(ES) č. 702/2007Nařízení Komise (ES) č. 702/2007 ze dne 21. června 2007 , kterým se mění nařízení (EHS) č. 2568/91 o charakteristikách olivového oleje a olivového oleje z pokrutin a o příslušných metodách analýzy
| Publikováno: | Úř. věst. L 161, 22.6.2007, s. 11-27 | Druh předpisu: | Nařízení |
| Přijato: | 21. června 2007 | Autor předpisu: | Evropská komise |
| Platnost od: | 25. června 2007 | Nabývá účinnosti: | 1. ledna 2008 |
| Platnost předpisu: | Zrušen předpisem (EU) 2022/2104 | Pozbývá platnosti: | 24. listopadu 2022 |
Text předpisu s celou hlavičkou je dostupný pouze pro registrované uživatele.
NAŘÍZENÍ KOMISE (ES) č. 702/2007
ze dne 21. června 2007,
kterým se mění nařízení (EHS) č. 2568/91 o charakteristikách olivového oleje a olivového oleje z pokrutin a o příslušných metodách analýzy
KOMISE EVROPSKÝCH SPOLEČENSTVÍ,
s ohledem na Smlouvu o založení Evropského společenství,
s ohledem na nařízení Rady (ES) č. 865/2004 ze dne 29. dubna 2004 o společné organizaci trhu s olivovým olejem a stolními olivami a o změně nařízení (EHS) č. 827/68 (1), a zejména na čl. 5 odst. 3 uvedeného nařízení,
vzhledem k těmto důvodům:
|
(1) |
Nařízení Komise (EHS) č. 2568/91 (2) definuje fyzikální a chemické charakteristiky olivového oleje a olivového oleje z pokrutin a stanoví metody hodnocení těchto charakteristik. Tyto metody, jakož i mezní hodnoty týkající se charakteristik olejů, musí být aktualizovány, přičemž je třeba zohlednit názory chemických odborníků a postupovat v souladu s dokončenými pracemi v rámci Mezinárodní rady pro olivový olej. |
|
(2) |
Chemičtí odborníci se především domnívají, že kvantifikace procentuálního podílu 2-glyceril monopalmitátu je přesnější pro detekci esterifikovaných olejů. Snížení mezní hodnoty pro stigmasta v panenských olivových olejích také umožňuje lepší oddělení panenských a rafinovaných olivových olejů. |
|
(3) |
Aby se poskytlo období pro přizpůsobení se novým normám a pro zavedení prostředků potřebných k jejich použití a v zájmu zabránění narušení obchodních operací je třeba použití tohoto nařízení odložit do 1. ledna 2008. Ze stejných důvodů je třeba stanovit, že olivové oleje a pokrutiny, které byly v souladu s právními předpisy vyrobeny a označeny etiketou ve Společenství nebo byly dovezeny do Společenství a propuštěny do volného oběhu před uvedeným datem, mohou být uváděny na trh až do vyčerpání zásob. |
|
(4) |
Opatření stanovená tímto nařízením jsou v souladu se stanoviskem Řídícího výboru pro olivový olej a stolní olivy, |
PŘIJALA TOTO NAŘÍZENÍ:
Článek 1
Nařízení (EHS) č. 2568/91 se mění takto:
|
1) |
V ustanovení čl. 2 odst. 1 se šestá odrážka nahrazuje tímto:
|
|
2) |
Přílohy se mění v souladu s přílohou tohoto nařízení. |
Článek 2
Toto nařízení vstupuje v platnost třetím dnem po vyhlášení v Úředním věstníku Evropské unie.
Toto nařízení se použije ode dne 1. ledna 2008.
Produkty, které však byly v souladu s právními předpisy vyrobeny a označeny etiketami ve Společenství nebo dovezeny do Společenství a propuštěny do volného oběhu před 1. lednem 2008, mohou být uváděny na trh až do vyčerpání zásob.
Toto nařízení je závazné v celém rozsahu a přímo použitelné ve všech členských státech.
V Bruselu dne 21. června 2007.
Za Komisi
Mariann FISCHER BOEL
členka Komise
(1) Úř. věst. L 161, 30.4.2004, s. 97.
(2) Úř. věst. L 248, 5.9.1991, s. 1. Nařízení naposledy pozměněné nařízením (ES) č. 1989/2003 (Úř. věst. L 295, 13.11.2003, s. 57).
PŘÍLOHA
Přílohy nařízení (EHS) č. 2568/91 se mění takto:
|
1) |
Obsah se mění takto:
|
|
2) |
Příloha I se nahrazuje tímto: „PŘÍLOHA I CHARAKTERISTIKY OLIVOVÉHO OLEJE Poznámky:
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
3) |
Dodatek 1 se mění takto:
|
|
4) |
V příloze II se název nahrazuje tímto: |
|
5) |
Příloha IV se nahrazuje tímto: „PŘÍLOHA IV STANOVENÍ OBSAHU VOSKU POMOCÍ KAPILÁRNÍ PLYNOVÉ CHROMATOGRAFIE 1. PŘEDMĚT Tato metoda popisuje postup stanovení obsahu vosku v olivových olejích. Vosky se dělí podle počtu atomů uhlíku. Tuto metodu lze použít zejména k rozlišení mezi olivovým olejem získaným lisováním a extrakcí (olivový olej z pokrutin). 2. PODSTATA METODY K tuku nebo oleji se přimísí vhodný vnitřní standard a poté se provádí chromatografická frakční destilace na hydratované silikagelové koloně. Získaná frakce eluovaná nejprve při testovacích podmínkách (jejíž polarita je menší než polarita triglyceridů) se přímo analyzuje pomocí kapilární plynové chromatografie. 3. PŘÍSTROJE A POMŮCKY 3.1 Kónická 25ml baňka. 3.2 Skleněná chromatografická plynová kolona o vnitřním průměru 15 mm a délce 30 až 40 cm vybavená ventilem. 3.3 Plynový chromatograf vhodný pro použití s kapilární kolonou a vybavený přímým vstřikováním, obsahující: 3.3.1 Termostatickou komoru pro kolony vybavenou programátorem teploty. 3.3.2 Studené nástřikové zařízení pro přímé zavádění do kolony. 3.3.3 Plameno-ionizační detekční čidlo a konverzní zesilovač. 3.3.4 Integrátor se zapisovačem, vhodný pro provoz s konverzním zesilovačem (3.3.3), s rychlostí odezvy nižší než 1 sekundu a s nastavitelnou rychlostí posunu papíru. (Lze též použít informační systémy, které umožňují získávání dat z plynové chromatografie pomocí PC.) 3.3.5 Kapilární kolona, ze skla nebo taveného křemene, dlouhá 8 až 12 m s vnitřním průměrem 0,25 až 0,32 mm, obsahující kapalnou fázi, o jednotné tloušťce 0,10 až 0,30 μm. (Kapalné fáze SE 52 nebo SE 54 vhodné k použití v obchodu.) 3.4 Mikrostříkačka na 10 μl pro přímý vstřik do kolony opatřená tvrzenou jehlou. 3.5 Elektrický vibrátor. 3.6 Rotační odpařovač. 3.7 Muflová pec. 3.8 Analytické váhy s přesností měření ± 0,1 mg. 3.9 Běžné laboratorní skleněné nádoby. 4. REAKČNÍ ČINIDLA 4.1 Silikagel s velikostí zrna od 60 do 200 μm. Silikagel se umístí alespoň na čtyři hodiny do pece o teplotě 500 °C. Nechá se ochladit a přidají se 2 % vody v poměru k odebranému množství silikagelu. Řádným protřepáním se směs homogenizuje. Před použitím se ponechá nejméně 12 hodin ve tmě. 4.2 n-hexan pro chromatografii. 4.3 Diethylether pro chromatografii. 4.4 n-heptan pro chromatografii. 4.5 Standardní roztok 0,1 % (m/V) laurylarachidátu v hexanu (vnitřní standard). (Lze též použít palmityl palmitát a myristyl stearát.) 4.5.1 Sudan 1 (1-fenyl-azo-2-naftol). 4.6 Nosný plyn: vodík nebo čisté helium pro plynovou chromatografii. 4.7 Pomocné plyny:
5. POSTUP 5.1 Příprava chromatografické kolony Provede se suspenze 15 g silikagelu (4.1) v n-hexanu (4.2) a zavede se do kolony (3.2). Po spontánní sedimentaci se tato dokončí pomocí elektrického vibrátoru (3.5), aby byla chromatografická vrstva co nejhomogennější. Provede se perkolace 30 ml n-hexanu za účelem odstranění případných nečistot. Pomocí vah (3.8) se naváží přesně 500 mg vzorku do baňky (3.1) a přidá se vhodné množství vnitřního standardu (4.5) v závislosti na předpokládaném obsahu vosku. Např. 0,1 mg laurylu arachidátu v případě olivového oleje a 0,25 až 0,5 mg v případě olivového oleje z pokrutin. Získaný vzorek se za pomoci dvou dávek 2 ml n-hexanu (4.2) převede do chromatografické kolony. Umožní se, aby hladina rozpouštědla poklesla tak, aby byla 1 mm nad horní úrovní absorbentu, a poté se provede perkolace 70 ml doplňkového n-hexanu za účelem odstranění n-alkanů, které jsou přirozeně přítomny. Poté se zahájí chromatografické eluování, odejme se 180 ml směsi n-hexanu/diethyletheru v poměru 99:1 při průtoku přibližně 15 kapek za 10 sekund. Eluování vzorku se musí provést při teplotě okolí 22 °C ± 4. Poznámky:
Získaná frakce se usuší v rotačním odpařovači (3.6), až je téměř všechno rozpouštědlo odstraněno. Poslední 2 ml rozpouštědla se odstraní za pomoci slabého proudu dusíku; poté se přidá 2–4 ml n-heptanu. 5.2 Analýza plynovou chromatografií 5.2.1 Přípravné práce Kolona se spojí s plynovým chromatografem (3.3), vstupní port se připojí ke kolonovému systému (on-column system) a výstupní port k detekčnímu činidlu. Poté se plynový chromatograf překontroluje (těsnost plynových vedení, funkce detekčního činidla a záznamníku atd.). Kapilární kolony, které mají být použity poprvé, je nutno nejprve kondicionovat. Kapilární kolonou se nechá protékat malé množství nosného plynu, poté se zapne plynový chromatograf a nechá se postupně zahřívat. Postupně se zahřívá, dokud není zhruba po 4 hodinách dosaženo teploty 350 °C. Tato teplota se udržuje nejméně dvě hodiny a poté se provede regulace pracovních podmínek (regulace průtoku plynu, zapálení plamene, připojení elektronického zapisovače (3.3.4), nastavení teploty kolonové komory, detekčního činidla atd.) a signál se nastaví na citlivost, která činí nejméně dvojnásobek nejvyšší úrovně uvažované pro provedení analýzy. Základní linie záznamu musí být rovná, bez jakýchkoli píků nebo driftů. Záporné drifty jsou známkou nedokonalé těsnosti spojů kolony, zatímco kladné svědčí o nedostatečné kondicionaci kolony. 5.2.2 Výběr pracovních podmínek Pracovní podmínky, které je třeba všeobecně dodržovat, jsou tyto:
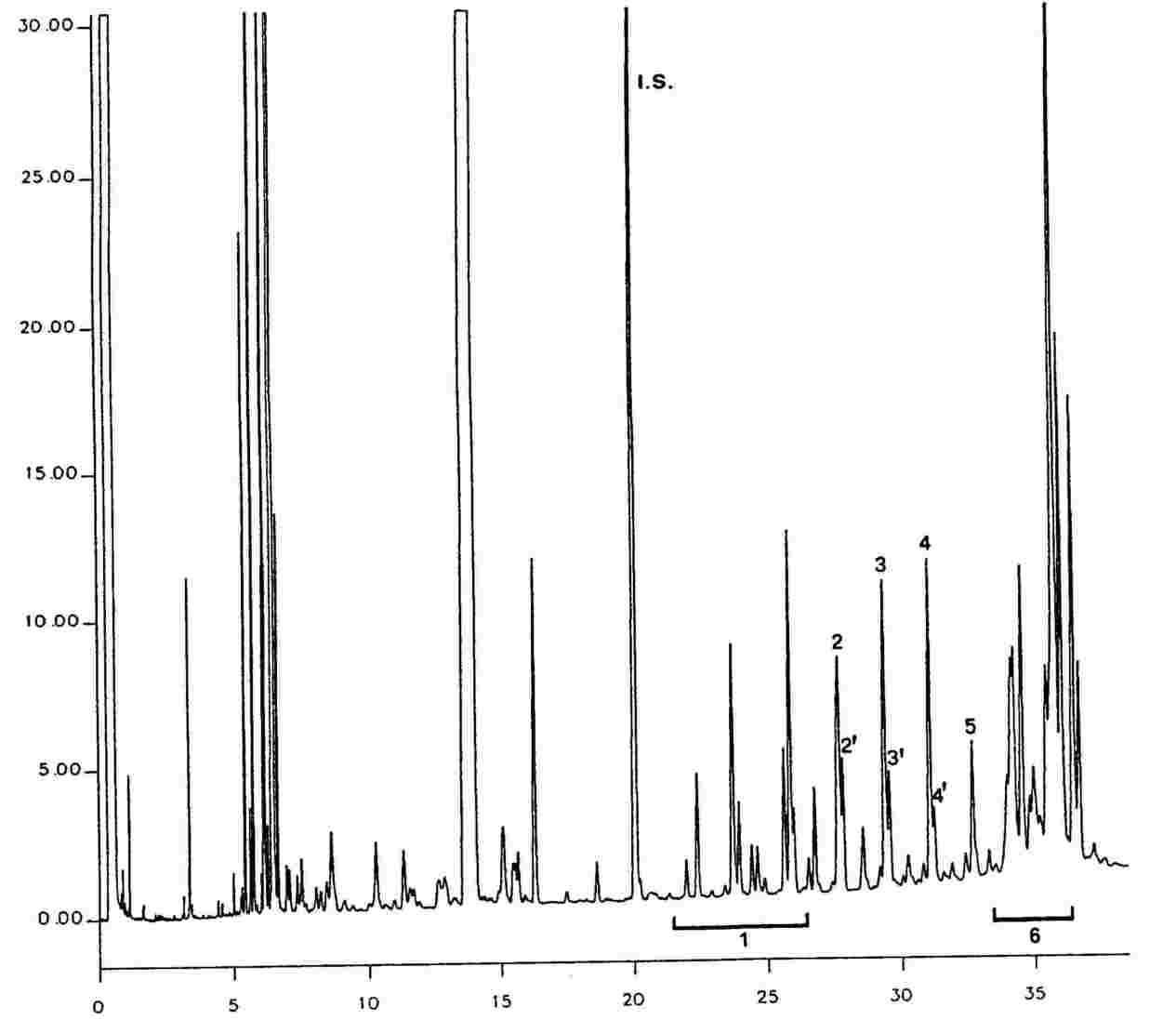
Tyto podmínky mohou být upraveny podle charakteristik kolony a plynového chromatografu s cílem oddělit všechny vosky, dosáhnout dostatečného rozpuštění píků (viz obrázek) a retenčního času vnitřního standardu C32, který musí být 18 ± 3 minuty. Nejreprezentativnější pík vosku musí dosáhnout nejméně 60 % z plného rozsahu. Parametry pro integraci píků se nastaví tak, aby došlo ke správnému vyhodnocení ploch uvažovaných píků. Poznámka: Vzhledem k tomu, že konečná teplota je vysoká, připouští se kladný drift, který nesmí přesáhnout 10 % z plného rozsahu. 5.3 Provedení analýzy Do mikrostříkačky na 10 μl se natáhne 1 μl roztoku; píst mikrostříkačky se povytáhne tak, aby se vyprázdnila jehla. Jehla se zavede přes septum vstřikové komory a přibližně po jedné nebo dvou sekundách se rychle nastříkne roztok; přibližně po pěti sekundách se jehla pomalu vytáhne. Zaznamenávání je prováděno, dokud vosky nejsou zcela eluovány. Základní linie záznamu musí vždy odpovídat požadavkům. 5.4 Identifikace píků Identifikace jednotlivých píků se provádí podle retenčních časů a porovnáním se směsmi vosků, jejichž retenční časy jsou známé a které byly analyzovány za stejných podmínek. Chromatogram vosků panenského olivového oleje je znázorněn na obrázku. 5.5 Kvantitativní vyhodnocení Pomocí integrátoru se vypočítají plochy píků odpovídajících vnitřnímu standardu a alifatickým esterům C40 až C46. Obsah vosku jednotlivých esterů vyjádřený v mg/kg tukové látky se vypočítá podle vzorce:
kde:
6. VYJÁDŘENÍ VÝSLEDKŮ Uvádí se souhrn obsahů jednotlivých vosků C40 až C46v mg/kg tukové látky (ppm). Poznámka: Sloučeniny, které je třeba kvantifikovat, se stanoví vzhledem k píkům esterů s počtem uhlíků mezi C40 a C46 podle příkladu chromatogramu vosků olivového oleje uvedeného na následujícím obrázku. Pokud se ester C46 objeví dvakrát, doporučuje se pro jeho identifikaci provést analýzu frakce vosků olivového oleje z pokrutin, v níž je pík C46 snadno rozpoznatelný, jelikož jasně převažuje. Výsledky se vyjádří s přesností na jedno desetinné místo. Obrázek Chromatogram vosků olivového oleje (9)
Dodatek Stanovení lineární rychlosti plynu Do plynového chromatografu nastaveného na normální pracovní podmínky se vstříkne 1 až 3 μl methanu nebo propanu. Změří se doba průchodu plynu kolonou od počátku vstřiku do okamžiku eluce píků (tM). Lineární rychlost proudění v cm/s je dána poměrem L/tM, kde L je délka kolony v cm a (tM) je čas změřený v sekundách. |
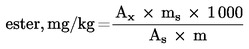
|
6) |
Příloha VII se nahrazuje tímto: „PŘÍLOHA VII STANOVENÍ PROCENTNÍHO PODÍLU 2-GLYCERIL MONOPALMITÁTU 1. PŘEDMĚT A ROZSAH POUŽITÍ Tato metoda popisuje analytický postup stanovení procentuálního podílu kyseliny palmitové ve 2. pozici triglyceridů hodnocením 2-glyceril monopalmitátu. Tato metoda se použije na tekuté rostlinné oleje při teplotě okolí (20 °C). 2. PODSTATA METODY Po přípravě se vzorek oleje podrobí účinku pankreatické lipázy. Parciální a specifická hydrolýza v pozici 1 a 3 molekuly triglyceridu způsobuje, že monoglyceridy se objeví na 2. pozici. Procentuální podíl 2-glyceril monopalmitátu v monoglycerické frakci se stanoví po silylaci pomocí kapilární plynové chromatografie. 3. PŘÍSTROJE A POMŮCKY 3.1 Kónická 25ml baňka. 3.2 Kádinky na 100, 250 a 300 ml. 3.3 Skleněná chromatografická kolona o vnitřním průměru 21–23 mm a délce 400 mm s vložkou ze sintrovaného skla a kohoutem. 3.4 Nedělené pipety na 10, 50, 100 a 200 ml. 3.5 Baňky na 100 a 250 ml. 3.6 Rotační odpařovač. 3.7 Centrifugační zkumavky s konickým dnem na 10 ml se zabroušenou skleněnou zátkou. 3.8 Odstředivka pro zkumavky o obsahu 10 a 100 ml. 3.9 Termostat umožňující udržet teplotu na 40 °C s přesností na 0,5 °C. 3.10 Dělené pipety na 1 a 2 ml. 3.11 Hypodermická stříkačka na 1 ml. 3.12 Mikrostříkačka na 100 μl. 3.13 Nálevka na 1 000 ml. 3.14 Plynový chromatograf pro kapilární kolony se studeným nástřikovým zařízením on column pro přímé zavádění vzorku do kolony a s termostatem, který je schopen udržovat žádanou teplotu s přesností na 1 °C. 3.15 Studené nástřikové zařízení on column pro přímé zavádění vzorku do kolony. 3.16 Plameno-ionizační detektor a elektrometr. 3.17 Integrátor se zapisovačem, vhodný pro elektrometr, s rychlostí odezvy nižší než 1 sekundu a s nastavitelnou rychlostí posunu papíru. 3.18 Kapilární kolona, ze skla nebo taveného křemene, dlouhá 8 až 12 m s vnitřním průměrem 0,25 až 0,32 mm, obsahující methylpolysiloxan nebo 5 %-fenyl-methylpolysiloxan, o tloušťce 0,10 až 0,30 μm, kterou je možno použít při 370 °C. 3.19 Mikrostříkačka na 10 μl pro přímý vstřik do kolony opatřená tvrzenou jehlou o délce min. 7,5 cm. 4. CHEMIKÁLIE 4.1 Silikagel s velikostí zrna od 0,063 až po 0,200 mm (70/280 mesh), který se připraví takto: silikagel se položí do porcelánové misky, suší se v sušárně po dobu 4 hodin při teplotě 160 °C, pak se nechá ochladit v exsikátoru při pokojové teplotě. Přidá se voda o objemu shodném s 5 % váhy silikagelu takto: do 500 ml baňky se naváží 152 g silikagelu a přidá se 8 g destilované vody, baňka se zazátkuje a jemně protřepá, aby se voda rozložila rovnoměrně. Nechá se odstát na alespoň 12 hodin před použitím. 4.2 n-hexan pro chromatografii. 4.3 Isopropanol. 4.4 Isopropanol, vodný roztok 1/1 (V/V). 4.5 Pankreatická lipáza. Použitá lipáza musí mít aktivitu mezi 2,0 A 10 jednotkami lipázy na mg (v obchodě existují pankreatické lipázy s aktivitou mezi 2 a 10 jednotkami na mg enzymu). 4.6 Tlumivý roztok trihydroxy-methyl-aminomethanu: 1 M vodný roztok upravený na pH 8 (kontrola potenciometrem) přidáním koncentrované kyseliny chlorovodíkové (1/1 V/V). 4.7 Cholát sodný (enzymatické kvality), vodný roztok o koncentraci 0,1 % (tento roztok se musí použít do 15 dnů po přípravě). 4.8 Chlorid vápenatý, vodný roztok o koncentraci 22 %. 4.9 Diethylether pro chromatografii. 4.10 Vyvíjecí rozpouštědlo: směs n-hexanu/diethyletheru (87/13) (V/V). 4.11 Hydroxid sodný, roztok o koncentraci 12 % hmotnostních. 4.12 Fenolftalein, 1 % roztok ethanolu. 4.13 Nosný plyn: vodík nebo helium, pro plynovou chromatografii. 4.14 Pomocné plyny: – vodík o minimální čistotě 99 %, bez vlhkosti a organických látek, a vzduch, pro plynovou chromatografii stejná čistota. 4.15 Silanizační činidla: směs pyridinu/hexametyldisilazanu, trimetylchlorosilanu 9/3/1 (V/V/V) (Roztoky připravené k použití jsou na trhu. Mohou se použít jiná silylační činidla, zejména bis-trimethylsilyl trifluoracetamid + 1 % trimetylchlorosilan, zředěná stejným objemem bezvodého pyridinu.) 4.16 Referenční vzorky: čisté monoglyceridy nebo směsi, které mají podobné procentuální složení jako vzorek. 5. POSTUP 5.1 Příprava vzorku 5.1.1 Oleje, které mají volnou kyselost nižší než 3 %, nemusí být neutralizovány před chromatografií na silikagelové koloně. Oleje, které mají volnou kyselost vyšší než 3 %, musí být neutralizovány podle bodu 5.1.1.1. 5.1.1.1 Do nálevky na 1 000 ml (3.13) se vleje 50 g oleje a 200 ml n-hexanu. Přidá se 100 ml isopropanolu a množství roztoku hydroxidu sodného o koncentraci 12 % (4.11) odpovídající volné kyselosti oleje, plus 5 % navíc. Jednu minutu se důkladně protřepe. Přidá se 50 ml destilované vody, znovu se protřepe a nechá se usadit. Po dělení se odstraní spodní mýdlová vrstva. Odstraní se také jakékoli mezilehlé vrstvy (sliz, nerozpustné hmoty). Hexanový roztok neutralizovaného oleje se promyje následnými dávkami 50 až 60 ml roztoku isopropanolu/vody 1/1 (V/V) (4.4), dokud růžové zabarvení fenolftaleinu nezmizí. Většina hexanu se odstraní destilací ve vakuu (např. v rotačním odpařovači) a olej se přelije do baňky na 100 ml (3.5). Olej se suší ve vakuu, dokud nebude rozpouštědlo zcela odstraněno. K tomu musí být kyselost oleje nižší než 0,5 %. 5.1.2 Do kónické 25 ml baňky (3.1) se převede olej připravený podle výše uvedeného způsobu a rozpustí se ve vyvíjecím rozpouštědle (10 ml, 4.10). Roztok se nechá odstát na alespoň 15 minut před chromatografií na silikagelové koloně. Pokud je roztok kalný, odstředí se, aby byly zabezpečeny optimální podmínky pro chromatografii. (Mohou se použít kartony silikagelu SPE na 500 mg, které jsou připraveny k použití.) 5.1.3 Příprava chromatografické kolony Do kolony se vlije (3.3) zhruba 30 ml vyvíjecího rozpouštědla (4.10) a pomocí skleněné tyčinky se do spodní části kolony zavede kousek vaty. Stlačí se, aby se odstranil vzduch. V kádince se připraví suspenze 25 g silikagelu (4.1) ve zhruba 80 ml vyvíjecího rozpouštědla a naleje se do kolony za pomoci nálevky. Ověří se, zda je celý silikagel zaveden do kolony; vymyje se vyvíjecím rozpouštědlem (4.10), otevře se kohoutek a hladina kapaliny se nechá dostoupit zhruba 2 mm nad horní úroveň silikagelu. 5.1.4 Kolonová chromatografie Do 25 ml baňky (3.1) se naváží přesně 1,0 g připraveného vzorku podle bodu 5.1. Vzorek se rozpustí v 10 ml vyvíjecího rozpouštědla (4.10). Roztok se vlije do připravené chromatografické kolony podle bodu 5.1.3. Zabrání se tomu, aby se plocha kolony hýbala. Otevře se kohoutek a roztok vzorku se nechá odkapat, než dosáhne hladiny silikagelu. Rozpustí se pomocí 150 ml vyvíjecího rozpouštědla. Průtok se upraví na 2 ml/min (tak, aby 150 ml proteklo do kolony přibližně za 60–70 minut). Do baňky na 250 ml, která byla předtím zvážena, se odebere eluát. Rozpouštědlo se ve vakuu odpaří a jeho zbylé stopy se odstraní pod proudem dusíku. Baňka se zváží a vypočítá se množství získaného extraktu. (V případě použití už hotových silikagelových patron SPE se postupuje takto: zavede se 1 ml roztoku (5.1.2) do předem připravených patron s 3 ml n-hexanu. Po perkolování roztoku se vyvíjí 4 ml n-hexanu/dietyleteru v objemovém poměru 9/1 (V/V). Eluát se odebere do 10 ml zkumavky a odpařuje se pod proudem dusíku až do vysušení. Suché reziduum se podrobí pankreatické lipáze (5.2). Základem je ověřit složení mastných kyselin před a po přechodu patronou SPE.) 5.2 Hydrolýza pankreatickou lipázou 5.2.1 Do centrifugační zkumavky se odváže 0,1 g oleje připraveného podle bodu 5.1. Přidá se 2 ml tlumivého roztoku (4.6), 0,5 ml roztoku cholátu sodného (4.7) a 0,2 ml roztoku chloridu vápenatého, přičemž po každém přidání se protřepe řádně směsí. Zkumavka se uzavře zábrusovou zátkou a umístí se do termostatu při teplotě 40 ± 0,5 °C. 5.2.2 Přidá se 20 mg lipázy, opatrně se protřepe (tak, aby se nenamočila zátka), a zkumavka se dá do termostatu přesně na 2 minuty, potom se vybere, během 1 minuty důkladně protřepává a nechá se vychladit. 5.2.3 Přidá se 1 ml dietyleteru, zazátkuje se a důkladně protřepe, potom se odstředí a pomocí mikrostříkačky se roztok etheru přenese do čisté a suché zkumavky. 5.3 Příprava silanizovaných derivátů a plynová chromatografie 5.3.1 Pomocí mikrostříkačky se 100 μl roztoku (5.2.3) zavede do 10 ml zkumavky s kónickým dnem. 5.3.2 Rozpouštědlo se odstraní pod slabým proudem dusíku, přidá se 200 μl silanizačního činidla (4.15), zkumavka se uzavře zátkou a nechá 20 minut odstát. 5.3.3 Po 20 minutách se přidá 1 až 5 ml n-hexanu (v závislosti na chromatografických podmínkách): výsledný roztok je připravený pro plynovou chromatografii. 5.4 Plynová chromatografie Pracovní podmínky jsou tyto:
5.4.1 Identifikace píků Identifikace jednotlivých monoglyceridů se provádí podle retenčních časů a porovnáním se standardními směsmi monoglyceridů, které byly analyzovány za stejných podmínek. 5.4.2 Kvantitativní vyhodnocení Plocha každého píku se vypočítá pomocí elektronického integrátoru. 6. VYJÁDŘENÍ VÝSLEDKŮ Procentuální obsah glyceryl monopalmitátu se vypočítá na základě vztahu mezi plochou odpovídajícího píku a součtem ploch píků všech monoglyceridů (viz obrázek 2), podle vzorce: Glycéril monopalmitate (%): (Glycéril monopalmitate = glyceril monopalmitát) kde:
Výsledek se uvádí s přesností na jedno desetinné místo. 7. ZPRÁVA O ANALÝZE Zpráva o analýze musí uvádět:
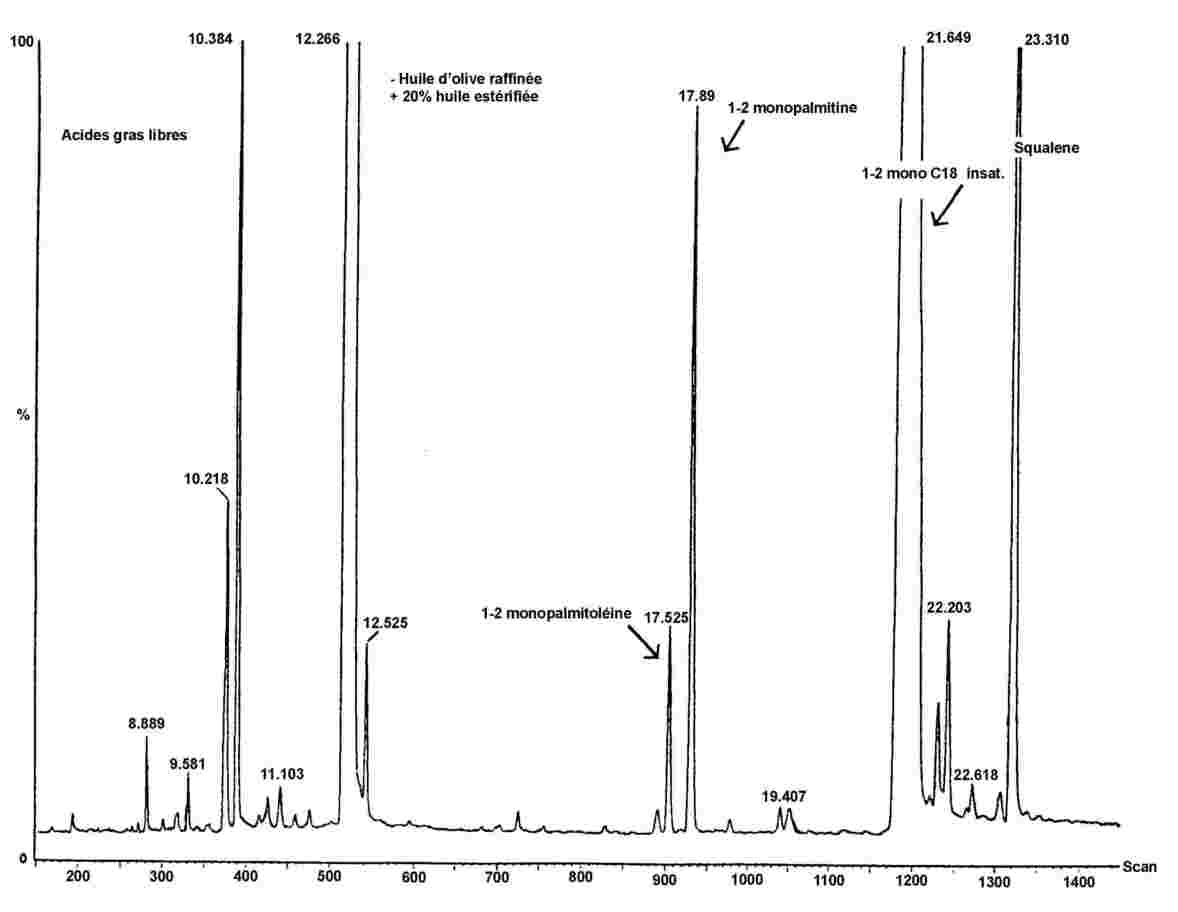
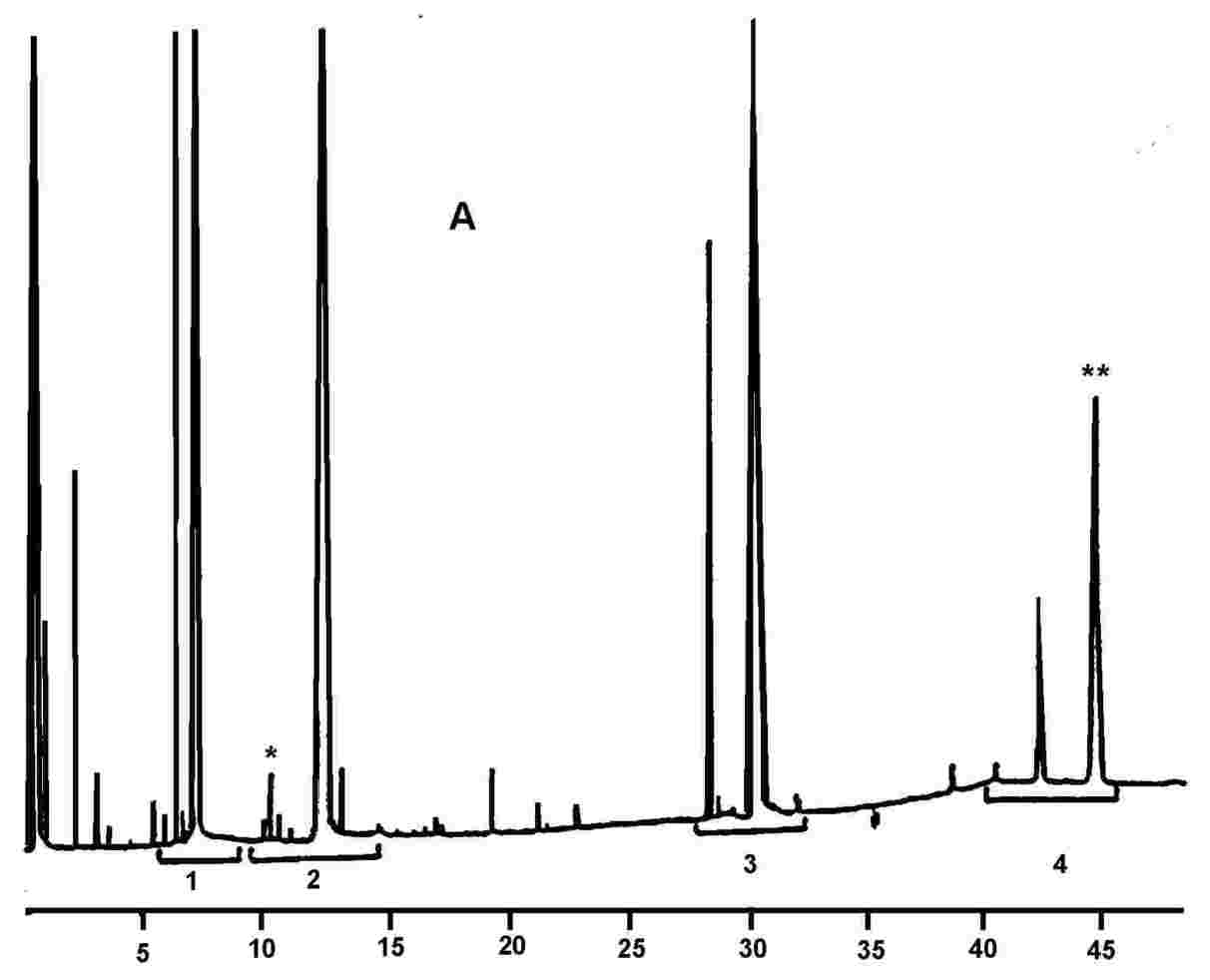
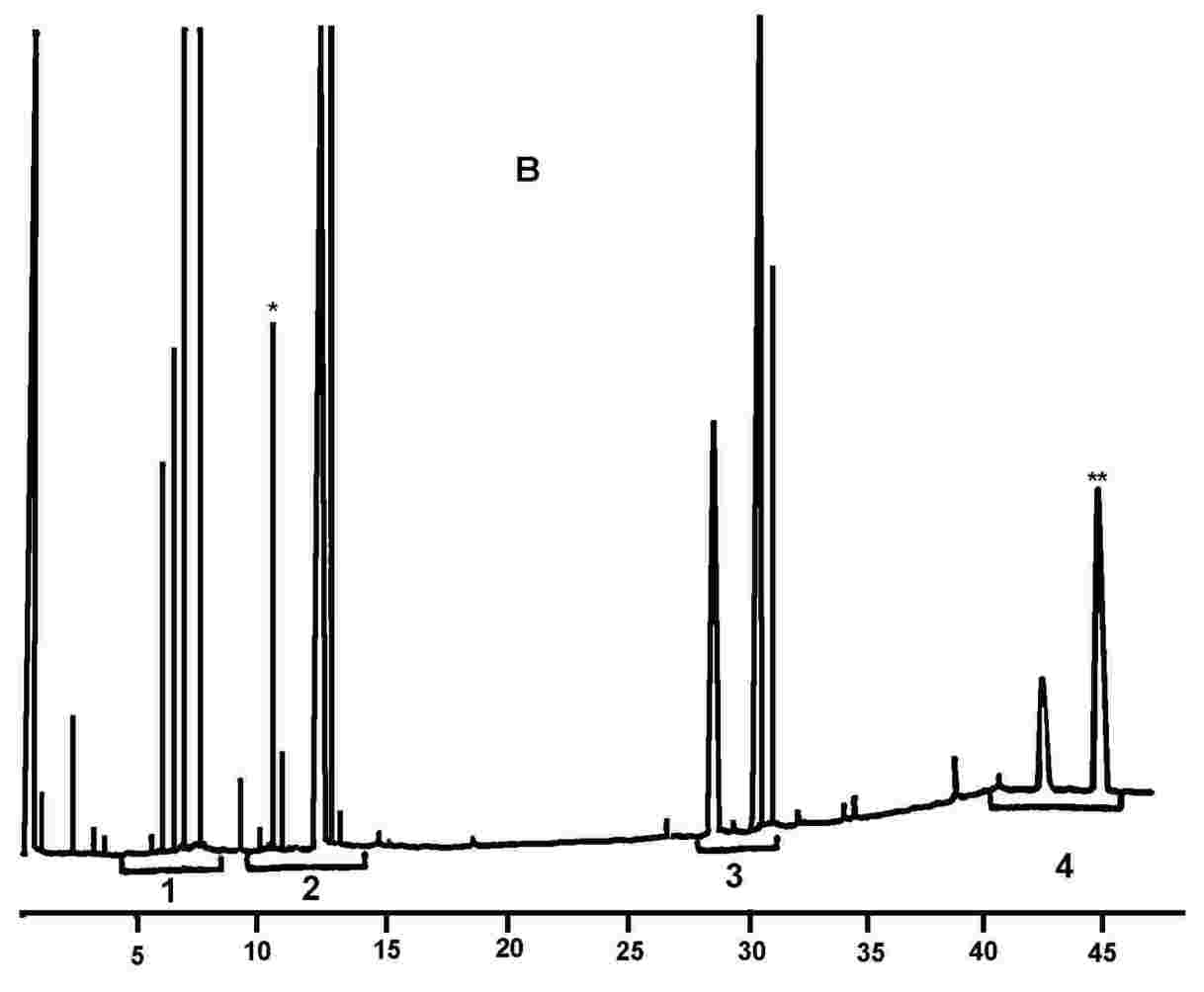
Obrázek 1 Chromatogram produktů z reakce silanizace, které byly získány lipázou na rafinovaném olivovém oleji s přidáním 20 % esterifikovaného oleje (100 %)
Obrázek 2 Chromatogram: A) neesterifikovaného olivového oleje po lipáze; po silanizaci; za těchto podmínek (kapilární kolona 8–12 m) je vosková frakce eluovaná zároveň s frakcí diglyceridu nebo krátce poté. Po lipáze by obsah triglyceridů neměl překročit 15 %
Chromatogram: B) esterifikovaného olivového oleje po lipáze; po silanizaci; za těchto podmínek (kapilární kolona 8–12 m) je vosková frakce eluovaná zároveň s frakcí diglyceridu nebo krátce poté. Po lipáze by obsah triglyceridů neměl překročit 15 %
8. POZNÁMKY Poznámka 1: PŘÍPRAVA LIPÁZY Lipázy s dostatečnou aktivitou jsou obchodně dostupné. Je ale rovněž možné je připravit v laboratoři takto: 5 kg čerstvé vepřové slinivky břišní se vychladí na 0 °C; okolní sádlo a vazivová tkáň se oddělí a v míchadle se rozmělní, dokud nevznikne tekutá mazlavá pasta. Tato pasta se protřepe po dobu 4 až 6 hodin s 2,5 l bezvodého acetonu a poté odstředí. Zbytek se extrahuje třikrát pomocí stejného množství bezvodého acetonu, potom dvakrát pomocí směsi acetonu a diethyletheru 1:1 (V/V) a dvakrát pomocí diethyletheru. Zbytek se po 48 hodin suší ve vakuu za účelem získání stabilního prášku, který je možné dlouho skladovat v ledničce chráněný před vlhkostí. Poznámka 2: KONTROLA AKTIVITY LIPÁZY Emulze olivového oleje se připraví takto: V míchadle se po 10 minut protřepává směs složená ze 165 ml roztoku arabské gumy o koncentraci 100 g/l, 15 g drceného ledu a 20 ml předem neutralizovaného oleje. Do 50 ml kádinky se zavede 10 ml této emulze, poté se postupně přidá 0,3 ml roztoku cholátu sodného o koncentraci 0,2 g/ml a 20 ml destilované vody. Kádinka se vloží do termostatu udržovaného na teplotě 37 °C; vloží se elektrody pH metru a spirálovité míchadlo. Pomocí byrety se po kapkách přidává roztok hydroxidu sodného 0,1 N, dokud hodnota pH nedosáhne 8,3. Přidá se objem vodní suspenze lipázy (0,1 g/ml lipázy). Jakmile pH metr začne ukazovat pH 8,3, zapnou se stopky a po kapkách se přikapává roztok hydroxidu sodného takovou rychlostí, aby se udržovala hodnota pH 8,3. Zaznamená se objem spotřebovaného roztoku za minutu. Údaje se zaznamenávají do grafového systému souřadnic tak, že osa nezávisle proměnných (x-ová osa) ponese časové údaje a na ose závisle proměnných se uvede počet mililitrů alkalického roztoku 0,1 N spotřebovaného na udržení konstantního pH. Výsledný graf musí být lineární. Aktivita lipázy vyjádřená v lipázových jednotkách na 1 mg je pak dána tímto vzorcem:
kde:
Jednotka lipázy je definovaná jako množství enzymu, které uvolní 10 mikro-ekvivalentů kyseliny za minutu.“ |
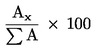
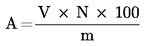
|
7) |
V příloze X „A“ se bod 6.2 nahrazuje tímto:
|
(1) Úhrn izomerů (ne)separovatelných prostřednictvím kapilární kolony.
(2) Nebo pokud je medián závad nejvýš 2,5 a medián ovocnosti roven 0.
(3) Oleje s obsahem vosku mezi 300 mg/kg a 350 mg/kg se zařazují do kategorie lampantového olivového oleje, pokud je celkový obsah alifatických alkoholů nejvýš 350 mg/kg nebo pokud je obsah erythrodiolu a uvaolu nejvýš 3,5 %.
(4) Oleje s obsahem vosku mezi 300 mg/kg a 350 mg/kg se zařazují do kategorie surového olivového oleje z pokrutin, pokud je celkový obsah alifatických alkoholů vyšší než 350 mg/kg a pokud je obsah erythrodiolu a uvaolu vyšší než 3,5 %.
(5) Obsah ostatních mastných kyselin (%): kyselina palmitová: 7,5–20,0; palmitoolejová: 0,3–3,5; heptadekanová: ≤ 0,3; heptadecenová: ≤ 0,3; stearová: 0,5–5,0; olejová: 55,0–83,0; linolová: 3,5–21,0.
(6) Úhrn: Delta-5-23-stigmastadien + chlerosterol + beta-sitosterol + sitostanol + delta-5-avenasterol + delta-5-24-stigmastadien.
(7) Oleje s obsahem vosku mezi 300 mg/kg a 350 mg/kg se zařazují do kategorie lampantového olivového oleje, pokud je celkový obsah alifatických alkoholů nejvýš 350 mg/kg nebo pokud je obsah erythrodiolu a uvaolu nejvýš 3,5 %.
(8) Oleje s obsahem vosku mezi 300 mg/kg a 350 mg/kg se zařazují do kategorie surového olivového oleje z pokrutin, pokud je celkový obsah alifatických alkoholů vyšší než 350 mg/kg a pokud je obsah erythrodiolu a uvaolu vyšší než 3,5 %.
(9) Po eluování esterů sterolů musí být chromatografická linie bez významných píků (triglyceridů).