermatitis Mc Graw-Hill New York, pp 7–18. |
|
(25) |
Griesinger C, Barroso J & Zuang V: ECVAM background document on the recent adaptations of the ECVAM performance standards for in vitro skin irritation testing in the context of the drafting process of an EU test method and an OECD draft test guideline. Ispra, 13. listopad 2008. |
Dodatek
Hodnocení charakteristik chování navrhovaných modelů rekonstruované lidské epidermis in vitro pro dráždění kůže
ÚVOD
Navrhované postupy pro použití v rámci této zkušební metody je nutné vyhodnotit s cílem stanovit jejich spolehlivost a přesnost pomocí látek zastupujících plný rozsah Draizeho škály dráždivosti. Při vyhodnocení pomocí 20 doporučených referenčních látek (tabulka 2) by navrhované postupy měly mít hodnoty spolehlivosti a přesnosti, které jsou srovnatelné s hodnotami validované referenční metody 1 (tabulka 3) (1). Standardy přesnosti a spolehlivosti, kterých by mělo být dosaženo, jsou uvedeny v částech II a III níže. Neklasifikované a klasifikované látky (kategorie 2 GHS OSN), představující platné chemické třídy, jsou zahrnuty s tím, že spolehlivost a chování (citlivost, specifičnost, míry falešné negativity a pozitivity a přesnost) navrhované zkušební metody lze srovnávat se spolehlivostí a dalšími parametry validované referenční metody 1. Spolehlivost zkušební metody společně s její schopností správně identifikovat dráždivé látky kategorie 2 GHS OSN by měla být stanovena před jejím použitím ke zkoušení nových látek.
STANDARDY CHOVÁNÍ
Standardy chování zahrnují následující tři prvky I) zásadní složky zkušební metody, II) referenční látky a III) definované hodnoty přesnosti a spolehlivosti (2). Tyto standardy chování jsou založeny na standardech chování definovaných po dokončení validační studie dráždění kůže ECVAM (3).
I) Zásadní složky zkušební metody
Všeobecné podmínky modelu
K sestavení epitelu je zapotřebí použít normální lidské keratinocyty. Pod funkčním stratum corneum by měly být přítomny vícenásobné vrstvy životaschopných epitelových buněk (bazální vrstva, stratum spinosum, stratum granulosum). Stratum corneum by mělo být vícevrstevnaté a obsahovat profil esenciálních lipidů k vytvoření funkční bariéry robustní natolik, aby vzdorovala rychlému průniku cytotoxických látek markeru, např. SDS nebo přípravku Triton X-100. Funkci bariéry lze hodnotit buď stanovením koncentrace, při níž látka markeru snižuje životaschopnost tkání o 50 % (IC50) po pevně stanovené expoziční době, nebo stanovením expoziční doby požadované ke snížení životaschopnosti buněk o 50 % (ET50) po nasazení látky markeru ve specifikované pevně stanovené koncentraci. Zadržovací vlastnosti modelu by měly zabránit průchodu materiálu v oblasti stratum corneum do životaschopné tkáně, což by způsobilo špatné modelování expozice kůže. Model kůže by neměl být kontaminován bakteriemi, viry, mykoplazmou ani mykózami.
Funkční podmínky modelu
Životaschopnost
MTT (4) je upřednostňovanou kvantitativní analýzou pro stanovení míry životaschopnosti. OD extrahovaného (rozpustitelného) barviva z tkáně ošetřené NC by měla být nejméně dvacetinásobně větší než OD extrakčního rozpouštědla samotného. Je nutné prokázat, že tkáň ošetřená NC je stabilní v kultuře (poskytnout podobná měření životaschopnosti) po dobu trvání lhůty zkušební expozice.
Bariérová funkce
Stratum corneum a jeho lipidové složení by mělo dostačovat na to, aby odolávalo rychlému průniku cytotoxických látek markeru, např. SDS nebo Triton X-100, podle odhadu na základě IC50 nebo ET50.
Morfologie
Histologické vyšetření rekonstruované kůže/epidermis by měl provádět vhodně kvalifikovaný personál a mělo by prokázat strukturu podobnou lidské kůži/epidermis (včetně vícevrstevnatého stratum corneum).
Reprodukovatelnost
Výsledky metody používající specifický model by měly prokázat reprodukovatelnost v průběhu času, nejlépe pomocí vhodné kontrolní (srovnávací) látky šarže (viz definice v bodě 1.2).
Kontroly kvality (QC) modelu
Každá šarže použitého epidermálního modelu by měla splňovat definovaná produkční kritéria uvolnění do oběhu, kdy mezi nejvýznamnější patří kritérium životaschopnosti a bariérové funkce. Rozsah přijatelnosti (horní a dolní mez) pro IC50 nebo ET50 je zapotřebí zjistit u dodavatele modelu kůže (nebo experimentátora, pokud se používá vlastní model). Bariérové vlastnosti tkání je nutno ověřit laboratorně po dodání tkání. Pro spolehlivou předpověď dráždivých účinků mohou být přijatelné pouze výsledky vyprodukované pomocí náležitých tkání. Jako příklad jsou dále uvedeny rozsahy přijatelnosti pro validované referenční metody.
Tabulka 1
Příklady kritérií kontroly kvality pro uvolnění šarže do oběhu
|
|
Dolní mez přijatelnosti |
Průměr rozsahu přijatelnosti |
Horní mez přijatelnosti |
|
Validovaná referenční metoda 1 (18 hodin ošetření SDS) |
IC50 = 1,0 mg/ml |
IC50 = 2,32 mg/ml |
IC50 = 3,0 mg/ml |
|
Validovaná referenční metoda 2 (1 % Triton X100) |
ET50 = 4,8 h |
ET50 = 6,7 h |
ET50 = 8,7 h |
II) Referenční látky
Referenční látky slouží k rozhodnutí o tom, zda spolehlivost a přesnost navrhované originální zkušební metody založené na rekonstruované lidské epidermis in vitro, kdy se prokázalo, že je strukturálně a funkčně dostatečně podobná validovaným referenčním metodám, nebo kdy představuje drobnou úpravu validované referenční metody, vykazuje srovnatelné chování s chováním validované referenční metody 1 (1). 20 referenčních látek uvedených v tabulce 2 zahrnuje látky představující různé příslušné chemické třídy a zároveň i látky kategorie 2 GHS OSN. Látky uvedené v tomto seznamu zahrnují 10 látek kategorie 2 GHS OSN, 3 látky volitelné kategorie 3 GHS OSN a 7 nekategorizovaných látek. Podle této zkušební metody se volitelná kategorie 3 nepovažuje za kategorii. Tyto referenční látky představují minimální počet látek, které je zapotřebí použít pro vyhodnocení přesnosti a spolehlivosti navrhované zkušební metody pro dráždění kůže založené na rekonstruované lidské epidermis. V situacích, kdy látka na seznamu není k dispozici, lze použít jiné látky, pro něž jsou k dispozici odpovídající referenční údaje in vivo. Bude-li to žádoucí, lze k minimálnímu seznamu referenčních látek přidat dodatečné látky zastupující další chemické třídy, pro něž jsou k dispozici odpovídající referenční údaje in vivo, pro další hodnocení přesnosti navrhované zkušební metody.
Tabulka 2
Referenční látky pro stanovení hodnot přesnosti a spolehlivosti pro modely dráždění kůže založené na rekonstruované lidské epidermis
|
Látka (1) |
Číslo CAS |
Číslo EINECS |
Skupenství |
Skóre in vivo |
Kategorie GHS in vitro |
Kategorie GHS in vivo |
|
1-brom-4-chlorbutan |
6940–78–9 |
230–089–3 |
kapalné |
0 |
kategorie 2 |
žádná kategorie |
|
diethyl-ftalát |
84–66–2 |
201–550–6 |
kapalné |
0 |
žádná kategorie |
žádná kategorie |
|
kyselina naftalen-1-octová |
86–87–3 |
201–705–8 |
pevné |
0 |
žádná kategorie |
žádná kategorie |
|
allyl-fenoxyacetát |
7493–74–5 |
231–335–2 |
kapalné |
0,3 |
žádná kategorie |
žádná kategorie |
|
isopropylalkohol |
67–63–0 |
200–661–7 |
kapalné |
0,3 |
žádná kategorie |
žádná kategorie |
|
4-(methylthio)benzaldehyd |
3446–89–7 |
222–365–7 |
kapalné |
1 |
kategorie 2 |
žádná kategorie |
|
methyl-stearát |
112–61–8 |
203–990–4 |
pevné |
1 |
žádná kategorie |
žádná kategorie |
|
heptyl-butyrát |
5870–93–9 |
227–526–5 |
kapalné |
1,7 |
žádná kategorie |
volitelná kategorie 3 |
|
hexyl-salicylát |
6259–76–3 |
228–408–6 |
kapalné |
2 |
žádná kategorie |
volitelná kategorie 3 |
|
triisobutyl-fosfát |
126–71–6 |
204–798–3 |
kapalné |
2 |
kategorie 2 |
volitelná kategorie 3 |
|
dekan-1-ol |
112–30–1 |
203–956–9 |
kapalné |
2,3 |
kategorie 2 |
kategorie 2 |
|
cyklamenaldehyd |
103–95–7 |
203–161–7 |
kapalné |
2,3 |
kategorie 2 |
kategorie 2 |
|
1-bromhexan |
111–25–1 |
203–850–2 |
kapalné |
2,7 |
kategorie 2 |
kategorie 2 |
|
2-(chlormethyl)-4-methoxy-3,5-dimethylpyridin-hydrochlorid |
86604–75–3 |
434–680–9 |
pevné |
2,7 |
kategorie 2 |
kategorie 2 |
|
α-terpineol |
98–55–5 |
202–680–6 |
kapalné |
2,7 |
kategorie 2 |
kategorie 2 |
|
dipropyldisulfid |
629–19–6 |
211–079–8 |
kapalné |
3 |
žádná kategorie |
kategorie 2 |
|
butyl-methakrylát |
97–88–1 |
202–615–1 |
kapalné |
3 |
kategorie 2 |
kategorie 2 |
|
5-terc-butyl-2-methylbenzen-1-thiol |
7340–90–1 |
438–520–9 |
kapalné |
3,3 |
kategorie 2 |
kategorie 2 |
|
3-fenyl-1-methylpiperazin |
5271–27–2 |
431–180–2 |
pevné |
3,3 |
kategorie 2 |
kategorie 2 |
|
heptanal |
111–71–7 |
203–898–4 |
kapalné |
4 |
kategorie 2 |
kategorie 2 |
Látky uvedené v tabulce 2 poskytují reprezentativní rozdělení 58 látek použitých v mezinárodní validační studii dráždění kůže ECVAM (1). Jejich výběr je založen na následujících kritériích:
|
— |
látky jsou komerčně dostupné, |
|
— |
jsou to zástupci plného rozsahu Draizeho škály dráždivosti (od nedráždivých až po silně dráždivé), |
|
— |
mají dobře definovanou chemickou strukturu, |
|
— |
jsou představitelem reprodukovatelnosti a předpovědní schopnosti validované metody, jak je určuje validační studie ECVAM, |
|
— |
jsou představitelem chemické funkčnosti používané ve validačním procesu, |
|
— |
nejsou spojeny s mimořádně toxickým profilem (např. karcinogenní nebo toxický pro reprodukční systém) a nejsou spojeny s neúnosnými náklady na likvidaci. |
III) Definované hodnoty přesnosti a spolehlivosti
Chování (citlivost, specifičnost, míra falešné negativity, míra falešné pozitivity a přesnost) navrhované zkušební metody by mělo být srovnatelné s parametry validované referenční metody 1 (tabulka 3), tj. citlivost by měla být rovna nebo vyšší (≥) než 80 %, specifičnost by měla být rovna nebo vyšší (≥) než 70 % a přesnost by měla být rovna nebo vyšší (≥) než 75 %. Výpočet daného chování je nutno provést s použitím všech klasifikací získaných pro 20 látek v různých účastnických laboratořích. Klasifikace pro každou látku v každé laboratoři by se měla získat pomocí průměrné hodnoty životaschopnosti v rámci různých prováděných zkoušek (minimálně tři platné zkoušky).
Tabulka 3
Chování validované referenční metody 1 (2)
|
Zkušební metoda |
Počet látek |
Citlivost |
Specifičnost |
Míra falešné negativity |
Míra falešné pozitivity |
Přesnost |
|
Validovaná referenční metoda 1 (3) |
58 |
87,2 % (4) |
71,1 % (5) |
12,8 % |
29,9 % |
74,7 % |
|
Validovaná referenční metoda 1 (3) |
20 |
90 % |
73,3 % |
10 % |
26,7 % |
81,7 % |
Spolehlivost navrhované zkušební metody by měla být srovnatelná se spolehlivostí validovaných referenčních metod.
Vnitrolaboratorní reprodukovatelnost
Hodnocení vnitrolaboratorní variability by mělo ukázat shodu klasifikací (kategorie 2/žádná kategorie) získaných v různých nezávislých zkouškách 20 referenčních látek v rámci jedné jediné laboratoře rovnou nebo vyšší (≥) než 90 %.
Mezilaboratorní reprodukovatelnost
Hodnocení mezilaboratorní reprodukovatelnosti není zásadně důležité, jestliže se navrhovaná zkušební metoda má používat pouze v jediné laboratoři. U metod, které se mají přenášet mezi laboratořemi, by shoda klasifikací (kategorie 2/žádná kategorie) získaná v různých nezávislých zkouškách 20 referenčních látek mezi nejlépe minimálně třemi laboratořemi měla být rovna nebo vyšší (≥) než 80 %.
LITERATURA
|
(1) |
Spielmann, H., Hoffmann, S., Liebsch, M., Botham, P., Fentem, J., Eskes, C., Roguet, R., Cotovió, J., Cole, T., Worth, A., Heylings, J., Jones, P., Robles, C., Kandárová, H., Gamer, A., Remmele, M., Curren, R., Raabe, H., Cockshott, A., Gerner, I. and Zuang, V. (2007) The ECVAM International Validation Study on In Vitro Tests for Acute Skin Irritation: Report on the Validity of the EPISKIN and EpiDerm Assays and on the Skin Integrity Function Test. ATLA 35, 559–601. |
|
(2) |
Dokument pokynu č. 34 OECD (2005) o hodnocení a mezinárodním přijetí nových či aktualizovaných zkušebních metod pro hodnocení nebezpečnosti. OECD, Paříž. |
|
(3) |
ECVAM (2007) Performance Standards for applying human skin models to in vitro skin irritation. K dispozici jako studijní dokumenty ke stažení (Download Study Documents) na adrese http://ecvam.jrc.ec.europa.eu. Vstup na stránky uskutečněn dne 27. října 2008. |
|
(4) |
Mosman, T. (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Journal of Immunological Methods 65, 55–63. |
|
(5) |
Eskes, C., Cole, T., Hoffmann, S., Worth, A., Cockshott, A., Gerner, I. & Zuang. V (2007) ECVAM International Validation Study on In Vitro Tests for Acute Skin Irritation: Selection of Test Chemicals. ATLA 35, 603–619. |
(1) Těchto 20 referenčních látek zahrnuje reprezentativní výběr z 58 látek, které byly původně použity pro validaci referenční metody 1 (EpiSkin™). Je k dispozici úplný seznam zkoušených látek a kritéria pro jejich výběr (5).
(2) Tabulka 3 uvádí chování validované referenční metody 1 s ohledem na vlastní schopnost správně identifikovat dráždivé látky (kategorie 2 GHS OSN) a neklasifikované látky (žádná kategorie včetně volitelné kategorie 3) pro 58 a 20 referenčních látek v prvním a druhém případě (tabulka 2).
(3) EpiSkin™.
(4) Na základě 13 dráždivých látek kategorie 2 GHS.
(5) Na základě 45 dráždivých látek kategorie 3 GHS nebo látek nezařazených do žádné kategorie GHS.
PŘÍLOHA IV
|
C.3 |
ZKOUŠKA INHIBICE RŮSTU SLADKOVODNÍCH ŘAS A SINIC |
1. METODA
Tato metoda je rovnocenná Pokynu OECD pro zkoušení 201 (2006) (1).
1.1 ÚVOD
Zkušební metody se pravidelně přezkoumávají a aktualizují s ohledem na vědecký pokrok. Zkušební metoda C.3 musela být přepracována tak, aby zahrnovala další druhy a splňovala požadavky na hodnocení nebezpečnosti a klasifikace chemických látek. Přepracování bylo dokončeno na základě rozsáhlých praktických zkušeností, vědeckého pokroku na poli studií toxicity řas a širokého uplatňování příslušných právních předpisů od přijetí původního znění.
1.2 DEFINICE
Pro účely této zkušební metody se používají následující definice a zkratky:
Biomasa: je suchá hmotnost živé hmoty přítomné v populaci vyjádřená ve vztahu k danému objemu; např. mg řas/litr zkušebního roztoku. „Biomasa“ je obvykle definována jako hmota, ale v rámci této zkoušky se toto slovo používá pro hmotu vztaženou na objem. V této zkoušce se obvykle měří náhrady biomasy, například počet buněk, fluorescence atd., a používání termínu „biomasa“ tedy odkazuje na tyto náhradní míry.
Variační koeficient: je bezrozměrná veličina proměnlivosti parametru, definovaná jako poměr směrodatné odchylky a střední hodnoty. Lze jej rovněž vyjádřit jako procentuální údaj. Střední variační koeficient průměrné specifické růstové rychlosti v kontrolních kulturách použitých k opakování se vypočítává následovně:
|
1) |
Vypočítejte % variační koeficient průměrné specifické růstové rychlosti z denních růstových rychlostí nebo růstových rychlostí po jednotlivých časových úsecích pro příslušné opakování. |
|
2) |
Vypočítejte střední hodnotu ze všech hodnot vypočítaných v bodě 1 pro získání středního variačního koeficientu denní specifické růstové rychlosti nebo specifické růstové rychlosti po jednotlivých časových úsecích v kontrolních kulturách použitých k opakování. |
ECx : je koncentrace zkoušené látky rozpuštěné ve zkušebním médiu, která pro danou expoziční dobu vede k x% (např. 50 %) snížení růstu testovacího organismu (musí se výslovně uvést, pokud se odchyluje od plné či normální doby trvání zkoušky). Pro jednoznačný popis hodnoty EC odvozené z růstové rychlosti nebo z výtěžku se v příslušných případech používají symboly „ErC“ a „EyC“.
Růstové médium: je úplné syntetické kultivační médium, v němž rostou zkušební řasy při expozici zkoušené látce. Zkoušená látka bude za normálních podmínek ve zkušebním médiu rozpuštěna.
Růstová rychlost (průměrná specifická růstová rychlost): je logaritmické zvýšení biomasy během období expozice.
Nejnižší koncentrace s pozorovanými účinky (Lowest Observed Effect Concentration, LOEC): je nejnižší zkušební koncentrací, při níž je pro danou expoziční dobu pozorován statisticky významný účinek látky na snížení růstu (na hladině spolehlivosti p < 0,05) ve srovnání s kontrolou. Všechny zkušební koncentrace vyšší než LOEC musí však mít stejné nebo vážnější škodlivé účinky, než jsou účinky pozorované při koncentraci LOEC. Nelze-li tyto dvě podmínky splnit, musí být podrobně vysvětleno, jak byla LOEC (a tedy i NOEC) zvolena.
Koncentrace bez pozorovaných účinků (No Observed Effect Concentration, NOEC): je zkušební koncentrace bezprostředně nižší než LOEC.
Proměnná odezvy: je proměnná pro odhad toxicity odvozená z jakýchkoliv naměřených parametrů, jež popisují biomasu, různými metodami výpočtu. U této metody jsou proměnnými odezvy růstová rychlost a výtěžek, které se odvozují přímo z měření biomasy nebo jakékoliv z uvedených náhrad.
Specifická růstová rychlost: je proměnná odezvy definovaná jako kvocient rozdílu přirozených logaritmů sledovaného parametru (v této zkušební metodě je jím biomasa) a příslušného časového období.
Výtěžek: je hodnota proměnné měření na konci expoziční doby minus hodnota proměnné měření na počátku expoziční doby pro vyjádření nárůstu biomasy během zkoušky.
1.3 POUŽITELNOST ZKUŠEBNÍ METODY
Tato zkušební metoda se nejsnadněji používá pro látky rozpustné ve vodě, které za podmínek zkoušky pravděpodobně ve vodě zůstanou. Pro zkoušení látek, které jsou těkavé, silně adsorbují, jsou zbarvené, mají nízkou rozpustnost ve vodě, nebo látek, které mohou nepříznivě ovlivňovat dostupnost živin či minerálů ve zkušebním médiu, mohou být potřebné určité úpravy popsaného postupu (např. uzavřený systém, klimatizace zkušebních nádob). Pokyny k některým vhodným úpravám jsou uvedeny v literatuře (2, 3 a 4).
1.4 PODSTATA ZKUŠEBNÍ METODY
Účelem této zkoušky je stanovit účinek látky na růst sladkovodních mikrořas a/nebo sinic. Exponenciálně rostoucí testovací organismy jsou vystaveny zkoušené látce v dávkových kulturách obvykle po dobu 72 hodin. Navzdory relativně krátké době trvání zkoušky lze posoudit účinky na několik generací.
Systémovou odezvou je snížení růstu v řadě kultur řas (zkušebních jednotek) vystavených různým koncentracím zkoušené látky. Odezva se vyhodnocuje jako funkce expoziční koncentrace v porovnání s průměrným růstem neexponovaných kontrolních kultur použitých k opakování. Pro plné vyjádření systémové odezvy na toxické účinky (optimální citlivost) se kultury ponechají růst exponenciálně bez omezení za dostatečných vyživovacích podmínek a nepřetržitého osvětlení po dostatečnou dobu, aby bylo možné změřit snížení specifické růstové rychlosti.
Růst a inhibice růstu se kvantifikují měřeními biomasy řas jakožto funkce času. Biomasa řas se definuje jako suchá hmotnost na objem, např. mg řas/litr zkušebního roztoku. Ovšem suchá hmotnost se měří obtížně, proto se používají náhradní parametry. Z těchto náhrad se nejčastěji používají počty buněk. Mezi další náhradní parametry patří buněčný objem, fluorescence, optická hustota atd. Je třeba znát přepočítávací faktor mezi naměřeným náhradním parametrem a biomasou.
Zkoumaným účinkem je inhibice růstu vyjádřená jako logaritmické zvýšení biomasy (průměrná specifická růstová rychlost) během expoziční doby. Z průměrných specifických růstových rychlostí zaznamenaných v řadě zkušebních roztoků se stanoví koncentrace způsobující stanovenou x% inhibici růstové rychlosti (např. 50 %) a vyjádří se jako ErCx (např. ErC50).
Při použití této metody v rámci práva EU by měl být výpočet výsledků založen na průměrné specifické růstové rychlosti, a to z důvodů uvedených v bodě 2.2. Další proměnnou odezvy použitou v této zkušební metodě je výtěžek, který může být nutný ke splnění specifických právních požadavků v některých zemích. Je definován jako biomasa na konci expoziční doby minus biomasa na počátku expoziční doby. Z výtěžku zaznamenaného v řadě zkušebních roztoků se vypočítá koncentrace způsobující stanovenou x% inhibici výtěžku (např. 50 %) a vyjádří se jako EyCx (např. EyC50).
Kromě toho se může statisticky určit nejnižší koncentrace s pozorovanými účinky (LOEC) a koncentrace bez pozorovaných účinků (NOEC).
1.5 INFORMACE O ZKOUŠENÉ LÁTCE
Užitečnými informacemi o zkoušené látce pro účely stanovení zkušebních podmínek mohou být strukturní vzorec, čistota, stálost na světle, stálost v podmínkách zkoušky, vlastnosti světelné absorpce, pK a a výsledky studií transformace včetně biologické rozložitelnosti ve vodě.
Měly by být známy rozpustnost zkoušené látky ve vodě, její rozdělovací koeficient oktanol/voda (P o/v) a tlak jejích par a měla by být k dispozici validovaná metoda kvantitativního stanovení látky ve zkušebních roztocích, a to s doloženou výtěžností a mezí detekce.
1.6 REFERENČNÍ LÁTKA
Jako prostředek kontroly zkušebního postupu lze zkoušet referenční látku (látky), například 3,5-dichlorfenol použitý v mezinárodní kroužkové zkoušce (4). Dichroman draselný lze rovněž použít jako referenční látku pro zelené řasy. Je žádoucí zkoušet referenční látku nejméně dvakrát ročně.
1.7 VALIDITA ZKOUŠKY
Má-li být zkouška platná, musí být splněna tato kritéria reprodukční schopnosti:
|
— |
Biomasa by se měla v kontrolních kulturách během 72hodinové zkušební doby zvýšit exponenciálně, a to nejméně šestnáctkrát. To odpovídá specifické růstové rychlosti 0,92 den–1. U nejčastěji používaných druhů je obvykle růstová rychlost výrazně vyšší (viz dodatek 1). Může se stát, že se toto kritérium nesplní, pokud se používají druhy, které rostou pomaleji než druhy uvedené na seznamu v dodatku 1. V takovém případě je třeba prodloužit zkušební dobu, aby bylo dosaženo nejméně šestnáctinásobného růstu kontrolních kultur, přičemž růst musí být během zkušebního období exponenciální. Zkušební dobu lze zkrátit až na minimální dobu 48 hodin, aby se během zkoušky udržel neomezený exponenciální růst, pokud bude dosaženo minimálního násobícího koeficientu 16. |
|
— |
Střední variační koeficient pro specifické růstové rychlosti v jednotlivých obdobích (dny 0–1, 1–2 a 2–3 při 72hodinové zkoušce) v kontrolních kulturách (viz bod 1.2 odstavec „variační koeficient“) nesmí překročit 35 %. Pro výpočet specifické růstové rychlosti v jednotlivých obdobích viz bod 2.2.1 druhý odstavec. Toto kritérium platí pro střední hodnotu variačních koeficientů vypočítaných pro kontrolní kultury použité k opakování. |
|
— |
Variační koeficient průměrných specifických růstových rychlostí během celého zkušebního období u kontrolních kultur použitých k opakování nesmí překročit 7 % u zkoušek s Pseudokirchneriella subcapitata a Desmodesmus subspicatus. U jiných méně často zkoušených druhů by hodnota neměla překročit 10 %. |
1.8 POPIS METODY
1.8.1 Přístroje a pomůcky
Zkušební nádoby a jiné přístroje a pomůcky, které přijdou do styku se zkušebními roztoky, by měly být celoskleněné nebo z jiného chemicky inertního materiálu. Jednotlivé díly je zapotřebí důkladně vymýt, aby bylo zajištěno, že žádné organické ani anorganické nečistoty nebudou moci narušovat růst řas nebo složení zkušebních roztoků.
Zkušebními nádobami budou za normálních podmínek skleněné baňky takových rozměrů, aby pojaly dostatečný objem kultury pro měření během zkoušky a umožnily dostatečný hmotnostní přenos CO2 z atmosféry (viz bod 1.8.9 druhý odstavec). Je třeba mít na paměti, že objem kapaliny musí být dostačující pro analytická stanovení (viz bod 1.8.11 pátý odstavec).
Kromě toho bude potřebné veškeré následující vybavení nebo jeho část:
|
— |
Kultivační zařízení: doporučuje se místnost nebo komora, v níž lze udržovat zvolenou inkubační teplotu na ±2 °C. |
|
— |
Přístroje na měření světla: je důležité mít na paměti, že metoda měření intenzity světla a zejména typ snímače (kolektoru) ovlivní naměřenou hodnotu. Měření by se mělo nejlépe provádět pomocí kulového (4 π) snímače (reaguje na přímé a odražené světlo ze všech úhlů nad a pod rovinou měření), nebo 2 π snímače (reaguje na světlo ze všech úhlů nad rovinou měření). |
|
— |
Zařízení pro stanovení biomasy řas. Počet buněk, který je nejčastěji používaným náhradním parametrem pro biomasu řas, lze určit pomocí elektronického počítače částic, mikroskopu s počítací komůrkou nebo průtokovým cytometrem. Další náhrady biomasy lze měřit průtokovým cytometrem, fluorimetrem, spektrofotometrem nebo kolorimetrem. Je užitečné vypočítat faktor pro konverzi počtu buněk na suchou hmotnost. Aby bylo možné zajistit užitečná měření při nízkých koncentracích biomasy při použití spektrofotometru, může vzniknout nutnost používat kyvety s dráhou světla nejméně 4 cm. |
1.8.2 Testovací organismy
Lze použít několik druhů nevázaných mikrořas a sinic. Kmeny uvedené v dodatku 1 se ukázaly jako vhodné na základě zkušebního postupu uvedeného v této zkušební metodě.
Jestliže se použijí jiné druhy, je třeba uvést ve zprávě kmen a/nebo původ. Musí se potvrdit, že lze udržovat exponenciální růst vybraných zkušebních řas po zkušební dobu za převažujících podmínek.
1.8.3 Růstové médium
Doporučují se dvě alternativní růstová média, médium OECD a AAP. Složení těchto médií je uvedeno v dodatku 2. Je třeba mít na paměti, že počáteční hodnota pH a kapacita pufru (regulující zvýšení pH) těchto dvou médií jsou odlišné. Proto mohou být výsledky zkoušek odlišné v závislosti na použitém médiu, zejména při zkoušení ionizujících látek.
Pro určité účely může být nezbytná úprava růstových médií, např. když se zkouší kovy a chelátotvorná činidla nebo se provádí zkoušky při různých hodnotách pH. Použití upraveného média musí být podrobně popsáno a zdůvodněno (3, 4).
1.8.4 Počáteční koncentrace biomasy
Počáteční biomasa musí být stejná ve všech zkušebních kulturách a dostatečně nízká, aby umožňovala exponenciální růst po celou inkubační dobu bez rizika vyčerpání živin. Počáteční biomasa by neměla překročit 0,5 mg/l suché hmotnosti. Doporučují se následující počáteční koncentrace buněk:
|
Pseudokirchneriella subcapitata |
5 × 103–104 |
buněk/ml |
|
Desmodesmus subspicatus |
2–5 × 103 |
buněk/ml |
|
Navicula pelliculosa |
104 |
buněk/ml |
|
Anabaena flos-aquae |
104 |
buněk/ml |
|
Synechococcus leopoliensis |
5 × 104–105 |
buněk/ml |
1.8.5 Koncentrace zkoušené látky
Koncentrační rozmezí, ve kterém lze očekávat účinky, lze určit na základě výsledků orientačních zkoušek. Pro závěrečnou definitivní zkoušku je zapotřebí vybrat alespoň pět koncentrací tvořících geometrickou řadu s faktorem nepřekračujícím 3,2. U zkušebních látek vykazujících plochou křivku závislosti odezvy na koncentraci může být opodstatněný vyšší faktor. Řady koncentrací by nejlépe měly pokrývat rozmezí způsobující 5–75 % inhibici růstové rychlosti řas.
1.8.6 Opakování a kontroly
Plán zkoušky by měl zahrnovat tři opakování s každou zkušební koncentrací. Jestliže se stanovení NOEC nepožaduje, plán zkoušky lze pozměnit tak, že se zvýší počet koncentrací a sníží počet opakování na příslušnou koncentraci. Počet kontrolních opakování musí být nejméně tři a v ideálním případě by měl být dvojnásobkem počtu opakování používaných pro každou zkušební koncentraci.
Pro analytická stanovení koncentrací zkušební látky lze připravit samostatnou sadu zkušebních roztoků (viz bod 1.8.11 čtvrtý a šestý odstavec).
Pokud se k rozpouštění zkoušené látky používá rozpouštědlo, musí být do plánu zkoušky zahrnuty dodatečné kontroly obsahující rozpouštědlo o stejné koncentraci, jaká se používá ve zkušebních kulturách.
1.8.7 Příprava očkovací kultury
K přizpůsobení zkušebních řas na podmínky zkoušky a zajištění toho, aby řasy byly v exponenciální růstové fázi, když se použijí pro naočkování zkušebních roztoků, se očkovací kultura připravuje ve zkušebním médiu 2 až 4 dny před zahájením zkoušky. Biomasu řas je třeba upravit, aby mohl v očkovací kultuře převážit do zahájení zkoušky exponenciální růst. Očkovací kultura se musí inkubovat za stejných podmínek jako zkušební kultury. Měřte nárůst biomasy v očkovací kultuře, aby bylo zajištěno, že je růst v rámci normálního rozmezí pro zkušební kmen za podmínek kultivace. Příklad postupu pro kultivaci řas je popsán v dodatku 3. Aby se zabránilo synchronnímu dělení buněk během zkoušky, může být potřebné provést druhý krok propagace očkovací kultury.
1.8.8 Příprava zkušebních roztoků
Všechny zkušební roztoky musí obsahovat stejné koncentrace růstového média a počáteční biomasy zkušebních řas. Zkušební roztoky zvolených koncentrací se obvykle připravují smísením zásobního roztoku zkoušené látky s růstovým médiem a očkovací kulturou. Zásobní roztoky se obyčejně připravují rozpuštěním látky ve zkušebním médiu.
Jako nosiče pro přidávání látek o nízké rozpustnosti ve vodě do zkušebního média mohou být použita rozpouštědla, např. aceton, terc-butylalkohol a dimethylformamid (2, 3). Koncentrace rozpouštědla by neměla překročit 100 µl/l a do všech kultur (včetně kontrol) ve zkušební řadě by měla být přidána stejná koncentrace rozpouštědla.
1.8.9 Inkubace
Zakryjte zkušební nádoby zátkami, které propouští vzduch. Nádoby se protřepou a umístí se do kultivačního zařízení. Během zkoušky je nezbytné udržovat řasy v suspensi a umožnit přenos CO2. Za tímto účelem je třeba provádět nepřetržité třepání nebo míchání. Kultury se udržují při teplotě 21–24 °C udržované s přesností na ±2 °C. U jiných druhů, než jsou druhy uvedené v seznamu v dodatku 1, např. u tropických druhů, mohou být vhodné vyšší teploty, pokud lze ovšem splnit kritéria validity. Doporučuje se umísťovat baňky náhodně a denně je v inkubátoru přemísťovat.
Hodnota pH kontrolního média by neměla během zkoušky vzrůst o více než 1,5. U kovů a sloučenin, které částečně ionizují při pH okolo pH zkoušky může být nezbytné omezit posun pH pro získání reprodukovatelných a dobře definovaných výsledků. Posun o < 0,5 pH je technicky proveditelný a lze jej dosáhnout zajištěním adekvátní rychlosti přesunu hmoty CO2 z okolního vzduchu do zkušebního roztoku, např. zvýšením rychlosti třepání. Další možností je snížit potřebu CO2 snížením počáteční biomasy nebo doby trvání zkoušky.
Povrch, kde jsou kultury inkubovány, by měl být trvale osvětlen jednotným fluorescenčním světlem, např. „chladným bílým“ nebo „denním“ světlem. Jednotlivé kmeny řas a sinic mají různé požadavky na osvětlení. Intenzita světla by měla být zvolena tak, aby vyhovovala použitému testovacímu organismu. Pro doporučené druhy zelených řas se intenzita světla na úrovni zkušebních roztoků zvolí v rozmezí 60–120 µE·m–2·s–1, přičemž se měří ve fotosynteticky efektivním rozsahu vlnových délek od 400 do 700 nm pomocí vhodného snímače. Některé druhy, zejména Anabaena flos-aquae, dobře rostou při nízkých intenzitách osvětlení a mohou být při vysokých intenzitách poškozeny. U takových druhů by měla být zvolena průměrná intenzita světla 40–60 µE·m–2·s–1. (Měří-li se intenzita luxmetry, ekvivalentní rozpětí 4 440 až 8 880 luxů pro chladné bílé světlo odpovídá přibližně doporučené světelné intenzitě 60–120 µE·m–2·s–1). Intenzita světla se nesmí lišit o více než ±15 % od průměrné intenzity světla nad plochou inkubace.
1.8.10 Doba trvání zkoušky
Zkouška obvykle trvá 72 hodin. Lze však použít kratších nebo delších dob trvání zkoušky, pokud lze splnit všechna kritéria validity uvedená v bodě 1.7.
1.8.11 Měření a analytická stanovení
Biomasa řas se během doby zkoušky určí v každé baňce nejméně jednou denně. Jestliže se měření provádí na malých objemech vyjmutých ze zkušebního roztoku pipetou, neměly by se nahrazovat.
Měření biomasy se provádí ručním počítáním buněk pod mikroskopem nebo pomocí elektronického počítače částic (počty buněk a/nebo biologický objem). Alternativní techniky, např. průtoková cytometrie, fluorescence chlorofylu in vitro nebo in vivo (6, 7) nebo optická hustota, lze použít za předpokladu, že je možné prokázat uspokojivou korelaci s biomasou v rozmezí, v němž se biomasa během zkoušky vyskytuje.
pH roztoků se měří na počátku a na konci zkoušky.
Pokud je dostupný analytický postup k určení zkoušené látky v rozmezí použitých koncentrací, zkušební roztoky je třeba analyzovat, aby se ověřily počáteční koncentrace a udržení expozičních koncentrací během zkoušky.
Analýza koncentrace zkoušené látky na počátku a konci zkoušky při nízké a vysoké zkušební koncentraci a koncentrace okolo očekávané EC50 mohou dostačovat, jestliže je pravděpodobné, že se budou během zkoušky expoziční koncentrace lišit o méně než 20 % od nominálních hodnot. Analýza všech zkušebních koncentrací na počátku a konci zkoušky se doporučuje v případě, kdy je nepravděpodobné, že koncentrace zůstanou v rozmezí 80 až 120 % nominální hodnoty. U těkavých, nestabilních nebo silně adsorbujících zkoušených látek se doporučuje provést dodatečný odběr vzorků k analýze v 24hodinových intervalech během expoziční doby s cílem lépe definovat ztrátu zkoušené látky. U těchto látek jsou zapotřebí dodatečná opakování. Ve všech případech je třeba provést určení koncentrací zkoušené látky při každé zkušební koncentraci pouze u jedné nádoby k opakování (nebo u sdruženého obsahu nádob jednoho opakování).
Zkušební média specificky připravená k analýze expozičních koncentrací během zkoušky by měla být exponována stejným způsobem jako zkušební média použitá ke zkoušení, tj. měla by být naočkována řasami a inkubována za identických podmínek. Jestliže se požaduje analýza koncentrace rozpuštěné zkušební látky, může být nezbytné oddělit řasy od média. Oddělení by se mělo nejlépe provádět odstřeďováním při nízké síle g dostatečné pro usazení řas.
Lze-li prokázat, že byla po celou dobu zkoušky koncentrace zkoušené látky uspokojivě udržována v rozmezí ±20 % nominální nebo naměřené počáteční koncentrace, může být analýza výsledků založena na nominálních nebo naměřených počátečních hodnotách. Je-li odchylka od nominální nebo naměřené počáteční koncentrace větší než ±20 %, analýza výsledků by měla být založena na geometrické střední koncentraci během expozice nebo na modelech popisujících pokles koncentrace zkoušené látky (3, 8).
Zkouška inhibice růstu řas je dynamičtější zkušební systém než většina jiných krátkodobých zkoušek vodní toxicity. V důsledku toho může být obtížné definovat skutečné expoziční koncentrace, zvláště u adsorbujících látek zkoušených při nízkých koncentracích. V takových případech vymizení látky z roztoku adsorpcí do zvyšující se biomasy řas neznamená, že se ztratila ze zkušebního systému. Při analýze výsledku zkoušky je třeba zkontrolovat, zda-li je pokles koncentrace zkoušené látky v průběhu zkoušky doprovázen poklesem inhibice růstu. Pokud tomu tak je, lze zvážit použití vhodného modelu popisujícího pokles koncentrace zkoušené látky (8). Pokud tomu tak není, může být vhodné založit analýzu výsledků na počátečních (nominálních či naměřených) koncentracích.
1.8.12 Další pozorování
Pro ověření normálního a zdravého vzhledu očkovací kultury a sledování jakéhokoliv abnormálního vzhledu řas (které může být způsobeno expozicí zkoušené látce) na konci zkoušky by mělo být provedeno mikroskopické pozorování.
1.8.13 Limitní zkouška
Za určitých okolností, např. pokud předběžná zkouška naznačuje, že zkoušená látka nemá žádné toxické účinky při koncentracích až do 100 mg l–1 nebo až do meze své rozpustnosti ve zkušebním médiu (podle toho, co je nižší), může být provedena limitní zkouška zahrnující porovnání odezvy v kontrolní skupině a odezvy v jedné exponované skupině (100 mg l–1 nebo koncentrace rovná mezi rozpustnosti). Důrazně se doporučuje podpořit tuto zkoušku analýzou expozičních koncentrací. Všechny dříve popsané zkušební podmínky a kritéria validity se vztahují i na limitní zkoušku s výjimkou toho, že by mělo být provedeno alespoň šest opakování s exponovanými vzorky. Proměnné odezvy v kontrolní a exponované skupině mohou být analyzovány pomocí statistického testu pro porovnání středních hodnot, např. Studentova t-testu. Jestliže rozptyly obou skupin nejsou shodné, je třeba provést t-test upravený pro rozdílné rozptyly.
1.8.14 Úprava pro intenzivně zbarvené látky
Ozářenost (intenzita světla) by měla dosahovat hodnot na nejvyšším konci rozsahu předepsaného v této zkušební metodě: 120 µE·m–2·s–1 nebo více.
Dráha světla by měla být zkrácena snížením objemu zkušebních roztoků (v rozmezí 5–25 ml).
Je nezbytné dostatečné míchání (například mírným protřepáváním), aby bylo dosaženo vysoké frekvence expozice řas vysoké ozářenosti na povrchu kultury.
2. DATA
2.1 VYNÁŠENÍ RŮSTOVÝCH KŘIVEK
Biomasa ve zkušebních nádobách může být vyjádřena v jednotkách náhradního parametru použitého pro měření (např. počet buněk, fluorescence).
Pro vynesení růstových křivek do grafu sestavte tabulku odhadovaných koncentrací biomasy ve zkušebních kulturách a kontrolách společně s koncentracemi zkušebního materiálu a časy měření zaznamenanými v rozlišení nejméně na celé hodiny. V tomto prvním stadiu mohou být užitečné jak logaritmické, tak lineární stupnice, ale logaritmické stupnice jsou povinné a obecně poskytují lepší zobrazení odchylek ve vzorci růstu během zkušebního období. Nezapomeňte, že exponenciální růst má při vynesení na logaritmické stupnici podobu přímky a že sklon (směrnice) přímky ukazuje specifickou růstovou rychlost.
Po vynesení prověřte, zda-li rostou kontrolní kultury exponenciálně očekávanou rychlostí po celou dobu zkoušky. Kriticky zhodnoťte všechny datové body a vzhled grafů a zkontrolujte, zda v hrubých datech a postupech nejsou chyby. Zejména zkontrolujte všechny datové body, u nichž se zdá, že se odchylují na základě systematické chyby. Pokud je zřejmé, že lze rozpoznat procesní chyby a/nebo je lze považovat za vysoce pravděpodobné, specifický datový bod se označí jako odlehlá hodnota a nebude zahrnut do následné statistické analýzy. (Nulová koncentrace řas v jedné ze dvou nebo tří nádob použitých k opakování může ukazovat na to, že nádoba nebyla správně naočkována nebo byla nesprávně vyčištěna.) Důvody k odmítnutí datového bodu jako odlehlé hodnoty musí být jasně uvedeny ve zkušebním protokolu. Přijatelnými důvody jsou pouze (vzácné) procesní chyby a nikoli jen malá preciznost. Statistické postupy pro rozpoznání odlehlé hodnoty mají u tohoto typu problému pouze omezené použití a nemohou nahradit odborné posouzení. Odlehlé hodnoty (takto označené) by nejlépe měly zůstat mezi datovými body zobrazenými v jakémkoliv následné grafickém či tabulárním zobrazení dat.
2.2 PROMĚNNÉ ODEZVY
Účelem této zkoušky je stanovit účinky zkoušené látky na růst řas. Tato zkušební metoda popisuje dvě proměnné odezvy, protože členské země mají odlišné preference a právní požadavky. Mají-li být výsledky zkoušky přijatelné ve všech členských zemích, účinky je třeba vyhodnotit pomocí obou proměnných odezvy a) a b) popsaných dále.
|
a) |
Průměrná specifická růstová rychlost: tato proměnná odezvy se vypočítává na základě logaritmického zvýšení biomasy během zkušebního období a vyjádří se na den. |
|
b) |
Výtěžek: tato proměnná odezvy je biomasa na konci zkoušky minus počáteční biomasa. |
Při použití této metody v rámci práva EU by měl být výpočet výsledků založen na průměrné specifické růstové rychlosti, a to z níže uvedených důvodů. Je třeba uvést, že hodnoty toxicity vypočítané pomocí těchto dvou proměnných odezvy nejsou srovnatelné a při používání výsledků zkoušky se musí vzít tento rozdíl na vědomí. Hodnoty ECx založené na průměrné specifické růstové rychlosti (ErCx) budou všeobecně vyšší než výsledky založené na výtěžku (EyCx), pokud se dodrží zkušební podmínky této zkušební metody, a to díky matematické základně příslušných přístupů. Tuto skutečnost nelze vykládat jako rozdíl v citlivosti mezi těmito dvěma proměnnými odezvy; hodnoty jsou jednoduše matematicky odlišné. Koncepce průměrné specifické růstové rychlosti je založena na obecném průběhu exponenciálního růstu řas v nelimitovaných kulturách, u nichž se toxicita odhaduje na základě účinků na růstovou rychlost, aniž by závisela na absolutní úrovni specifické růstové rychlosti kontroly, na sklonu křivky závislosti koncentrace-odezva nebo na době trvání zkoušky. Naproti tomu výsledky založené na proměnné odezvy „výtěžek“ jsou závislé na všech těchto ostatních proměnných. EyCx závisí na specifické růstové rychlosti druhů řas použitých v každé zkoušce a na maximální specifické růstové rychlosti, která se může u jednotlivých druhů, a dokonce i u různých kmenů řas lišit. Tato proměnná odezvy by se neměla používat pro srovnávání citlivosti na toxické látky mezi druhy řas nebo dokonce různými kmeny. Přestože se z vědeckého hlediska dává přednost použití průměrné specifické růstové rychlosti pro odhad toxicity, odhady toxicity založené na výtěžku jsou rovněž do této zkušební metody zahrnuty, aby uspokojily současné právní požadavky v některých zemích.
2.2.1 Průměrná růstová rychlost
Průměrná specifická růstová rychlost pro konkrétní období se vypočítá jako logaritmické zvýšení biomasy z rovnice pro každou jednotlivou nádobu s kontrolními a exponovanými vzorky:
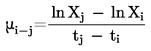 (den–1)
(den–1)
kde:
|
µi–j: |
je průměrná specifická růstová rychlost od doby i do doby j, |
|
Xi: |
je biomasa v době i, |
|
Xj: |
je biomasa v době j. |
Pro každou exponovanou skupinu a kontrolní skupinu vypočítejte střední hodnotu růstové rychlosti a odhady rozptylu.
Vypočítejte průměrnou specifickou růstovou rychlost po celou dobu trvání zkoušky (obvykle dny 0 až 3), přičemž namísto naměřené počáteční hodnoty použijte jako počáteční hodnotu spíše nominálně naočkovanou biomasu, protože se tímto způsobem obvykle dosáhne větší preciznosti. Jestliže zařízení použité pro měření biomasy umožňuje dostatečně precizní určení nízké biomasy inokula (např. průtokovým cytometrem), pak lze použít naměřenou počáteční koncentraci biomasy. Rovněž se vyhodnotí růstová rychlost po jednotlivých časových úsecích vypočtená jako specifické růstové rychlosti pro každý den v průběhu zkoušky (dny 0–1, 1–2 a 2–3) a prozkoumá se, zda-li růstová rychlost kontroly zůstává konstantní (viz kritéria validity, bod 1.7). Významně nižší specifická růstová rychlost v den jedna v porovnání s celkovou průměrnou specifickou růstovou rychlostí může poukazovat na fázi iniciace. Zatímco v kontrolních kulturách lze fázi iniciace minimalizovat a prakticky vyloučit správnou kultivací předkultury, fáze iniciace v exponovaných kulturách může být známkou obnovy po počátečním toxickém stresu nebo známkou snížené expozice vyvolané ztrátou zkoušené látky (včetně sorpce na biomasu řas) po počáteční expozici. Odpovídajícím způsobem lze vyhodnocovat růstovou rychlost v jednotlivých časových úsecích s cílem zhodnotit účinky zkoušené látky, které se vyskytly během expoziční doby. Významné rozdíly mezi růstovou rychlostí v jednotlivých časových úsecích a průměrnou růstovou rychlostí jsou známkou odchylky od konstantního exponenciálního růstu a vyžadují důkladné prozkoumání růstových křivek.
Vypočítejte procentuální inhibici růstové rychlosti u jednotlivých opakování s exponovanými vzorky z rovnice:
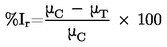
kde:
|
%I r: |
procentuální inhibice průměrné specifické růstové rychlosti, |
|
µ C: |
střední hodnota průměrné specifické růstové rychlosti (µ) v kontrolní skupině, |
|
µ T: |
průměrná specifická růstová rychlost u opakování s exponovaným vzorkem. |
Jestliže se k přípravě zkušebních roztoků používají rozpouštědla, pro výpočet procentuální inhibice by se měly namísto kontrol bez rozpouštědel používat kontroly s rozpouštědly.
2.2.2 Výtěžek
Výtěžek se vypočítá jako biomasa na konci zkoušky minus počáteční biomasa pro každou jednotlivou nádobu s kontrolními a exponovanými vzorky. Pro každou zkušební koncentraci a kontrolu vypočítejte střední hodnotu výtěžku a odhady rozptylu. Procentuální inhibici výtěžku (%I y) lze vypočítat pro každé opakování s exponovanými vzorky následovně:
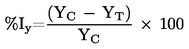
kde:
|
% I y: |
procentuální inhibice výtěžku, |
|
Y C: |
střední hodnota výtěžku v kontrolní skupině, |
|
Y T: |
hodnota výtěžku u opakování s exponovaným vzorkem. |
2.3 VYNÁŠENÍ KŘIVKY ZÁVISLOSTI KONCENTRACE-ODEZVA
Vyneste procentuální hodnotu inhibice proti logaritmu koncentrace zkušební látky a vynesené body pečlivě prozkoumejte, přičemž neberte v úvahu žádný takový datový bod, který byl v první fázi vyloučen jako odlehlá hodnota. Podle oka nebo pomocí počítačové interpolace veďte datovými body tenkou čáru, abyste získali první dojem o vztahu koncentrace a odezvy, poté přistupte k podrobnější metodě, nejlépe počítačové statistické metodě. V závislosti na zamýšleném použití dat, jakosti (preciznosti) a množství dat, jakož i na dostupnosti nástrojů pro analýzu dat může být rozhodnuto (a někdy je to dostatečně odůvodněno) o tom, že se analýza dat v tomto stadiu zastaví a klíčové hodnoty EC50 a EC10 (a/nebo EC20) se jednoduše odečtou z křivky vytvořené podle oka (rovněž viz následující bod o stimulačních účincích). Platnými důvody pro nepoužití statistické metody mohou být:
|
— |
Údaje nejsou vhodné pro počítačové metody, které neposkytnou žádné spolehlivější výsledky než ty, které lze získat odborným posouzením – v takových situacích mohou dokonce některé počítačové programy selhat při získání spolehlivého řešení (iterace nemusí konvergovat atd.). |
|
— |
Stimulační růstové odezvy nelze odpovídajícím způsobem zpracovat pomocí dostupných počítačových programů (viz níže). |
2.4 STATISTICKÉ POSTUPY
Cílem je získat kvantitativní vztah mezi koncentrací a odezvou pomocí regresní analýzy. Je možné použít váženou lineární regresi po provedení linearizující transformace dat odezvy – například do probitových nebo logitových či Weibullových jednotek (9), ale dává se přednost nelineárním regresním postupům, s jejichž pomocí se lépe zpracovávají nevyhnutelné nepravidelnosti dat a odchylky od hladkých distribucí. Při přiblížení se k nule či úplné inhibici mohou být takové nepravidelnosti zesíleny transformací a rušit při analýze (9). Je zapotřebí uvést, že standardní metody analýzy používající probitové, logitové nebo Weibullovy transformace jsou určeny k použití s kvantálními daty (např. mortalita nebo přežití) a musí se upravit, aby mohly být použity s daty růstu či biomasy. Speciální postupy pro stanovení hodnoty ECx ze spojitých dat lze nalézt v literatuře (10, 11 a 12). Použití nelineární regresní analýzy je dále podrobně popsáno v dodatku 4.
Pro každou proměnnou odezvy, která se má analyzovat, použijte vztah koncentrace a odezvy pro výpočet bodových odhadů hodnot ECx. Je-li to možné, je zapotřebí určit 95% intervaly spolehlivosti pro každý odhad. Jakost shodnosti dat odezvy s regresním modelem je zapotřebí vyhodnotit buď graficky, nebo statisticky. Regresní analýza by se měla provádět pomocí odezev jednotlivých opakování, nikoliv pomocí středních hodnot exponovaných skupin. Pokud však je vynesení nelineární křivky obtížné či neúspěšné kvůli příliš velkému rozptylu dat, problém lze obejít provedením regrese na skupinových středních hodnotách, což je praktický způsob pro snížení vlivu podezřelých odlehlých hodnot. Použití této možnosti je zapotřebí zaznamenat ve zkušebním protokolu jako odchylku od normálního postupu v důsledku skutečnosti, že křivka, která vyhovuje jednotlivých opakováním, nevedla k uspokojivému výsledku.
Odhady EC50 a intervaly spolehlivosti lze rovněž získat lineární interpolací s bootstrapem (13), pokud se dostupné regresní modely/metody pro příslušná data nehodí.
Pro odhad LOEC, a tedy i NOEC a pro účinky zkoušené látky na růstovou rychlost je nezbytné porovnat střední hodnoty exponovaných vzorků pomocí metod analýzy rozptylu (ANOVA). Střední hodnota pro každou koncentraci musí být poté porovnána se střední hodnotou pro kontrolní skupinu, a to vhodnou metodou vícenásobného porovnání nebo metodou zkoušky trendu. Užitečné mohou být Dunnettův nebo Williamsův test (14, 15, 16, 17, 18). Je nezbytné vyhodnotit, zda je splněn předpoklad homogenity rozptylu nezbytný pro ANOVA. Toto hodnocení lze provádět graficky nebo formálním testem (18). Mezi vhodné testy patří Levenův nebo Bartlettův test. Nesplnění předpokladu o homogenitě rozptylů lze někdy napravit logaritmickou transformací dat. Jestliže je heterogenita rozptylu extrémní a nelze ji napravit transformací, pak je třeba zvážit analýzu takovými metodami, jako jsou testy sestupného trendu Jonckheere. Další pokyny pro stanovení NOEC lze nalézt v (12).
Vědecký rozvoj poslední doby vedl k doporučení opustit koncepci NOEC a nahradit ji odhady bodů ECx na základě regrese. Vhodná hodnota pro x nebyla pro tuto zkoušku pomocí řas stanovena. Zdá se, že vhodné je rozmezí od 10 do 20 % (v závislosti na zvolené proměnné odezvy) a nejlépe by se měly ve zkušebním protokolu uvádět jak EC10, tak EC20.
2.5 STIMULACE RŮSTU
Při nízkých koncentracích bývá někdy pozorována stimulace růstu (negativní inhibice). To může být důsledkem buď hormese („toxická stimulace“), nebo přidání stimulujících růstových faktorů se zkušebním materiálem k minimálnímu použitému médiu. Nezapomeňte, že přidání anorganických živin by neměl mít žádný přímý účinek, protože zkušební médium by mělo po celou dobu zkoušky udržovat přebytek živin. Stimulaci nízkými dávkami lze ve výpočtech EC50 obvykle ignorovat, pokud není extrémní. Je-li však extrémní nebo má-li být vypočtena hodnota ECx pro nízké x, může být zapotřebí speciálních postupů. K vymazání stimulačních odezev z analýzy dat by nemělo pokud možno dojít, a pokud dostupný software pro stanovení křivky nemůže drobnou stimulaci přijmout, lze použít lineární interpolaci s bootstrapem. Jestliže je stimulace extrémní, lze zvážit použití modelu předpokládajícího hormesi (19).
2.6 NETOXICKÁ INHIBICE RŮSTU
Zkušební materiály absorbující světlo mohou způsobit snížení růstové rychlosti, protože stínění snižuje množství dostupného světla. Takové účinky fyzikální povahy je zapotřebí oddělit od toxických účinků, a to úpravou zkušebních podmínek, přičemž takové fyzikální účinky je zapotřebí uvádět ve zkušebním protokolu samostatně. Pokyny lze nalézt v literatuře (2 a 3).
3. ZPRÁVY
3.1 PROTOKOL O ZKOUŠCE
Protokol o zkoušce musí obsahovat tyto údaje:
Zkoušená látka:
|
— |
fyzikální povaha a relevantní fyzikálně-chemické vlastnosti včetně meze rozpustnosti ve vodě, |
|
— |
údaje o chemické identifikaci včetně čistoty. |
Testovací druhy:
|
— |
kmen, dodavatel nebo zdroj a použité podmínky kultivace. |
Zkušební podmínky:
|
— |
datum zahájení zkoušky a délka jejího trvání, |
|
— |
popis plánu zkoušky: zkušební nádoby, objemy kultur, hustota biomasy na počátky zkoušky, |
|
— |
složení média, |
|
— |
zkušební koncentrace a opakování (například počet opakování, počet zkušebních koncentrací a použitá geometrická progrese), |
|
— |
popis přípravy zkušebních roztoků včetně použití rozpouštědel atd., |
|
— |
kultivační zařízení, |
|
— |
intenzita a kvalita světla (zdroj, homogenita), |
|
— |
teplota, |
|
— |
zkušební koncentrace: nominální zkušební koncentrace a jakékoliv výsledky analýz pro stanovení koncentrace zkoušené látky ve zkušebních nádobách. Ve zkušebním protokolu je třeba uvést výtěžnost metody a mez kvantifikace ve zkušební matrici, |
|
— |
všechny odchylky od této zkušební metody, |
|
— |
metoda pro stanovení biomasy a důkaz o korelaci mezi naměřeným parametrem a suchou hmotností. |
Výsledky:
|
— |
hodnoty pH na počátku a konci zkoušky u všech expozic, |
|
— |
biomasa pro každou baňku v každém měřicím bodě a metoda pro měření biomasy, |
|
— |
růstové křivky (vynášení biomasy vůči času), |
|
— |
vypočítané proměnné odezvy pro všechna opakování s exponovanými vzorky, spolu se středními hodnotami a variačním koeficientem pro opakování, |
|
— |
grafické znázornění vztahu koncentrace a účinku, |
|
— |
odhady toxicity pro proměnné odezvy, např. EC50, EC10, EC20, a související intervaly spolehlivosti. Jestliže se počítají LOEC a NOEC, uvedou se jejich hodnoty a statistické metody použité k jejich stanovení, |
|
— |
jestliže byla použita ANOVA, uvede se velikost účinku, který lze detekovat (např. nejmenší významný rozdíl), |
|
— |
jakákoliv stimulace růstu zjištěná v jakémkoli exponovaném vzorku, |
|
— |
jakékoliv jiné pozorované účinky, např. morfologické změny řas, |
|
— |
diskuse výsledků včetně jakéhokoli vlivu na výsledek zkoušky v důsledku odchylek od této zkušební metody. |
4. LITERATURA
|
(1) |
OECD TG 201 (2006) Freshwater Alga and Cyanobacteria, Growth Inhibition Test. |
|
(2) |
ISO 1998: Water quality – Guidance for algal growth inhibition tests with poorly soluble materials, volatile compounds, metals and waster water. ISO/DIS 14442. |
|
(3) |
OECD 2000: Guidance Document on Aquatic Toxicity Testing of Difficult Substances and mixtures. Environmental Health and Safety Publications. Series on Testing and Assessment, no. 23. |
|
(4) |
ISO 1998: Jakost vod – Odběr vzorků – Část 16: Pokyny pro biologické zkoušení vzorků. ISO 5667–16. |
|
(5) |
ISO 1993: Jakost vod – Zkouška inhibice růstu sladkovodních zelených řas. ISO 8692. |
|
(6) |
Mayer, P., Cuhel, R., Nyholm, N. (1997). A simple in vitro fluorescence method for biomass measurements in algal growth inhibition tests. Water Research 31: 2525–2531. |
|
(7) |
Slovacey, R.E., Hanna, P.J. In vivo fluorescence determinations of phytoplancton chlorophyll, Limnology & Oceanography 22,5 (1977), 919–925 |
|
(8) |
Simpson, S.L., Roland, M.G.E., Stauber, J.L., Batley, G.E. (2003) Effect of declining toxicant concentrations on algal bioassay endpoints. Environ. Toxicol. Chem. 22, 2073–2079. |
|
(9) |
Christensen, E.R., Nyholm, N. (1984): Ecotoxicological Assays with Algae: Weibull Dose-Response Curves. Env. Sci. Technol. 19, 713–718. |
|
(10) |
Nyholm, N. Sørensen, P.S., Kusk, K.O., Christensen, E.R. (1992): Statistical treatment of data from microbial toxicity tests. Environ. Toxicol. Chem. 11, 157–167. |
|
(11) |
Bruce, R.D., Versteeg, D.J. (1992). A statistical procedure for modelling continuous toxicity data. Env. Toxicol. Chem. 11:1485–1494. |
|
(12) |
OECD (2004). Guidance Document on Statistical Analysis of Ecotoxicity Data. |
|
(13) |
Norberg-King T.J. (1988) An interpolation estimate for chronic toxicity: The ICp approach. National Effluent Toxicity Assessment Center Technical Report 05–88. USEPA, Duluth, MN. |
|
(14) |
Dunnett, C.W. (1955) A multiple comparisons procedure for comparing several treatments with a control. J. Amer. Statist. Assoc. 50: 1096–1121. |
|
(15) |
Dunnett, C.W. (1964) New tables for multiple comparisons with a control. Biometrics 20: 482–491. |
|
(16) |
Williams, D.A. (1971) A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics 27: 103–117. |
|
(17) |
Williams, D.A. (1972) The comparison of several dose levels with a zero dose control. Biometrics 28: 510–531. |
|
(18) |
Draper, N.R. and Smith, H. (1981). Applied Regression Analysis, second edition. Wiley, New York. |
|
(19) |
Brain P. and Cousens R. (1989). An equation to describe dose-responses where there is stimulation of growth at low doses. Weed Research, 29, 93–96. |
Dodatek 1
Kmeny, u nichž byla prokázána vhodnost pro zkoušku
Zelené řasy
|
— |
Pseudokirchneriella subcapitata (dříve známá jako Selenastrum capricornutum), ATCC 22662, CCAP 278/4, 61.81 SAG |
|
— |
Desmodesmus subspicatus (dříve známá jako Scenedesmus subspicatus) 86.81 SAG |
Rozsivky
|
— |
Navicula pelliculosa, UTEX 664 |
Sinice
|
— |
Anabaena flos-aquae, UTEX 1444, ATCC 29413, CCAP 1403/13A |
|
— |
Synechococcus leopoliensis, UTEX 625, CCAP 1405/1 |
Zdroje kmenů
Doporučené kmeny jsou k dispozici v jednodruhových kulturách z následujících sbírek (v abecedním pořadí):
|
|
|
|
|
|
|
|
|
|
|
|
Vzhled a charakteristiky doporučených druhů
|
|
P. subcapitata |
D. subspicatus |
N. pelliculosa |
A. flos-aquae |
S. leopoliensis |
|
Vzhled |
zakřivené, zkroucené jednotlivé buňky |
oválné, většinou jednotlivé buňky |
tyčinky |
řetězce oválných buněk |
tyčinky |
|
Velikost (d × š) µm |
8–14 × 2–3 |
7–15 × 3–12 |
7,1 × 3,7 |
4,5 × 3 |
6 × 1 |
|
Buněčný objem (µm3/buňka) |
40–60 (1) |
60–80 (1) |
40–50 (1) |
30–40 (1) |
2,5 (2) |
|
Hmotnost buněčné sušiny (mg/buňka) |
2–3 × 10–8 |
3–4 × 10–8 |
3–4 × 10–8 |
1–2 × 10–8 |
2–3 × 10–9 |
|
Růstová rychlost (3) (den–1) |
1,5–1,7 |
1,2–1,5 |
1,4 |
1,1–1,4 |
2,0–2,4 |
Specifická doporučení ohledně kultivace doporučených zkušebních druhů a nakládání s nimi
Pseudokirchneriella subcapitata a Desmodesmus subspicatus
Tyto zelené řasy se všeobecně snadno udržují v různých kultivačních médiích. Informace o vhodných médiích lze získat od sbírek kultur. Buňky jsou normálně samostatné a měření hustoty buněk lze snadno provádět pomocí elektronického počítače částic nebo mikroskopu.
Anabaena flos-aquae
Pro uchovávání zásobní kultury lze použít různá růstová média. Zvláště důležité je zabránit tomu, aby vsázková kultura překročila při obnově fázi logaritmického růstu; zpětné získání je v tomto bodě obtížné.
Anabaena flos-aquae vytváří shluky vnořených řetězců buněk. Velikost těchto shluků se může měnit s kultivačními podmínkami. Může vzniknout potřeba tyto shluky rozrušit, pokud se ke stanovení biomasy použije počítání pod mikroskopem nebo elektronický počítač částic.
Pro snížení variability počtu mohou být v dílčích vzorcích použity k rozrušení řetězců ultrazvukové vibrace. Delší ultrazvukové vibrace, než jsou nutné k rozrušení řetězců do kratších délek, mohou buňky zničit. Intenzita ultrazvukových vibrací a doba trvání musí být shodné pro každou expozici.
Spočítejte na hemocytometru dostatek políček (nejméně 400 buněk), což pomůže variabilitu kompenzovat. Tím se zlepší spolehlivost mikroskopického stanovení hustoty.
Pro stanovení celkového buněčného objemu sinic rodu Anabaena po rozrušení buněčných řetězců opatrnými ultrazvukovými vibracemi lze použít elektronický počítač částic. Ultrazvuková energie se musí nastavit tak, aby se zabránilo narušení buněk.
Aby se zajistilo dobré promíchání a homogenita suspense řas použitých k naočkování zkušebních nádob, použijte vířivou míchačku nebo podobnou odpovídající metodu.
Zkušební nádoby je nutno umístit na stůl orbitální nebo reciproké třepačky při přibližně 150 otáčkách za minutu. Případně lze použít občasné promíchávání ke snížení sklonu sinic rodu Anabaena vytvářet shluky. Jestliže ke shlukování dojde, musí se postupovat pečlivě k získání reprezentativních vzorků pro měření biomasy. Pro rozrušení shluků sinic před odběrem vzorků může být nezbytné intenzivní promíchávání.
Synechococcus leopoliensis
Pro uchovávání zásobní kultury lze použít různá růstová média. Informace o vhodných médiích lze získat od sbírek kultur.
Synechococcus leopoliensis vyrůstají jako jednotlivé buňky tyčinkového tvaru. Buňky jsou velmi malé, což ztěžuje použití počítání pod mikroskopem pro měření biomasy. Užitečné jsou elektronické počítače částic vybavené pro počítání částic až do velikosti přibližně 1 µm. Rovněž jsou využitelná fluorometrická měření in vitro.
Navicula pelliculosa
Pro uchovávání zásobní kultury lze použít různá růstová média. Informace o vhodných médiích lze získat od sbírek kultur. Je nutné mít na paměti, že v médiu se vyžaduje přítomnost křemičitanu.
Navicula pelliculosa může za určitých růstových podmínek vytvářet shluky. Buňky mají někdy sklon se shlukovat v povrchovém filmu kvůli produkci lipidů. Za těchto okolností se musí přijmout zvláštní opatření, když se odebírají dílčí vzorky pro stanovení biomasy s cílem získat reprezentativní vzorky. Může být nutné intenzivní protřepávání, např. použití vířivé míchačky.
(1) Měřeno elektronickým počítačem částic.
(2) Vypočítáno z velikosti.
(3) Nejčastěji pozorovaná růstová rychlost v médiu OECD při světelné intenzitě přibližně 70 µE·m–2·s–1 a teplotě 21 °C.
Dodatek 2
Růstová média
Lze použít jedno ze dvou následujících růstových médií:
Médium OECD: původní médium podle Pokynu OECD pro zkoušení 201, rovněž podle normy ISO 8692.
Médium US. EPA AAP, rovněž podle ASTM.
Při přípravě těchto médií je zapotřebí používat chemické látky analytické čistoty nebo na úrovni činidel a deionizovanou vodu.
Složení média AAP (US. EPA) a média podle Pokynu OECD pro zkoušení 201
|
Složka |
EPA |
OECD |
||
|
|
mg/l |
mM |
mg/l |
mM |
|
NaHCO3 |
15,0 |
0,179 |
50,0 |
0,595 |
|
NaNO3 |
25,5 |
0,300 |
|
|
|
NH4Cl |
|
|
15,0 |
0,280 |
|
MgCl2·6H2O |
12,16 |
0,0598 |
12,0 |
0,0590 |
|
CaCl2·2H2O |
4,41 |
0,0300 |
18,0 |
0,122 |
|
MgSO4·7H2O |
14,6 |
0,0592 |
15,0 |
0,0609 |
|
K2HPO4 |
1,044 |
0,00599 |
|
|
|
KH2PO4 |
|
|
1,60 |
0,00919 |
|
FeCl3·6H2O |
0,160 |
0,000591 |
0,0640 |
0,000237 |
|
Na2EDTA·2H2O |
0,300 |
0,000806 |
0,100 |
0,000269 (1) |
|
H3BO3 |
0,186 |
0,00300 |
0,185 |
0,00299 |
|
MnCl2·4H2O |
0,415 |
0,00201 |
0,415 |
0,00210 |
|
ZnCl2 |
0,00327 |
0,000024 |
0,00300 |
0,0000220 |
|
CoCl2·6H2O |
0,00143 |
0,000006 |
0,00150 |
0,00000630 |
|
Na2MoO4·2H2O |
0,00726 |
0,000030 |
0,00700 |
0,0000289 |
|
CuCl2·2H2O |
0,000012 |
0,00000007 |
0,00001 |
0,00000006 |
|
pH |
7,5 |
8,1 |
||
Při zkoušce s rozsivkou Navicula pelliculosa musí být obě média doplněna Na2SiO3·9H20 pro dosažení koncentrace 1,4 mg Si/l.
pH média se získá při rovnováze mezi uhličitanovým systémem média a parciálním tlakem CO2 v atmosférickém vzduchu. Přibližný vztah mezi pH při 25 °C a molární koncentrací hydrogenuhličitanu je:
pHrovn = 11,30 + log [HCO3]
S 15 mg/l NaHCO3 je pHrovn = 7,5 (médium US. EPA) a s 50 mg/l NaHCO3 je pHrovn = 8,1 (médium OECD).
Prvkové složení zkušebních médií
|
Prvek |
EPA |
OECD |
|
|
mg/l |
mg/l |
|
C |
2,144 |
7,148 |
|
N |
4,202 |
3,927 |
|
P |
0,186 |
0,285 |
|
K |
0,469 |
0,459 |
|
Na |
11,044 |
13,704 |
|
Ca |
1,202 |
4,905 |
|
Mg |
2,909 |
2,913 |
|
Fe |
0,033 |
0,017 |
|
Mn |
0,115 |
0,115 |
Příprava média OECD
|
Živina |
Koncentrace v zásobním roztoku |
|
Zásobní roztok č. 1: makroživiny |
|
|
NH4Cl MgCl2·6H2O CaCl2·2H2O MgSO4·7H2O KH2PO4 |
1,5 g·l–1 1,2 g·l–1 1,8 g·l–1 1,5 g·l–1 0,16 g·l–1 |
|
Zásobní roztok č. 2: železo |
|
|
FeCl3·6H2O Na2EDTA·2H2O |
64 mg·l–1 100 mg·l–1 |
|
Zásobní roztok č. 3: stopové prvky |
|
|
H3BO3 MnCl2·4H2O ZnCl2 CoCl2·6H2O CuCl2·2H2O Na2MoO4·2H2O |
185 mg·l–1 415 mg·l–1 3 mg·l–1 1,5 mg·l–1 0,01 mg·l–1 7 mg·l–1 |
|
Zásobní roztok 4: hydrogenuhličitan |
|
|
NaHCO3 |
50 g·l–1 |
|
Na2SiO3·9H20 |
|
Vysterilizujte zásobní roztoky membránovou filtrací (střední průměr pórů 0,2 µm) nebo v autoklávu (120 °C, 15 minut). Roztoky skladujte v temnu při 4 °C.
Zásobní roztoky 2 a 4 neošetřujte v autoklávu, ale sterilizujte je membránovou filtrací.
Připravte růstové médium přidáním odpovídajícího objemu zásobních roztoků 1 až 4 do vody:
Do 500 ml sterilizované vody přidejte:
|
— |
10 ml zásobního roztoku 1, |
|
— |
1 ml zásobního roztoku 2, |
|
— |
1 ml zásobního roztoku 3, |
|
— |
1 ml zásobního roztoku 4. |
Doplňte do 1 000 ml sterilizovanou vodou.
Nechte stát dostatečně dlouhou dobu na to, aby se vytvořila rovnováha média s atmosférickým CO2, v případě potřeby pomocí probublávání sterilním filtrovaným vzduchem po několik hodin.
Příprava média AAP
|
A1.1 |
Přidejte 1 ml každého zásobního roztoku v A1.2.1–A1.2.7 do přibližně 900 ml deionizované nebo destilované vody a poté nařeďte do 1 l. |
|
A1.2 |
Zásobní roztoky makroživin se připravují rozpuštěním následujících látek v 500 ml deionizované nebo destilované vody. Činidla A1.2.1, A1.2.2, A1.2.3 a A1.2.4 lze spojit do jednoho zásobního roztoku: |
|
A1.2.1 |
NaNO3—12,750 g. |
|
A1.2.2 |
MgCl2·6H2O—6,082 g. |
|
A1.2.3 |
CaCl2·2H2O—2,205 g. |
|
A1.2.4 |
Zásobní roztok mikroživin – (viz A1.3). |
|
A1.2.5 |
MgSO4·7H2O—7,350 g. |
|
A1.2.6 |
K2HPO4—0,522 g. |
|
A1.2.7 |
NaHCO3—7,500 g. |
|
A1.2.8 |
Na2SiO3·9H2O – viz poznámka A1.1. Poznámka A1.1 – Používejte pouze pro testovací druhy rozsivek. Může být přidán přímo (202,4 mg) nebo prostřednictvím zásobního roztoku, aby bylo dosaženo konečné koncentrace 20 mg/l Si v médiu. |
|
A1.3 |
Zásobní roztok mikroživin se připravuje rozpuštěním následujících látek v 500 ml deionizované nebo destilované vody: |
|
A1.3.1 |
H3BO3—92,760 mg. |
|
A1.3.2 |
MnCl2·4H2O—207,690 mg. |
|
A1.3.3 |
ZnCl2—1,635 mg. |
|
A1.3.4 |
FeCl3·6H2O—79,880 mg. |
|
A1.3.5 |
CoCl2·6H2O—0,714 mg. |
|
A1.3.6 |
Na2MoO4·2H2O—3,630 mg. |
|
A1.3.7 |
CuCl2·2H2O—0,006 mg. |
|
A1.3.8 |
Na2EDTA·2H2O—150,000 mg. [dinatrium-ethylendiamintetraacetát dihydrát]. |
|
A1.3.9 |
Na2SeO4·5H2O—0,005 mg, viz poznámka A1.2. Poznámka A1.2 – Používejte pouze v médiu pro zásobní kultury druhů rozsivek. |
|
A1.4 |
Upravte pH na 7,5 ± 0,1 přidáním 0,1 N nebo 1,0 N NaOH nebo HCl. |
|
A1.5 |
Odfiltrujte média do sterilní nádoby prostřednictvím 0,22μm membránového filtru, pokud se má používat počítač částic, nebo 0,45μm filtru, pokud se počítač částic používat nemá. |
|
A1.6 |
Médium skladujte až do použití v temnu při teplotě přibližně 4 °C. |
(1) Molární poměr EDTA k železu mírně překračuje jednu. Tím se zabrání srážení železa a současně se minimalizuje chelatace iontů těžkých kovů.
Dodatek 3
Příklad postupu kultivace řas
Obecné poznámky
Účelem kultivace následujícím postupem je získání kultur řas pro zkoušky toxicity.
Musí být použity vhodné metody k tomu, aby bylo zajištěno, že kultury řas nebudou infikovány bakteriemi. Vhodné mohou být axenické kultury, avšak musí se vytvořit a používat jednodruhové kultury.
Všechny operace se musí provádět za sterilních podmínek, aby nedošlo ke kontaminaci bakteriemi a jinými řasami.
Zařízení a materiály
Viz zkušební metoda: Přístroje a pomůcky.
Postupy při získávání kultur řas
Příprava živných roztoků (média):
Všechny soli živin média se připravují jako koncentrované zásobní roztoky a skladují se v temnu a chladu. Tyto roztoky jsou sterilizovány filtrací nebo autoklávem.
Médium se připraví přidáním správného množství zásobního roztoku do sterilní destilované vody, přičemž se dbá na to, aby nedošlo k žádné infekci. K získání pevného média se přidává 0,8 procent agaru.
Kmenová kultura:
Kmenové kultury jsou malé kultury řas, které se pravidelně přenášejí do čerstvého média, kde slouží jako výchozí zkušební materiál. Nejsou-li kultury pravidelně používány, vyočkovávají se na šikmý agar. Poté se nejméně jednou za dva měsíce přenášejí do čerstvého média.
Kmenové kultury se pěstují v Erlenmeyerových baňkách obsahujících vhodné médium (objem přibližně 100 ml). Jsou-li řasy kultivovány při teplotě 20 °C a stálém osvětlení, musí se přenášet každý týden.
Při přeočkování se přenese sterilní pipetou do baňky s čerstvým médiem takové množství „staré“ kultury, aby byla počáteční koncentrace u rychle rostoucího druhu řas asi stokrát menší než koncentrace kultury staré.
Růstovou rychlost druhu řas lze odečíst z růstové křivky. Je-li známa, lze z ní odhadnout hustotu, při níž musí být kultura přenesena do čerstvého média. K tomu musí dojít před fází odumírání kultury.
Předkultura:
Účelem předkultur je poskytnout dostatečné množství řas potřebných pro naočkování zkušebních kultur. Předkultura se kultivuje za zkušebních podmínek a použije se ještě během exponenciálního růstu, to znamená obvykle po inkubační lhůtě o délce 2 až 4 dní. Obsahují-li kultury řas deformované nebo abnormální buňky, musí se odstranit.
Dodatek 4
Datová analýza pomocí nelineární regrese
Obecné poznámky
Odezva ve zkoušce růstu řas a jiných zkouškách mikrobiálního růstu – růstu biomasy je svojí povahou spojitá nebo metrická proměnná – procesní rychlost, jestliže se používá růstové rychlosti, a její integrál během doby, pokud se zvolí biomasa. Obě jsou popsány příslušnou střední odezvou neexponovaných kontrol použitých k opakování, jež ukazují maximální odezvu při daných podmínkách – přičemž světlo a teplota jsou primární určující faktory ve zkoušce růstu řas. Systém je distribuovaný nebo homogenní a na biomasu lze pohlížet jako na kontinuum při zanedbání jednotlivých buněk. Distribuce rozptylu typu odezvy pro takový systém závisí výlučně na experimentálních faktorech (obvykle popsaných lognormálními nebo normálními distribucemi chyb). To je v kontrastu k typickým odezvám biologických zkoušek s kvantálními daty, u nichž se často předpokládá, že tolerance (obvykle binominálně distribuovaná) jednotlivých organismů je dominantní složkou rozptylu. Zde jsou kontrolní odezvy nula nebo základní úroveň.
V nekomplikované situaci normalizovaná nebo relativní odezva, r, monotónně klesá z 1 (nulová inhibice) na 0 (stoprocentní inhibice). Je třeba mít na paměti, že všechny odezvy mají přidruženou chybu a že očividně negativní inhibice lze vypočítat pouze jako výsledek náhodné chyby.
Regresní analýza
Modely
Cílem regresní analýzy je kvantitativně popsat křivku závislosti koncentrace a odezvy ve formě matematické regresní funkce Y = f (C) nebo častěji F (Z), kde Z = log C. Použití inverzní funkce C = f–1(Y) dovoluje vypočítat hodnoty ECx včetně EC50, EC10 a EC20 a jejich 95% intervaly spolehlivosti. Prokázalo se, že vztah koncentrace a odezvy získané ve zkouškách inhibice růstu řas úspěšně popisuje několik jednoduchých matematických funkcí. Funkce například obsahují logistickou rovnici, nesymetrickou Weibullovu rovnici a lognormální distribuční funkci, které jsou všechny esovitými křivkami asymptoticky se blížícími jedné pro C → 0 a nule pro C → nekonečno.
Použití modelů spojité prahové funkce (např. Kooijmanův model „pro inhibici populačního růstu“, Kooijman et al. 1996) byl nedávno navržen jako alternativa k modelům asymptotickým. Tento model nepředpokládá žádné účinky při koncentracích pod určitým prahem, EC0+, který se odhaduje extrapolací vztahu koncentrace a odezvy pro protnutí osy koncentrace pomocí jednoduché spojité funkce, která není v počátečním bodě diferencovatelná.
Je třeba mít na paměti, že analýza může být prostou minimalizací reziduálních součtů čtverců (za předpokladu konstantního rozptylu) nebo vážených čtverců, pokud se kompenzuje heterogenita rozptylu.
Postup
Postup lze nastínit následovně: zvolte příslušnou funkční rovnici, Y = f (C), a uzpůsobte ji podle údajů pomocí nelineární regrese. Je lepší použít měření z každé jednotlivé baňky než střední hodnoty opakování, aby se z dat získalo co nejvíce informací. Jestliže je rozptyl vysoký, praktická zkušenost naopak naznačuje, že střední hodnoty opakování mohou poskytovat spolehlivější matematický odhad, méně ovlivněný náhodnými chybami v datech, než tomu je u každého zachovaného jednotlivého datového bodu.
Vyneste vytvořenou křivku a naměřená data a prozkoumejte, zda-li byla křivka vynesena správně. Analýza reziduálních hodnot může být pro tento účel mimořádně užitečným nástrojem. Jestliže zvolený funkční vztah pro vyjádření odezvy koncentrace nepopisuje celou křivku nebo její některou zásadně důležitou část, například odezvu při propadu nízkých koncentrací, zvolte jinou variantu vložení křivky – např. nesymetrickou křivku, jako je Weibullova funkce, namísto křivky symetrické. Negativní inhibice mohou být problém například u lognormální distribuční funkce, která podobně vyžaduje alternativní regresní funkci. Takovým negativním hodnotám se nedoporučuje přiřazovat nulu nebo malou kladnou hodnotu, protože se tím deformuje distribuce chyb. Může být vhodné nasadit na části křivky samostatnou křivku, například v části s nízkou inhibicí pro odhad hodnot ECnízké x. Vypočítejte ze vsazené rovnice („inverzním odhadem“ C = f–1 (Y)) odhady charakteristického bodu ECx a uveďte minimálně EC50 a jeden nebo dva odhady ECnízké x. Zkušenost z praktického zkoušení ukázala, že preciznost zkoušky růstu řas obyčejně umožňuje přiměřeně přesný odhad na úrovni 10 % inhibice, jsou-li datové body dostačující – pokud se nevyskytne stimulace při nízkých koncentracích jakožto zkreslující faktor. Preciznost odhadu EC20 je často značně lepší než přesnost odhadu EC10, protože EC20 je obvykle umístěna na přibližně lineární části centrální křivky odezvy koncentrace. Někdy lze EC10 interpretovat jen obtížně kvůli růstové stimulaci. Proto zatímco je EC10 normálně zjistitelná s dostatečnou přesností, rovněž se vždy doporučuje uvádět EC20.
Váhové faktory
Experimentální rozptyl není obecně konstantní a typicky zahrnuje proporcionální složku, proto je výhodné běžně provádět váženou regresi. Váhové faktory pro takovou analýzu se obvykle považují za nepřímo úměrné k rozptylu:
w i = 1/Var(r i)
Mnoho regresních programů má možnost vážené regresní analýzy s váhovými faktory uvedenými v tabulce. Váhové faktory by se měly pohodlně normalizovat vynásobením n/Σ w i (n je počet datových bodů) tak, aby se jejich součet rovnal jedné.
Normalizace odezev
Normalizování střední kontrolní odezvou způsobuje určité principiální problémy a vede ke vzniku poněkud komplikované struktury rozptylů. Dělením odezev střední kontrolní odezvou pro získání procentuální hodnoty inhibice se zavádí dodatečná chyba způsobená chybou kontrolní střední hodnoty. Není-li tato chyba zanedbatelně malá, musí se opravit váhové faktory v regresi a intervaly spolehlivosti na kovarianci s kontrolou (17). Je nutné mít na paměti, že je důležitá vysoká preciznost u odhadované střední kontrolní odezvy pro minimalizaci celkového rozptylu u relativní odezvy. Tento rozptyl je následující:
(i v dolním indexu znamená úroveň koncentrace i a 0 v dolním indexu odkazuje na kontroly)
Yi = relativní odezva = ri /r 0 = 1 – I = f (Ci )
s rozptylem:
Var (Yi ) = Var (ri /r 0) δ (δYi / δ ri )2·Var(ri ) + (δ Yi / δ r 0)2·Var (r 0)
a jelikož
(δ Yi / δ ri ) = 1/r 0 a (δ Yi / δ r 0) = ri /r 0 2
s normálně distribuovanými daty a opakováními m i a m0:
Var(ri ) = σ2/mi
se pak celkový rozptyl relativní odezvy Yi vypočítá:
Var(Yi ) = σ2/(r 0 2 mi ) + ri 2·σ2/r 0 4 m 0
Chyba kontrolní střední hodnoty je nepřímo úměrná druhé odmocnině počtu zprůměrovaných kontrolních opakování, přičemž někdy je odůvodněné zanést historická data, čímž se chyba značně sníží. Alternativním postupem je data nenormalizovat a nevynášet absolutní odezvy včetně dat kontrolní odezvy, nýbrž zavést hodnoty kontrolní odezvy jakožto dodatečný parametr, který bude upraven lineární regresí. Zatímco obvyklá regresní rovnice je dvouparametrická, tato metoda vyžaduje vnesení 3 parametrů, a proto potřebuje více datových bodů než nelineární regrese prováděná s daty, která jsou normalizována pomocí předem stanovené kontrolní odezvy.
Inverzní intervaly spolehlivosti
Výpočet nelineárních regresních intervalů spolehlivosti inverzním odhadem je poněkud složité a není to dostupná standardní možnost v běžných balících statistických počítačových programů. Přibližné intervaly spolehlivosti lze získat pomocí standardních programů nelineární regrese s opakovanou parametrizací (Bruce a Versteeg, 1992), což zahrnuje přepisování matematické rovnice za použití požadovaných odhadů bodů, např. EC10 a EC50 jakožto parametrů, které se mají odhadnout. Nechť je funkce I = f (α, β, koncentrace); definiční vztahy f (α, β, EC10) = 0,1 a f (α, β, EC50) = 0,5 se použijí k nahrazení f (α, β, koncentrace) ekvivalentní funkcí g (EC10, EC50, koncentrace).
Přímější výpočet (Andersen et al., 1998) se provádí zachováním původní rovnice a použitím Taylorova rozšíření okolo středních hodnot ri a r 0.
V poslední době se staly oblíbenými metody „bootstrap“. Takové metody používají naměřených údajů a častého opětovného odběru vzorků určovaného generátorem náhodných čísel pro odhad empirické distribuce rozptylů.
Literatura
Kooijman, S.A.L.M., Hanstveit, A.O., Nyholm, N. (1996): No-effect concentrations in algal growth inhibition tests. Water Research, 30, 1625–1632.
Bruce, R.D.; Versteeg, D.J.(1992): A Statistical Procedure for Modelling Continuous Ecotoxicity Data. Env. Toxicol. Chem.11, 1485–1494.
Andersen, J.S., Holst, H., Spliid, H., Andersen, H., Baun, A., Nyholm, N. (1998): Continuous ecotoxicological data evaluated relative to a control response. Journal of Agricultural, Biological and Environmental Statistics, 3, 405–420.
PŘÍLOHA V
|
C.25 |
AEROBNÍ MINERALIZACE V POVRCHOVÉ VODĚ – SIMULAČNÍ ZKOUŠKA BIOLOGICKÉHO ROZKLADU |
1. METODA
Tato metoda je rovnocenná Pokynu OECD pro zkoušení 309 (2004) (1).
1.1 ÚVOD
Cílem této zkoušky je změřit časový průběh biologického rozkladu zkoušené látky při nízké koncentraci v aerobní přírodní vodě a kvantifikovat pozorování ve formě výrazů pro kinetickou rychlost. Tato simulační zkouška je dávková zkouška v laboratorní třepací lahvi pro stanovení rychlostí aerobního biologického rozkladu organických látek ve vzorcích přírodní povrchové vody (sladkovodní, brakické nebo mořské). Je založena na ISO/DIS 14592–1 (2) a rovněž obsahuje prvky ze zkušebních metod C.23 a C.24 (3) (4). Volitelně při dlouhých zkušebních časech je dávkové zkoušení nahrazeno polospojitou zkouškou s cílem zabránit zhoršení zkušebního mikrokosmu. Základním cílem simulační zkoušky je určit mineralizaci zkoušené látky v povrchové vodě a mineralizace tvoří základnu pro vyjádření kinetiky rozkladu. Ovšem volitelným sekundárním cílem zkoušky je získat informace o primárním rozkladu a tvorbě významnějších transformačních produktů. Určení transformačních produktů, a bude-li to možné, kvantitativní stanovení jejich koncentrací jsou zvláště důležité pro látky, které jsou velmi pomale mineralizované (např. s poločasy pro celkový zbytkový 14C překračující 60 dnů). Pro určení a kvantitativní stanovení významnějších transformačních produktů by se vzhledem k analytickým omezením měly obvykle používat vyšší koncentrace zkoušené látky (např. > 100 μg/l).
Nízká koncentrace v této zkoušce znamená koncentraci (např. méně než 1 μg/l až 100 μg/l), která je dostatečně nízká na to, aby zajistila, že kinetika biologického rozkladu získaná ve zkoušce odráží kinetiku očekávanou v životním prostředí. V porovnání s celkovou hmotností biologicky rozložitelných uhlíkových substrátů dostupných v přírodní vodě používané pro zkoušku slouží zkoušená látka přítomná v nízké koncentraci jako sekundární substrát. To znamená, že očekávaná kinetika biologického rozkladu je prvního řádu („nerůstová“ kinetika) a že zkoušená látka může být rozkládána „spolumetabolismem“. Z kinetiky prvního řádu vyplývá, že rychlost rozkladu (mg/l/den) je přímo úměrná koncentraci substrátu, která s časem klesá. Při opravdové kinetice prvního řádu je specifická rychlostní konstanta rozkladu, k, nezávislá na času a koncentraci. To znamená, že k se znatelně nemění během doby experimentu a nemění se s rostoucí koncentrací během experimentů. Z definice je specifická rychlostní konstanta rozkladu rovna relativní změně koncentrace za čas: k = (1/C) · (dC/dt). Ačkoliv se za předepsaných podmínek normálně očekává kinetika prvního řádu, mohou nastat určité okolnosti, za kterých se projeví kinetiky jiných řádů. Odchylky od kinetiky prvního řádu lze pozorovat například tehdy, jestliže rychlost biologické transformace omezuje spíše než biologická reakční rychlost jev spojený s přenosem hmoty, například rychlost difuse. Ovšem údaje lze téměř vždy popsat kinetikou pseudoprvního řádu a přijmout rychlost závislou na koncentraci jako konstantní.
Informace o biologické rozložitelnosti zkoušené látky při vyšších koncentracích (např. ze standardních screeningových zkoušek) společně s informacemi o abiotické rozložitelnosti, transformačních produktech a relevantních fyzikálně-chemických vlastnostech by měly být dostupné před zkouškou jako pomůcka pro vypracování plánu experimentu a interpretaci výsledků. Použití zkoušených látek značených isotopem 14C a stanovení fázové distribuce 14C na konci zkoušky umožňuje stanovení úplné biologické rozložitelnosti. Při použití neznačené zkoušené látky lze úplný biologický rozklad pouze odhadovat, pokud se zkouší vyšší koncentrace a jsou známy všechny významnější transformační produkty.
1.2 DEFINICE
Primární biologický rozklad: strukturální změna (transformace) chemické látky působením mikroorganismů, která má za následek ztrátu chemické identity.
Funkční biologický rozklad: strukturální změna (transformace) chemické látky působením mikroorganismů, která má za následek ztrátu specifické vlastnosti.
Úplný aerobní biologický rozklad: rozložení chemické látky působením mikroorganismů za přítomnosti kyslíku na oxid uhličitý, vodu a minerální soli jakýchkoli jiných přítomných prvků (mineralizace) a tvorba nové biomasy a organických mikrobiálních produktů biologické syntézy.
Mineralizace: rozložení chemické látky nebo organické hmoty působením mikroorganismů za přítomnosti kyslíku na oxid uhličitý, vodu a minerální soli jakýchkoli jiných přítomných prvků.
Fáze iniciace: doba od počátku zkoušky do dosažení adaptace rozkládajících mikroorganismů a zvýšení stupně biologického rozkladu chemické látky nebo organické hmoty na detekovatelnou úroveň (např. 10 % maximálního teoretického biologického rozkladu nebo nižší v závislosti na přesnosti techniky měření).
Maximální úroveň biologického rozkladu: stupeň biologického rozkladu chemické látky nebo organické hmoty při zkoušce uváděný v procentech, při jehož překročení neprobíhá během zkoušky žádný další biologický rozklad.
Primární substrát: soubor přírodního uhlíku a zdrojů energie, který zajišťuje růst a udržování mikrobiální biomasy.
Sekundární substrát: substrátová složka přítomná v tak nízké koncentraci, že svými rozkladem dodává pouze nevýznamná množství uhlíku a energie kompetentním mikroorganismům v porovnání s uhlíkem a energií dodávanou rozkladem hlavních složek substrátu (primárních substrátů).
Rychlostní konstanta rozkladu: rychlostní konstanta kinetiky prvního nebo pseudoprvního řádu, k (d–1), která ukazuje rychlost rozkladných procesů. U dávkového experimentu se k odhaduje z počáteční části rozkladové křivky získané po ukončení fáze iniciace.
Poločas, t 1/2 (d): termín používaný pro charakteristiku rychlosti reakce prvního řádu. Je to časový interval, který odpovídá poklesu koncentrace na polovinu. Poločas a rychlostní konstanta rozkladu jsou ve vzájemném vztahu vyjádřeném rovnicí t 1/2 = ln2/k.
Poločas rozkladu, DT50 (d): termín používaný pro kvantifikaci výsledků zkoušek biologického rozkladu. Je to časový interval včetně fáze iniciace potřebný k dosažení 50 % biologického rozkladu.
Mez detekce (LOD) a mez kvantifikace (LOQ): Mez detekce (LOD) je koncentrace látky, pod níž nelze totožnost látky rozlišit od analytických artefaktů. Mez kvantifikace (LOQ) je koncentrace látky, pod níž nelze koncentraci určit s přijatelnou přesností.
Rozpuštěný organický uhlík (dissolved organic carbon, DOC): ta část organického uhlíku ve vzorku vody, kterou nelze odstranit stanovenou fázovou separací, například odstřeďováním po dobu 15 minut při zrychlení 40 000 ms–2 nebo membránovou filtrací pomocí membrán s póry o průměru 0,2 μm až 0,45 μm.
Celková aktivita organického 14C (total organic 14C activity, TOA): Celková aktivita 14C spojená s organickým uhlíkem.
Aktivita rozpuštěného organického 14C (dissolved organic 14C activity, DOA): Celková aktivita 14C spojená s rozpuštěným organickým uhlíkem.
Aktivita částicového organického 14C (particulate organic 14C activity, POA): Celková aktivita 14C spojená s částicovým organickým uhlíkem.
1.3 POUŽITELNOST ZKUŠEBNÍ METODY
Tato simulační zkouška je použitelná pro netěkavé nebo slabě těkavé organické látky zkoušené při nízkých koncentracích. Při použití baněk otevřených do atmosféry (např. se zátkou z vaty) lze látky s Henryho konstantou nižší než 1 Pa·m3/mol (přibližně 10–5 atm·m3/mol) považovat za prakticky netěkavé. Při použití uzavřených baněk s dostatečným volným prostorem nad látkou je možné zkoušet mírně těkavé látky (s Henryho konstantami < 100 Pa·m3/mol nebo < 10–3 atm·m3/mol) beze ztrát ve zkušebním systému. Při odstraňování CO2 může dojít ke ztrátám látek značených isotopem 14C, pokud nejsou přijata správná předběžná opatření. V takových situacích může být nezbytné jímat CO2 v interním absorbéru s alkálií nebo použít externí systém pro absorpci CO2 (přímé stanovení 14CO2, viz dodatek 3). Pro stanovení kinetiky biologického rozkladu musí být koncentrace zkoušené látky nižší než její rozpustnost ve vodě. Je však nutné uvést, že hodnoty rozpustnosti ve vodě uváděné v literatuře mohou být významně vyšší než rozpustnost zkoušené látky v přírodních vodách. Případně lze stanovit rozpustnost zkoušených látek se zvláště špatnou rozpustností ve vodě pomocí zkušebních přírodních vod.
Tuto metodu lze použít pro simulování biologického rozkladu v povrchové vodě bez hrubých částic („pelagická zkouška“) nebo ve zkalené povrchové vodě, která by například mohla existovat v blízkosti rozhraní voda/sediment („zkouška se suspendovanými sedimenty“).
1.4 PODSTATA ZKOUŠKY
Zkouška se provádí v dávce inkubací zkoušené látky buď pouze s povrchovou vodou („pelagická zkouška“), nebo povrchovou vodou doplněnou suspendovanými pevnými látkami/sedimenty o suché hmotnosti 0,01 až 1 g/l („zkouška se suspendovanými sedimenty“) pro simulaci vodní hmoty se suspendovanými pevnými látkami nebo resuspendovanými sedimenty. Koncentrace suspendovaných pevných látek/sedimentů v nižším rozmezí tohoto intervalu je typická pro většinu povrchových vod. Zkušební baňky se inkubují ve tmě za teploty okolí za aerobních podmínek a míchání. Pro stanovení kinetiky rozkladu je zapotřebí použít nejméně dvou odlišných koncentrací zkoušené látky. Koncentrace by se měly navzájem lišit faktorem 5 až 10 a měly by představovat očekávané rozmezí koncentrací v životním prostředí. Maximální koncentrace zkoušené látky by neměla překročit 100 μg/l, ale dává se přednost maximálním koncentracím nižším než 10 μg/l, aby se zajistilo, že biologický rozklad dodržuje kinetiku prvního řádu. Nejnižší koncentrace by neměla překročit 10 μg/l, ale dává se přednost nejnižším zkušebním koncentracím 1–2 μg/l nebo méně než 1 μg/l. Normálně lze odpovídající analýzy takové nízké koncentrace provést pomocí komerčně dostupných látek značených isotopem 14C. Kvůli analytickým omezením je často nemožné změřit koncentraci zkoušené látky s požadovanou přesností, pokud se zkoušená látka používá v koncentraci ≤ 100 µg/l (viz bod 1.7.2 druhý odstavec). Vyšší koncentrace zkoušené látky (> 100 μg/l a někdy > 1 mg/l) lze použít pro indentifikaci a stanovení množství významnějších transformačních produktů, nebo není-li k dispozici specifická analytická metoda s nízkou mezí detekce. Jestliže se zkouší vysoké koncentrace zkoušené látky, nemusí být použití výsledků pro odhad rozkladové konstanty prvního řádu a poločasu možné, protože rozklad pravděpodobně nebude probíhat podle kinetiky prvního řádu.
Rozklad je sledován v příslušných časových intervalech měřením zbytkového 14C nebo zbytkové koncentrace zkoušené látky při použití specifické chemické analýzy. Značení nejstabilnější části molekuly isotopem 14C zaručuje určení celkové mineralizace, zatímco značení méně stabilní části molekuly isotopem 14C společně s využitím specifické analýzy umožňuje hodnotit pouze primární biologický rozklad. Ovšem nejstabilnější část nemusí nutně obsahovat relevantní funkční část molekuly (kterou lze vztahovat ke konkrétní vlastnosti, například toxicitě, bioakumulaci atd.). V tomto případě může být vhodné použít zkoušenou látku, která je značena isotopem 14C, ve funkční části ke sledování eliminace konkrétní vlastnosti.
1.5 INFORMACE O ZKOUŠENÉ LÁTCE
V této zkoušce lze použít jak zkoušené látky značené radioisotopy, tak látky neznačené. Doporučuje se metoda značení isotopem 14C, přičemž značena by obvykle měla být nejstabilnější část (části) molekuly (viz také bod 1.4). U látek obsahujících více než jeden aromatický kruh by se isotopem 14C měly nejlépe označit jeden či více uhlíků v každém kruhu. Dále by se isotopem 14C měl nejlépe označit jeden či více uhlíků na obou stranách snadno rozložitelných vazeb. Chemická a/nebo radiochemická čistota zkoušené látky by měla být > 95 %. U radioisotopově značených látek se dává přednost specifické aktivitě přibližně 50 μCi/mg (1,85 MBq) nebo více, aby se usnadnila měření 14C u zkoušek prováděných s nízkými počátečními koncentracemi. O zkoušené látce by měly být k dispozici následující informace:
|
— |
rozpustnost ve vodě (metoda A.6), |
|
— |
rozpustnost v organickém(ých) rozpouštědle(ch) (látky používané s rozpouštědlem nebo s nízkou rozpustností ve vodě), |
|
— |
disociační konstanta (pK a), jestliže látka podléhá protonaci nebo deprotonaci (Pokyn OECD pro zkoušení 112) (5), |
|
— |
tlak par (metoda A.4) a Henryho konstanta, |
|
— |
chemická stálost ve vodě a ve tmě (hydrolýza) (metoda C.7). |
Zkouší-li se v mořské vodě látky špatně rozpustné ve vodě, může být rovněž užitečné znát vysolovací konstantu (neboli „Sečenovovu konstantu“) Ks , která je definována výrazem: log (S/S') = Ks C m, kde S je rozpustnost látky ve sladké vodě a S' v mořské vodě, a C m je molární koncentrace soli.
Jestliže se zkouška provádí jako „zkouška se suspendovanými sedimenty“, měly by být k dispozici rovněž následující informace:
|
— |
rozdělovací koeficient oktanol-1-ol/voda (metoda A.8), |
|
— |
adsorpční koeficient (metoda C.18). |
Mezi další užitečné informace patří:
|
— |
koncentrace v životním prostředí, je-li známá nebo odhadnutá, |
|
— |
toxicita zkoušené látky pro mikroorganismy (metoda C.11), |
|
— |
snadná a/nebo vlastní biologická rozložitelnost (metody C.4 A–F, C.12, C.9, Pokyn OECD pro zkoušení 302 (5)), |
|
— |
aerobní nebo anaerobní biologická rozložitelnost v půdě a studie transformace v systémech voda/sediment (metody C.23, C.24). |
1.6 REFERENČNÍ LÁTKA
Jako referenční látka by se měla používat látka, která se za aerobních podmínek obvykle snadno rozkládá (např. anilin nebo benzoát sodný). Očekávaný časový interval pro rozklad anilinu a benzoátu sodného je obvykle méně než 2 týdny. Účelem referenčních látek je zajistit, aby byla mikrobiální aktivita zkušební vody v určitých mezích, tj., aby voda obsahovala aktivní mikrobiální populaci.
1.7 KRITÉRIA JAKOSTI
1.7.1 Výtěžnost
Bezprostředně po přidání zkoušené látky je třeba ověřit každou počáteční zkušební koncentraci měřením aktivity 14C nebo – v případě neznačených látek – chemickými analýzami vždy nejméně v duplikátních vzorcích. Takto se získají informace o použitelnosti a opakovatelnosti analytické metody a o homogenitě distribuce zkoušené látky. V následných analýzách údajů se obvykle namísto nominální koncentrace použije spíše naměřená počáteční aktivita 14C nebo koncentrace zkoušené látky, neboť se jejich použitím kompenzují ztráty způsobené sorpcí a chybami při dávkování. U zkoušené látky značené isotopem 14C se hodnota výtěžnosti na konci experimentu udává pomocí hmotnostní bilance (viz bod 1.8.9.4 poslední odstavec). V ideálním případě by se hmotnostní bilance radioisotopově značené látky měla pohybovat od 90 % do 110 %, zatímco analytická přesnost by měla vést k počáteční výtěžnosti od 70 % do 110 % pro neznačené zkoušené látky. Tato rozmezí je zapotřebí vykládat jako cíle a neměla by se používat jako kritéria pro přijetí zkoušky. Případně lze analytickou přesnost stanovit pro zkoušenou látku při nižší koncentraci, než je koncentrace počáteční, a pro významnější transformační produkty.
1.7.2 Opakovatelnost a citlivost analytické metody
Opakovatelnost analytické metody (včetně účinnosti první extrakce) pro kvantitativní stanovení zkoušené látky a případně transformačních produktů by se měla ověřit pěti opakovanými analýzami jednotlivých extraktů povrchové vody.
Mez detekce (LOD) analytické metody pro zkoušenou látku a pro transformační produkty by měla být pokud možno alespoň 1 % počátečního množství použitého ve zkušebním systému. Mez kvantifikace (LOQ) by se měla rovnat nebo být menší než 10 % použité koncentrace. Chemické analýzy mnoha organických látek a jejich transformačních produktů často vyžadují, aby se zkoušená látka používala v relativně vysoké koncentraci, tj. > 100 μg/l.
1.8 POPIS ZKUŠEBNÍ METODY
1.8.1 Vybavení
Zkoušku lze provádět v Erlenmeyerových baňkách nebo válcových nádobách vhodné kapacity (např. 0,5 nebo 1,0 litru) uzavřených silikonovou nebo pryžovou zátkou nebo v baňkách na sérum s víky těsnícími proti úniku CO2 (např. opatřené septou z butylové pryže). Další možností je provádět zkoušku pomocí více baněk a používat jako vzorek celý objem vždy nejméně dvou baněk v každém intervalu odběru vzorku (viz bod 1.8.9.1 poslední odstavec). Pro netěkavé zkoušené látky, které nejsou radioisotopově označeny, nejsou nutné plynotěsné zátky nebo víka; vhodné jsou volné vatové zátky, které brání kontaminaci ze vzduchu (viz bod 1.8.9.1 druhý odstavec). Lehce těkavé látky by měly být zkoušeny v systému biometrického typu s jemným mícháním povrchu vody. Aby se zajistilo, že nedojde k bakteriální kontaminaci, lze nádoby před použitím případně sterilizovat ohřevem nebo autoklávováním. Dále se používá toto standardní laboratorní vybavení:
|
— |
desková třepačka nebo magnetické míchačky pro nepřetržité míchání zkušebních baněk, |
|
— |
odstředivka, |
|
— |
pH-metr, |
|
— |
turbidimetr pro nefelometrická měření turbidity (zákalu), |
|
— |
pec nebo mikrovlnná pec pro stanovení suché hmotnosti, |
|
— |
membránový filtrační přístroj, |
|
— |
autokláv nebo pec pro tepelnou sterilizaci skleněného nádobí, |
|
— |
zařízení pro manipulaci s látkami značenými isotopem 14C, |
|
— |
zařízení pro kvantifikaci aktivity 14C ve vzorcích z roztoků pro s jímaným CO2 a, bude-li to nutné, ze vzorků sedimentů, |
|
— |
analytické vybavení pro určení zkoušené (a referenční) látky, jestliže se použije specifická chemická analýza (např. plynový chromatograf, chromatograf pro vysokotlakou kapalinovou chromatografii). |
1.8.2 Zásobní roztoky zkoušené látky
Pro přípravu zásobních roztoků zkoušených a referenčních látek se používá deionizovaná voda (viz bod 1.8.7 první odstavec). Deionizovaná voda by měla být zbavená látek, které mohou být toxické pro mikroorganismy a podíl rozpuštěného organického uhlíku (DOC) by neměl být vyšší než 1 mg/l (6).
1.8.3 Odběr a přeprava povrchové vody
Místo odběru povrchové vody by mělo být vybráno podle účelu zkoušky v dané situaci. Při výběru míst odběru musí být přihlédnuto k historii případných zemědělských, průmyslových nebo domácích vstupů. Jestliže je známo, že bylo vodní prostředí v předchozích čtyřech letech kontaminováno zkoušenou látkou nebo jejími strukturními analogy, nemělo by se používat k odběru zkušební vody, s výjimkou případů, kdy je zkoumání rychlostí rozkladu v dříve exponovaných místech výslovným účelem zkoušejícího. V místě odběru by se měly změřit pH a teplota vody. Dále je třeba zaznamenat hloubku odběru a vzhled vzorku vody (např. barva a turbidita) (viz bod 3). Měly by se měřit koncentrace kyslíku a/nebo oxidačně-redukční potenciál ve vodě a v povrchové vrstvě sedimentu s cílem prokázat aerobní podmínky, s výjimkou případů, kdy je jejich existence patrná ze vzhledu a historické zkušenosti s daným místem. Povrchová voda by se měla přepravovat v důkladně vyčištěném zásobníku. Během přepravy by teplota vzorku neměla významně překračovat teplotu použitou při zkoušce. Doporučuje se chlazení na 4 °C, pokud trvání přepravy překračuje 2 až 3 hodiny. Vzorek vody se nesmí zmrazit.
1.8.4 Skladování a příprava povrchové vody
Zkouška by měla být nejlépe zahájena do jednoho dne po odběru vzorku. Skladování vody, je-li nutné, by se mělo minimalizovat a nesmí v žádném případě překračovat maximální hranici 4 týdnů. Vzorek vody je zapotřebí skladovat až do použití při teplotě 4 °C za provzdušňování. Před použitím je třeba odstranit hrubé částice, např. filtrací přes nylonový filtr s velikostí ok okolo 100 μm nebo přes hrubý papírový filtr, případně sedimentací.
1.8.5 Příprava vody doplněné sedimentem (nepovinné)
Pro zkoušku se suspendovanými sedimenty se do baněk obsahujících přírodní vodu (přefiltrovanou za účelem odstranění hrubých částic, jak je popsáno v bodě 1.8.4) přidává povrchový sediment, aby vznikla suspenze; koncentrace suspendovaných pevných látek by měla být v rozmezí 0,01 až 1 g/l. Povrchový sediment by měl pocházet ze stejného místa, z něhož byl odebrán vzorek vody. V závislosti na konkrétním vodním prostředí může být povrchový sediment buď charakterizován vysokým obsahem organického uhlíku (2,5–7,5 %) a jemnou strukturou, nebo nízkým obsahem organického uhlíku (0,5–2,5 %) a hrubou strukturou (3). Povrchový sediment lze připravit následovně: odeberte několik jader sedimentu pomocí zkumavky z průhledného plastu, bezprostředně po odběru vzorku oddělte horní aerobní vrstvy (od povrchu do hloubky maximálně 5 mm) a smiste je. Výsledný vzorek sedimentu by měl být přepravován v zásobníku s velkým vzduchovým prostorem nad sedimentem, aby se sediment udržel za aerobních podmínek (chladit na 4 °C, jestliže doba přepravy překračuje 2 až 3 hodiny). Vzorek sedimentu by měl být suspendován ve zkušební vodě v poměru 1:10 a do použití skladován při teplotě 4 °C za provzdušňování. Skladování sedimentu, je-li nutné, by se mělo minimalizovat a nesmí v žádném případě překračovat maximální hranici 4 týdnů.
1.8.6 Polospojitý postup (nepovinné)
Jestliže nastává dlouhá fáze iniciace před tím, než je možné měřit významný rozklad zkoušené látky, může být nezbytná prodloužená inkubace (několik měsíců). Pokud je tato skutečnost z předchozího zkoušení látky známa, zkoušku lze zahájit používáním polospojitého postupu, který umožňuje pravidelné obnovování části zkušební vody nebo suspenze (viz dodatek 2). Alternativně lze normální dávkovou zkoušku změnit na polospojitou zkoušku, pokud nebylo dosaženo rozkladu zkoušené látky přibližně během 60 dnů zkoušení s využitím dávkového postupu (viz bod 1.8.8.3 druhý odstavec).
1.8.7 Přidání zkoušené (nebo referenční) látky
U látek s vysokou rozpustností ve vodě (> 1 mg/l) a nízkou těkavostí (Henryho konstanta < 1 Pa·m3/mol nebo < 10–5 atm·m3/mol) lze připravit zásobní roztok v deionizované vodě (viz bod 1.8.2), vhodný objem zásobního roztoku se přidává do zkušebních nádob pro dosažení požadované koncentrace. Objem jakéhokoliv přidaného zásobního roztoku je třeba omezit na co nejnižší možnou míru (< 10 % konečného objemu kapaliny, je-li to možné). Dalším postupem, který lze považovat za alternativu pro používání organických rozpouštědel, je rozpustit zkoušenou látku ve větším objemu zkušební vody.
Je-li to nevyhnutelné, měly by se zásobní roztoky netěkavých látek se špatnou rozpustností ve vodě připravit za použití těkavého organického rozpouštědla, avšak množství rozpouštědla přidaného do zkušebního systému by nemělo překročit 1 % obj. a nemělo by mít nepříznivé účinky na mikrobiální aktivitu. Rozpouštědlo by nemělo ovlivňovat stálost zkoušené látky ve vodě. Rozpouštědlo by mělo být odstraněno na mimořádně malé množství tak, aby významně nezvyšovalo koncentraci DOC zkušební vody nebo suspenze. Toto je třeba zkontrolovat analýzou specifickou pro danou látku, nebo je-li to možné, analýzou DOC (6). Musí se vynaložit úsilí na omezení přenášeného rozpouštědla na absolutně nezbytné množství a zajistit, aby množství zkoušené látky bylo možné rozpustit v konečném objemu zkušební vody. Pro zavedení zkoušené látky do zkušebních nádob lze používat i další metody, které jsou popsané v literatuře (7) a (8). Používá-li se k aplikaci zkoušené látky organické rozpouštědlo, měly by se kontroly s rozpouštědlem obsahující zkušební vodu (bez přídavků) a zkušební vodu s přidanou referenční látkou exponovat podobně jako aktivní zkušební nádoby doplněné zkoušenou látkou v nosném rozpouštědle. Cílem kontrol s rozpouštědlem je prozkoumat na základě rozkladu referenční látky možné nežádoucí účinky na mikrobiální populaci způsobené rozpouštědlem.
1.8.8 Zkušební podmínky
1.8.8.1 Zkušební teplota
Inkubace by měla probíhat (nejlépe) v temnu nebo v difuzním světle při kontrolované (±2 °C) teplotě, která může být teplotou terénu nebo standardní teplotou 20 až 25 °C. Teplotou terénu může být buď skutečná teplota vzorku v době odběru, nebo průměrná teplota terénu na místě odběru.
1.8.8.2 Promíchávání
Je nutné zajistit promíchávání nepřetržitým protřepáváním nebo mícháním, aby se částice a mikroorganismy udržovaly v suspenzi. Promíchávání rovněž umožňuje přenos kyslíku do kapaliny z volného prostoru nad kapalinou tak, aby bylo možné odpovídajícím způsobem udržovat aerobní podmínky. Umístěte baňky na deskovou třepačku (promíchávání přibližně při 100 ot/min) nebo použijte magnetické míchání. Promíchávání musí být nepřetržité. Třepání nebo míchání by však mělo být co nejjemnější, přičemž je třeba udržovat homogenní suspenzi.
1.8.8.3 Doba trvání zkoušky
Doba trvání zkoušky by obecně neměla překročit 60 dnů, pokud se nepoužije polospojitý postup s pravidelným obnovováním zkušební suspenze (viz bod 1.8.6 a dodatek 2). Ovšem doba trvání může být u dávkové zkoušky prodloužena na maximálně 90 dnů, jestliže rozklad zkoušené látky začal během prvních 60 dnů. Rozklad se sleduje ve vhodných časových intervalech určováním zbytkové aktivity 14C nebo uvolněného 14CO2 (viz bod 1.8.9.4) a/nebo chemickou analýzou (bod 1.8.9.5). Doba inkubace musí být dostatečně dlouhá pro vyhodnocení procesu rozkladu. Rozsah rozkladu by měl nejlépe překročit 50 %; u pomalu rozložitelných látek musí být rozsah rozkladu dostatečný (obvykle větší než 20 % rozklad) k tomu, aby byl možný odhad kinetické rychlostní konstanty rozkladu.
Musí se provádět pravidelná měření pH a koncentrace kyslíku ve zkušebním systému, pokud nejsou k dispozici předchozí zkušenosti z podobných zkoušek se vzorky vody a sedimentů odebraných ze stejného místa, přičemž v takovém případě by tato měření nebyla potřebná. Za určitých podmínek by mohl metabolismus primárních substrátů při mnohem vyšších koncentracích ve vodě nebo v sedimentu vést k tak velkému uvolňování CO2, a tak značnému vyčerpání kyslíku, že by to významně změnilo experimentální podmínky během zkoušky.
1.8.9 Postup
1.8.9.1 Příprava baněk pro pelagickou zkoušku
Do zkušebních baněk se převede vhodný objem zkušební vody, a to až do přibližně jedné třetiny objemu baňky a ne méně než přibližně 100 ml. Jestliže se používá více baněk (umožňujících zkoušení na celém objemu baňky v každém čase odběru vzorku), je vhodný objem zkušební vody rovněž přibližně 100 ml, protože malé objemy vzorku mohou ovlivnit délku fáze iniciace. Zkoušená látka se přidá ze zásobního roztoku, jak je popsáno v bodech 1.8.2 a 1.8.7. Pro určení kinetiky rozkladu a výpočet kinetické rychlostní konstanty rozkladu by se měly použít nejméně dvě odlišné koncentrace zkoušené látky lišící se faktorem 5 až 10. Obě zvolené koncentrace by měly být nižší než 100 μg/l a nejlépe v rozmezí < 1–10 μg/l.
Baňky se uzavřou zátkami nebo víčky nepropustnými pro vzduch a CO2. U netěkavých zkušebních chemických látek neznačených isotopem 14C jsou vhodné volné vatové zátky, které brání kontaminaci ze vzduchu (viz bod 1.8.1), pokud je ovšem o všech hlavních produktech rozkladu známo, že jsou netěkavé, a pokud se používá nepřímé stanovení CO2 (viz dodatek 3).
Baňky se inkubují při zvolené teplotě (viz bod 1.8.8.1). Vzorky pro chemickou analýzu nebo měření 14C je třeba odebrat na počátku zkoušky (tj. předtím, než začne biologický rozklad; viz bod 1.7.1) a poté ve vhodných časových intervalech v průběhu zkoušky. Odběr vzorků lze provádět odběrem dílčích vzorků (např. 5 ml alikvotních objemů) z každého opakování nebo odběrem vždy celého objemu baňky v každém čase odběru vzorku. Mineralizace zkoušené látky může být určena buď přímo, nebo nepřímo (viz dodatek 3). Obvykle se vyžaduje minimálně pět bodů pro odběr vzorku během fáze rozkladu (tj. po ukončené fázi iniciace) s cílem odhadnout spolehlivou rychlostní konstantu, s výjimkou případů, kdy je možné odůvodnit, že u rychle rozložitelných látek jsou dostatečné tři body odběru vzorku. U látek, které nepodléhají rychlému rozkladu, lze snadno provést více měření během fáze rozkladu, a proto by se k odhadu k mělo používat více datových bodů. Pro odběr vzorků nelze uvést žádný pevný časový harmonogram, protože rychlost biologického rozkladu se v jednotlivých případech liší; ovšem doporučuje se odebírat vzorky jednou týdně, pokud je rozklad pomalý. Jestliže zkoušená látka podléhá rychlému rozkladu, odběr vzorků by se měl během prvních tří dnů provádět jednou denně a poté každý druhý nebo třetí den. Za určitých okolností, například u velmi rychle hydrolyzujících látek, může být nezbytné odebírat vzorky v hodinových intervalech. Doporučuje se, aby se před zkouškou provedla předběžná studie s cílem určit vhodné intervaly odběru vzorků. Pokud musí být vzorky k dispozici pro další specifickou analýzu, je užitečné odebrat více vzorků a poté na konci experimentu zvolit ty, které se mají analyzovat, v opačném pořadí, tj. poslední vzorky se analyzují první (viz bod 1.8.9.5 druhý odstavec, kde jsou pokyny ke stálosti vzorků během skladování).
1.8.9.2 Počet baněk a vzorků
Nasaďte dostatečný počet zkušebních baněk v tomto rozsahu:
|
— |
zkušební baňky; nejméně dvojí baňky pro každou koncentraci zkoušené látky (nejlépe minimálně 3) nebo více zkušebních baněk pro každou koncentraci, jestliže se jako vzorek použije vždy celý objem baňky v každém čase odběru vzorků (označeno symbolem FT), |
|
— |
zkušební baňky pro výpočet hmotnostní bilance; nejméně dvojí baňky pro každou zkušební koncentraci (označeno symbolem FM), |
|
— |
slepé zkoušky, bez zkoušené látky; nejméně jedna zkušební baňka pro slepou zkoušku obsahující pouze zkušební vodu (označeno symbolem FB), |
|
— |
referenční kontrola; dvojí baňky s referenční látkou (např. anilinem nebo benzoátem sodným v koncentraci 10 μg/l) (označeno symbolem FC). Cílem referenční kontroly je potvrdit minimum mikrobiální aktivity. Je-li to výhodné, lze použít radioisotopově značenou referenční látku, a to i tehdy, je-li rozklad zkoušené látky sledován chemickými analýzami, |
|
— |
sterilní kontrola; jedna nebo dvě baňky obsahující sterilizovanou zkušební vodu ke zkoumání možného abiotického rozkladu nebo jiného nebiologického odstranění zkoušené látky (označeno symbolem FS). Biologickou aktivitu lze zastavit autoklávováním (121 °C, 20 minut) zkušební vody nebo přidáním toxické látky (např. azidu sodného (NaN3) v množství 10–20 g/l, chloridu rtuťnatého (HgCl2) v množství 100 mg/l nebo formalinu v množství 100 mg/l) nebo zářením gama. Použije-li se HgCl2, měl by se zneškodnit jako toxický odpad. U vody s velkým množstvím přidaného sedimentu se sterilních podmínek dosahuje nesnadno; v tomto případě se doporučuje opakované autoklávování (např. třikrát). Je třeba vzít v úvahu, že sorpční charakteristiky sedimentu se mohou autoklávováním pozměnit, |
|
— |
kontroly s rozpouštědlem obsahující zkušební vodu a zkušební vodu s referenční látkou; dvojí baňky ošetřené stejným množství rozpouštědla a podrobené stejnému postupu jako v případě aplikace zkoušené látky. Cílem je zkoumat pomocí určení rozkladu referenční látky možné nežádoucí účinky rozpouštědla. |
V plánu zkoušky by měl zkoušející vzít v úvahu relativní důležitost většího počtu opakování oproti zvýšenému počtu časů odběru vzorků. Přesný počet požadovaných baněk bude záviset na metodě použité pro měření rozkladu (viz bod 1.8.9.1 třetí odstavec, bod 1.8.9.4 a dodatek 3).
V každém čase odběru vzorku by se měly odebrat dva dílčí vzorky (např. 5 ml alikvotních objemů) z každé zkušební baňky. Jestliže se používá více baněk, aby bylo možné použít jako vzorek vždy celý objem baňky, v každém čase odběru vzorků by se měly odebrat minimálně dvě baňky (viz bod 1.8.9.1 první odstavec).
1.8.9.3 Příprava baněk pro zkoušku se suspendovanými sedimenty [nepovinné]
Přidejte do zkušebních nádob potřebná množství zkušební vody a případně sedimentu (viz bod 1.8.5). Příprava baněk pro zkoušku se suspendovanými sedimenty je stejná jako pro pelagickou zkoušku (viz body 1.8.9.1 a 1.8.9.2). Nejlépe použijte lahvičky na sérum nebo baňky podobných tvarů. Umístěte uzavřené baňky horizontálně do třepačky. Je zřejmé, že otevřené baňky pro netěkavé látky neznačené isotopem 14C by se měly umístit do svislé polohy; v tomto případě se doporučuje magnetické míchání a používání magnetických tyčinek pokrytých sklem. Je-li to nezbytné, přivádějte do lahví vzduch, aby se udržely správné aerobní podmínky.
1.8.9.4 Radiochemická stanovení
Uvolněný 14CO2 se měří nepřímo a přímo (viz dodatek 3). Nepřímo se 14CO2 určuje jako rozdíl mezi počáteční aktivitou 14C ve zkušební vodě nebo suspenzi a celkovou zbytkovou aktivitou v době odběru vzorku po okyselení vzorku na pH 2–3 a odvedení CO2. Odstraní se tak anorganický uhlík a naměřená zbytková aktivita se odvodí z organického materiálu. Nepřímé stanovení 14CO2 by se nemělo používat, jestliže během transformace zkoušené látky vznikají významnější těkavé transformační produkty (viz dodatek 3). Je-li to možné, uvolňování 14CO2 by se mělo měřit přímo (viz dodatek 3) v každém čase odběru vzorků v nejméně jedné zkušební baňce; tento postup umožňuje kontrolovat jak hmotnostní bilanci, tak proces biologického rozkladu, ale je omezen na zkoušky prováděné s uzavřenými baňkami.
Jestliže je uvolněný 14CO2 během zkoušky měřen přímo, vyčlení se pro tento účel na počátku zkoušky více baněk. Přímé stanovení 14CO2 se doporučuje tehdy, jestliže během transformace zkoušené látky vznikají významnější těkavé transformační produkty. V každém bodě měření se dodatečné zkušební baňky okyselí na pH 2 až 3 a 14CO2 se jímá v interním či externím absorbéru (viz dodatek 3).
Případně lze koncentrace zkoušené látky značené isotopem 14C a významnějších transformačních produktů stanovit radiochromatograficky (např. chromatografií na tenké vrstvě, RAD-TLC) nebo pomocí HPLC s radiochemickou detekcí.
Případně lze určit fázovou distribuci zbývající radioaktivity (viz dodatek 1) a zbytkovou zkoušenou látku a transformační produkty.
Na konci zkoušky by se měla určit hmotnostní bilance, a to přímým měřením 14CO2 pomocí samostatných zkušebních baněk, z nichž se v průběhu zkoušky neodebírají žádné vzorky (viz dodatek 3).
1.8.9.5 Specifická chemická analýza
Jestliže je k dispozici citlivá specifická analytická metoda, lze primární biologický rozklad hodnotit měřením celkové zbytkové koncentrace zkoušené látky namísto využívání metod značení radioisotopem. Jestliže se použije zkoušená látka značená radioisotopem (pro měření celkové mineralizace), lze souběžně provádět specifické chemické analýzy s cílem získat užitečné dodatečné informace a ověřit postup. Specifické chemické analýzy lze rovněž použít pro měření transformačních produktů vzniklých během rozkladu zkoušené látky, přičemž se toto doporučuje u látek, které jsou mineralizovány s poločasy překračujícími 60 dnů. Měla by se změřit a v protokolu o zkoušce uvést koncentrace zkoušené látky a transformačních produktů v každém čase odběru vzorků (jako koncentrace a jako procentuální podíl použité koncentrace). Obecně je zapotřebí identifikovat transformační produkty zjištěné při ≥ 10 % použité koncentrace v jakoukoliv dobu odběru vzorku; v opačném případě je nutné přiměřeně odůvodnit, proč nebyly identifikovány. Transformační produkty, jejichž koncentrace během studie trvale rostou, by rovněž měly být určeny, a to i tehdy, pokud jejich koncentrace nepřekračuje shora uvedenou mez, neboť tyto transformační produkty mohou být známkou perzistentnosti. Analýzy transformačních produktů ve sterilních kontrolách by se měly vzít v potaz v případě, že se rychlá abiotická přeměna zkoušené látky (např. hydrolýza) považuje za možnou. Potřeba kvantitativního stanovení a identifikace transformačních produktů by se měla posuzovat případ od případu a odůvodnění by měla být uvedena v protokolu o zkoušce. Extrakční postupy s organickými rozpouštědly by se měly používat podle pokynů uvedených v příslušném analytickém postupu.
Všechny vzorky by se měly skladovat vzduchotěsně při teplotách od 2 do 4 °C, pokud se analýza provádí do 24 hodin (nejlépe). Při delším skladování je zapotřebí vzorky zmrazovat na teplotu nižší než –18 °C nebo je chemicky konzervovat. Okyselení jako metoda pro konzervaci vzorků se nedoporučuje, protože okyselené vzorky mohou být nestálé. Jestliže se vzorky neanalyzují do 24 hodin a jsou-li skladovány po delší dobu, je zapotřebí provést studii stability při skladování, aby se prokázala stálost dotčených chemických látek při teplotách skladování nižších než –18 °C nebo při konzervaci. Jestliže analytická metoda vyžaduje extrakci rozpouštědla nebo extrakci tuhé fáze (SPE), je třeba provést tuto extrakci bezprostředně po odběru vzorků nebo po skladování vzorku v chlazeném stavu po dobu maximálně 24 hodin.
V závislosti na citlivosti analytické metody mohou být nezbytné větší objemy vzorků než objemy uvedené v bodě 1.8.1. Zkoušku lze snadno provést při zkušebních objemech jeden litr v baňkách o objemu 2 až 3 litry, což umožňuje odebírat vzorky o objemu přibližně 100 ml.
2. DATA A ZPRÁVY
2.1 ZPRACOVÁNÍ VÝSLEDKŮ
2.1.1 Vynášení dat
Časy odběru vzorku se zaokrouhlí na celé hodiny (s výjimkou případů, kdy se látka významně rozkládá v řádu minut až hodin), ne však na celé dny. Odhady zbytkové aktivity zkoušené látky (pro látky značené isotopem 14C) nebo zbytkové koncentrace (pro neznačené látky) se vynesou proti času jak v lineárním, tak v semilogaritmickém tvaru (viz obrázky 1a a 1b). Jestliže došlo k rozkladu, výsledky z baněk FT je třeba porovnat s výsledky z baněk FS. Jestliže se střední hodnoty výsledků z baněk se zkoušenou látkou (FT) a sterilních baněk (FS) odchylují o méně než 10 %, lze předpokládat, že pozorovaný rozklad je převážně abiotický. Jestliže je rozklad v baňkách FS nižší, lze hodnoty použít pro korekci hodnot získaných pomocí baněk FT (odečtením), odhadnout tak rozsah biologického rozkladu. Provádí-li se nepovinné analýzy významnějších transformačních produktů, je zapotřebí získat kromě grafického znázornění úbytku zkoušené látky také grafické znázornění jejich tvorby a úbytku.
Trvání fáze iniciace t L se odhadne z křivky rozkladu (semilogaritmický graf) extrapolací její lineární části na nulový rozklad nebo případně určením doby přibližně 10 % rozkladu (viz obrázky 1a a 1b). Ze semilogaritmického grafu se odhadne rychlostní konstanta prvního řádu, k, a její standardní chyba lineární regresí ln (zbytková aktivita 14C nebo koncentrace zkoušené látky) ve vztahu k času. Zejména při měřeních 14C je nutné používat pouze údaje patřící do počáteční lineární části křivky po ukončení fáze iniciace a dávat přednost výběru spíše malého počtu reprezentativních údajů před výběrem velkého počtu více nejistých dat. Nejistota zde zahrnuje chyby vlastní doporučenému přímému použití naměřených zbytkových aktivit 14C (viz dále). Někdy může být důležité vypočítat dvě odlišné rychlostní konstanty, pokud rozklad postupuje ve dvou fázích. Za tímto účelem se definují dvě odlišné fáze rozkladové křivky. Rychlostní konstanta, k, a poločas, t ½ = ln2/k, by se měly vypočítat pro každou z jednotlivých baněk k opakování, pokud se odebírají dílčí vzorky ze stejné baňky, nebo pomocí průměrných hodnot, pokud se použije jako vzorek celý objem baňky v každé době odběru vzorku (viz bod 1.8.9.2 poslední odstavec). Použije-li se první uvedený postup, je třeba v protokolu o zkoušce uvést rychlostní konstantu a poločas pro každou z jednotlivých baněk k opakování a vyjádřit je rovněž jako průměrnou hodnotu se standardní chybou. Jestliže se používaly vysoké koncentrace zkoušené látky, křivka rozkladu se může značně odchylovat od přímky (semilogaritmický graf) a kinetika prvního řádu nemusí platit. Definování poločasu proto nemá žádným smysl. Ovšem při omezeném rozpětí dat lze použít kinetiku pseudoprvního řádu a poločas rozkladu DT50 (čas pro dosažení 50 % rozkladu) odhadnout. Je však třeba mít na paměti, že časový průběh rozkladu nad zvolené datové rozpětí nelze předpovídat pomocí parametru DT50, který slouží pouze k popisu dané datové množiny. Analytické nástroje pro usnadnění statistických výpočtů a sestavení křivky jsou snadno dostupné a doporučuje se používat software tohoto typu.
Jestliže se provádí specifické chemické analýzy, je třeba odhadnout rychlostní konstanty a poločasy primárního rozkladu, jak je shora uvedeno pro celkovou mineralizaci. Jestliže je limitujícím procesem primární rozklad, lze někdy použít datové body z celého průběhu rozkladu. Je to proto, že měření jsou přímá, na rozdíl od měření aktivity 14C.
Jestliže se použijí látky značené isotopem 14C, hmotnostní bilance by měla být vyjádřena v procentech použité počáteční koncentrace nejméně na konci zkoušky.
2.1.2 Zbytková aktivita
Při biologickém rozkladu té části organické látky, která je značená isotopem 14C, se hlavní část 14C přemění na 14CO2, zatímco další část se využije pro růst biomasy a/nebo syntézu mimobuněčných metabolitů. Proto zcela „úplný“ biologický rozklad látky nedává 100 % přeměnu jejího uhlíku na 14CO2. Isotop 14C zabudovaný do produktů vytvářených biologickou syntézou se následně v důsledku „sekundární mineralizace“ pomalu uvolňuje jako 14CO2. Z těchto důvodů vykazují grafy zbytkové aktivity organického 14C (měřeno po odstranění CO2) nebo vzniklého 14CO2 v závislosti na čase i po dokončení rozkladu „chvostování“. Tím se komplikuje kinetická interpretace dat, a proto je k odhadu rychlostní konstanty rozkladu zapotřebí obvykle používat pouze počáteční část křivky (po skončení fáze iniciace a před dosažením přibližně 50 % rozkladu). Při rozkladu zkoušené látky je celková zbytková aktivita organického 14C vždy vyšší než aktivita 14C spojená se zbývající nedotčenou zkoušenou látkou. Jestliže se zkoušená látka rozkládá reakcí prvního řádu a konstantní frakce α se mineralizuje na CO2, počáteční sklon křivky úbytku 14C (celkový organický 14C vzhledem k času) bude α-násobkem sklonu příslušné křivky koncentrace zkoušené látky (nebo přesněji řečeno té části zkoušené látky, která je značená isotopem 14C). Při použití nekorigovaného měření celkové aktivity organického 14C bude proto vypočítaná rychlostní konstanta rozkladu konzervativní. V literatuře (2) (9) (10) (11) byly popsány postupy pro odhad koncentrací zkoušené látky z naměřených radiochemických aktivit na základě různých zjednodušujících předpokladů. Takové postupy se nejsnáze používají u rychle rozložitelných látek.
2.2 INTERPRETACE VÝSLEDKŮ
Jestliže se zjistí, že k nezávisí na rostoucí koncentraci (tj. pokud je vypočítaná konstanta k přibližně stejná při různých koncentracích zkoušené látky), lze předpokládat, že rychlostní konstanta prvního řádu je reprezentativní pro použité zkušební podmínky, tj. pro zkoušenou látku, vzorek vody a zkušební teplotu. Odborníci musí vyhodnotit, do jaké míry lze výsledky generalizovat či extrapolovat na jiné systémy. Jestliže se zkoušená látka použije ve vysoké koncentraci, a rozklad proto neprobíhá podle kinetiky prvního řádu, nelze data použít pro přímý odhad rychlostní konstanty prvního řádu nebo odpovídajícího poločasu. Ovšem data odvozená ze zkoušky s vysokou koncentrací zkoušené látky mohou být přesto použitelná pro odhad stupně celkové mineralizace a/nebo pro detekci a kvantifikaci transformačních produktů.
Jestliže jsou známy rychlosti jiných ztrátových procesů než biologického rozkladu (například hydrolýza nebo těkání), mohou být odečteny od čisté ztrátové rychlosti pozorované během zkoušky, čímž se získá přibližný odhad rychlosti biologického rozkladu. Údaje o hydrolýze mohou být například získány ze sterilní kontroly nebo ze souběžného zkoušení pomocí vyšší koncentrace zkoušené látky.
Nepřímé a přímé stanovení 14CO2 (bod 1.8.9.4 a dodatek 3) lze použít pouze pro měření rozsahu mineralizace zkoušené látky na CO2. K analýze koncentrací zkoušené látky značené isotopem 14C a tvorby významnějších transformačních produktů lze použít radiochromatografii (RAD-TLC) nebo HPLC (bod 1.8.9.4 třetí odstavec). Aby byl možný přímý odhad poločasu, je nezbytné, aby nebyly přítomny žádné významnější transformační produkty (definované jako ≥ 10 % použitého množství zkoušené látky). Jestliže jsou přítomny významnější transformační produkty odpovídající této definici, požaduje se podrobné hodnocení dat. To může zahrnovat opakované zkoušení a/nebo identifikaci transformačních produktů (viz bod 1.8.9.5 první odstavec), pokud nelze osud transformačních produktů přiměřeně vyhodnotit na základě zkušenosti (např. informace o cestě rozkladu). Protože se poměr uhlíku zkoušené látky přeměněného na CO2 mění (do velké míry závisí na koncentraci zkoušené látky a dalších dostupných substrátech, na zkušebních podmínkách a mikrobiálním společenství), tato zkouška nedovoluje přímý odhad úplného biologického rozkladu, jako tomu je u zkoušky na úbytek rozpuštěného organického uhlíku (DOC); výsledek je však podobný výsledku získanému zkouškou respirometrií. Stupeň mineralizace tak bude rovný minimální úrovni úplného biologického rozkladu nebo bude nižší. Pro získání ucelenějšího obrazu úplného biologického rozkladu (mineralizace a zabudování do biomasy) by se měla analýza fázové distribuce 14C provádět na konci zkoušky (viz dodatek 1). 14C obsažený v souboru částic bude obsahovat 14C zabudovaný do bakteriální biomasy a 14C sorbovaný do organických částic.
2.3 VALIDITA ZKOUŠKY
Jestliže se referenční látka nerozloží během očekávaného časového intervalu (u anilinu a benzoátu sodného je to obvykle méně než dva týdny), validita zkoušky je zpochybněna a musí být dále ověřena, případně by se zkouška měla opakovat s novým vzorkem vody. V kroužkové zkoušce této metody pořádané ISO, které se účastnilo sedm laboratoří z celé Evropy, se upravené rychlostní konstanty rozkladu pro anilin pohybovaly od 0,3 do 1,7 dne–1 s průměrem 0,8 d–1 při teplotě 20 °C a standardní chybě ±0,4 d–1 (t ½ = 0,9 dne). Typické fáze iniciace trvaly 1 až 7 dnů. Ve zkoumaných vodách byla uváděna bakteriální biomasa odpovídající 103 až 104 jednotek tvořících kolonie (CFU) na ml. Rychlosti rozkladu ve středoevropských vodách bohatých na živiny byly vyšší než ve skandinávských oligotrofních vodách, což může být způsobeno odlišným trofickým stavem nebo expozicí chemickým látkám.
Celková výtěžnost (hmotnostní bilance) na konci experimentu by měl být v rozmezí 90 % až 110 % pro radioisotopově značené látky, zatímco počáteční výtěžnost na počátku experimentu by se měla pohybovat v rozmezí 70 % až 110 % pro neznačené látky. Ovšem tato rozmezí je zapotřebí vykládat pouze jako cílová a neměla by se používat jako kritéria pro přijetí zkoušky.
3. PROTOKOL O ZKOUŠCE
V protokolu o zkoušce, který bude rovněž obsahovat nejméně následující informace, musí být jasně uveden typ studie, tj. pelagická nebo zkouška se suspendovanými sedimenty:
Zkoušená látka a referenční látka(y):
|
— |
obecné názvy, chemické názvy (doporučují se názvy podle IUPAC a/nebo CAS), čísla CAS, strukturní vzorce (uvádějící polohu 14C, pokud se použije radioisotopově značená látka) a relevantní fyzikálně-chemické vlastnosti zkoušené a referenční látky (viz body 1.5 a 1.6), |
|
— |
chemické názvy, čísla CAS, strukturní vzorce (uvádějící polohu 14C, pokud se použije radioisotopově značená látka) a relevantní fyzikálně-chemické vlastnosti látek použitých jako standardy k identifikaci a kvantifikaci transformačních produktů, |
|
— |
čistota zkoušené a referenční látky (obsah nečistot), |
|
— |
popřípadě radiochemická čistota značené chemické látky a specifická aktivita. |
Povrchová voda:
O vzorku vody musí být poskytnuty tyto minimální informace:
|
— |
lokalita a popis místa odběru vzorku, pokud možno včetně dřívější kontaminace, |
|
— |
datum a čas odběru vzorku, |
|
— |
živiny (celkový obsah N, amonia, dusitanů, dusičnanů, celkový fosfor, rozpuštěný orthofosforečnan), |
|
— |
hloubka odběru, |
|
— |
vzhled vzorku (např. barva a turbidita), |
|
— |
DOC a TOC, |
|
— |
BOD, |
|
— |
teplota a pH v místě a době odběru, |
|
— |
kyslík nebo oxidačně-redukční potenciál (povinné pouze v případě, že nejsou zřejmé aerobní podmínky), |
|
— |
slanost nebo vodivost (v případě mořské vody a brakické vody), |
|
— |
suspendované pevné látky (v případě zakaleného vzorku), |
|
— |
podle možnosti další významné informace o lokalitě odběru vzorku v době jeho odběru (např. aktuální či dřívější údaje o průtoku řek nebo mořských proudech v blízkosti hlavních výpustí a typ výpustí, meteorologické podmínky předcházející době odběru vzorku), |
a nepovinně:
|
— |
mikrobiální biomasa (např. AODC (přímé počítání za použití akridinové oranže) nebo jednotky tvořící kolonie), |
|
— |
anorganický uhlík, |
|
— |
koncentrace chlorofylu a jakožto specifický odhad biomasy řas. |
Dále by se měly poskytovat následující informace o sedimentu, pokud se provádí zkouška se suspendovanými sedimenty:
|
— |
hloubka odběru sedimentu, |
|
— |
vzhled sedimentu (například barevný, blátivý, bahnitý nebo písečný), |
|
— |
struktura (např. % hrubého písku, jemného písku, bahna a jílu), |
|
— |
suchá hmotnost v g/l suspendovaných pevných látek, koncentrace TOC nebo hmotnostní ztráta při vznícení jako měřítko obsahu organické hmoty, |
|
— |
pH, |
|
— |
kyslíkový nebo oxidačně-redukční potenciál (povinné pouze v případě, že nejsou zřejmé aerobní podmínky). |
Zkušební podmínky:
|
— |
zpoždění mezi odběrem a použitím v laboratorní zkoušce, skladování vzorků a předúprava vzorku, data provádění studií, |
|
— |
množství použité zkoušené látky, zkušební koncentrace a referenční látka, |
|
— |
metoda aplikace zkoušené látky včetně použití rozpouštědel, |
|
— |
objem použité povrchové vody a sedimentu (používá-li se) a objem vzorků odebraných v každém intervalu pro analýzu, |
|
— |
popis použitého zkušebního systému, |
jestliže zkouška nemusí být prováděna v temnu, informace o podmínkách „difuzního světla“,
|
— |
informace o použité metodě nebo metodách k vytvoření sterilních kontrol (např. teplota, čas a počet autoklávování), |
|
— |
inkubační teplota, |
|
— |
informace o analytických technikách a metodě nebo metodách použitých pro radiochemická měření a pro kontrolu hmotnostní bilance a měření fázové distribuce (provádí-li se), |
|
— |
počet opakování. |
Výsledky:
|
— |
výtěžnost v procentech (viz bod 1.7.1), |
|
— |
opakovatelnost a citlivost používaných analytických metod včetně meze detekce (LOD) a meze kvantifikace (LOQ) (viz bod 1.7.2), |
|
— |
všechna naměřená data (včetně časových bodů odběru vzorků) a vypočítané hodnoty v tabulkové formě a křivky rozkladu; pro každou zkušební koncentraci a pro každou baňku k opakování je nutné v protokolu o zkoušce uvést lineární korelační koeficient pro sklon logaritmického grafu, odhadovanou fázi iniciace a rychlostní konstantu prvního řádu nebo pseudoprvního řádu (je-li to možné) a odpovídající poločas rozkladu (nebo doba poločasu t 50), |
|
— |
v protokolu o zkoušce zaznamenejte relevantní hodnoty jako průměry výsledků zjištěných u jednotlivých opakování, např. délka fáze iniciace, rychlostní konstanta rozkladu a poločas rozkladu (nebo t 50), |
|
— |
na základě vzhledu křivky rozkladu a možného vlivu zkušební koncentrace je třeba systém kategorizovat buď jako nepřizpůsobený, nebo přizpůsobený, |
|
— |
výsledky kontroly konečné hmotnostní bilance a výsledky měření pro stanovení fázových distribucí (jsou-li k dispozici), |
|
— |
podíl mineralizovaného 14C, a použijí-li se specifické analýzy, konečná úroveň primárního rozkladu, |
|
— |
případně identifikace, molární koncentrace a procentuální podíl aplikovaných významnějších transformačních produktů (viz bod 1.8.9.5 první odstavec), |
|
— |
případně navrhovaná cesta transformace, |
|
— |
diskuse výsledků. |
4. LITERATURA
|
(1) |
OECD TG 309 (2004) Aerobic Mineralisation in surface water – Simulation Biodegradation Test. |
|
(2) |
ISO/DIS 14592–1 (1999) Water quality – Evaluation of the aerobic biodegradability of organic compounds at low concentrations – Part 1: Shake flask batch test with surface water or surface water/sediment suspensions. |
|
(3) |
Zkušební metoda C.23 Aerobní a anaerobní transformace v půdě. |
|
(4) |
Zkušební metoda C.24 Aerobní a anaerobní transformace v systémech voda/sediment. |
|
(5) |
OECD (1993). Guidelines for the Testing of Chemicals. OECD, Paris. |
|
(6) |
ISO 8245 (1999). Water quality – Guidelines on the determination of total organic carbon (TOC) and dissolved organic carbon (DOC). |
|
(7) |
ISO 10634 (1995). Water quality – Guidance for the preparation and treatment of poorly water-soluble organic compounds for the subsequent evaluation of their biodegradability in an aqueous medium. |
|
(8) |
OECD, předloha (2000). Guidance Document on aquatic toxicity testing of difficult substances and mixtures. Environmental Health and Safety Publications. Series on Testing and Assessment. No 22 (předpokládané zveřejnění v létě roku 2000). |
|
(9) |
Simkins, S., Alexander, M. (1984). Models for mineralization kinetics with the variables of substrate concentration and population density. Appl. Environ. Microbiol. 47, 394–401. |
|
(10) |
Ingerslev, F., Nyholm, N. (2000). Shake-flask test for determination of biodegradation rates of 14C-labeled chemicals at low concentrations in surface water systems. Ecotoxicol. Environ. Saf. 45, 274–283. |
|
(11) |
ISO/CD 14592–1 (1999). Ring test report: Water Quality – Evaluation of the aerobic biodegradability of organic compounds at low concentrations part 1 – report of 1998/1999 ring-test. Shake flask batch test with surface water or surface water/sediment suspensions. |
Dodatek 1
Fázová distribuce 14C
Pro kontrolu postupu je zapotřebí doplnit rutinní měření zbytkové celkové aktivity organického 14C (TOA) měřeními hmotnostní bilance zahrnující přímé stanovení uvolněného 14CO2 po jímání v absorbéru (viz dodatek 3). Sama o sobě je pozitivní tvorba 14CO2 přímým důkazem o biologickém rozkladu, na rozdíl od abiotického rozkladu nebo jiných ztrátových mechanismů, jakými jsou těkání a sorpce. Dodatečné užitečné informace charakterizující chování při biologické rozložitelnosti lze získat z měření distribuce TOA mezi rozpuštěným stavem (aktivita rozpuštěného organického 14C, DOA) a částicovým stavem (aktivita částicového organického 14C, POA) po separaci částic membránovou filtrací nebo odstřeďováním. POA obsahuje zkoušenou látku sorbovanou na mikrobiální biomasu a na další částice a dále z uhlíku zkoušené látky, který byl použit k syntéze nového buněčného materiálu, a tím začleněn do frakce částicové biomasy. Tvorbu rozpuštěného organického materiálu s 14C lze odhadnout jako DOA na konci biologického rozkladu (plató na křivce závislosti rozkladu na čase).
Fázová distribuce zbytkového 14C ve zvolených vzorcích se odhadne filtrováním vzorků na membránovém filtru s velikostí pórů 0,22 μm nebo 0,45 μm a z materiálu, který neabsorbuje významná množství zkoušené látky (vhodné mohou být polykarbonátové filtry). Jestliže sorpce zkoušené látky na filtr není zanedbatelně nízká (tuto skutečnost je nutné ověřit před experimentem), lze použít místo filtrace vysokorychlostní odstřeďování (2 000 g, 10 minut).
Dále je třeba s filtrátem či centrifugátem pracovat tak, jak je uvedeno v dodatku 3 pro nefiltrované vzorky. Membránové filtry se rozpustí ve vhodné scintilační kapalině a počítá se jako obvykle, obecně pouze za použití metody externího standardního poměru pro korekci na zhášení, nebo se použije oxidační činidlo na vzorek. Pokud bylo použito odstřeďování, vytvořené pelety částicové frakce se resuspendují v 1–2 ml destilované vody a převedou se do scintilační lahvičky. Následně se provedou dvě promytí pomocí 1 ml destilované vody a promývací voda se převede do lahvičky. V případě potřeby lze suspenzi ukotvit do gelu za účelem počítání scintilací v roztoku.
Dodatek 2
Polospojitý postup
Pro dosažení významného rozkladu obtížně rozložitelných látek může být vyžadována prodloužená inkubace až po několik měsíců. Doba trvání zkoušky by obecně neměla překročit 60 dnů, pokud nebudou zachovány charakteristiky původního vzorku vody obnovením zkušební suspenze. Ovšem doba trvání zkoušky může být prodloužena na maximálně 90 dnů bez obnovení zkušební suspenze, jestliže rozklad zkoušené látky začal během prvních 60 dnů.
Během inkubace po dlouhá období může být různorodost mikrobiální komunity snížena v důsledku různých ztrátových mechanismů a možného vyčerpání významných živin a primárních uhlíkových substrátů ze vzorku vody. Proto se doporučuje, aby se pro adekvátní určení rychlosti rozkladu pomalu se rozkládajících látek používala polospojitá zkouška. Zkouška by se měla zahájit použitím polospojitého postupu, pokud se na základě předchozí zkušenosti očekává, že pro dosažení 20% rozkladu látky je nezbytná inkubační doba tří měsíců. Alternativně lze normální dávkovou zkoušku změnit na polospojitou zkoušku, pokud nebylo dosaženo rozkladu zkoušené látky přibližně během 60 dnů zkoušení s využitím dávkového postupu. Polospojitý postup lze zastavit a ve zkoušce pokračovat jakožto v dávkovém experimentu, když je zaznamenán významný rozklad (např. > 20 %).
Při polospojité zkoušce se každé dva týdny nahrazuje přibližně jedna třetina objemu zkušební suspenze čerstvě odebranou vodou, do níž byla přidána zkoušená látka v počáteční koncentraci. Podobně se do vody, která má být použita k obnovení, přidá sediment v počáteční koncentraci (mezi 0,01 a 1 g/l), pokud se provádí nepovinná zkouška se suspendovanými sedimenty. Při provádění zkoušky se suspendovanými částicemi sedimentu je důležité, aby byl plně suspendovaný systém zachován také během obnovování vody a aby doba zdržení byla shodná pro pevné látky a vodu, protože jinak se může ztratit zamýšlená podobnost s homogenním vodným systémem bez zakotvených fází. Z těchto důvodů se při použití polospojitého postupu dává přednost počáteční koncentraci suspendovaných sedimentů s nižším rozpětím stanoveného intervalu.
U předepsaného přídavku zkoušené látky se předpokládá, že počáteční koncentrace zkoušené látky není překročena částečným obnovením zkušební suspenze, a proto se zabrání přizpůsobení, které je často pozorováno při vysokých koncentracích zkoušených látek. Protože postup obsahuje opakovanou inokulaci i náhradu vyčerpaných živin a primárních substrátů, obnoví se původní mikrobiální různorodost a dobu trvání zkoušky lze v zásadě prodloužit do nekonečna. Při použití polospojitého postupu je důležité mít na paměti, že zbytková koncentrace zkoušené látky se musí korigovat na množství zkoušené látky přidané a odstraněné v každém postupu obnovení. U sloučenin, které se málo sorbují, lze celkovou koncentraci a koncentraci rozpuštěné zkoušené látky používat zaměnitelně. Sorpce je za stanovených podmínek (0,1–1 g pevných látek/l) pro látky s log K o/v < 3 (platí pro neutrální, lipofilní sloučeniny) nevýznamná (< 5 %). To je ilustrováno následujícím příkladem výpočtu. 0,1 g/l pevných látek zhruba odpovídá 10 mg uhlíku na litr (uhlíková frakce, f C = 0,01). Předpokládejme, že:
Log K o/v (zkoušené látky) = 3
K ou = 0,42 × K o/v
Rozdělovací koeficient K d = f C × K ou
rozpuštěná frakce celkové koncentrace (C-voda(C v)/C-celkové(C c) je pak:
C v/C c = 1/(1 + K d × SS) = 1(1 + K oc × f C × SS) = 1/(1 + 0,42 × 103 × 0,01 × 0,1 × 10–3) = 0,999
Dodatek 3
Stanovení 14co2
Nepřímé stanovení 14CO2
Pro rutinní měření je nepřímá metoda obvykle nejméně časově náročnou a nejpřesnější metodou, pokud je zkoušená látka netěkavá a netransformuje se na těkavé transformační produkty. Nefiltrované vzorky, např. o objemu 5 ml, se jednoduše převedou do scintilačních lahviček. Vhodná aktivita ve vzorcích je na počátku 5 000 dpm–10 000 dpm (80–170 Bq) a minimální počáteční aktivita je přibližně 1 000 dpm. CO2 by měl být odstraněn po okyselení na pH 2 až 3 pomocí 1 či 2 kapek koncentrované H3PO4 nebo HCl. Odstraňování CO2 lze provádět probubláváním vzduchem přibližně po ½–1 hodinu. Eventuálně lze lahvičky intenzivně protřepávat po dobu 1 až 2 hodin (například na třepačce s mikrodeskou) nebo jemně protřepávat přes noc. Účinnost procesu odstraňování CO2 se musí zkontrolovat (prodloužením doby provzdušňování nebo protřepávání). Poté by se měla přidat scintilační kapalina vhodná pro počítání vodných vzorků, vzorek by se měl homogenizovat vířivou míchačkou a radioaktivita určit počítáním scintilací v roztoku, přičemž je nutné odečíst aktivitu pozadí zjištěnou ve slepých zkouškách (FB). Pokud nebude zkušební voda velmi zabarvená ani nebude obsahovat vysokou koncentraci částic, vzorky budou obvykle vykazovat jednotné zhášení a postačí provedení korekcí na zhášení pomocí externího standardu. Je-li zkušební voda vysoce zbarvená, může být nutná korekce na zhášení přidáním interního standardu. Je-li koncentrace částic vysoká, nemusí být možné získat homogenní roztok či gel, případně může být odchylka zhášení mezi vzorky velká. V takovém případě lze použít níže popsanou metodu počítání pro zkušební suspenze. Provádí-li se zkouška jako zkouška se suspendovanými sedimenty, měření 14CO2 by se mohlo provádět nepřímo odběrem homogenního 10ml vzorku zkušební vody/suspenze a oddělením fází pomocí odstřeďování při vhodných otáčkách (např. při 40 000 m/s2 po dobu 15 minut). Vodní fáze by poté měla být ošetřena shora popsaným postupem. Aktivita 14C v částicové fázi (POA) by se měla určit resuspendací sedimentu v malém objemu destilované vody, převedením do scintilačních lahviček a přidáním scintilační kapaliny pro vytvoření gelu (za tímto účelem jsou k dispozici speciální scintilační kapaliny). V závislosti na povaze částic (např. jejich obsahu organického materiálu) může být před přidáním scintilační kapaliny vzorek přes noc vyluhován tkáňovým rozpustidlem a poté homogenizován vířivou míchačkou. Alternativně lze POA určit spalováním v nadbytku kyslíku použitím oxidačního činidla na vzorek. Při počítání je třeba vždy zahrnout interní standard a může být nezbytné provádět korekce na zhášení pomocí přidání interního standardu pro každý jednotlivý vzorek.
Přímé stanovení 14CO2
Měří-li se uvolňovaný 14CO2 přímo, je třeba nasadit na počátku zkoušky více baněk, používat jako vzorek vždy celý objem zkušební baňky v každém měřicím bodě, okyselit zkušební baňky na pH 2 až 3 a jímat 14CO2 v interním (umístěný v každé zkušební baňce na počátku zkoušky) nebo externím absorbéru. Jako absorpční médium lze používat alkálii (např. 1 N roztok NaOH nebo peleta NaOH), ethanolamin nebo na ethanolaminu založený absorbér a komerčně dostupné absorbéry. Pro přímé měření 14CO2 je třeba baňky uzavřít např. septou z butylové pryže.
Obrázek 1a
Příklad aritmetického vynášení dat (zbytková aktivita vzhledem k času)
Obrázek 1b
Příklad semilogaritmického vynesení dat (ln zbytkové aktivity vzhledem k času)
PŘÍLOHA VI
|
C.26 |
ZKOUŠKA INHIBICE RŮSTU LEMNA SPP. |
1. METODA
Tato metoda je rovnocenná Pokynu OECD pro zkoušení 221 (2006) (1). Mezi orgány EU panuje široká shoda v tom, že zkouška na Lemna spp. je u intenzivně zbarvených látek vhodnou alternativou ke zkoušce na řasách (2, 3).
1.1 ÚVOD
Tato zkušební metoda je určena k hodnocení toxicity látek pro sladkovodní rostliny rodu Lemna (okřehek). Je založena na existujících pokynech (4, 5, 6, 7, 8, 9), ale obsahuje úpravy těchto metod, aby odrážela nejnovější výsledky výzkumu a konzultace v řadě klíčových záležitostí. Navrhovaná metoda byla validována mezinárodní kroužkovou zkouškou (10).
Tato zkušební metoda popisuje zkoušení toxicity pomocí Lemna gibba a Lemna minor, které byly obě rozsáhle studovány a jsou předmětem shora uvedených norem. Taxonomie Lemna spp. je obtížná a je komplikována existencí široké škály fenotypů. Ačkoliv se u Lemna může vyskytnout genetická variabilita v odezvě na toxické látky, není v současnosti dostatek údajů o tomto zdroji variability, aby mohl být pro použití v rámci této metody doporučen specifický klon. Je třeba uvést, že zkouška se neprovádí axenicky, ale v jednotlivých stupních během zkušebního procesu se provádí opatření pro udržení kontaminace jinými organismy na minimu.
Podrobně se popisuje zkoušení s obnovením (semistatické a průtokové) a bez obnovení (statické) zkušebního roztoku. V závislosti na cílech zkoušky a právních požadavcích se doporučuje zvážit použití semistatických a průtokových metod, například pro látky, které se z roztoku rychle ztrácí v důsledku odpaření, fotodegradace, srážení nebo biologického rozkladu. Další pokyny jsou uvedeny v (11).
1.2 DEFINICE
Pro účely této zkušební metody se používají následující definice a zkratky:
Biomasa: je suchá hmotnost živé hmoty přítomné v populaci. V této zkoušce se obvykle měří náhrady biomasy, například počet lístků nebo plocha lístků a používání termínu „biomasa“ rovněž odkazuje na tyto náhradní míry.
Chloróza: je žloutnutí tkáně lístků.
Klon: je organismus nebo buňka vzniklá z jediného jedince asexuální reprodukcí. Jedinci ze stejného klonu jsou proto geneticky identičtí.
Kolonie: znamená soubor mateřských a dceřiných lístků (obvykle 2 až 4) navzájem spolu spojených. Někdy se označuje jako rostlina.
ECx : je koncentrace zkoušené látky rozpuštěné ve zkušebním médiu, která pro danou expoziční dobu vede k x% (např. 50%) snížení růstu Lemna (musí se výslovně uvést, pokud se odchyluje od plné či normální doby trvání zkoušky). Pro jednoznačný popis hodnoty EC odvozené z růstové rychlosti nebo z výtěžku se pro růstovou rychlost používá symbol „ErC“ a pro výtěžek „EyC“ následovaný proměnnou měření, např. ErC (počet lístků).
Průtoková zkouška: je zkouškou, při níž jsou zkušební roztoky průběžně vyměňovány.
Lístek: je individuální/samostatnou listovitou strukturou rostliny okřehku. Je to nejmenší jednotka, tj. jedinec, schopná reprodukce.
Hrbatost: znamená lístky mající hrbolatý či napuchlý vzhled.
Růst: je zvýšením proměnné měření, např. počtu lístků, suché hmotnosti, hmotnosti za mokra nebo plochy lístků, během doby zkoušky.
Růstová rychlost (průměrná specifická růstová rychlost): je logaritmické zvýšení biomasy během období expozice.
Nejnižší koncentrace s pozorovanými účinky (Lowest Observed Effect Concentration, LOEC): je nejnižší zkušební koncentrací, při níž je pro danou expoziční dobu pozorován statisticky významný účinek látky na snížení růstu (na hladině spolehlivosti p < 0,05) ve srovnání s kontrolou. Všechny zkušební koncentrace vyšší než LOEC musí však mít stejné nebo vážnější škodlivé účinky, než jsou účinky pozorované při koncentraci LOEC. Nelze-li tyto dvě podmínky splnit, musí být podrobně vysvětleno, jak byla LOEC (a tedy i NOEC) zvolena.
Proměnné měření: jsou jakýmkoliv druhem proměnných, které se měří pro vyjádření zkoumaného účinku pomocí jedné či více různých proměnných odezvy. V této metodě jsou proměnnými měření počet lístků, plocha lístků, čerstvá hmotnost a suchá hmotnost.
Monokultura: je kultura s jedním druhem rostliny.
Nekróza: je mrtvá (tj. bílá či vodou nasáklá) tkáň lístku.
Koncentrace bez pozorovaných účinků (No Observed Effect Concentration, NOEC): je zkušební koncentrace bezprostředně nižší než LOEC.
Fenotyp: je pozorovatelnou charakteristikou organismu určenou interakcí jeho genů s prostředím.
Proměnné odezvy: jsou proměnné pro odhad toxicity odvozené z jakýchkoliv naměřených proměnných, jež popisují biomasu, různými metodami výpočtu. U této metody jsou proměnnými odezvy růstová rychlost a výtěžek, které se odvozují z proměnných měření jako např. počtu lístků, plochy lístků, čerstvé hmotnosti nebo suché hmotnosti.
Semistatická (obnovovací) zkouška: je zkouškou, při níž je zkušební roztok během zkoušky ve stanovených intervalech pravidelně nahrazován.
Statická zkouška: je zkušební metoda bez obnovování zkušebního roztoku během zkoušky.
Zkoumaný účinek: popisuje obecný faktor, který bude jakožto cíl zkoušky změněn zkoušenou chemickou látkou vzhledem ke kontrole. Při této metodě je zkoumaným účinkem inhibice růstu, kterou lze vyjádřit různými proměnnými odezvy, jež jsou založeny na jedné či více proměnných měření.
Zkušební médium: je úplné syntetické růstové médium, v němž rostou zkušební rostliny při expozici zkoušené látce. Zkoušená látka bude za normálních podmínek ve zkušebním médiu rozpuštěna.
Výtěžek: je hodnota proměnné měření pro vyjádření biomasy na konci expoziční doby minus proměnná měření na počátku expoziční doby.
1.3 PODSTATA ZKOUŠKY
Exponenciálně rostoucí kultury rostlin rodu Lemna se po dobu sedmi dnů nechají růst jako monokultury v různých koncentracích zkoušené látky. Cílem zkoušky je kvantifikovat účinky související s látkou na vegetativní růst během tohoto období, a to na základě hodnocení zvolených proměnných měření. Primární proměnnou měření je počet lístků. Rovněž se měří nejméně jedna další proměnná měření (celková plocha lístků, suchá hmotnost nebo čerstvá hmotnost), protože některé látky mohou ovlivňovat jiné proměnné měření mnohem více než počet lístků. Pro kvantifikaci účinků souvisejících s látkou se růst ve zkušebních roztocích porovnává s růstem kontrol a stanoví se koncentrace způsobující stanovenou x% inhibici růstu (např. 50%) a vyjádří jako ECx (např. EC50).
Zkoumaným účinkem je inhibice růstu vyjádřená jako logaritmické zvýšení proměnné měření (průměrná specifická růstová rychlost) během expoziční doby. Z průměrných specifických růstových rychlostí zaznamenaných v řadě zkušebních roztoků se stanoví koncentrace způsobující stanovenou x% inhibici růstové rychlosti (např. 50%) a vyjádří se jako ErCx (např. ErC50).
Další proměnnou odezvy použitou v této zkušební metodě je výtěžek, který může být nutný ke splnění specifických právních požadavků v některých zemích. Je definován jako proměnné měření na konci expoziční doby minus proměnné měření na počátku expoziční doby. Z výtěžku zaznamenaného v řadě zkušebních roztoků se vypočítá koncentrace způsobující stanovenou x% inhibici výtěžku (např. 50%) a vyjádří se jako EyCx (např. EyC50).
Kromě toho se může statisticky určit nejnižší koncentrace s pozorovanými účinky (LOEC) a koncentrace bez pozorovaných účinků (NOEC).
1.4 INFORMACE O ZKOUŠENÉ LÁTCE
Měla by být k dispozici analytická metoda s odpovídající citlivostí pro kvantifikaci látky ve zkušebním médiu.
Užitečnými informacemi o zkoušené látce pro účely stanovení zkušebních podmínek mohou být strukturní vzorec, čistota, rozpustnost ve vodě, stálost ve vodě na světle, pK a, K o/v, tlak par a biologická rozložitelnost. Pro výpočet Henryho konstanty, která bude ukazovat, zda-li jsou pravděpodobné významné ztráty zkoušené látky během doby zkoušky, lze použít rozpustnost ve vodě a tlak par. To pomáhá zjistit, zda-li by se měly podniknout konkrétní opatření ke kontrole takových ztrát. Jestliže nejsou informace o rozpustnosti a stálosti zkoušené látky jisté, doporučuje se, aby byly hodnoceny za podmínek zkoušky, tj. v růstovém médiu, při teplotě a režimu osvětlení, které mají být při zkoušce použity.
Pokud je zvláště důležitá kontrola pH zkušebního média, např. zkouší-li se kovy nebo látky, které jsou hydrolyticky nestálé, doporučuje se přídavek pufru do růstového média (viz bod 1.7.4 první odstavec). Další pokyny pro zkoušení látek s fyzikálně-chemickými vlastnostmi, které způsobují jejich obtížnou testovatelnost, je uveden v (11).
1.5 REFERENČNÍ LÁTKA
Jako prostředek kontroly zkušebního postupu lze zkoušet referenční látku (látky), například 3,5-dichlorfenol použitý v mezinárodní kruhové zkoušce (10). Doporučuje se zkoušet referenční látku nejméně dvakrát ročně nebo – pokud se zkoušení provádí s menší četností – souběžně se stanovováním toxicity zkoušené látky.
1.6 VALIDITA ZKOUŠKY
Má-li být zkouška platná, musí být doba do zdvojnásobení počtu lístků v kontrole menší než 2,5 dnů (60 hodin), což odpovídá přibližně sedminásobnému zvýšení za sedm dnů a průměrné specifické růstové rychlosti 0,275 d–1. Při využití médií a zkušebních podmínek popsaných v této zkušební metodě lze tohoto kritéria dosáhnout použitím statické zkoušky (8). Rovněž se předpokládá, že toto kritérium bude splnitelné za podmínek semistatické a průtokové zkoušky. Výpočet doby zdvojnásobení je uveden v bodě 2.1.
1.7 POPIS METODY
1.7.1 Přístroje a pomůcky
Veškeré vybavení přicházející od styku se zkušebními médii by mělo být vyrobeno ze skla nebo jiného chemicky inertního materiálu. Skleněné pomůcky použité pro kultivační a zkušební účely je zapotřebí vyčistit od chemických kontaminantů, které by mohly unikat do zkušebního média, a musí být sterilní. Zkušební nádoby by měly být dostatečně široké, aby mohly lístky jednotlivých kolonií v kontrolních nádobách růst, aniž by se na konci zkoušky překrývaly. Není důležité, zda se kořeny dotýkají dna zkušebních nádob, ale doporučuje se, aby v každé zkušební nádobě byla minimální hloubka 20 mm a minimální objem 100 ml. Volba zkušebních nádob není zásadně důležitá, pokud budou splněny tyto požadavky. Jako vhodné se potvrdily skleněné kádinky, krystalizační misky nebo skleněné Petriho misky vhodných rozměrů. Zkušební nádoby musí být zakryty, aby se minimalizovalo odpařování a náhodná kontaminace, přičemž však musí být umožněna nezbytná výměna vzduchu. Vhodné zkušební nádoby a zejména kryty musí zabránit stínění nebo změnám spektrálních charakteristik světla.
Kultury a zkušební nádoby by se neměly přechovávat společně. Toho se nejlépe dosáhne pomocí samostatných komor, inkubátorů či místností pro růst za podmínek prostředí. Osvětlení a teplota musí být regulovatelné a musí se udržovat na stálé úrovni (viz bod 1.7.8).
1.7.2 Testovací organismus
Organismem použitým pro tuto zkoušku je Lemna gibba nebo Lemna minor. Krátké popisy druhů okřehků, které byly použity pro zkoušky toxicity, jsou uvedeny v dodatku 1. Rostlinný materiál lze získat ze sbírek kultur, z jiné laboratoře nebo z terénu. Jestliže se sběr provádí z terénu, rostliny by se měly kultivovat ve stejném médiu, jaké se používá ke zkoušení, minimálně po dobu osmi týdnů před použitím. Terénní místa použitá pro sběr výchozích kultur musí být prostá zřejmých zdrojů kontaminace. Pokud se získávají z jiné laboratoře nebo ze sbírek kultur, měly by se podobně uchovávat minimálně po tři týdny. Ve zkušebním protokolu je vždy třeba uvést zdroj rostlinného materiálu a druhů a klonu (je-li znám) použitých ke zkoušení.
Měly by se používat monokultury, které jsou očividně prosté kontaminace jinými organismy, například řasami a prvoky. Zdravé rostliny L. minor obsahují kolonie skládající se ze dvou až pěti lístků, zatímco zdravé kolonie L. gibba mohou obsahovat až sedm lístků.
Kvalita a jednotnost rostlin použitých pro zkoušku bude mít významný vliv na výsledek zkoušky, a proto by se měla volit s velkou péčí. Měly by se používat mladé, rychle rostoucí rostliny bez viditelných lézí či odbarvení (chlorózy). Kultury dobré kvality se vyznačují vysokým výskytem kolonií obsahujících nejméně dva lístky. Velký počet jednotlivých lístků je ukazatelem environmentálního stresu, např. nedostatku živin, a rostlinný materiál z takovýchto kultur by se ke zkoušení neměl používat.
1.7.3 Kultivace
Pro snížení četnosti udržování kultury (např. pokud se na určité období neplánují žádné zkoušky s Lemna) lze kultury udržovat za sníženého osvětlení a teploty (4–10 °C). Podrobnosti o kultivaci jsou uvedeny v dodatku 2. Očividné známky kontaminace řasami nebo jinými organismy vyžadují povrchovou sterilizaci dílčího vzorku lístků Lemna a následný přenos do čerstvého média (viz dodatek 2). V tomto případě je třeba zbývající kontaminovanou kulturu zlikvidovat.
Nejméně sedm dnů před zkoušením se asepticky převede dostatečný počet kolonií do čerstvého sterilního média a kultivuje se 7 až 10 dnů za podmínek zkoušky.
1.7.4 Zkušební médium
Pro Lemna minor a Lemna gibba se doporučují odlišná média, jak je dále uvedeno. Je třeba pečlivě zvážit zahrnutí pufru pH do zkušebního média (MOPS (3-(N-morfolino)propansulfonová kyselina, číslo CAS: 1132–61–2; číslo EINECS: 214–478–5) do média pro L. minor a NaHCO3 do média pro L. gibba), jestliže existuje podezření, že by mohl reagovat se zkoušenou látkou a ovlivňovat expresi její toxicity. Steinbergovo médium (12) je rovněž přijatelné, pokud jsou splněna kritéria validity.
Pro kultivaci a zkoušení s L. minor se doporučuje úprava švédského standardního (SIS) růstového média pro Lemna. Složení tohoto média je uvedeno v dodatku 3.
Ke kultivaci a zkoušení s L. gibba se doporučuje růstové médium 20X AAP, popsané v dodatku 3.
Steinbergovo médium, popsané v dodatku 3, je rovněž vhodné pro L. minor, ale může se používat i pro L. gibba, pokud jsou splněna kritéria validity.
1.7.5 Zkušební roztoky
Zkušební roztoky se obvykle připravují ředěním zásobního roztoku. Zásobní roztoky zkoušené látky se obyčejně připravují rozpuštěním látky v růstovém médiu.
Nejvyšší zkušební koncentrace zkoušené látky by obvykle neměla překročit rozpustnost látky ve vodě za zkušebních podmínek. Je však třeba poznamenat, že Lemna spp. plují na povrchu a mohou být vystaveny působení látek, které se hromadí na rozhraní voda-vzduch (např. látky špatně rozpustné ve vodě nebo hydrofobní či povrchově aktivní látky). Za takových podmínek dochází k expozici jiným látkám, než jaké se nachází v roztoku, a zkušební koncentrace mohou v závislosti na vlastnostech zkoušené látky překračovat rozpustnost ve vodě. Pro zkoušené látky s nízkou rozpustností ve vodě může být nezbytné připravit koncentrovaný zásobní roztok nebo disperzi látky pomocí organického rozpouštědla nebo dispergátoru, aby se usnadnilo přidávání přesných množství zkoušené látky do zkušebního média, a napomohlo se tak její dispergaci a rozpuštění. Měla by být vynaložena maximální snaha takové látky nepoužívat. Používání pomocných rozpouštědel nebo dispergátorů by nemělo způsobovat žádnou fytotoxicitu. Příkladem běžně používaných rozpouštědel, která nezpůsobují fytotoxicitu při koncentracích až do 100 μl·l–1, jsou aceton a dimethylformamid. Jestliže se použije rozpouštědlo nebo dispergátor, jeho konečná koncentrace by se měla zapsat ve zkušební zprávě a udržovat na minimu (≤ 100 μl·l–1) a všechny exponované vzorky a kontroly by měly obsahovat stejnou koncentraci rozpouštědla nebo dispergátoru. Další pokyny k používání dispergátorů jsou uvedeny v (11).
1.7.6 Zkušební a kontrolní skupiny
Předběžná znalost toxicity zkoušené látky pro Lemna, např. z orientační zkoušky, pomůže při volbě vhodných zkušebních koncentrací. Při definitivní zkoušce toxicity se zpravidla použije alespoň pět zkušebních koncentrací tvořících geometrickou řadu. Faktor mezi zkušebními koncentracemi by nejlépe neměl překročit 3,2, ale lze použít vyšší hodnotu, jestliže je křivka závislosti koncentrace a odezvy plochá. Použití méně než pěti koncentrací by mělo být zdůvodněno. V rámci každé zkušební koncentrace se použijí nejméně tři opakování.
Při volbě rozsahu zkušebních koncentrací (pro orientační a/nebo definitivní zkoušku toxicity) je zapotřebí zvážit následující:
|
— |
Pro stanovení ECx by se zkušební koncentrace měly pohybovat pod a nad hodnotu ECx, aby se zajistila vhodná úroveň spolehlivosti. Například pokud se odhaduje EC50, nejvyšší zkušební koncentrace by měla být vyšší než hodnota EC50. Jestliže hodnota EC50 leží mimo rozmezí zkušebních koncentrací, související intervaly spolehlivosti budou velké a správné hodnocení statistické vhodnosti modelu nemusí být možné. |
|
— |
Jestliže je cílem odhadnout LOEC/NOEC, nejnižší zkušební koncentrace by měla být dostatečně nízká, aby růst nebyl výrazně nižší než růst kontroly. Dále by nejvyšší zkušební koncentrace měla být dostatečně vysoká, aby byl růst výrazně nižší než růst kontroly. Pokud tomu tak není, zkouška se bude muset opakovat v odlišném rozmezí koncentrací (s výjimkou případů, kdy je nejvyšší koncentrace na mezi rozpustnosti nebo je maximální požadovanou limitní koncentrací, např. 100 mg.l–1). |
Každá zkouška by měla zahrnovat kontroly, pro něž se použije stejné živné médium, stejný počet lístků a kolonií, stejné podmínky okolního prostředí a postupy jako pro zkušební nádoby, avšak neobsahují zkoušenou látku. Jestliže se použije pomocné rozpouštědlo nebo dispergátor, měla by být zahrnuta i další kontrolní expozice s rozpouštědlem/dispergátorem přítomným ve stejné koncentraci jako v nádobách se zkoušenou látkou. Počet kontrolních nádob použitých k opakování (a případných nádob s rozpouštědly) by měl být nejméně roven počtu nádob použitých pro každou zkušební koncentraci; v ideálním případě by měl být dvojnásobný.
Jestliže se stanovení NOEC nepožaduje, plán zkoušky lze pozměnit tak, že se zvýší počet koncentrací a sníží počet opakování na příslušnou koncentraci. Počet kontrolních opakování však musí být nejméně tři.
1.7.7 Expozice
Kolonie tvořené 2 až 4 viditelnými lístky se převedou z očkovací kultury a za aseptických podmínek se náhodně přiřadí do zkušebních nádob. Každá nádoba by měla celkem obsahovat 9 až 12 lístků. Počet lístků a kolonií by měl být v každé zkušební nádobě stejný. Zkušenosti získané s touto metodou a údaje z kruhové zkoušky ukazují, že použití tří opakování na expozici, přičemž pro každé opakování se zpočátku použije od 9 do 12 lístků, dostačuje ke zjištění rozdílů v inhibici růstu přibližně o 4 až 7 % vypočítané pomocí růstové rychlosti (10 až 15 % počítáno pomocí výtěžku) mezi jednotlivými expozicemi (10).
K minimalizaci vlivu prostorových rozdílů v intenzitě světla nebo v teplotě je potřebné náhodné uspořádání zkušebních nádob v inkubátoru. Při provádění pozorování se rovněž vyžaduje uspořádání do bloků nebo náhodné přemísťování nádob (nebo častější přemísťování).
Jestliže předběžná zkouška stálosti ukazuje, že během doby trvání zkoušky (7 dnů) nelze udržet koncentraci zkoušené látky (tj. měřená koncentrace poklesne pod 80 % naměřené počáteční koncentrace), doporučuje se semistatická zkouška. V tomto případě by měly být kolonie nejméně dvakrát během zkoušky (např. v den 3 a 5) vystaveny čerstvě připraveným zkušebním a kontrolním roztokům. Četnost expozice čerstvému médiu závisí na stálosti zkoušené látky; pro udržení téměř konstantních koncentrací vysoce nestálých či těkavých látek může být zapotřebí vyšší četnost. Za určitých okolností může být nutný průtokový postup (11, 13).
Scénář expozice prostřednictvím aplikace na lístek (rozprašování) není do této zkušební metody zahrnut, avšak tomuto tématu se věnuje literatura (14).
1.7.8 Inkubační podmínky
Pomocí kontinuálního teplého nebo chladného bílého fluorescenčního osvětlení je třeba zajistit světelnou intenzitu zvolenou z rozmezí 85–135 μE·m–2·s–1, přičemž se tato intenzita měří při fotosynteticky aktivním záření (400 až 700 nm) v bodech nacházejících se ve stejné vzdálenosti od zdroje světla jako lístky Lemna (ekvivalent 6 500 až 10 000 luxů). Jakékoliv odchylky od zvolené světelné intenzity nad zkušební plochou by neměly překročit ±15 %. Naměřenou hodnotu bude ovlivňovat metoda detekce světla a měření, zejména typ čidla. Kulová čidla (která reagují na světlo ze všech úhlů nad a pod rovinou měření) a „kosinová“ čidla (která reagují na světlo ze všech úhlů nad rovnou měření) jsou vhodnější než jednosměrná čidla, neboť poskytují vyšší naměřené hodnoty pro vícebodový světelný zdroj zde popsaného typu.
Teplota ve zkušebních nádobách by měla být (24 ± 2) °C. Hodnota pH kontrolního média by neměla během zkoušky vzrůst o více než 1,5. Ovšem odchylka o více než 1,5 nebude znamenat neplatnost zkoušky, pokud je možné prokázat, že byla splněna kritéria validity. Zvláštní pozornost je zapotřebí věnovat posunu pH ve zvláštních případech, jako jsou zkoušky nestálých látek nebo kovů. Další pokyny naleznete v (11).
1.7.9 Doba trvání
Zkouška je ukončena za 7 dnů poté, co byly rostliny převedeny do zkušebních nádob.
1.7.10 Měření a analytická stanovení
Na počátku zkoušky se určí a zaznamená počet lístků ve zkušebních nádobách, přičemž se pečlivě dbá na to, aby byly započítány vystupující, jasně viditelné lístky. Na počátku zkoušky, dále nejméně jednou během každých 3 dnů během expoziční doby (tj. nejméně dvakrát během celkové doby 7 dnů) a při ukončení zkoušky se musí určit počty lístků, které mají normální či abnormální vzhled. Je zapotřebí zaznamenat ve zkušební zprávě změny ve vývoji rostlin, např. velikost lístku, vzhled, známky nekrózy, chlorózy nebo hrbatosti, rozpadávání kolonií nebo ztráta vzplývavosti a v délce a vzhledu kořenu. Je rovněž nutné zaznamenat významné vlastnosti zkušebního média (např. přítomnost nerozpuštěného materiálu, růst řas ve zkušební nádobě).
Kromě určení počtu lístků během zkoušky se také hodnotí účinky zkoušené látky na jednu (nebo více) následujících proměnných měření:
|
i) |
celková plocha lístků, |
|
ii) |
suchá hmotnost, |
|
iii) |
čerstvá hmotnost. |
Celková plocha lístků má tu výhodu, že ji lze u každé zkušební a kontrolní nádoby určit na počátku zkoušky, v jejím průběhu i na jejím konci. Suchá nebo čerstvá hmotnost by se měla určit na počátku zkoušky ze vzorku očkovací kultury, jaké se obvykle používá k zahájení zkoušky, a na konci zkoušky pak na základě rostlinného materiálu z každé zkušební a kontrolní nádoby. Jestliže se plocha lístků neměří, dává se přednost suché hmotnosti před čerstvou hmotností.
Celková plocha lístků, suchá hmotnost a čerstvá hmotnost se mohou určit následovně:
|
i) |
Celková plocha lístků: Celková plocha lístků všech kolonií může být stanovena analýzou obrazu. Pomocí videokamery lze zachytit siluetu zkušební nádoby a rostlin (tj. umístíte nádobu na svítící skříň) a výsledný obraz se digitalizuje. Plochu lístků ve zkušební nádobě lze poté stanovit kalibrací pomocí plochých tvarů o známé ploše. Je třeba dbát na vyloučení rušení způsobeného hranou zkušební nádoby. Alternativní, ale pracnější způsob spočívá v pořízení fotografie zkušebních nádob a rostlin, vyříznutí výsledné siluety kolonií a určení jejich plochy pomocí analyzátoru lístkové plochy nebo milimetrového papíru. Mohou být vhodné i další postupy (například poměr hmotnosti papíru mezi plochou siluety kolonií a jednotkovou plochou). |
|
ii) |
Suchá hmotnost: Z každé ze zkušebních nádob se shromáždí všechny kolonie a propláchnou se destilovanou nebo deionizovanou vodou. Nadbytečná voda se odstraní savým papírem a pak se vzorky suší při 60 °C do konstantní hmotnosti. Měly by být zahrnuty jakékoliv úlomky kořenů. Suchá hmotnost by se měla vyjádřit s přesností na nejméně 0,1 mg. |
|
iii) |
Čerstvá hmotnost: Všechny kolonie se převedou do předem zvážených polystyrenových zkumavek (nebo vyrobených z jiného interního materiálu) s malými (1 mm) otvory v zakulacených dnech. Zkumavky se poté odstřeďují při 3 000 ot/min po dobu 10 minut při pokojové teplotě. Zkumavky obsahující nyní osušené kolonie se opětovně zváží a čerstvá hmotnost se vypočítá odečtením hmotnosti prázdné zkumavky. |
1.7.10.1 Četnost měření a analytická stanovení
Jestliže se používá plán statické zkoušky, pH každé expozice by se mělo změřit na počátku a na konci zkoušky. Jestliže se používá plán semistatické zkoušky, pH by se mělo měřit v každé vsázce „čerstvého“ zkušebního roztoku před každým obnovením a rovněž i v odpovídajících „spotřebovaných“ roztocích.
Intenzita osvětlení by se měla měřit v růstové komoře, inkubátoru nebo místnosti v bodech nacházejících se ve stejné vzdálenosti od zdroje světla jako lístky Lemna. Měření je třeba provádět nejméně jednou během zkoušky. Teplota média v náhradní nádobě držené za stejných podmínek v růstové komoře, inkubátoru nebo místnosti by se měla zaznamenávat nejméně jednou denně.
Během zkoušky se koncentrace zkoušené látky určují ve vhodných intervalech. Ve statických zkouškách je minimálním požadavkem určení koncentrací na počátku a na konci zkoušky.
U semistatických zkoušek, u nichž se neočekává, že koncentrace zkoušené látky zůstane v rozmezí ±20 % nominální koncentrace, je nezbytné analyzovat všechny čerstvě připravené zkušební roztoky a tytéž roztoky při každém obnovení (viz bod 1.7.7 třetí odstavec). Ovšem u těch zkoušek, u nichž nejsou naměřené počáteční koncentrace zkoušené látky v rozmezí ±20 % nominální hodnoty, avšak u nichž lze dostatečně prokázat, že počáteční koncentrace jsou reprodukovatelné a stálé (tj. od 80 do 120 % počátečních koncentrací), mohou být chemická stanovení provedena pouze na nejvyšších a nejnižších zkušebních koncentracích. Ve všech případech je třeba provést určení koncentrací zkoušené látky před obnovením při každé zkušební koncentraci vždy pouze u jedné nádoby k opakování (nebo u sdruženého obsahu nádob jednoho opakování).
U průtokových zkoušek je vhodný podobný vzorkovací režim, jaký je popsán u semistatických zkoušek včetně analýzy na počátku, uprostřed a na konci zkoušky, ale měření „spotřebovaného“ roztoku není v tomto případě vhodné. U tohoto typu zkoušky je zapotřebí kontrolovat denně průtok ředicí vody a zkoušené látky nebo zásobního roztoku zkoušené látky.
Lze-li prokázat, že byla po celou dobu zkoušky koncentrace zkoušené látky uspokojivě udržována v rozmezí ±20 % nominální nebo naměřené počáteční koncentrace, může být analýza výsledků založena na nominálních nebo naměřených počátečních hodnotách. Je-li odchylka od nominální nebo naměřené počáteční koncentrace větší než ±20 %, analýza výsledků by měla být založena na geometrické střední koncentraci během expozice nebo na modelech popisujících pokles koncentrace zkoušené látky (11).
1.7.11 Limitní zkouška
Za určitých okolností, např. pokud předběžná zkouška naznačuje, že zkoušená látka nemá žádné toxické účinky při koncentracích až do 100 mg l–1 nebo až do meze své rozpustnosti ve zkušebním médiu (podle toho, co je nižší), může být provedena limitní zkouška zahrnující porovnání odezvy v kontrolní skupině a odezvy v jedné exponované skupině (100 mg l–1 nebo koncentrace rovná mezi rozpustnosti). Důrazně se doporučuje podpořit tuto zkoušku analýzou expozičních koncentrací. Všechny dříve popsané zkušební podmínky a kritéria validity se vztahují i na limitní zkoušku s výjimkou toho, že počet opakování s exponovanými vzorky by se měl zdvojnásobit. Růst v kontrolní a exponované skupině může být analyzován pomocí statistického testu pro porovnání středních hodnot, např. Studentova t-testu.
2. DATA A ZPRÁVY
2.1 DOBA ZDVOJNÁSOBENÍ
Pro stanovení doby zdvojnásobení (T d) počtu lístků a dodržování tohoto kritéria validity ve studii (bod 1.6) se používá následující vzorec s údaji získanými z kontrolních nádob:
T d = ln 2/μ
kde μ je průměrná specifická růstová rychlost určená podle bodu 2.2.1 prvního a druhého odstavce.
2.2 PROMĚNNÉ ODEZVY
Účelem této zkoušky je stanovit účinky zkoušené látky na vegetativní růst Lemna. Tato zkušební metoda popisuje dvě proměnné odezvy, protože členské země mají odlišné preference a právní požadavky. Mají-li být výsledky zkoušky přijatelné ve všech členských zemích, účinky je třeba vyhodnotit pomocí obou proměnných odezvy a) a b) popsaných dále.
|
a) |
Průměrná specifická růstová rychlost: tato proměnná odezvy se počítá na základě změn logaritmů počtů lístků a dále na základě změn logaritmů jiného parametru měření (celková plocha lístků, suchá hmotnost nebo čerstvá hmotnost) v čase (vyjádřeno na den) v kontrolách a každé exponované skupině. Někdy se označuje jako relativní růstová rychlost (15). |
|
b) |
Výtěžek: tato proměnná odezvy se počítá na základě změn počtu lístků a dále na základě změn jiného parametru měření (celková plocha lístků, suchá hmotnost nebo čerstvá hmotnost) v kontrolách a každé exponované skupině až do konce zkoušky. |
Je třeba uvést, že hodnoty toxicity vypočítané pomocí těchto dvou proměnných odezvy nejsou srovnatelné a při používání výsledků zkoušky se musí vzít tento rozdíl na vědomí. Hodnoty ECx založené na průměrné specifické růstové rychlosti (ErCx) budou všeobecně vyšší než výsledky založené na výtěžku (EyCx), pokud se dodrží zkušební podmínky této zkušební metody, a to díky matematické základně příslušných přístupů. Tuto skutečnost nelze vykládat jako rozdíl v citlivosti mezi těmito dvěma proměnnými odezvy; hodnoty jsou jednoduše matematicky odlišné. Koncepce průměrné specifické růstové rychlosti je založena na obecném průběhu exponenciálního růstu okřehku v nelimitovaných kulturách, u nichž se toxicita odhaduje na základě účinků na růstovou rychlost, aniž by závisela na absolutní úrovni specifické růstové rychlosti kontroly, na sklonu křivky závislosti koncentrace-odezva nebo na době trvání zkoušky. Naproti tomu výsledky založené na proměnné odezvy „výtěžek“ jsou závislé na všech těchto ostatních proměnných. EyCx závisí na specifické růstové rychlosti druhů okřehku použitých v každé zkoušce a na maximální specifické růstové rychlosti, která se může u jednotlivých druhů, a dokonce i u různých klonů lišit. Tato proměnná odezvy by se neměla používat pro srovnávání citlivosti na toxické látky mezi druhy okřehku nebo dokonce různými klony. Přestože se z vědeckého hlediska dává přednost použití průměrné specifické růstové rychlosti pro odhad toxicity, odhady toxicity založené na výtěžku jsou rovněž do této zkušební metody zahrnuty, aby uspokojily současné právní požadavky v některých zemích.
Odhady toxicity by měly být založeny na počtu lístků a na jedné další proměnné měření (celková plocha lístků, suchá hmotnost nebo čerstvá hmotnost), protože některé látky mohou ovlivňovat jiné proměnné měření mnohem více než počet lístků. Pouhým určováním počtu lístků by se tento účinek nezjistil.
Počet lístků a jakákoliv další zaznamenaná proměnná měření, tj. celková plocha lístků, suchá hmotnost nebo čerstvá hmotnost, se pro každé měření uvedou v tabulce společně s koncentracemi zkoušené látky. Následná analýza dat, např. odhad LOEC, NOEC nebo ECx, by měla být založena na hodnotách pro jednotlivá opakování, a nikoliv na vypočítaných středních hodnotách pro každou exponovanou skupinu.
2.2.1 Průměrná specifická růstová rychlost
Průměrná specifická růstová rychlost pro konkrétní období se vypočítá jako logaritmické zvýšení proměnných růstu – počtu lístků a jedné další proměnné měření (celková plocha lístků, suchá hmotnost nebo čerstvá hmotnost) – pomocí následujícího vzorce pro každé opakování kontrolních roztoků a exponovaných roztoků:
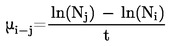
kde:
|
— |
μi–j: |
průměrná specifická růstová rychlost od doby i do doby j, |
|
— |
Ni: |
proměnná měření ve zkušební nebo kontrolní nádobě v době i, |
|
— |
Nj: |
proměnná měření ve zkušební nebo kontrolní nádobě v době j, |
|
— |
t: |
čas od i do j. |
Pro každou exponovanou skupinu a kontrolní skupinu vypočítejte střední hodnotu růstové rychlosti a odhady rozptylu.
Průměrná specifická růstová rychlost by se měla vypočítat pro celou dobu trvání zkoušky (doba „i“ ve shora uvedeném vzorci je počátkem zkoušky a doba „j“ je ukončením zkoušky). Pro každou zkušební koncentraci a kontrolu vypočítejte střední hodnotu průměrné specifické růstové rychlosti a odhady rozptylu. Dále je zapotřebí vyhodnocovat růstovou rychlost v jednotlivých časových úsecích s cílem zhodnotit účinky zkoušené látky, které se vyskytly během expoziční doby (např. kontrolou logaritmicky transformovaných růstových křivek). Významné rozdíly mezi růstovou rychlostí v jednotlivých časových úsecích a průměrnou růstovou rychlostí jsou známkou odchylky od konstantního exponenciálního růstu a vyžadují důkladné prozkoumání růstových křivek. V tomto případě by konzervativní přístup spočíval v porovnání specifických růstových rychlostí exponovaných kultur během doby maximální inhibice se specifickými růstovými rychlostmi kontrol pro totéž období.
Procentuální inhibici růstové rychlosti (I r) lze potom vypočítat pro každou zkušební koncentraci (exponovaná skupina) podle následujícího vzorce:
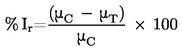
kde:
|
— |
% I r: |
procentuální inhibice průměrné specifické růstové rychlosti, |
|
— |
μ C: |
střední hodnota μ v kontrolní skupině, |
|
— |
μ T: |
střední hodnota μ v exponované skupině. |
2.2.2 Výtěžek
Účinky na výtěžek se určují na základě dvou proměnných měření, tj. počtu lístků a jedné další proměnné měření (celková plocha lístků, suchá hmotnost nebo čerstvá hmotnost), přítomných v každé zkušební nádobě na počátku a konci zkoušky. Pro suchou hmotnost nebo čerstvou hmotnost se počáteční biomasa určuje na základě vzorku lístků odebraných ze stejné vsázky použité k naočkování zkušebních nádob (viz bod 1.7.3 druhý odstavec). Pro každou zkušební koncentraci a kontrolu vypočítejte střední hodnotu výtěžku a odhady rozptylu. Střední procentuální inhibici výtěžku (%I y) lze vypočítat pro každou exponovanou skupinu následovně:
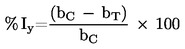
kde:
|
— |
% I y: |
procentuální snížení výtěžku, |
|
— |
b C: |
konečná biomasa minus počáteční biomasa pro kontrolní skupinu, |
|
— |
b T: |
konečná biomasa minus počáteční biomasa pro exponovanou skupinu. |
2.2.3 Vynášení křivek závislosti koncentrace-odezva
Je zapotřebí vynést křivky závislosti koncentrace na odezvě vyjadřující vztah střední procentuální inhibice proměnné odezvy (I r nebo I y vypočítané podle pokynů v bodě 2.2.1 posledním odstavci nebo v bodě 2.2.2) a logaritmu koncentrace zkoušené látky.
2.2.4 Odhad ECx
Odhady ECx (např. EC50) by měly být založeny jak na průměrné specifické růstové rychlosti (ErCx), tak na výtěžku (EyCx) a obě tyto hodnoty by zase měly vycházet z počtu lístků a jedné další proměnné měření (celková plocha lístků, suchá hmotnost nebo čerstvá hmotnost). Důvodem je skutečnost, že existují zkoušené látky, které mají odlišný dopad na počet lístků a na další proměnné měření. Požadovanými parametry toxicity jsou proto čtyři hodnoty ECx pro každou vypočítanou hodnotu inhibice x: ErCx (počet lístků), ErCx (celková plocha lístků, suchá hmotnost nebo čerstvá hmotnost), EyCx (počet lístků) a EyCx (celková plocha lístků, suchá hmotnost nebo čerstvá hmotnost),
2.3 STATISTICKÉ POSTUPY
Cílem je získat kvantitativní vztah mezi koncentrací a odezvou pomocí regresní analýzy. Je možné použít váženou lineární regresi po provedení linearizující transformace dat odezvy, například do probitových nebo logitových či Weibullových jednotek (16), ale dává se přednost nelineárním regresním postupům, s jejichž pomocí se lépe zpracovávají nevyhnutelné nepravidelnosti dat a odchylky od hladkých distribucí. Při přiblížení se k nule či úplné inhibici mohou být takové nepravidelnosti zesíleny transformací a rušit při analýze (16). Je zapotřebí uvést, že standardní metody analýzy používající probitové, logitové nebo Weibullovy transformace jsou určeny k použití s kvantálními daty (např. mortalita nebo přežití) a musí se upravit, aby mohly být použity s daty růstové rychlosti či výtěžku. Speciální postupy pro stanovení hodnot ECx ze spojitých dat lze nalézt v literatuře (17, 18 a 19).
Pro každou proměnnou odezvy, která se má analyzovat, použijte vztah koncentrace a odezvy pro výpočet bodových odhadů hodnot ECx. Je-li to možné, je zapotřebí určit 95% intervaly spolehlivosti pro každý odhad. Jakost shodnosti dat odezvy s regresním modelem je zapotřebí vyhodnotit buď graficky, nebo statisticky. Regresní analýza by se měla provádět pomocí odezev jednotlivých opakování, nikoliv pomocí středních hodnot exponovaných skupin.
Odhady EC50 a intervaly spolehlivosti lze rovněž získat lineární interpolací s bootstrapem (20), pokud se dostupné regresní modely/metody pro příslušná data nehodí.
Pro odhad LOEC, a tedy i NOEC je nezbytné porovnat střední hodnoty exponovaných vzorků pomocí metod analýzy rozptylu (ANOVA). Střední hodnota pro každou koncentraci musí být poté porovnána se střední hodnotou pro kontrolní skupinu, a to vhodnou metodou vícenásobného porovnání nebo metodou zkoušky trendu. Užitečné mohou být Dunnettův nebo Williamsův test (21, 22, 23, 24). Je nezbytné vyhodnotit, zda je splněn předpoklad homogenity rozptylu nezbytný pro ANOVA. Toto hodnocení lze provádět graficky nebo formálním testem (25). Mezi vhodné testy patří Levenův a Bartlettův test. Nesplnění předpokladu o homogenitě rozptylů lze někdy napravit logaritmickou transformací dat. Jestliže je heterogenita rozptylů extrémní a nelze ji napravit transformací, pak je třeba zvážit analýzu takovými metodami, jako jsou testy sestupného trendu Jonckheere. Další pokyny pro stanovení NOEC lze nalézt v (19).
Vědecký vývoj poslední doby vedl k doporučení opustit koncepci NOEC a nahradit ji odhady bodů ECx na základě regrese. Vhodná hodnota pro x nebyla pro tuto zkoušku s Lemna stanovena. Nicméně se zdá, že vhodné je rozmezí od 10 do 20 % (v závislosti na zvolené proměnné odezvy) a nejlépe by se měly uvádět jak EC10, tak EC20.
3. ZPRÁVY
3.1 PROTOKOL O ZKOUŠCE
Protokol o zkoušce musí obsahovat tyto údaje:
Zkoušená látka:
|
— |
fyzikální povaha a fyzikálně-chemické vlastnosti včetně meze rozpustnosti ve vodě, |
|
— |
údaje o chemické identifikaci (např. číslo CAS) včetně čistoty. |
Testovací druhy:
|
— |
vědecký název, klon (je-li znám) a zdroj. |
Zkušební podmínky:
|
— |
použitý zkušební postup (statický, semistatický nebo průtokový), |
|
— |
datum zahájení zkoušky a délka jejího trvání, |
|
— |
zkušební médium, |
|
— |
popis experimentálního provedení: zkušební nádoby a kryty, objemy roztoků, počty kolonií a lístků na zkušební nádobu na počátku zkoušky, |
|
— |
zkušební koncentrace (nominální, popř. naměřené) a počet opakování na každou koncentraci, |
|
— |
metody přípravy zásobních a zkušebních roztoků včetně použití jakýchkoliv rozpouštědel nebo dispergátorů, |
|
— |
teplota během zkoušky, |
|
— |
zdroj světla, intenzita světla a homogenita, |
|
— |
hodnoty pH zkušebních a kontrolních médií, |
|
— |
koncentrace zkoušené látky a metoda analýzy s příslušnými údaji pro hodnocení jakosti (validační studie, směrodatné odchylky nebo intervaly spolehlivosti analýz), |
|
— |
metody pro určení počtu lístků a dalších proměnných měření, např. suchá hmotnost, čerstvá hmotnost nebo plocha lístků, |
|
— |
všechny odchylky od této zkušební metody. |
Výsledky:
|
— |
hrubá data: počet lístků a dalších proměnných měření v každé zkušební a kontrolní nádobě při každém pozorování a každé analýze, |
|
— |
střední hodnoty a směrodatné odchylky pro každou proměnnou měření, |
|
— |
růstové křivky pro každou koncentraci (doporučuje se s logaritmicky transformovanou proměnnou měření, viz bod 2.2.1 druhý odstavec), |
|
— |
doba zdvojnásobení/růstová rychlost v kontrole na základě počtu lístků, |
|
— |
vypočítané proměnné odezvy pro všechna opakování s exponovanými vzorky, spolu se středními hodnotami a variačním koeficientem pro opakování, |
|
— |
grafické znázornění vztahu koncentrace a účinku, |
|
— |
odhady toxických účinků pro proměnné odezvy, např. EC50, EC10, EC20, a související intervaly spolehlivosti. Jestliže se počítají LOEC a/nebo NOEC, uvedou se jejich hodnoty a statistické metody použité k jejich stanovení, |
|
— |
jestliže byla použita ANOVA, uvede se velikost účinku, který lze detekovat (např. nejmenší významný rozdíl), |
|
— |
jakákoliv stimulace růstu zjištěná v jakémkoli exponovaném vzorku, |
|
— |
jakékoliv vizuální známky fytotoxicity, jakož i pozorování zkušebních roztoků, |
|
— |
diskuse výsledků včetně jakéhokoliv vlivu na výsledek zkoušky v důsledku odchylek od této zkušební metody. |
4. LITERATURA
|
(1) |
OECD TG 221 (2006) Lemna Sp. Growth Inhibition Test. |
|
(2) |
O použití studií na Lemna spp. u zbarvených látek pojednává oddíl 13.5.3 publikace EU Manual of Decisions z července 2006, která je dostupná na internetových stránkách http://ecb.jrc.ec.europa.eu/new-chemicals. |
|
(3) |
Guidance on information requirements and chemical safety assessment – kapitola R.7b: Endpoint specific guidance; tabulka 7.8.3: Summary of difficult substance testing issues, dostupné na internetových stránkách http://guidance.echa.europa.eu/docs/guidance_document/information_requirements_en.htm?time=1234958685#A. |
|
(4) |
ASTM International. (2003). Standard Guide for Conducting Static Toxicity Test With Lemna gibba G3. E 1415–91 (Reapproved 1998). s. 733–742. In: Annual Book of ASTM Standards, Vol. 11.05 Biological Effects and Environmental Fate; Biotechnology; Pesticides, ASTM, West Conshohocken, PA. |
|
(5) |
USEPA – United States Environmental Protection Agency. (1996). OPPTS 850.4400 Aquatic Plant Toxicity Test Using Lemna spp., „Public draft“. EPA 712-C-96–156. 8 stran. |
|
(6) |
AFNOR – Association Française de Normalisation. (1996). XP T 90–337: Détermination de l’inhibition de la croissance de Lemna minor. 10 stran. |
|
(7) |
SSI – Swedish Standards Institute. (1995). Water quality – Determination of growth inhibition (7-d) Lemna minor, duckweed. SS 02 82 13. 15 stran (švédsky). |
|
(8) |
Environment Canada. (1999). Biological Test Method: Test for Measuring the Inhibition of Growth Using the Freshwater Macrophyte, Lemna minor. EPS 1/RM/37, 120 stran. |
|
(9) |
Environment Canada. (1993) Proposed Guidelines for Registration of Chemical Pesticides: Non-Target Plant Testing and Evaluation. Canadian Wildlife Service, Technical Report Series No. 145. |
|
(10) |
Sims I., Whitehouse P., and Lacey R. (1999) The OECD Lemna Growth Inhibition Test. Development and Ring-testing of draft OECD Test Guideline. R&D Technical Report EMA 003. WRc plc – Environment Agency. |
|
(11) |
OECD (2000). Guidance Document on Aquatic Toxicity Testing of Difficult Substances and Mixtures. OECD Environmental Health and Safety Publications, Series on Testing and Assessment No.23. |
|
(12) |
ISO DIS 20079. Water Quality – Determination of the Toxic Effect of Water Constituents and Waste Water to Duckweed (Lemna minor) – Duckweed Growth Inhibition Test. |
|
(13) |
Walbridge C. T. (1977). A flow-through testing procedure with duckweed (Lemna minor L.). Environmental Research Laboratory – Duluth, Minnesota 55804. US EPA Report No. EPA-600/3–77 108. September 1977. |
|
(14) |
Lockhart W. L., Billeck B. N. and Baron C. L. (1989). Bioassays with a floating plant (Lemna minor) for effects of sprayed and dissolved glyphosate. Hydrobiologia, 118/119, 353–359. |
|
(15) |
Huebert, D.B. and Shay J.M. (1993) Considerations in the assessment of toxicity using duckweeds. Environmental Toxicology and Chemistry, 12, 481–483. |
|
(16) |
Christensen, E.R., Nyholm, N. (1984): Ecotoxicological Assays with Algae: Weibull Dose-Response Curves. Env. Sci. Technol. 19, 713–718. |
|
(17) |
Nyholm, N. Sørensen, P.S., Kusk, K.O. and Christensen, E.R. (1992): Statistical treatment of data from microbial toxicity tests. Environ. Toxicol. Chem. 11, 157–167. |
|
(18) |
Bruce R.D., Versteeg D.J. (1992) A statistical procedure for modelling continuous toxicity data. Environmental Toxicology and Chemistry, 11, 1485–1494. |
|
(19) |
OECD. (2004). Guidance Document on Statistical Analysis of Ecotoxicity Data. |
|
(20) |
Norberg-King T.J. (1988) An interpolation estimate for chronic toxicity: The ICp approach. National Effluent Toxicity Assessment Center Technical Report 05–88. USEPA, Duluth, MN. |
|
(21) |
Dunnett, C.W. (1955) A multiple comparisons procedure for comparing several treatments with a control. J. Amer. Statist. Assoc., 50, 1096–1121. |
|
(22) |
Dunnett, C.W. (1964) New tables for multiple comparisons with a control. Biometrics, 20, 482–491. |
|
(23) |
Williams, D.A. (1971) A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics, 27: 103–117. |
|
(24) |
Williams, D.A. (1972) The comparison of several dose levels with a zero dose control. Biometrics, 28: 510–531. |
|
(25) |
Brain P., Cousens R. (1989). An equation to describe dose-responses where there is stimulation of growth at low doses. Weed Research, 29, 93–96. |
Dodatek 1
Popis Lemna spp.
Vodní rostlina označovaná jako okřehek, Lemna spp., patří do čeledi Lemnaceae, která zahrnuje celosvětově rozšířené druhy ve čtyřech rodech. Jejich vzhled a taxonomie byly popsány vyčerpávajícím způsobem (1, 2). Lemna gibba a L. minor jsou představitelé druhů mírných oblastí a běžně se používají pro zkoušky toxicity. Oba druhy mají plovoucí nebo ponořenou lodyžku diskového tvaru (lístek) a ze středu spodní části každého lístku vychází velmi tenký kořínek. Lemna spp. zřídkakdy kvetou a rostliny se množí vegetativně vytvářením nových lístků (3). Při srovnání se staršími rostlinami jsou mladší rostliny světlejší, mají kratší kořínky a skládají se ze dvou až tří lístků různých velikostí. Díky své malé velikosti, jednoduché struktuře, asexuálnímu rozmnožování a krátké generační době jsou rostliny rodu Lemna velmi vhodné pro laboratorní zkoušení (4, 5).
Z důvodu pravděpodobných mezidruhových rozdílů v citlivosti jsou platná pouze srovnání citlivosti v rámci druhu.
Příklady druhů Lemna, které byly použity ke zkoušení: odkazy na druhy
Lemna aequinoctialis: Eklund, B. (1996). The use of the red alga Ceramium strictum and the duckweed Lemna aequinoctialis in aquatic ecotoxicological bioassays. Licentiate in Philosophy Thesis 1996:2. Dep. of Systems Ecology, Stockholm University.
Lemna major: Clark, N. A. (1925). The rate of reproduction of Lemna major as a function of intensity and duration of light. J. Phys. Chem., 29: 935–941.
Lemna minor: United States Environmental Protection Agency (USEPA). (1996). OPPTS 850.4400 Aquatic Plant Toxicity Test Using Lemna spp., „Public draft“. EPA 712-C-96–156. 8pp.
Association Française de Normalisation (AFNOR). (1996). XP T 90–337: Détermination de l’inhibition de la croissance de Lemna minor. 10pp.
Švédský normalizační institut (SIS). (1995). Water quality – Determination of growth inhibition (7-d) Lemna minor, duckweed. SS 02 82 13. 15 stran (švédsky).
Lemna gibba: ASTM International. (2003). Standard Guide for Conducting Static Toxicity Test With Lemna gibba G3. E 1415–91 (Reapproved 1998). s. 733–742.
United States Environmental Protection Agency (USEPA). (1996). OPPTS 850.4400 Aquatic Plant Toxicity Test Using Lemna spp., „Public draft“. EPA 712-C-96–156. 8 stran.
Lemna paucicostata: Nasu, Y., Kugimoto, M. (1981). Lemna (duckweed) as an indicator of water pollution. I. The sensitivity of Lemna paucicostata to heavy metals. Arch. Environ. Contam. Toxicol., 10:1959–1969.
Lemna perpusilla: Clark, J. R. et al. (1981). Accumulation and depuration of metals by duckweed (Lemna perpusilla). Ecotoxicol. Environ. Saf., 5:87–96.
Lemna trisulca: Huebert, D. B., Shay, J. M. (1993). Considerations in the assessment of toxicity using duckweeds. Environ. Toxicol. and Chem., 12:481–483.
Lemna valdiviana: Hutchinson, T.C., Czyrska, H. (1975). Heavy metal toxicity and synergism to floating aquatic weeds. Verh.-Int. Ver. Limnol., 19:2102–2111.
Zdroje druhů Lemna
|
University of Toronto Culture Collection of Algae and Cyanobacteria |
|
Department of Botany, University of Toronto |
|
Toronto, Ontario, Kanada, M5S 3 B2 |
|
Tel.: +1–416–978–3641 |
|
Fax:+1–416–978–5878 |
|
e-mail: [email protected] |
|
http://www.botany.utoronto.ca/utcc |
|
North Carolina State University |
|
Forestry Dept |
|
Duckweed Culture Collection |
|
Campus Box 8002 |
|
Raleigh, NC 27695–8002 |
|
USA |
|
Tel.: 001 (919) 515–7572 |
|
Institutionen före tillämpad miljövetenskap (ITM) Stockholms universitet |
|
SE-106 91 Stockholm |
|
ŠVÉDSKO |
|
Tel.: +46 86747240 |
|
Fax: +46 86747636 |
|
Umweltbundesamt (UBA) |
|
FG III 3.4 |
|
Schichauweg 58 |
|
12307 Berlin |
|
Německo |
|
e-mail: [email protected] |
|
http://www.umweltbundesamt.de/contact.htm |
Literatura
|
(1) |
Hillman, W.S. (1961). The Lemnaceae or duckweeds: A review of the descriptive and experimental literature. The Botanical Review, 27:221–287. |
|
(2) |
Landolt, E. (1986). Biosystematic investigations in the family of duckweed (Lemnaceae). Vol. 2. Geobotanischen Inst. ETH, Stiftung Rubel, Zürich, Switzerland. |
|
(3) |
Björndahl, G. (1982). Growth performance, nutrient uptake and human utilization of duckweeds (Lemnaceae family). ISBN 82–991150–0–0. The Agricultural Research Council of Norway, University of Oslo. |
|
(4) |
Wang, W. (1986). Toxicity tests of aquatic pollutants by using common duckweed. Environmental Pollution, Ser B, 11:1–14. |
|
(5) |
Wang, W. (1990). Literature review on duckweed toxicity testing. Environmental Research, 52:7–22. |
Dodatek 2
Uchovávání zásobní kultury
Zásobní kultury lze uchovávat za nízkých teplot (4–10 °C) po delší dobu, aniž by bylo zapotřebí je znovu zakládat. Růstové médium pro Lemna může být stejné jako médium používané ke zkoušení, ale pro zásobní kultury lze použít jiná média bohatá na živiny.
Pravidelně se odebere několik mladých, světlezelených rostlin a aseptickým postupem se umístí do nových kultivačních nádob obsahujících čerstvé médium. Za chladnějších podmínek, které se zde navrhují, lze provádět tvorbu dílčích kultur v intervalech až do tří měsíců.
Je třeba používat chemicky čisté (kyselinou promyté) a sterilní skleněné kultivační nádoby a pracovat aseptickými manipulačními postupy. V případě kontaminace zásobní kultury např. řasami či kvasinkami, je nezbytné přijmout opatření na odstranění kontaminujících organismů. V případě řas a většiny dalších kontaminujících organismů toho lze dosáhnout povrchovou sterilizací. Odebere se vzorek kontaminovaného rostlinného materiálu a odříznou se kořeny. Materiál se poté intenzivně protřepává v čisté vodě, poté následuje ponoření do 0,5 % (obj.) roztoku chlornanu sodného po dobu 30 sekund až 5 minut. Rostlinný materiál se dále opláchne sterilní vodou a převede se jako řada vsázek do kultivačních nádob obsahujících čerstvé růstové médium. Mnoho lístků v důsledku tohoto ošetření odumře, zejména použijí-li se delší expoziční doby, ale některé z přeživších rostlin budou obvykle zbaveny kontaminace. Ty lze poté použít pro přeočkování nových kultur.
Dodatek 3
Média
Pro L. minor a L. gibba se doporučují různá růstová média. Pro L. minor se doporučuje upravené švédské standardní (SIS) médium, zatímco pro L. gibba se doporučuje médium 20X AAP. Složení obou médií je uvedeno níže. Při přípravě těchto médií je zapotřebí používat chemické látky analytické čistoty nebo na úrovni činidel a deionizovanou vodu.
Švédské standardní (SIS) růstové médium pro Lemna
|
— |
Zásobní roztoky I–V se sterilizují v autoklávu (120 °C, 15 minut) nebo membránovou filtrací (přibližná velikost pórů 0,2 µm). |
|
— |
Zásobní roztok VI (a případně VII) se sterilizují pouze membránovou filtrací, neautoklávují se. |
|
— |
Sterilní zásobní roztoky se skladují v chladu a temnu. Zásobní roztoky I až V je třeba zlikvidovat po šesti měsících, zatímco zásobní roztoky VI (a případně VII) mají životnost jeden měsíc.
|
||||||||||||||||||||||||||||||||||||||||||||||||||
|
— |
Pro přípravu jednoho litru média SIS se do 900 ml deionizované vody přidá:
Poznámka: Pro některé zkoušené látky možná bude zapotřebí další zásobní roztok VII (pufr MOPS) (viz bod 1.4 poslední odstavec). |
|
— |
pH se upraví na 6,5 ± 0,2 přidáním 0,1M nebo 1M-HCl nebo NaOH a objem se doplní deionizovanou vodou do jednoho litru. |
Růstové médium 20X AAP
Zásobní roztoky se připravují ve sterilní destilované nebo deionizované vodě.
Sterilní zásobní roztoky se skladují v chladu a temnu. Za těchto podmínek budou mít zásobní roztoky životnost nejméně 6 až 8 týdnů.
Pro médium 20X AAP se připraví pět zásobních roztoků živin (A1, A2, A3, B a C), přičemž se použijí chemické látky na úrovni činidel. Růstové médium se vytvoří přidáním 20 ml každého zásobního roztoku živin do přibližně 850 ml deionizované vody. pH se upraví na 7,5 ± 0,1 přidáním 0,1M nebo 1M-HCl nebo NaOH a objem se doplní do jednoho litru deionizovanou vodou. Médium se poté filtruje přes (přibližně) 0,2μm membránový filtr do sterilního zásobníku.
Růstové médium určené ke zkoušení by se mělo připravovat 1 až 2 dny před použitím, aby se mohlo stabilizovat pH. pH růstového média by se před použitím mělo zkontrolovat a znovu upravit, bude-li to nezbytné, přídavkem 0,1M nebo 1M-NaOH či HCl, jak je popsáno výše.
|
Zásobní roztok č. |
Látka |
Koncentrace v zásobním roztoku (g.l–1) (1) |
Koncentrace v připraveném médiu (mg.l–1) (1) |
Připravené médium |
|
|
Prvek |
Koncentrace (mg·l–1) (1) |
||||
|
A1 |
NaNO3 MgCl2·6H2O CaCl2·2H2O |
26 12 4,4 |
510 240 90 |
Na; N Mg Ca |
190; 84 58,08 24,04 |
|
A2 |
MgSO4·7H2O |
15 |
290 |
S |
38,22 |
|
A3 |
K2HPO4·3H2O |
1,4 |
30 |
K; P |
9,4; 3,7 |
|
B |
H3BO3 MnCl2·4H2O FeCl3·6H2O Na2EDTA·2H2O ZnCl2 CoCl2·6H2O Na2MoO4·2H2O CuCl2·2H2O |
0,19 0,42 0,16 0,30 3,3 mg·l–1 1,4 mg·l–1 7,3 mg·l–1 0,012 mg·l–1 |
3,7 8,3 3,2 6,0 66 μg·l–1 29 μg·l–1 145 μg·l–1 0,24 μg·l–1 |
B Mn Fe — Zn Co Mo Cu |
0,65 2,3 0,66 — 31 μg·l–1 7,1 μg·l–1 58 μg·l–1 0,080 μg·l–1 |
|
C |
NaHCO3 |
15 |
300 |
Na; C |
220; 43 |
Poznámka pod čarou: Teoreticky správná konečná koncentrace hydrogenuhličitanu (při níž není nutná výrazná úprava pH) je 15 mg/l, nikoliv 300 mg/l. Ovšem historické používání média 20X AAP včetně kruhové zkoušky pro tuto metodu je založeno na 300 mg/l. (I. Sims, P. Whitehouse and R. Lacey. (1999) The OECD Lemna Growth Inhibition Test. Development and Ring-testing of draft OECD Test Guideline. R&D Technical Report EMA 003. WRc plc – Environment Agency).
STEINBERGOVO médium (podle ISO 20079)
Koncentrace a zásobní roztoky
|
— |
Upravené Steinbergovo médium se v normě ISO 20079 používá pouze pro Lemna minor (protože uvedená norma povoluje pouze Lemna minor), ale zkoušky ukázaly, že by se mohlo dosáhnout dobrých výsledků i s Lemna gibba. |
|
— |
Při přípravě média je zapotřebí používat chemické látky analytické čistoty nebo na úrovni činidel a deionizovanou vodu. |
|
— |
Připravte živné médium ze zásobních roztoků nebo desetinásobně koncentrovaného média, které umožňuje maximální koncentraci média bez srážení. |
Tabulka 1
STEINBERGOVO médium se stabilizovaným pH (upraveno podle Altenburgera)
|
Látka |
Živné médium |
||
|
Makroprvky |
molekulová hmotnost |
mg/l |
mmol/l |
|
KNO3 |
101,12 |
350,00 |
3,46 |
|
Ca(NO3)2 · 4H2O |
236,15 |
295,00 |
1,25 |
|
KH2PO4 |
136,09 |
90,00 |
0,66 |
|
K2HPO4 |
174,18 |
12,60 |
0,072 |
|
MgSO4 · 7H2O |
246,37 |
100,00 |
0,41 |
|
Mikroprvky |
molekulová hmotnost |
µg/l |
µmol/l |
|
H3BO3 |
61,83 |
120,00 |
1,94 |
|
ZnSO4 · 7H2O |
287,43 |
180,00 |
0,63 |
|
Na2MoO4 · 2H2O |
241,92 |
44,00 |
0,18 |
|
MnCl2 · 4H2O |
197,84 |
180,00 |
0,91 |
|
FeCl3 · 6H2O |
270,21 |
760,00 |
2,81 |
|
dinatrium-EDTA dihydrát |
372,24 |
1 500,00 |
4,03 |
Tabulka 2
Zásobní roztoky (makroprvky)
|
g/l |
||
|
Zásobní roztok 1: |
|||
|
KNO3 |
17,50 |
||
|
KH2PO4 |
4,5 |
||
|
K2HPO4 |
0,63 |
||
|
Zásobní roztok 2: |
|||
|
MgSO4 · 7H2O |
5,00 |
||
|
Zásobní roztok 3: |
|||
|
Ca(NO3)2 · 4H2O |
14,75 |
||
Tabulka 3
Zásobní roztoky (mikroprvky)
|
mg/l |
||
|
Zásobní roztok 4: |
|||
|
H3BO3 |
120,0 |
||
|
Zásobní roztok 5: |
|||
|
ZnSO4 · 7H2O |
180,0 |
||
|
Zásobní roztok 6: |
|||
|
Na2MoO4 · 2H2O |
44,0 |
||
|
Zásobní roztok 7: |
|||
|
MnCl2 · 4H2O |
180,0 |
||
|
Zásobní roztok 8: |
|||
|
FeCl3 · 6H2O |
760,00 |
||
|
Ethylendiamintetraoctan dvojsodný-dihydrát |
1 500,00 |
||
|
— |
Zásobní roztoky 2 a 3 a odděleně 4 až 7 lze sdružovat (při zohlednění požadovaných koncentrací). |
|
— |
K dosažení delší životnosti autoklávujte zásobní roztoky při teplotě 121 °C po dobu 20 minut nebo alternativně proveďte sterilní filtraci (0,2 μm). U zásobního roztoku 8 se důrazně doporučuje sterilní filtrace (0,2 μm). |
Příprava konečné koncentrace STEINBERGOVA média (upraveného)
|
— |
Přidejte 20 ml zásobních roztoků 1, 2 a 3 (viz tabulka 2) do přibližně 900 ml deionizované vody, aby se zabránilo srážení. |
|
— |
Přidejte 1,0 ml zásobních roztoků 4, 5, 6, 7 a 8 (viz tabulka 3). |
|
— |
pH by mělo být 5,5 ± 0,2 (upravte přidáním minimálního objemu roztoku NaOH nebo HCl). |
|
— |
Doplňte vodou do 1 000 ml. |
|
— |
Jestliže se zásobní roztoky sterilizují a použije-li se vhodná voda, žádná další sterilizace není zapotřebí. Pokud se sterilizuje konečné médium, zásobní roztok 8 se přidá po autoklávování (při teplotě 121 °C po dobu 20 min). |
Příprava 10násobně koncentrovaného STEINBERGOVA média (upraveného) pro přechodné uložení
|
— |
Přidejte 20 ml zásobních roztoků 1, 2 a 3 (viz tabulka 2) do přibližně 30 ml vody, aby se zabránilo srážení. |
|
— |
Přidejte 1,0 ml zásobních roztoků 4, 5, 6, 7 a 8 (viz tabulka 3). Doplňte vodou do 100 ml. |
|
— |
Jestliže se zásobní roztoky sterilizují a použije-li se vhodná voda, žádná další sterilizace není zapotřebí. Pokud se sterilizuje konečné médium, zásobní roztok 8 se přidá po autoklávování (při teplotě 121 °C po dobu 20 minut). |
|
— |
pH média (konečná koncentrace) by mělo být 5,5 ± 0,2. |
(1) Pokud není uvedeno jinak.