2006/86/ESSměrnice Komise 2006/86/ES ze dne 24. října 2006 , kterou se provádí směrnice Evropského parlamentu a Rady 2004/23/ES, pokud jde o požadavky na sledovatelnost, oznamování závažných nežádoucích reakcí a účinků a některé technické požadavky na kódování, zpracování, konzervaci, skladování a distribuci lidských tkání a buněk (Text s významem pro EHP)
| Publikováno: | Úř. věst. L 294, 25.10.2006, s. 32-50 | Druh předpisu: | Směrnice |
| Přijato: | 24. října 2006 | Autor předpisu: | Evropská komise |
| Platnost od: | 14. listopadu 2006 | Nabývá účinnosti: | 14. listopadu 2006 |
| Platnost předpisu: | Ano | Pozbývá platnosti: | |
Text předpisu s celou hlavičkou je dostupný pouze pro registrované uživatele.
SMĚRNICE KOMISE 2006/86/ES
ze dne 24. října 2006,
kterou se provádí směrnice Evropského parlamentu a Rady 2004/23/ES, pokud jde o požadavky na sledovatelnost, oznamování závažných nežádoucích reakcí a účinků a některé technické požadavky na kódování, zpracování, konzervaci, skladování a distribuci lidských tkání a buněk
(Text s významem pro EHP)
KOMISE EVROPSKÝCH SPOLEČENSTVÍ,
s ohledem na Smlouvu o založení Evropského společenství,
s ohledem na směrnici Evropského parlamentu a Rady 2004/23/ES ze dne 31. března 2004 o stanovení jakostních a bezpečnostních norem pro darování, odběr, vyšetřování, zpracování, konzervaci, skladování a distribuci lidských tkání a buněk (1), a zejména na článek 8, čl. 11 odst. 4 a čl. 28 písm. a), c), g), a h) uvedené směrnice,
vzhledem k těmto důvodům:
|
(1) |
Směrnice 2004/23/ES stanoví jakostní a bezpečnostní normy pro darování, odběr, vyšetřování, zpracování, konzervaci, skladování a distribuci lidských tkání a buněk určených k použití u člověka a přípravků získaných z lidských tkání a buněk určených k použití u člověka s cílem zajistit vysokou úroveň ochrany lidského zdraví. |
|
(2) |
Aby se předcházelo přenosu nemocí prostřednictvím lidských tkání a buněk určených k použití u člověka a aby se zaručila rovnocenná úroveň jakosti a bezpečnosti, vyzývá směrnice 2004/23/ES ke stanovení specifických technických požadavků pro každou z etap postupu použití lidských tkání a buněk, včetně norem a specifikací pro systém jakosti tkáňových zařízení. |
|
(3) |
Členské státy by měly v souladu se směrnicí 2004/23/ES stanovit systém akreditace a jmenování tkáňových zařízení a postupů přípravy tkání a buněk a udělování příslušného oprávnění nebo povolení, aby se zajistila vysoká úroveň ochrany lidského zdraví. Pro tento systém je třeba stanovit technické požadavky. |
|
(4) |
Požadavky na akreditaci a jmenování tkáňových zařízení a udělení příslušného oprávnění nebo povolení by se měly vztahovat na organizaci a řízení, pracovníky, vybavení, materiály, zařízení/prostory, dokumentaci, záznamy a přezkoumání jakosti. Akreditovaná a jmenovaná tkáňová zařízení nebo tkáňová zařízení, jimž bylo uděleno oprávnění nebo povolení, by měla splňovat dodatečné požadavky na zvláštní činnosti, které provádějí. |
|
(5) |
Norma týkající se kvality vzduchu při zpracování tkání a buněk je klíčovým faktorem, který může mít vliv na riziko kontaminace tkání nebo buněk. Obecně se vyžaduje kvalita vzduchu, v němž jsou počet částic a počet mikrobiálních kolonií rovnocenné počtům ve třídě čistoty A, jak je definována v příloze 1 Evropských pokynů pro správnou výrobní praxi a ve směrnici Komise 2003/94/ES (2). V určitých situacích však není kvalita vzduchu s počtem částic a počtem mikrobiálních kolonií rovnocennými počtům v normě třídy A indikována. Za takových okolností by mělo být prokázáno a zdokumentováno, že zvolené prostředí zaručuje jakost a bezpečnost, jež jsou vyžadovány pro daný druh tkáně a buněk, daný proces a dané použití u člověka. |
|
(6) |
Oblast působnosti této směrnice by měla zahrnovat jakost a bezpečnost lidských tkání a buněk při kódování, zpracování, konzervaci, skladování a distribuci do zdravotnických zařízení, v němž se použijí v lidském těle. Neměla by však být rozšířena na použití těchto tkání a buněk u člověka (např. při implantaci, perfuzi, inseminaci nebo přenosu embryí). Ustanovení této směrnice týkající se sledovatelnosti a oznamování závažných nežádoucích reakcí a účinků se rovněž vztahují na darování, odběr a vyšetřování lidských tkání a buněk, které jsou upraveny směrnicí Komise 2006/17/ES (3). |
|
(7) |
Použití tkání a buněk u člověka může pro příjemce znamenat riziko přenosu nemoci a další případné nežádoucí účinky. Aby bylo možné sledovat a snížit tyto účinky, měly by se stanovit specifické požadavky na sledovatelnost a postup Společenství pro oznamování závažných nežádoucích reakcí a účinků. |
|
(8) |
Podezřelé závažné nežádoucí reakce, ať u dárce, či u příjemce, a závažné nežádoucí účinky od darování až po distribucí tkání a buněk, jež mohou mít vliv na jakost a bezpečnost tkání a buněk a mohou být způsobeny odběrem (včetně hodnocení a výběru dárce), vyšetřováním, zpracováním, konzervací, skladováním a distribucí lidských tkání a buněk, se neprodleně ohlašují příslušnému orgánu. |
|
(9) |
Během odběru u živých dárců či po něm nebo během použití u člověka či po něm mohou být zjištěny závažné nežádoucí reakce. Měly by být oznámeny patřičnému tkáňovému zařízení pro účely následného šetření a oznámení příslušnému orgánu. To by organizaci provádějící odběr či organizaci odpovědné za použití u člověka nemělo bránit v tom, aby přímo uvědomily příslušný orgán, pokud si to přejí. Tato směrnice by měla vymezit minimální údaje, jež je třeba oznamovat příslušnému orgánu, aniž je tím dotčena schopnost členských států zachovávat či zavádět na svém území přísnější a ochranná opatření, která jsou v souladu s ustanoveními Smlouvy. |
|
(10) |
S cílem co nejvíce snížit výdaje na přenos, zabránit překrývání a zvýšit administrativní účinnost by se měly k provádění úkolů týkajících se přenosu a zpracování informací používat moderní technologie a řešení elektronické veřejné správy. Tyto technologie by měly využívat standardní formát pro výměnu informací za použití vhodného systému pro správu referenčních údajů. |
|
(11) |
Aby se usnadnila sledovatelnost a zjednodušily informace o hlavních charakteristikách a vlastnostech tkání a buněk, je nutné stanovit základní údaje, jež mají být zahrnuty do jednotného evropského kódu. |
|
(12) |
V této směrnici jsou dodržována základní práva a zachovávány zásady uznávané zejména Listinou základních práv Evropské unie. |
|
(13) |
Opatření stanovená touto směrnicí jsou v souladu se stanoviskem výboru zřízeného článkem 29 směrnice 2004/23/ES, |
PŘIJALA TUTO SMĚRNICI:
Článek 1
Oblast působnosti
1. Tato směrnice se vztahuje na kódování, zpracování, konzervaci, skladování a distribuci:
|
a) |
lidských tkání a buněk určených k použití u člověka a |
|
b) |
přípravků získaných z lidských tkání a buněk určených k použití u člověka, pokud se na takové přípravky nevztahují jiné směrnice. |
2. Ustanovení článků 5 až 9 této směrnice, která se týkají sledovatelnosti a oznamování závažných nežádoucích reakcí a účinků, se rovněž vztahují na darování, odběr, vyšetřování lidských tkání a buněk.
Článek 2
Definice
Pro účely této směrnice se:
|
a) |
„reprodukčními buňkami“ rozumějí všechny tkáně a buňky určené k účelům asistované reprodukce; |
|
b) |
„darováním mezi partnery“ rozumí darování reprodukčních buněk mezi mužem a ženou, kteří prohlásí, že mají intimní fyzický vztah; |
|
c) |
„systémem jakosti“ rozumí organizační struktura, stanovené povinnosti, postupy, procesy a zdroje k provádění řízení jakosti; zahrnuje všechny činnosti, které přímo či nepřímo přispívají k jakosti; |
|
d) |
„řízením jakosti“ rozumějí koordinované činnosti, které mají řídit a kontrolovat organizaci z hlediska jakosti; |
|
e) |
„standardními pracovními postupy“ (SOP) rozumějí písemné pokyny popisující jednotlivé etapy specifického postupu včetně materiálů a metod, které mají být použity, a očekávaný konečný přípravek; |
|
f) |
„validací“ (nebo „kvalifikací“ v případě zařízení nebo prostředí) rozumí vytvoření dokumentovaného důkazu, který poskytuje vysokou záruku, že určitý proces, standardní pracovní postup, části zařízení nebo prostředí konzistentně vytvoří produkt, který splňuje předem stanovené specifikace a atributy jakosti; |
|
g) |
„sledovatelností“ rozumí schopnost zjistit místo, kde se nachází, a identifikovat tkáň/buňku během každé etapy od odběru, přes zpracování, vyšetřování a skladování až po distribuci příjemci nebo likvidaci, což zahrnuje také schopnost identifikovat dárce a tkáňové zařízení nebo výrobní zařízení, které tkáň/buňky přijímá, zpracovává nebo skladuje, a schopnost identifikovat příjemce ve zdravotnickém zařízení používajícím/zdravotnických zařízeních používajících tkáň/buňky u příjemce/příjemců; |
|
h) |
„kritickým“ rozumí mající možný vliv na jakost nebo bezpečnost buněk a tkání nebo mající s nimi kontakt; |
|
i) |
„organizací provádějící odběr“ rozumí zdravotnické zařízení nebo jednotka nemocnice nebo jiný subjekt, který provozuje odběr lidských tkání a buněk a který nemusí být akreditován, určen, oprávněn nebo povolen jako tkáňové zařízení; |
|
j) |
„organizacemi odpovědnými za použití u člověka“ rozumějí zdravotnické zařízení nebo jednotka nemocnice nebo jiný subjekt, který používá lidské tkáně a buňky u člověka. |
Článek 3
Požadavky na akreditaci a jmenování tkáňových zařízení a udělení příslušného oprávnění nebo povolení
Tkáňové zařízení musí splňovat požadavky stanovené v příloze I.
Článek 4
Požadavky na akreditaci a jmenování postupů přípravy tkání a buněk a udělení příslušného oprávnění nebo povolení
Postupy přípravy v tkáňových zařízeních musejí splňovat požadavky stanovené v příloze II.
Článek 5
Oznamování závažných nežádoucích reakcí
1. Členské státy zajistí, aby:
|
a) |
organizace provádějící odběr měly zavedeny postupy pro vedení záznamů o odebraných tkáních a buňkách a pro to, aby mohly neprodleně uvědomovat tkáňová zařízení o závažných nežádoucích reakcích u žijících dárců, jež by mohly ovlivnit jakost a bezpečnost tkání a buněk; |
|
b) |
organizace odpovědné za použití tkání a buněk u člověka měly zavedeny postupy pro vedení záznamů o použitých tkáních a buňkách a pro to, aby mohly neprodleně uvědomovat tkáňová zařízení o závažných nežádoucích reakcích pozorovaných během klinického použití nebo po něm, které mohou souviset s jakostí a bezpečností tkání nebo buněk; |
|
c) |
tkáňová zařízení, která distribuují tkáně a buňky pro použití u člověka, poskytovala organizaci odpovědné za použití tkání a buněk u člověka informace o tom, jak má tato organizace oznamovat závažné nežádoucí reakce, jak je uvedeno v písmenu b). |
2. Členské státy zajistí, aby tkáňová zařízení:
|
a) |
měla zavedeny postupy pro to, aby mohla neprodleně sdělovat příslušnému orgánu všechny náležité dostupné informace o podezřelých závažných nežádoucích reakcích, jak je uvedeno v odst. 1 písm. a) a b); |
|
b) |
měla zavedeny postupy pro to, aby mohla neprodleně sdělovat příslušnému orgánu závěry vyšetřování pro účely analýzy příčin a následného výstupu. |
3. Členské státy zajistí, aby:
|
a) |
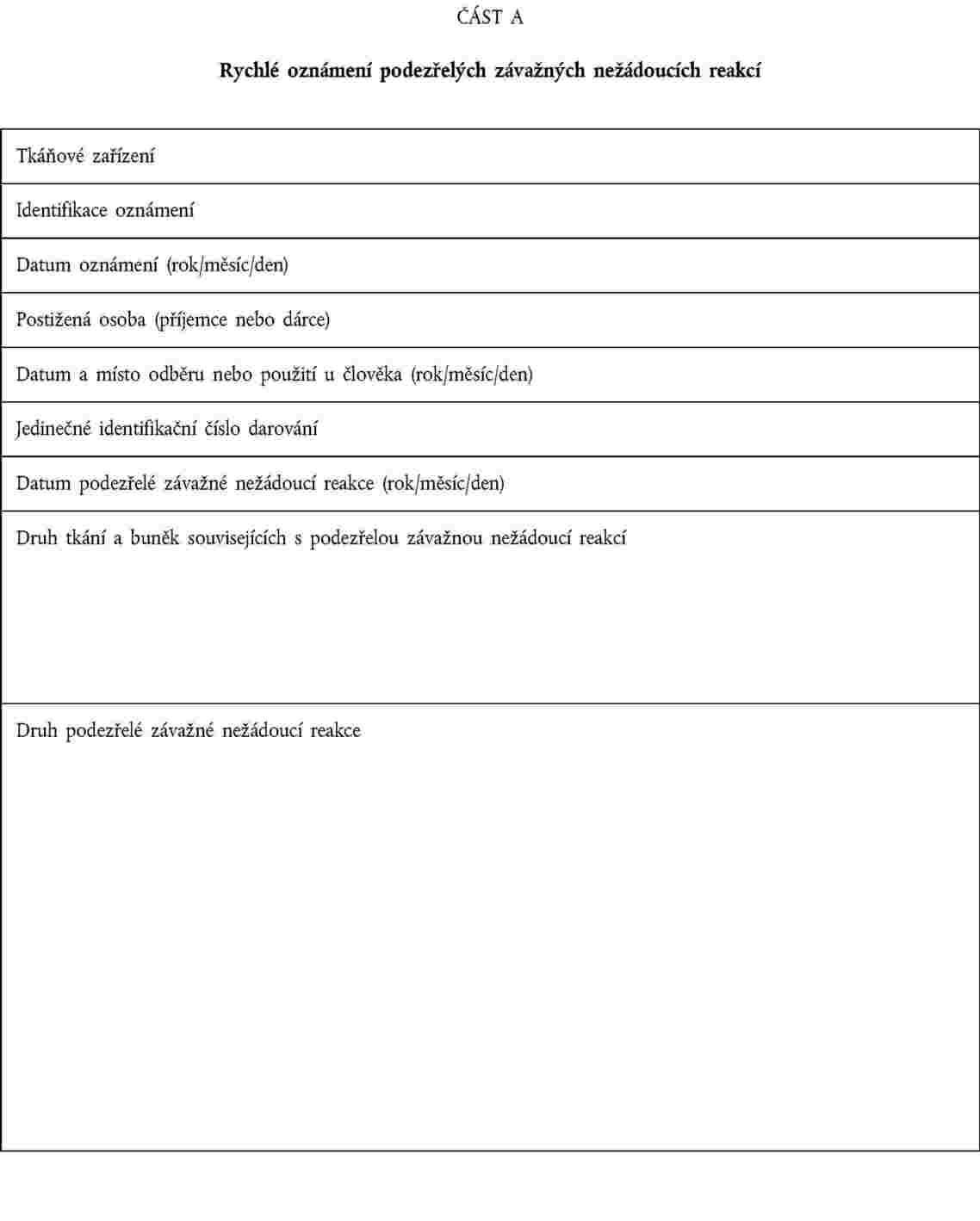
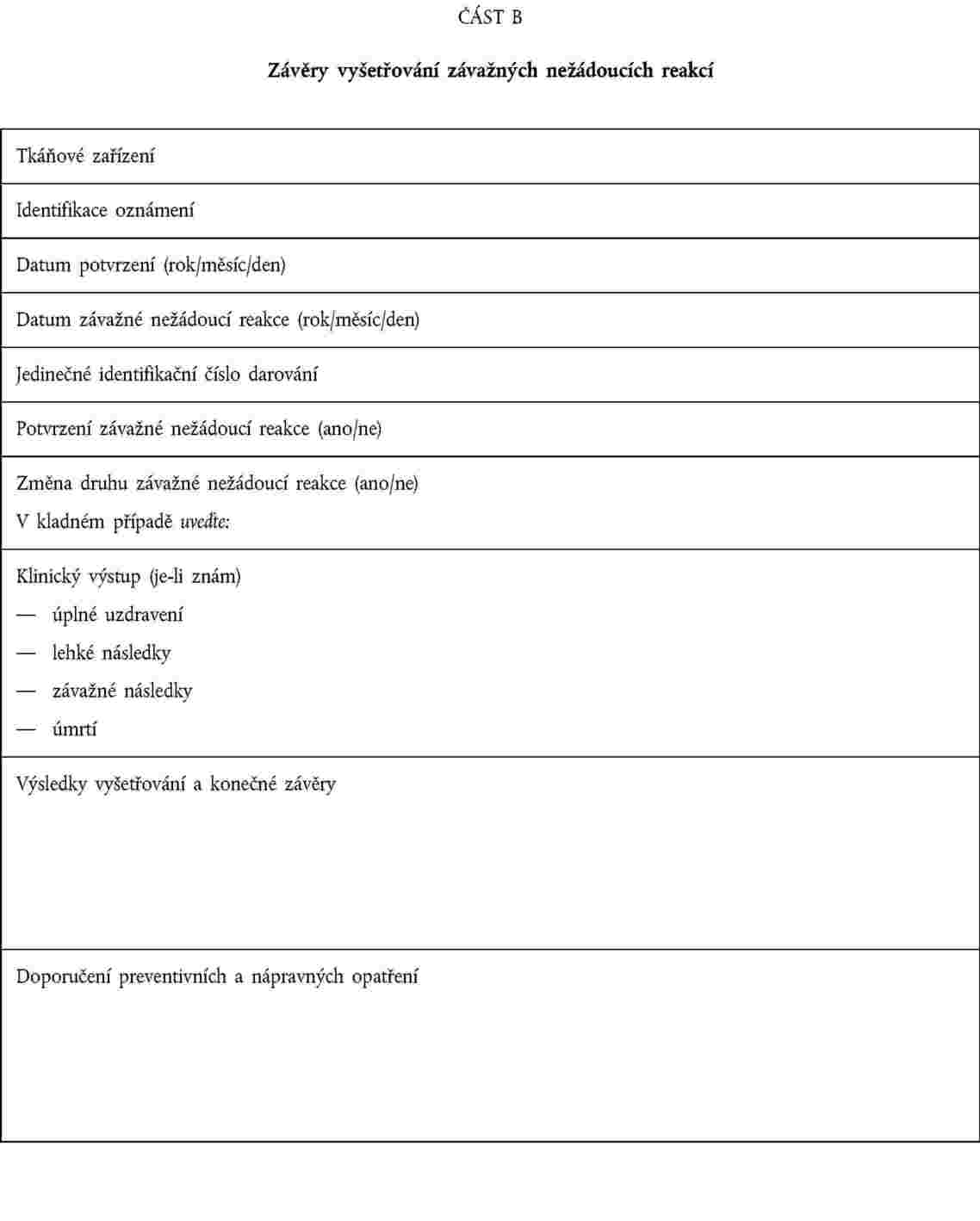
odpovědná osoba uvedená v článku 17 směrnice 2004/23/ES oznámila příslušnému orgánu informace obsažené v oznámení podle části A přílohy III; |
|
b) |
tkáňová zařízení oznámila příslušnému orgánu opatření přijatá ohledně ostatních dotčených tkání a buněk, které byly distribuovány pro účely použití u člověka; |
|
c) |
tkáňová zařízení oznámila příslušnému orgánu závěry vyšetřování, přičemž poskytnou alespoň informace podle části B přílohy III. |
Článek 6
Oznamování závažných nežádoucích účinků
1. Členské státy zajistí, aby:
|
a) |
organizace provádějící odběr a tkáňová zařízení měly zavedeny postupy pro vedení záznamů a pro to, aby mohly neprodleně uvědomovat tkáňová zařízení o závažných nežádoucích účincích, které se objeví během odběru a mohly by ovlivnit jakost a/nebo bezpečnost lidských tkání a buněk; |
|
b) |
organizace odpovědné za použití tkání a buněk u člověka měly zavedeny postupy, aby mohly neprodleně uvědomovat tkáňová zařízení o závažných nežádoucích účincích, které by mohly ovlivnit jakost a bezpečnost tkání nebo buněk; |
|
c) |
tkáňová zařízení poskytovala organizaci odpovědné za použití tkání a buněk u člověka informace o tom, jak má tato organizace oznamovat závažné nežádoucí účinky, které by mohly ovlivnit jakost a bezpečnost tkání nebo buněk. |
2. V případě asistované reprodukce se jakákoli špatná identifikace či záměna gamet nebo embryí považuje za závažný nežádoucí účinek. Všechny osoby či organizace provádějící odběr nebo organizace odpovědné za použití tkání a buněk u člověka, které provádějí asistovanou reprodukci, oznámí takovéto účinky dodavatelským tkáňovým zařízením pro účely vyšetřování a oznámení příslušnému orgánu.
3. Členské státy zajistí, aby tkáňová zařízení:
|
a) |
měla zavedeny postupy pro to, aby mohla neprodleně sdělovat příslušnému orgánu všechny náležité dostupné informace o podezřelých závažných nežádoucích účincích, jak je uvedeno v odst. 1 písm. a) a b); |
|
b) |
měla zavedeny postupy pro to, aby mohla neprodleně sdělovat příslušnému orgánu závěry vyšetřování pro účely analýzy příčin a následného výstupu. |
4. Členské státy zajistí, aby:
|
a) |
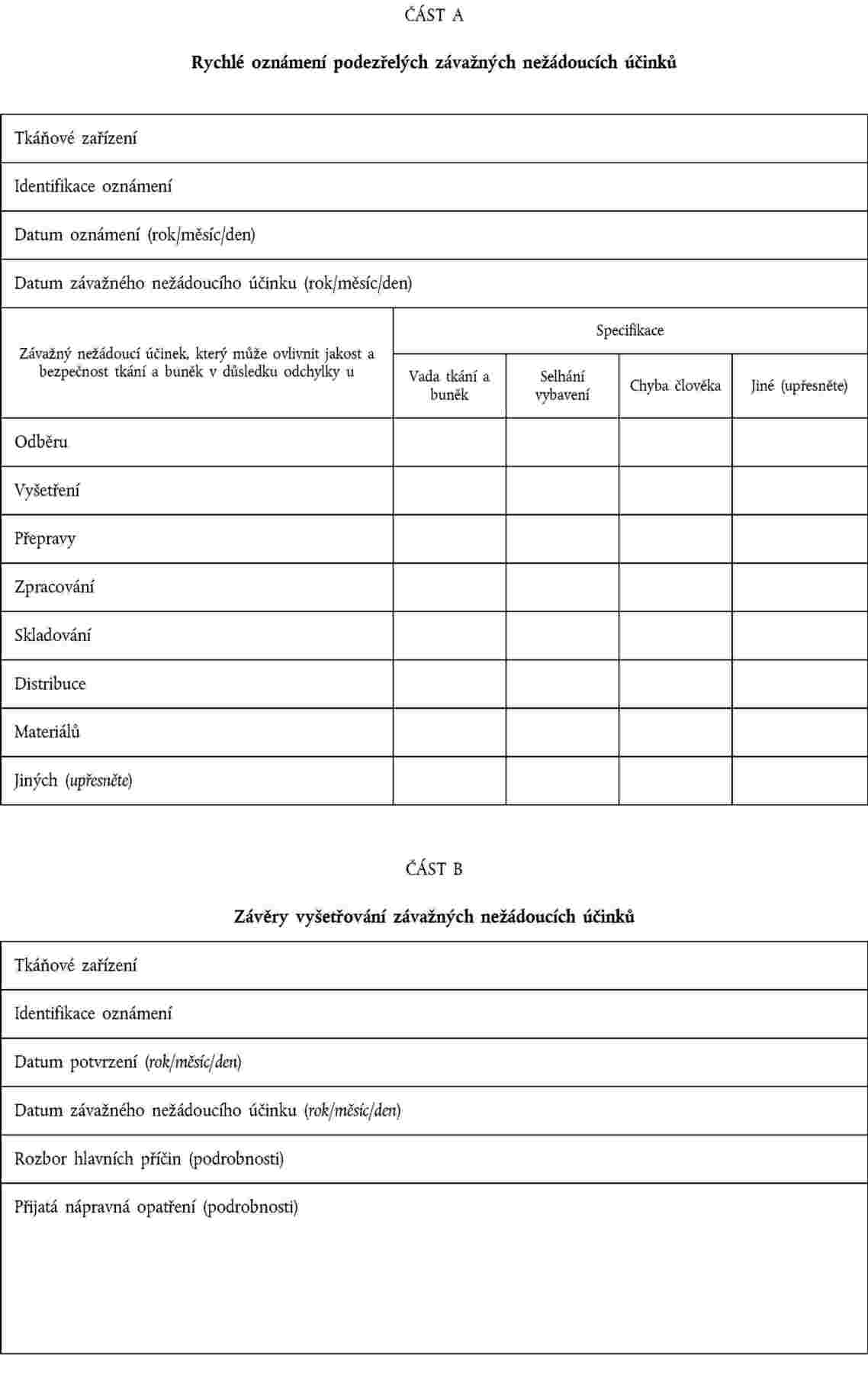
odpovědná osoba uvedená v článku 17 směrnice 2004/23/ES oznámila příslušnému orgánu informace obsažené v oznámení podle části A přílohy IV; |
|
b) |
tkáňová zařízení vyhodnotila závažné nežádoucí účinky, aby bylo možné v daném procesu určit příčiny, jimž lze předejít; |
|
c) |
tkáňová zařízení oznámila příslušnému orgánu závěry vyšetřování, přičemž poskytnou alespoň informace podle části B přílohy IV. |
Článek 7
Výroční zprávy
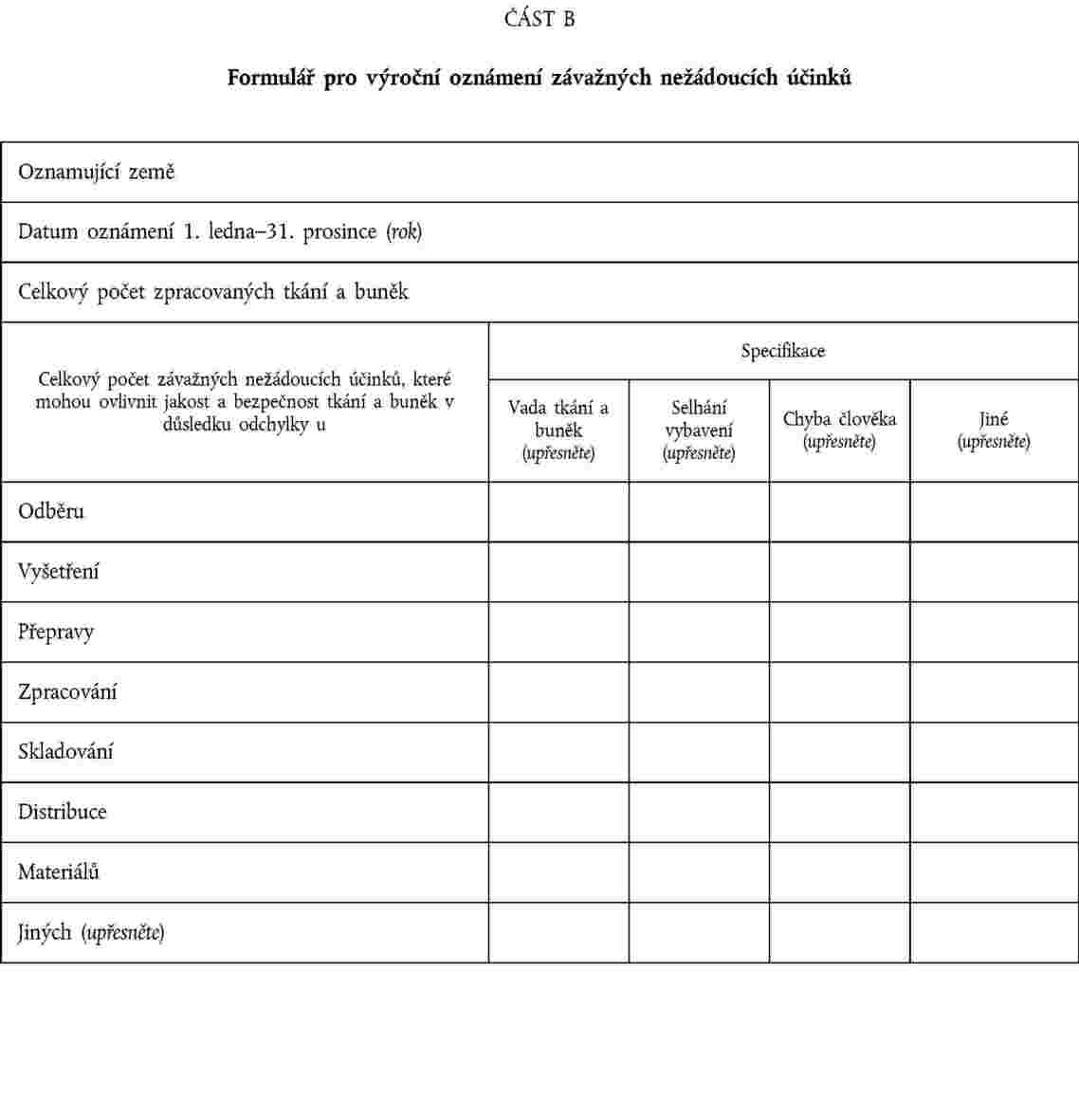
1. Do 30. června následujícího roku předloží členské státy Komisi výroční zprávu o oznámeních, která příslušný orgán obdržel o závažných nežádoucích reakcích a účincích. Komise předloží příslušným orgánům členských států shrnutí obdržených zpráv. Příslušný orgán tuto zprávu zpřístupní tkáňovým zařízením.
2. Přenos údajů odpovídá specifikacím týkajícím se formátu pro výměnu údajů stanoveným v částech A a B přílohy V a zajistí veškeré informace nezbytné pro identifikaci odesílatele a pro zachování jeho referenčních údajů.
Článek 8
Výměna informací mezi příslušnými orgány a jejich sdělování Komisi
Členské státy zajistí, aby si jejich příslušné orgány sdělovaly náležité informace, pokud jde o závažné nežádoucí reakce a účinky, aby se zaručilo, že budou přijata odpovídající opatření, a aby tyto informace sdělovaly Komisi.
Článek 9
Sledovatelnost
1. Tkáňová zařízení mají účinné a přesné systémy pro jednoznačnou identifikaci a označení obdržených a distribuovaných buněk/tkání.
2. Tkáňová zařízení a organizace odpovědné za použití u člověka uchovají údaje stanovené v příloze VI alespoň po dobu 30 let, a to na vhodném a čitelném médiu.
Článek 10
Evropský kódovací systém
1. Veškerým darovaným materiálům se v tkáňovém zařízení přidělí jedinečný evropský identifikační kód, aby se zajistila řádná identifikace dárce a sledovatelnost všech darovaných materiálů a zajistily informace o hlavních charakteristikách a vlastnostech tkání a buněk. Kód obsahuje alespoň informace uvedené v příloze VII.
2. Odstavec 1 se nevztahuje na darování reprodukčních buněk mezi partnery.
Článek 11
Provedení
1. Členské státy uvedou v účinnost právní a správní předpisy nezbytné pro dosažení souladu s touto směrnicí nejpozději do 1. září 2007. Neprodleně sdělí Komisi znění uvedených předpisů a srovnávací tabulku mezi ustanoveními uvedených předpisů a ustanoveními této směrnice.
Členské státy uvedou v účinnost právní a správní předpisy nezbytné pro dosažení souladu s článkem 10 této směrnice do 1. září 2008.
Tyto předpisy přijaté členskými státy musí obsahovat odkaz na tuto směrnici nebo musí být takový odkaz učiněn při jejich úředním vyhlášení. Způsob odkazu si stanoví členské státy.
2. Členské státy sdělí Komisi znění hlavních ustanovení vnitrostátních právních předpisů, které přijmou v oblasti působnosti této směrnice.
Článek 12
Vstup v platnost
Tato směrnice vstupuje v platnost dvacátým dnem po vyhlášení v Úředním věstníku Evropské unie.
Článek 13
Určení
Tato směrnice je určena členským státům.
V Bruselu dne 24. října 2006.
Za Komisi
Markos KYPRIANOU
člen Komise
(1) Úř. věst. L 102, 7.4.2004, s. 48.
(2) http://pharmacos.eudra.org/F2/eudralex/vol-4/home.htm a Úř. věst. L 262, 14.10.2003, s. 22.
(3) Úř. věst. L 38, 9.2.2006, s. 40.
PŘÍLOHA I
Požadavky na akreditaci a jmenování tkáňových zařízení a udělení příslušného oprávnění nebo povolení, jak je uvedeno v článku 3
A. ORGANIZACE A ŘÍZENÍ
|
1. |
Musí být jmenována odpovědná osoba, která má kvalifikaci a pověření, jak stanoví článek 17 směrnice 2004/23/ES. |
|
2. |
Tkáňové zařízení musí mít organizační strukturu a provozní postupy odpovídající činnostem, k nimž žádá o akreditaci, jmenování, oprávnění nebo povolení; musí být vypracováno organizační schéma, které jasně vymezuje strukturu zodpovědnosti a ohlašovací povinnosti. |
|
3. |
Každé tkáňové zařízení musí mít přístup k určenému registrovanému lékaři, jenž mu bude poskytovat poradenství a dohlížet na jeho lékařské činnosti, jako je výběr dárců, přezkoumání klinických výstupů použitých tkání a buněk nebo podle potřeby interakce s klinickými uživateli. |
|
4. |
Musí být zaveden zdokumentovaný systém řízení jakosti, který se uplatňuje na činnosti, k nimž se žádá o akreditaci, jmenování, oprávnění nebo povolení, a to v souladu s normami stanovenými v této směrnici. |
|
5. |
Musí být zaručeno, že jsou určena a minimalizována rizika spojená s používáním biologického materiálu a s manipulací s tímto materiálem, přičemž je zachována odpovídající jakost a bezpečnost pro určený účel použití tkání a buněk. Tato rizika se týkají postupů, prostředí a zdravotního stavu pracovníků v daném tkáňovém zařízení. |
|
6. |
Dohody mezi tkáňovými zařízeními a třetími stranami musejí být v souladu s článkem 24 směrnice 2004/23/ES. Dohody se třetími stranami musejí upřesňovat podmínky vztahu a povinnosti, jakož i protokoly, které je třeba dodržovat, aby byla splněna požadovaná specifikace plnění dohody. |
|
7. |
Musí být zaveden zdokumentovaný systém, na nějž dohlíží zodpovědná osoba, kterým se potvrzuje, že tkáně a/nebo buňky splňují příslušné specifikace bezpečnosti a jakosti pro propuštění a distribuci. |
|
8. |
V případě ukončení činností zahrnují uzavřené dohody a postupy přijaté v souladu s čl. 21 odst. 5 směrnice 2004/23/ES údaje o sledovatelnosti a materiál týkající se jakosti a bezpečnosti buněk a tkání. |
|
9. |
Musí být zaveden zdokumentovaný systém, který zaručuje identifikaci každé jednotky tkáně či buňky ve všech etapách činností, k nimž se žádá o akreditaci, jmenování, oprávnění nebo povolení. |
B. PRACOVNÍCI
|
1. |
Pracovníků musí být v tkáňovém zařízení dostatečný počet a musejí být kvalifikováni pro úkony, které provádějí. Způsobilost pracovníků musí být ve vhodných intervalech, upřesněných v systému jakosti, vyhodnocována. |
|
2. |
Všichni pracovníci musejí mít jasný, zdokumentovaný a aktuální popis práce. Jejich úkoly, povinnosti a zodpovědnost musejí být jasně zdokumentovány a pochopeny. |
|
3. |
Pracovníkům musí být poskytnuta počáteční či základní odborná příprava, aktualizovaná příprava v případě, že se změní postupy nebo se vyvíjejí vědecké poznatky, a odpovídající příležitosti pro odborný rozvoj. Program odborné přípravy musí zajistit a zdokumentovat, že každý jednotlivec:
|
C. VYBAVENÍ A MATERIÁLY
|
1. |
Veškeré vybavení a materiál musejí být navrženy a udržovány způsobem vhodným pro jejich určený účel a musejí pro příjemce a/nebo pracovníky představovat minimální riziko. |
|
2. |
Veškeré kritické vybavení a technické prostředky musejí být identifikovány a validovány, pravidelně kontrolovány a preventivně udržovány v souladu s pokyny výrobce. Pokud vybavení nebo materiály mají vliv na kritické parametry zpracování či skladování (např. teplota, tlak, počty částic, úrovně mikrobiální kontaminace), musejí být identifikovány a podle potřeby podléhat příslušnému sledování, systémům varování, poplachů a nápravných opatření, aby byly zjištěny poruchy a vady a bylo zajištěno, že jsou kritické parametry nepřetržitě udržovány v přijatelných mezích. Veškeré vybavení, kterým se měří kritické parametry, musí být kalibrováno podle sledovatelného etalonu, je-li k dispozici. |
|
3. |
Nové a opravené vybavení musí být při instalaci vyzkoušeno a před použitím validováno. Výsledky zkoušek se zdokumentují. |
|
4. |
Údržba, servisní služby, čištění, dezinfekce a sanitace se u veškerého kritického vybavení provádějí pravidelně a jsou náležitě zaznamenávány. |
|
5. |
Musejí být k dispozici postupy pro provoz každé součásti kritického vybavení, podrobně popisující opatření, jež mají být přijata v případě poruchy či selhání. |
|
6. |
Postupy u činností, k nimž se žádá o akreditaci, jmenování, oprávnění nebo povolení, musejí podrobně uvádět specifikace všech kritických materiálů a reakčních činidel. Zejména je třeba definovat specifikace přídatných látek (např. roztoků) a obalových materiálů. Kritická reakční činidla a materiály musejí splňovat zdokumentované požadavky a specifikace a případně požadavky uvedené ve směrnici Rady 93/42/EHS ze dne 14. června 1993 o zdravotnických prostředcích (1) a směrnici Evropského parlamentu a Rady 98/79/ES ze dne 27. října 1998 o diagnostických zdravotnických prostředcích in vitro (2). |
D. PROVOZOVNY A PROSTORY
|
1. |
Tkáňové zařízení musí mít vhodné provozovny pro provádění činností, k nimž se žádá o akreditaci, jmenování, oprávnění nebo povolení, a to v souladu s normami stanovenými v této směrnici. |
|
2. |
Pokud tyto činnosti zahrnují zpracování tkání a buněk při vystavení prostředí, musejí probíhat v prostředí s přesně stanovenou kvalitou a čistotou vzduchu, aby se minimalizovalo riziko kontaminace, včetně křížové kontaminace mezi tkáněmi a buňkami z různých darování. Je nutné validovat a sledovat účinnost těchto opatření. |
|
3. |
Pokud bod 4 nestanoví jinak, jsou-li tkáně či buňky vystaveny během zpracování prostředí, aniž by následně byly podrobeny procesu mikrobiální inaktivace, je vyžadována kvalita vzduchu, v němž jsou počet částic a počet mikrobiálních kolonií rovnocenné počtům ve třídě čistoty A, jak je definována v příloze 1 platných Evropských pokynů pro správnou výrobní praxi a ve směrnici 2003/94/ES, a prostředí pozadí vhodné pro zpracování příslušných tkání nebo buněk, které však odpovídá alespoň třídě čistoty D správné výrobní praxe, pokud jde o počet částic a počet mikrobiálních kolonií. |
|
4. |
Méně přísné nároky na prostředí, než jaké jsou uvedeny v bodě 3, jsou přijatelné v případě, že:
|
|
5. |
Je nutné jasně stanovit prostředí v bodě 4 písm. a), b), c) a d). Musí být prokázáno a zdokumentováno, že zvolené prostředí splňuje požadavky na jakost a bezpečnost, alespoň s ohledem na určený účel, způsob použití a imunitní stav příjemce. V každém příslušném oddělení tkáňového zařízení se musí poskytovat vhodný oděv a vybavení pro osobní ochranu a hygienu a písemné pokyny pro hygienu a oblékání. |
|
6. |
Pokud činnosti, k nimž se žádá o akreditaci, jmenování, oprávnění nebo povolení, zahrnují skladování tkání a buněk, musejí se definovat podmínky skladování nezbytné k tomu, aby si tkáně a buňky uchovaly požadované vlastnosti, včetně parametrů jako teplota, vlhkost nebo kvalita vzduchu. |
|
7. |
Kritické parametry se musejí kontrolovat, sledovat a zaznamenávat (např. teplota, vlhkost, kvalita vzduchu), aby se prokázalo, že jsou dodržovány stanovené podmínky skladování. |
|
8. |
Musejí být zajištěna skladovací zařízení, která jasně oddělují a odlišují tkáně a buňky před propuštěním v karanténě od těch, které jsou již propuštěny, a těch, které byly odmítnuty, aby se zabránilo záměně a jejich křížové kontaminaci. Pro uchovávání určitých tkání a buněk odebraných podle zvláštních kritérií musejí být v místech skladování tkání a buněk v karanténě i v místech skladování propuštěných tkání a buněk vyhrazeny fyzicky oddělené prostory či skladovací zařízení nebo musí být uvnitř zařízení zabezpečeno oddělení. |
|
9. |
Tkáňové zařízení musí mít písemnou koncepci a postupy pro kontrolovaný přístup, čištění a údržbu, nakládání s odpadem a pro obnovu poskytování služeb při mimořádné situaci. |
E. DOKUMENTACE A ZÁZNAMY
|
1. |
Pro činnosti, k nimž se žádá o akreditaci, jmenování, oprávnění nebo povolení, musí být zaveden systém, který zajistí jasně vymezenou a efektivní dokumentaci, správné záznamy a registry a schválené standardní pracovní postupy. Dokumenty musejí být pravidelně přezkoumávány a musejí splňovat normy stanovené v této směrnici. Systém musí zaručovat, že je práce prováděna podle daných norem a že veškeré kroky, tj. kódování, způsobilost dárce, odběr, zpracování, konzervaci, skladování, přepravu, distribuci či likvidaci, včetně aspektů týkajících se kontroly a zabezpečení jakosti, jsou sledovatelné. |
|
2. |
U každé kritické činnosti musí být identifikovány a zdokumentovány použité materiály, vybavení a zúčastnění pracovníci. |
|
3. |
Veškeré změny dokumentace v tkáňovém zařízení musejí být přezkoumány, opatřeny datem, schváleny, zdokumentovány a řádně provedeny oprávněnými pracovníky. |
|
4. |
Postup kontroly dokumentů musí být stanoven tak, aby poskytoval historii přezkoumání jednotlivých dokumentů a jejich změn a zajišťoval, že jsou používány pouze aktuální verze dokumentů. |
|
5. |
Záznamy musejí spolehlivě a pravdivě zobrazovat výsledky. |
|
6. |
Záznamy musejí být čitelné a nesmazatelné, mohou být psané ručně či převedené do jiného validovaného systému, např. do elektronické podoby či na mikrofilm. |
|
7. |
Aniž je dotčen čl. 9 odst. 2, veškeré záznamy, včetně primárních údajů, které jsou kriticky důležité pro bezpečnost a jakost tkání a buněk, se uchovají tak, aby k nim byl zajištěn přístup alespoň po dobu deseti let od data ukončení použitelnosti, klinického použití či likvidace. |
|
8. |
Záznamy musejí splňovat požadavky na důvěrnost podle článku 14 směrnice 2004/23/ES. Přístup do registrů a k údajům musí být povolen pouze osobám, které mají oprávnění od odpovědné osoby, a příslušnému orgánu pro účely inspekce a kontrolních opatření. |
F. PŘEZKOUMÁNÍ JAKOSTI
|
1. |
U činností, k nimž se žádá o akreditaci, jmenování, oprávnění nebo povolení, musí být zaveden systém auditu. Kvalifikované a způsobilé osoby musejí audit provádět nezávisle a alespoň jednou za dva roky, aby se ověřil soulad se schválenými protokoly a regulačními požadavky. Zjištění a nápravná opatření se musejí zdokumentovat. |
|
2. |
Odchylky od požadovaných norem pro jakost a bezpečnost se musejí vyšetřit a zdokumentovat, což zahrnuje rovněž rozhodnutí o případných nápravných a preventivních opatřeních. O osudu tkání a buněk, které nesplňují požadavky, se musí rozhodnout v souladu s písemnými postupy a za dohledu odpovědné osoby, toto rozhodnutí se musí zaznamenat. Všechny dotčené tkáně a buňky musejí být identifikovány a musí o nich být podána zpráva. |
|
3. |
Nápravná opatření musejí být zdokumentována, zahájena a dokončena včas a účinným způsobem. Po provedení preventivních a nápravných opatření by měla být posouzena jejich účinnost. |
|
4. |
Tkáňová zařízení musejí mít zavedeny postupy pro přezkoumání výkonu systému řízení jakosti, aby se zajistilo nepřetržité a systematické zlepšování. |
(1) Úř. věst. L 169, 12.7.1993, s. 1. Směrnice naposledy pozměněná nařízením Evropského parlamentu a Rady (ES) č. 1882/2003 (Úř. věst. L 284, 31.10.2003, s. 1).
(2) Úř. věst. L 331, 7.12.1998, s. 1. Směrnice ve znění nařízení (ES) č. 1882/2003.
PŘÍLOHA II
Požadavky pro udělení příslušného oprávnění pro postupy přípravy tkání a buněk v tkáňových zařízeních, jak je uvedeno v článku 4
Po vyhodnocení kritérií pro výběr dárce a postupů odběru, protokolů pro každou etapu postupu, kritérií pro řízení jakosti a závěrečných množstevních kritérií a kritérií pro jakost buněk a tkání udělí příslušný orgán každému postupu přípravy tkání a buněk oprávnění. Takové hodnocení musí splňovat alespoň požadavky stanovené v této příloze.
A. PŘIJETÍ V TKÁŇOVÉM ZAŘÍZENÍ
Při přijetí odebraných tkání a buněk v tkáňovém zařízení musejí tkáně a buňky splňovat požadavky definované ve směrnici 2006/17/ES.
B. ZPRACOVÁNÍ
Pokud činnosti, k nimž se žádá o akreditaci, jmenování, oprávnění nebo povolení, zahrnují zpracování tkání a buněk, musejí postupy v tkáňovém zařízení splňovat tato kritéria:
|
1. |
Kritické postupy zpracování musejí být validovány a nesmějí tkáně či buňky činit klinicky neúčinnými či škodlivými pro příjemce. Tato validace musí vycházet ze studií provedených zařízením nebo z údajů ze zveřejněných studií nebo, v případě osvědčených postupů zpracování, ze zpětného hodnocení klinických výsledků u tkání dodaných daným zařízením. |
|
2. |
Musí být prokázáno, že pracovníci mohou validovaný proces provádět v prostředí tkáňového zařízení shodně a účinně. |
|
3. |
Ve standardních pracovních postupech musejí být postupy zdokumentovány a musejí být v souladu s validovanou metodou a normami stanovenými v této směrnici, a to v souladu s přílohou I částí E body 1 až 4. |
|
4. |
Musí být zajištěno, že se veškeré procesy provádějí v souladu se schválenými standardními pracovními postupy. |
|
5. |
Je-li na tkáně nebo buňky uplatněn postup mikrobiální inaktivace, musí být tento postup specifikován, zdokumentován a validován. |
|
6. |
Před provedením jakékoli důležité změny ve zpracování musí být pozměněný postup validován a zdokumentován. |
|
7. |
Postupy zpracování musejí být podrobovány pravidelnému kritickému hodnocení, aby se zajistilo, že budou i nadále dosahovat určených výsledků. |
|
8. |
Postupy pro vyřazování tkání a buněk musejí bránit kontaminaci tkání a buněk z jiných darování a přípravků, prostředí pro zpracování a pracovníků. Tyto postupy musejí odpovídat vnitrostátním předpisům. |
C. SKLADOVÁNÍ A PROPOUŠTĚNÍ PŘÍPRAVKŮ
Pokud činnosti, k nimž se žádá o akreditaci, jmenování, oprávnění nebo povolení, zahrnují skladování a propouštění tkání a buněk, musejí postupy v tkáňovém zařízení s oprávněním splňovat tato kritéria:
|
1. |
Pro každý typ podmínek skladování musí být určena maximální doba skladování. Zvolená doba musí mimo jiné odrážet případné zhoršení požadovaných vlastností tkáně či buňky. |
|
2. |
Musí být zaveden systém evidence tkání a/nebo buněk v karanténě, aby se zaručilo, že nemohou být propuštěny, dokud nesplňují veškeré požadavky této směrnice. Musí existovat standardní pracovní postup, v němž jsou podrobně popsány okolnosti, povinnosti a postupy pro propouštění tkání a buněk k distribuci. |
|
3. |
Systém identifikace tkání a buněk v celém rámci jakékoli etapy zpracování v tkáňovém zařízení musí jasně rozlišovat přípravky propuštěné od přípravků nepropuštěných (v karanténě) a přípravků vyřazených. |
|
4. |
Záznamy musejí prokazovat, že byly před propuštěním tkání a buněk splněny všechny patřičné specifikace, zejména že byly všechny formuláře prohlášení, náležité lékařské záznamy, záznamy o zpracování a výsledky zkoušek ověřeny podle písemného postupu osobou pověřenou tímto úkolem odpovědnou osobou uvedenou v článku 17 směrnice 2004/23/ES. Pokud se k uvolnění výsledků z laboratoře použije počítač, musí záznam o kontrole uvádět, kdo je za uvolnění odpovědný. |
|
5. |
Je třeba provést zdokumentované posouzení rizik, které schválí odpovědná osoba uvedená v článku 17 směrnice 2004/23/ES, aby se po zavedení nových kritérií pro výběr či testování dárce nebo výrazně změněné etapy zpracování, jimiž se posílí bezpečnost a jakost, určil osud všech skladovaných tkání a buněk. |
D. DISTRIBUCE A STAŽENÍ
Pokud činnosti, k nimž se žádá o akreditaci, jmenování, oprávnění nebo povolení, zahrnují distribuci tkání a buněk, musejí postupy v tkáňovém zařízení s oprávněním splňovat tato kritéria:
|
1. |
Musejí být definovány kritické podmínky přepravy, např. teplota a lhůta, aby se zachovaly požadované vlastnosti tkáně a buňky. |
|
2. |
Nádoba/balení musejí být bezpečné a musejí zaručovat, že jsou tkáně a buňky uchovávány za přesně stanovených podmínek. Veškeré nádoby a balení musejí být validovány jako vhodné pro daný účel. |
|
3. |
Pokud distribuci provádí třetí strana na základě smlouvy, musí existovat zdokumentovaná dohoda, aby se zaručilo, že budou dodrženy požadované podmínky. |
|
4. |
V tkáňovém zařízení jsou zaměstnanci oprávnění posoudit nezbytnost stažení z oběhu a zahájit a koordinovat potřebné kroky. |
|
5. |
Musí být zaveden účinný postup stažení z oběhu včetně popisu povinností a kroků, jež se mají provést. Musejí zahrnovat oznámení příslušnému orgánu. |
|
6. |
Kroky učiněné v předem vymezeném období musejí zahrnovat sledování všech příslušných tkání a buněk a v případě potřeby také zpětné vysledování. Účelem šetření je identifikovat dárce, který mohl přispět k vyvolání reakce u příjemce, a stáhnout dostupné tkáně a buňky od takového dárce, jakož i informovat odběratele a příjemce tkání a buněk odebraných od tohoto dárce pro případ, že mohli být ohroženi. |
|
7. |
Musejí být zavedeny postupy pro vyřizování žádostí o tkáně a buňky. Je třeba zdokumentovat pravidla přidělování tkání a buněk určitým pacientům nebo zdravotnickým institucím a na žádost je těmto stranám zpřístupnit. |
|
8. |
Musí být zaveden zdokumentovaný systém pro manipulaci s vrácenými přípravky, případně včetně kritérií pro jejich přijetí do inventáře. |
E. KONEČNÉ OZNAČENÍ PRO DISTRIBUCI
|
1. |
Primární nádoba pro tkáně/buňky musí uvádět:
Pokud některé z informací v písmenech d) a e) nelze na štítku primární nádoby uvést, musejí být poskytnuty na odděleném listu, který primární nádobu provází. Tento list musí být k primární nádobě přibalen tak, aby se zaručilo, že od sebe nebudou odděleny. |
|
2. |
Na štítku nebo v průvodní dokumentaci se musejí uvést tyto informace:
|
F. VNĚJŠÍ OZNAČOVÁNÍ PŘEPRAVNÍHO KONTEJNERU
Pro přepravu musí být primární nádoba umístěna do přepravního kontejneru, na jehož štítku musejí být uvedeny alespoň tyto informace:
|
a) |
identifikace původního tkáňového zařízení včetně adresy a telefonního čísla; |
|
b) |
identifikace cílové organizace odpovědné za použití u člověka včetně adresy a telefonního čísla; |
|
c) |
prohlášení, že balení obsahuje lidské tkáně/buňky a nápis „MANIPULOVAT OPATRNĚ“; |
|
d) |
pokud jsou pro fungování transplantátu potřebné živé buňky, např. kmenové buňky, gamety a embrya, musí se doplnit nápis „NEOZAŘOVAT“; |
|
e) |
doporučené přepravní podmínky (např. uchovejte v chladu, ve svislé poloze atd.); |
|
f) |
bezpečnostní pokyny/metoda chlazení (pokud je to možné). |
PŘÍLOHA III
OZNAMOVÁNÍ ZÁVAŽNÝCH NEŽÁDOUCÍCH REAKCÍ
PŘÍLOHA IV
OZNAMOVÁNÍ ZÁVAŽNÝCH NEŽÁDOUCÍCH ÚČINKŮ
PŘÍLOHA V
FORMULÁŘ PRO VÝROČNÍ OZNÁMENÍ

PŘÍLOHA VI
Informace o minimálním rozsahu údajů o dárci a příjemci, které se mají uchovat, jak vyžaduje článek 9
A. VE TKÁŇOVÝCH ZAŘÍZENÍCH
Identifikace dárce
Identifikace darování zahrnující alespoň:
|
— |
identifikaci organizace provádějící odběr nebo tkáňového zařízení, |
|
— |
jedinečné identifikační číslo darování, |
|
— |
datum odběru, |
|
— |
místo odběru. |
|
— |
druh darování (např. jedna tkáň či více tkání; autologní či alogenní; žijící či zemřelý dárce) |
Identifikace přípravku zahrnující alespoň:
|
— |
identifikaci tkáňového zařízení, |
|
— |
druh tkáně a buňky/přípravku (základní nomenklatura), |
|
— |
číslo kolekce (je-li použitelné), |
|
— |
číslo frakce (je-li použitelné), |
|
— |
datum ukončení doby použitelnosti, |
|
— |
stav tkáně/buňky (tj. v karanténě, vhodné k použití atd.), |
|
— |
popis a původ přípravků, použitých kroků při zpracování, materiálů a přídatných látek, které přicházejí do styku s tkáněmi a buňkami a mají vliv na jejich jakost a/nebo bezpečnost, |
|
— |
identifikaci zařízení, které vydává konečný štítek. |
Identifikace použití u člověka zahrnujícího alespoň:
|
— |
datum distribuce/likvidace, |
|
— |
identifikace klinického lékaře a konečného uživatele/zařízení. |
B. V ORGANIZACÍCH ODPOVĚDNÝCH ZA POUŽITÍ U ČLOVĚKA
|
a) |
identifikace dodavatelského tkáňového zařízení; |
|
b) |
identifikace klinického lékaře a konečného uživatele/zařízení; |
|
c) |
druh tkání a buněk; |
|
d) |
identifikace přípravku; |
|
e) |
identifikace příjemce; |
|
f) |
datum použití. |
PŘÍLOHA VII
Informace obsažené v evropském kódovacím systému
|
a) |
Identifikace darování:
|
|
b) |
Identifikace přípravku:
|