(EHS) č. 2568/91NAŘÍZENÍ KOMISE (EHS) č. 2568/91 ze dne 11. července 1991 o charakteristikách olivového oleje a olivového oleje z pokrutin a o příslušných metodách analýzy
| Publikováno: | Úř. věst. L 248, 5.9.1991, s. 1-83 | Druh předpisu: | Nařízení |
| Přijato: | 11. července 1991 | Autor předpisu: | Evropská komise |
| Platnost od: | 6. září 1991 | Nabývá účinnosti: | 6. září 1991 |
| Platnost předpisu: | Zrušen předpisem (EU) 2022/2104 | Pozbývá platnosti: | 24. listopadu 2022 |
Text aktualizovaného znění s celou hlavičkou je dostupný pouze pro registrované uživatele.
Tento dokument slouží výhradně k informačním účelům a nemá žádný právní účinek. Orgány a instituce Evropské unie nenesou za jeho obsah žádnou odpovědnost. Závazná znění příslušných právních předpisů, včetně jejich právních východisek a odůvodnění, jsou zveřejněna v Úředním věstníku Evropské unie a jsou k dispozici v databázi EUR-Lex. Tato úřední znění jsou přímo dostupná přes odkazy uvedené v tomto dokumentu
|
NAŘÍZENÍ KOMISE (EHS) č. 2568/91 ze dne 11. července 1991 o charakteristikách olivového oleje a olivového oleje z pokrutin a o příslušných metodách analýzy (Úř. věst. L 248 5.9.1991, s. 1) |
Ve znění:
Opraveno:
|
(*) |
Tento akt nebyl nikdy publikován v češtině. |
NAŘÍZENÍ KOMISE (EHS) č. 2568/91
ze dne 11. července 1991
o charakteristikách olivového oleje a olivového oleje z pokrutin a o příslušných metodách analýzy
Článek 1
1. Oleje s charakteristikami, které odpovídají charakteristikám stanoveným v bodech 1 a 2 přílohy I tohoto nařízení, jsou považovány za panenské olivové oleje ve smyslu bodu 1 písm. a) a b) přílohy nařízení č. 136/66/EHS.
2. Olej s charakteristikami, které odpovídají charakteristikám stanoveným v bodu 3 přílohy I tohoto nařízení, je považován za lampantový olivový olej ve smyslu bodu 1 písm. c) přílohy nařízení č. 136/66/EHS.
3. Olej s charakteristikami, které odpovídají charakteristikám stanoveným v bodu 4 přílohy I tohoto nařízení, je považován za rafinovaný olivový olej ve smyslu bodu 2 přílohy nařízení č. 136/66/EHS.
4. Olej s charakteristikami, které odpovídají charakteristikám stanoveným v bodu 5 přílohy I tohoto nařízení, je považován za olivový olej obsahující směs rafinovaného olivového oleje a panenského olivového oleje ve smyslu bodu 3 přílohy nařízení č. 136/66/EHS.
5. Olej s charakteristikami, které odpovídají charakteristikám stanoveným v bodu 6 přílohy I tohoto nařízení, je považován za surový olivový olej z pokrutin ve smyslu bodu 4 přílohy nařízení č. 136/66/EHS.
6. Olej s charakteristikami, které odpovídají charakteristikám stanoveným v bodu 7 přílohy I tohoto nařízení, je považován za rafinovaný olivový olej z pokrutin ve smyslu bodu 5 přílohy nařízení č. 136/66/EHS.
7. Olej s charakteristikami, které odpovídají charakteristikám stanoveným v bodu 8 přílohy I tohoto nařízení, je považován za olivový olej z pokrutin ve smyslu bodu 6 přílohy nařízení č. 136/66/EHS.
Článek 2
1. Charakteristiky olivových olejů uvedených v příloze I se stanoví v souladu s těmito metodami analýzy:
a) stanovení volných mastných kyselin, vyjádřených v procentech kyseliny olejové, metodou uvedenou v příloze II;
b) stanovení peroxidového čísla metodou uvedenou v příloze III;
c) stanovení obsahu vosků metodou uvedenou v příloze IV;
d) stanovení složení a obsahu sterolů a triterpenických dialkoholů kapilární plynovou chromatografií metodou uvedenou v příloze V;
e) stanovení procentního podílu 2-glyceril-monopalmitátu metodou uvedenou v příloze VII;
f) spektrofotometrická analýza metodou uvedenou v příloze IX;
g) stanovení složení mastných kyselin metodou uvedenou v příloze X;
h) stanovení těkavých halogenovaných rozpouštědel metodou uvedenou v příloze XI;
i) hodnocení organoleptických charakteristik panenského olivového oleje metodou uvedenou v příloze XII;
j) stanovení stigmastadienolů metodou uvedenou v příloze XVII;
k) stanovení obsahu triglyceridů s ECN 42 metodou uvedenou v příloze XVIII;
l) stanovení složení a obsahu sterolů a stanovení alkoholových sloučenin kapilární plynovou chromatografií metodou uvedenou v příloze XIX;
m) stanovení obsahu vosků, methylesterů mastných kyselin a ethylesterů mastných kyselin metodou uvedenou v příloze XX.
▼M28 —————
2. Ověřování organoleptických charakteristik panenského oleje provádí vnitrostátní orgány nebo jejich zástupci v rámci zkušebních komisí schválených členskými státy.
Organoleptické vlastnosti oleje uvedeného v prvním pododstavci se považují za odpovídající deklarované kategorii v případě, že zkušební komise schválená členským státem potvrdí jeho zařazení.
V případě, že zkušební komise nepotvrdí deklarovanou kategorii oleje vzhledem k jeho organoleptickým charakteristikám, nechají vnitrostátní orgány nebo jejich zástupci na žádost zainteresované strany bez odkladu provést dvě oponentní hodnocení jinými schválenými zkušebními komisemi. Alespoň jedna z těchto komisí musí být schválena členským státem, z nějž pochází výrobce oleje. Dotčené charakteristiky se považují za odpovídající deklarovaným charakteristikám, pokud obě oponentní hodnocení potvrdí deklarovanou kvalitu. Pokud tomu tak není, prohlásí se klasifikace za neodpovídající uvedeným charakteristikám, nehledě na typ nedostatků zjištěných během hodnocení, a náklady na oponentní hodnocení ponese zainteresovaná strana.
3. Pokud vnitrostátní orgány nebo jejich zástupci ověřují vlastnosti oleje postupem podle odstavce 1, provádí se odběr vzorků v souladu s mezinárodními normami EN ISO 661 o přípravě zkušebních vzorků a EN ISO 5555 o odběru vzorků. Pokud jsou však takové oleje ve spotřebitelských obalech baleny po šaržích, pak odchylně od bodu 6.8 normy EN ISO 5555 se odběr vzorků provádí v souladu s přílohou Ia tohoto nařízení. V případě, že odběr vzorků oleje ve velkém balení nemůže být proveden v souladu s normou EN ISO 5555, odeberou se vzorky podle pokynů příslušného orgánu členského státu.
Aniž by byla dotčena ustanovení normy EN ISO 5555 a kapitoly 6 normy EN ISO 661, odebrané vzorky se co nejrychleji umístí z dosahu světla a vysokých teplot a nejpozději pět pracovních dní po odběru se odešlou k analýze do laboratoře, jinak musí být uchovávány tak, aby se neznehodnotily nebo nepoškodily během přepravy nebo skladování, než budou do laboratoře zaslány.
4. Pro účely ověření podle odstavce 3 se analýzy uvedené v přílohách II, III, IX, XII a XX, jakož i případné kontrolní analýzy stanovené vnitrostátními právními předpisy, v případě balených výrobků provádějí před datem minimální trvanlivosti. V případě odběru vzorků olejů ve velkých baleních se tyto analýzy provádějí nejpozději šestý měsíc po měsíci, v kterém byl vzorek odebrán.
Pro ostatní analýzy uvedené v tomto nařízení neplatí žádné lhůty.
Jestliže výsledky analýz neodpovídají vlastnostem deklarované třídy olivového oleje nebo olivového oleje z pokrutin, dotčená strana je o tom informována nejpozději jeden měsíc před koncem lhůty stanovené v prvním odstavci, kromě případu, kdy byl vzorek odebrán méně než dva měsíce před datem minimální trvanlivosti.
5. Ke stanovení charakteristik olivového oleje metodami uvedenými v prvním pododstavci odstavce 1 se výsledky analýz porovnávají přímo s mezními hodnotami danými tímto nařízením.
Článek 2a
1. Pro účely tohoto článku se pojmem „olivový olej uváděný na trh“ rozumí celkové množství olivového oleje a olivového oleje z pokrutin příslušného členského státu, které se v uvedeném členském státě spotřebuje nebo které se z uvedeného členského státu vyveze.
2. Členské státy zajistí, aby kontroly shody byly prováděny selektivně, na základě analýzy rizik a s přiměřenou četností s cílem zajistit, aby olivový olej uváděný na trh byl v souladu s deklarovanou kategorií.
3. Kritéria pro posouzení rizika mohou zahrnovat:
a) kategorii oleje, období produkce, cenu oleje v porovnání s jinými rostlinnými oleji, úkony mísení a balení, skladovací zařízení a podmínky, zemi původu, zemi určení, způsob přepravy nebo objem šarže;
b) postavení hospodářských subjektů v marketingovém řetězci, objem nebo hodnotu výrobků jimi uváděných na trh, sortiment olejů jimi uváděných na trh, druh vykonávané hospodářské činnosti jako např. lisování, skladování, rafinace, mísení, balení nebo maloobchodní prodej;
c) zjištění při předchozích kontrolách včetně počtu a druhu zjištěných nedostatků, obvyklou kvalitu olejů uváděných na trh, výkonnost používaného technického vybavení;
d) spolehlivost systému zajištění jakosti či systému vlastní kontroly hospodářských subjektů v souvislosti s dodržováním obchodních norem;
e) místo, kde se provádí kontrola, zejména, jde-li o první bod vstupu do Unie, poslední bod výstupu z Unie nebo o místo produkce, balení a naložení oleje či jeho prodeje konečnému spotřebiteli;
f) jakékoliv další informace, které by mohly poukazovat na riziko nedodržování norem.
4. Členské státy předem stanoví:
a) kritéria pro hodnocení rizika nedodržování norem u šarží;
b) na základě analýzy rizik pro každou kategorii rizik minimální počet hospodářských subjektů či šarží a/nebo množství, u nichž budou provedeny kontroly shody.
Ročně se provádí alespoň jedna kontrola shody na každých tisíc tun olivového oleje uváděného na trh v členském státě.
5. Členské státy ověří dodržování shody:
a) provedením analýz uvedených v příloze I v jakémkoli pořadí nebo
b) v pořadí uvedeném ve vývojovém diagramu v příloze Ib, dokud není dosaženo jednoho z rozhodnutí uvedených ve vývojovém diagramu.
▼M19 —————
Článek 3
Pokud se zjistí, že určitý olej neodpovídá popisu kategorie, do níž patří, uloží daný členský stát, aniž jsou dotčeny jakékoli další sankce, účinné, přiměřené a odrazující sankce, jejichž výše se stanoví podle závažnosti zjištěné nesrovnalosti.
Pokud jsou při kontrolách zjištěny závažné nesrovnalosti, zvýší členské státy četnost kontrol ve vztahu k etapě uvádění na trh, kategorii oleje, původu nebo jiným kritériím.
Článek 4
1. The Member States may approve assessment panels so that national authorities or their representatives can assess and verify organoleptic characteristics.
The terms of approval shall be set by Member States and ensure that:
— the requirements of Annex XII.4 are met,
— the panel head is given training recognised for this purpose by the Member State,
— continued approval depends on performance in annual checks arranged by the Member State.
Member States shall notify to the Commission a list of approved panels and the action taken under this paragraph.
2. Pokud členský stát narazí na potíže při sestavování zkušebních komisí na svém území, může povolat zkušební komisi schválenou v jiném členském státě.
3. Každý členský stát vypracuje seznam zkušebních komisí sestavených profesionálními nebo mezioborovými organizacemi za podmínek stanovených v odstavci 1 a zajistí dodržování těchto podmínek.
▼M19 —————
Článek 6
1. Obsah oleje u pokrutin z oliv a jiných zbytků po extrakci olivového oleje (podpoložky 2306 90 11 a 2306 90 19 ) se stanoví metodou uvedenou v příloze XV.
2. Obsah oleje, na který odkazuje odstavec 1, se vyjadřuje v procentech hmotnosti oleje v sušině.
Článek 7
Pokud jde o přítomnost kontaminujících látek, použijí se příslušné předpisy Společenství.
Pokud jde o halogenovaná rozpouštědla, zavádějí se pro všechny kategorie olivového oleje tyto limity:
— maximální obsah každého zjištěného halogenovaného rozpouštědla: 0,1 mg/kg,
— maximální celkový obsah zjištěných halogenovaných rozpouštědel: 0,2 mg/kg.
Článek 7a
Fyzické či právnické osoby a skupiny fyzických či právnických osob vlastnící z profesionálních či obchodních důvodů olivový olej a olivový olej z pokrutin od extrakce v lisovně až po fázi plnění, musí pro každou z kategorií těchto olejů vést evidenci vstupů a výstupů.
Členský stát zajistí, aby byla povinnost stanovená v prvním odstavci řádně dodržována.
Článek 8
1. Členské státy sdělí Komisi opatření přijatá k provádění tohoto nařízení. Rovněž Komisi uvědomí o veškerých následných změnách.
2. Nejpozději 31. května každého roku předají členské státy Komisi zprávu o provádění tohoto nařízení v předchozím kalendářním roce. Zpráva musí obsahovat alespoň výsledky kontrol shody provedené u olivového oleje podle vzoru stanoveného v příloze XXI.
3. Oznámení uvedená v tomto nařízení se provádějí v souladu s nařízením Komise (ES) č. 792/2009 ( 1 ).
Článek 9
Nařízení (EHS) č. 1058/77 se zrušuje.
Článek 10
1. Toto nařízení vstupuje v platnost třetím dnem po vyhlášení v Úředním věstníku Evropských společenství.
Metoda uvedená v příloze XII se však použije ode dne ►M1 1. listopadu 1992 ◄ , s výjimkou případů, kdy se jedná o intervenční opatření.
Tato metoda se nepoužije u panenského olivového oleje stočeného před 1. listopadem 1992.
2. Toto nařízení se nepoužije na olivový olej a olivový olej z pokrutin stočený do obalů před vstupem tohoto nařízení v platnost a uvedený na trh do 31. října 1992.
Toto nařízení je závazné v celém rozsahu a přímo použitelné ve všech členských státech.
PŘÍLOHY
OBSAH
|
Příloha I |
Charakteristiky olivového oleje |
|
Příloha Ia |
Odběr vzorků ze šarží olivového oleje a olivového oleje z pokrutin ve spotřebitelských obalech |
|
Příloha Ib |
Vývojový diagram pro ověření souladu vzorku olivového oleje s deklarovanou kategorií |
|
Příloha II |
Stanovení volných mastných kyselin, metoda za studena |
|
Příloha III |
Stanovení peroxidového čísla |
|
Příloha IV |
Stanovení obsahu vosku pomocí kapilární plynové chromatografie |
|
Příloha VII |
Stanovení procentního podílu 2-glyceryl monopalmitátu |
|
Příloha IX |
Spektrofotometrická analýza v ultrafialové oblasti spektra |
|
Příloha X |
Stanovení methylesterů mastných kyselin plynovou chromatografií |
|
Příloha XI |
Stanovení těkavých halogenovaných rozpouštědel v olivovém oleji |
|
Příloha XII |
Metoda mezinárodní rady pro olivový olej pro organoleptické hodnocení panenského olivového oleje |
|
Příloha XV |
Stanovení obsahu oleje v olivových pokrutinách |
|
Příloha XVI |
Stanovení jodového čísla |
|
Příloha XVII |
Metoda pro stanovení stigmastadienolů v rostlinných olejích |
|
Příloha XVIII |
Stanovení rozdílu mezi skutečným a teoretickým obsahem triacylglycerolů s ekvivalentním počtem uhlíkových atomů 42 (ECN 42) |
|
Příloha XIX |
Stanovení složení a obsahu sterolů a alkoholových sloučenin kapilární plynovou chromatografií |
|
Příloha XX |
Metoda stanovení obsahu vosků, methylesterů mastných kyselin a ethylesterů mastných kyselin kapilární plynovou chromatografií |
|
Příloha XXI |
Výsledky kontrol shody provedených u olivového oleje uvedené v čl. 8 odst. 2 |
PŘÍLOHA I
CHARAKTERISTIKY OLIVOVÉHO OLEJE
Jakostní charakteristiky
|
Kategorie |
Kyselost (%) (*) |
Peroxidové číslo (mEq O2/kg) |
K232 |
K268 nebo K270 |
Delta-K |
Organoleptické hodnocení |
Ethylestery mastných kyselin (mg/kg) |
|
|
Medián vad (Md) (*) |
Medián znaku ovocná chuť a vůně (Mf) |
|||||||
|
1. Extra panenský olivový olej |
≤ 0,80 |
≤ 20,0 |
≤ 2,50 |
≤ 0,22 |
≤ 0,01 |
Md = 0,0 |
Mf > 0,0 |
≤ 35 |
|
2. Panenský olivový olej |
≤ 2,0 |
≤ 20,0 |
≤ 2,60 |
≤ 0,25 |
≤ 0,01 |
Md ≤ 3,5 |
Mf > 0,0 |
— |
|
3. Lampantový olivový olej |
> 2,0 |
— |
— |
— |
— |
Md > 3,5 (1) |
— |
— |
|
4. Rafinovaný olivový olej |
≤ 0,30 |
≤ 5,0 |
— |
≤ 1,25 |
≤ 0,16 |
|
— |
— |
|
5. Olivový olej složený z rafinovaného a panenského olivového oleje |
≤ 1,00 |
≤ 15,0 |
— |
≤ 1,15 |
≤ 0,15 |
|
— |
— |
|
6. Surový olivový olej z pokrutin |
— |
— |
— |
— |
— |
|
— |
— |
|
7. Rafinovaný olivový olej z pokrutin |
≤ 0,30 |
≤ 5,0 |
— |
≤ 2,00 |
≤ 0,20 |
|
— |
— |
|
8. Olivový olej z pokrutin |
≤ 1,00 |
≤ 15,0 |
— |
≤ 1,70 |
≤ 0,18 |
|
— |
— |
|
(1) Medián vad může být nejvýše roven 3,5, je-li medián znaku ovocná chuť a vůně je roven 0,0. |
||||||||
Charakteristiky týkající se čistoty
|
Kategorie |
Složení mastných kyselin (1) |
Úhrn transizomerů kyseliny olejové (%) |
Úhrn transizomerů kyseliny linolové + linolenové (%) |
Stigmastadieny (mg/kg) (2) |
Rozdíl: ECN42 (HPLC) a ECN42 (teoretický výpočet) |
2-glyceryl monopalmitát (%) |
|||||
|
Myristová (%) |
Linolenová (%) |
Arachidová (%) |
Eikosanová (%) |
Behenová (%) |
á (%) |
||||||
|
1. Extra panenský olivový olej |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,20 |
≤ 0,20 |
≤ 0,05 |
≤ 0,05 |
≤ 0,05 |
≤ |0,20| |
≤ 0,9, pokud celkové množství palmitové kyseliny v % ≤ 14,00 % |
|
≤ 1,0, pokud celkové množství palmitové kyseliny v % > 14,00 % |
|||||||||||
|
2. Panenský olivový olej |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,20 |
≤ 0,20 |
≤ 0,05 |
≤ 0,05 |
≤ 0,05 |
≤ |0,20| |
≤ 0,9, pokud celkové množství palmitové kyseliny v % ≤ 14,00 % |
|
≤ 1,0, pokud celkové množství palmitové kyseliny v % > 14,00 % |
|||||||||||
|
3. Lampantový olivový olej |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,20 |
≤ 0,20 |
≤ 0,10 |
≤ 0,10 |
≤ 0,50 |
≤ |0,30| |
≤ 0,9, pokud celkové množství palmitové kyseliny v % ≤ 14,00 % |
|
≤ 1,1, pokud celkové množství palmitové kyseliny v % > 14,00 % |
|||||||||||
|
4. Rafinovaný olivový olej |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,30 |
— |
≤|0,30| |
≤ 0,9, pokud celkové množství palmitové kyseliny v % ≤ 14,00 % |
|
≤ 1,1, pokud celkové množství palmitové kyseliny v % > 14,00 % |
|||||||||||
|
5. Olivový olej složený z rafinovaného a panenského olivového oleje |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,20 |
≤ 0,20 |
≤ 0,20 |
≤ 0,30 |
— |
≤ |0,30| |
≤ 0,9, pokud celkové množství palmitové kyseliny v % ≤ 14,00 % |
|
≤ 1,0, pokud celkové množství palmitové kyseliny v % > 14,00 % |
|||||||||||
|
6. Surový olivový olej z pokrutin |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,30 |
≤ 0,20 |
≤ 0,20 |
≤ 0,10 |
— |
≤ |0,60| |
≤ 1,4 |
|
7. Rafinovaný olivový olej z pokrutin |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,30 |
≤ 0,20 |
≤ 0,40 |
≤ 0,35 |
— |
≤ |0,50| |
≤ 1,4 |
|
8. Olivový olej z pokrutin |
≤ 0,03 |
≤ 1,00 |
≤ 0,60 |
≤ 0,50 |
≤ 0,30 |
≤ 0,20 |
≤ 0,40 |
≤ 0,35 |
— |
≤ |0,50| |
≤ 1,2 |
|
(1) Obsah ostatních mastných kyselin (%): palmitová: 7,50–20,00; palmitolejová: 0,30–3,50; heptadekanová: ≤ 0,40; heptadecenová ≤ 0,60; stearová: 0,50–5,00; olejová: 55,00–83,00; linolová: 2,50–21,00. (2) Celkové množství izomerů, které by mohly (nebo nemohly) být separovány kapilární kolonou. |
|||||||||||
|
Kategorie |
Složení sterolů |
Steroly celkem (mg/kg) |
Erythrodiol a uvaol (%) (**) |
Vosky (mg/kg) (**) |
|||||
|
Cholesterol (%) |
Brassikasterol (%) |
Kampesterol (1) (%) |
Stigmasterol (%) |
Zjevný β–sitosterol (2) (%) |
Delta-7-stigmastenol (1) (%) |
||||
|
1. Extra panenský olivový olej |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< kamp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
C42 + C44 + C46 ≤ 150 |
|
2. Panenský olivový olej |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< kamp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
C42 + C44 + C46 ≤ 150 |
|
3. Lampantový olivový olej |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
— |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 (3) |
C40 + C42 + C44 + C46 ≤ 300 (3) |
|
4. Rafinovaný olivový olej |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< kamp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
C40 + C42 + C44 + C46 ≤ 350 |
|
5. Olivový olej složený z rafinovaného a panenského olivového oleje |
≤ 0,5 |
≤ 0,1 |
≤ 4,0 |
< kamp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 000 |
≤ 4,5 |
C40 + C42 + C44 + C46 ≤ 350 |
|
6. Surový olivový olej z pokrutin |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
— |
≥ 93,0 |
≤ 0,5 |
≥ 2 500 |
> 4,5 (4) |
C40 + C42 + C44 + C46 > 350 (4) |
|
7. Rafinovaný olivový olej z pokrutin |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
< kamp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 800 |
> 4,5 |
C40 + C42 + C44 + C46 > 350 |
|
8. Olivový olej z pokrutin |
≤ 0,5 |
≤ 0,2 |
≤ 4,0 |
< kamp. |
≥ 93,0 |
≤ 0,5 |
≥ 1 600 |
> 4,5 |
C40 + C42 + C44 + C46 > 350 |
|
(1) Viz dodatek k této příloze. (2) Zjevný β-sitosterol: Delta-5,23-stigmastadienol + chlerosterol + beta-sitosterol + sitostanol + delta-5-avenasterol + delta-5,24-stigmastadienol. (3) Oleje s obsahem vosků od 300 mg/kg do 350 mg/kg jsou považovány za lampantové olivové oleje, pokud je celkový obsah alifatických alkoholů nejvýše roven 350 mg/kg nebo pokud obsah erythrodiolu a uvaolu je nejvýše roven 3,5 %. (4) Oleje s obsahem vosků od 300 mg/kg do 350 mg/kg jsou považovány za surový olej z pokrutin, pokud je celkový obsah alifatických alkoholů vyšší než 350 mg/kg a pokud je obsah erythrodiolu a uvaolu větší než 3,5 %. |
|||||||||
Poznámky:
a) Výsledky zkoušek musí být uvedeny na stejný počet desetinných míst, jaký je předepsán pro každou charakteristiku. Poslední desetinné místo se přitom musí zaokrouhlit nahoru, pokud je číslice na dalším desetinném místě vyšší než 4.
b) Pokud jakákoli charakteristika neodpovídá uvedeným hodnotám, může být olivový olej zařazen do jiné kategorie nebo označen jako nesplňující požadavky pro účely tohoto nařízení.
c) V případě lampantového olivového oleje se mohou obě jakostní charakteristiky označené hvězdičkou (*) současně lišit od mezních hodnot stanovených pro tuto kategorii.
d) Charakteristiky olejů označené dvěma hvězdičkami (**) znamenají, že u surového oleje z pokrutin se od uvedených hodnot mohou lišit obě mezní hodnoty současně. V případě olivového oleje z pokrutin a rafinovaného olivového oleje z pokrutin se od uvedených hodnot může lišit jedna mezní hodnota.
Dodatek
Rozhodovací schémata
Kampesterol – rozhodovací schéma pro panenské a extra panenské olivové oleje:
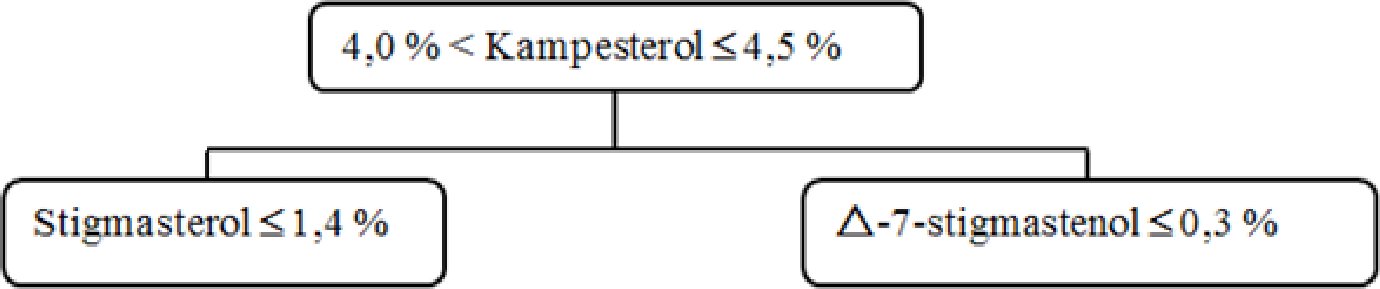
Ostatní parametry musí být v souladu s mezními hodnotami stanovenými v tomto nařízení.
Delta-7-stigmastenol – rozhodovací schéma pro:
— extra panenské a panenské olivové oleje
—
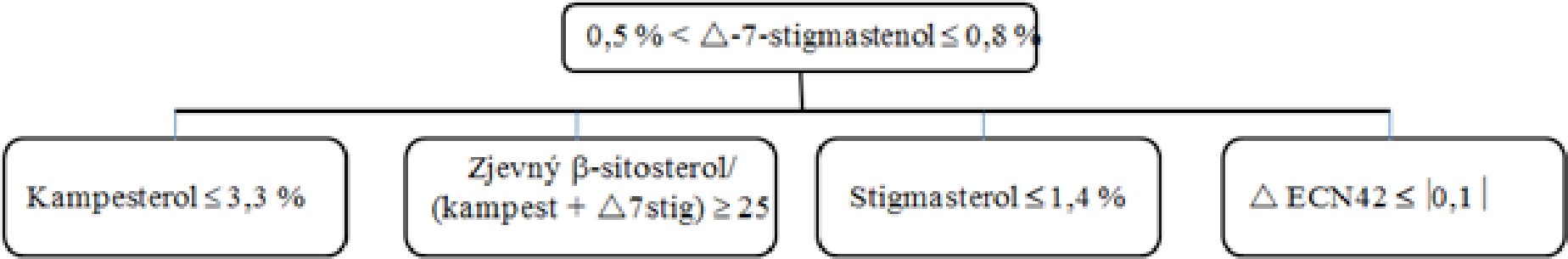
Ostatní parametry musí být v souladu s mezními hodnotami stanovenými v tomto nařízení.
— olivové oleje z pokrutin (surové a rafinované)
—

Ostatní parametry musí být v souladu s mezními hodnotami stanovenými v tomto nařízení.
PŘÍLOHA Ia
ODBĚR VZORKŮ ZE ŠARŽÍ OLIVOVÉHO OLEJE A OLIVOVÉHO OLEJE Z POKRUTIN VE SPOTŘEBITELSKÝCH OBALECH
Tato metoda odběru vzorků se použije pro šarže olivového oleje nebo olivového oleje z pokrutin stočené do spotřebitelských obalů. Použijí se různé metody odběru vzorků v závislosti na tom, zda objem spotřebitelských obalů přesahuje 5 litrů či nikoli.
„Šarže“ se skládá z více prodejních jednotek, které jsou vyprodukovány, vyrobeny a zabaleny za takových okolností, že olej obsažený v každé prodejní jednotce se vzhledem ke všem svým analytickým charakteristikám považuje za homogenní. Individualizace šarže musí být provedena v souladu se směrnicí Evropského parlamentu a Rady 2011/91/EU ( 2 ).
„Dílem“ se rozumí množství oleje obsažené ve spotřebitelském obalu a odebrané z namátkou vybraného místa v dané šarži.
1. OBSAH DÍLČÍHO VZORKU
1.1 Spotřebitelský obal o objemu nejvýše 5 litrů
„Dílčím vzorkem“ u spotřebitelského obalu o objemu nejvýše 5 litrů se rozumí počet dílů odebraných ze šarže v souladu s tabulkou 1.
Tabulka 1
Minimální velikost dílčího vzorku musí obsahovat
|
V případě spotřebitelských obalů o objemu |
Dílčí vzorek musí obsahovat olej z |
|
(a) nejméně 1 litr |
(a) 1 spotřebitelského obalu; |
|
(b) méně než 1 litr |
(b) minimálního počtu obalů, jejichž celkový objem je nejméně 1 litr |
Počet obalů uvedených v tabulce 1, které tvoří dílčí vzorek, může každý členský stát zvýšit podle vlastní potřeby (např. organoleptické hodnocení provedené jinou laboratoří než tou, která provedla chemické analýzy, kontrolní analýza atd.).
1.2 Spotřebitelský obal o objemu vyšším než 5 litrů
„Dílčím vzorkem“ u spotřebitelského obalu o objemu vyšším než 5 litrů se rozumí reprezentativní část všech dílů získaných redukcí jejich množství a v souladu s tabulkou 2. Dílčí vzorek se musí skládat z různých příkladů.
„Příkladem“ dílčího vzorku se rozumí každý obal, ze kterého se dílčí vzorek skládá.
Tabulka 2
Minimální počet dílů, které je nutno vybrat
|
Počet balení v šarži |
Minimální počet dílů, které je nutno vybrat |
|
Do 10 |
1 |
|
11 až 150 |
2 |
|
151 až 500 |
3 |
|
501 až 1 500 |
4 |
|
1 501 až 2 500 |
5 |
|
> 2 500 na 1 000 obalů |
1 díl navíc |
Za účelem zmenšení objemu vzorků odebraných ze spotřebitelských obalů se obsah odebraných dílů pro přípravu dílčího vzorku homogenizuje. Části jednotlivých dílů se nalijí do společné nádoby pro homogenizaci mícháním tak, aby byly co nejlépe chráněny před přístupem vzduchu.
Obsah dílčího vzorku se musí nalít do série obalů s minimální kapacitou 1,0 litru, z nichž každý představuje příklad dílčího vzorku.
Počet dílčích vzorků může každý členský stát zvýšit podle vlastní potřeby (např. organoleptické hodnocení provedené jinou laboratoří, než která provedla chemické analýzy, kontrolní analýza atd.).
Každý obal musí být naplněn tak, aby byla vzduchová vrstva v jeho vrchní části co nejmenší, a pak se odpovídajícím způsobem uzavře a utěsní s cílem zabránit neoprávněné manipulaci s produktem.
Tyto příklady musí být označeny tak, aby byla zajištěna jejich správná identifikace.
2. ANALÝZY A VÝSLEDKY
2.1 Každý dílčí vzorek se musí dále rozdělit na laboratorní vzorky v souladu s bodem 2.5 normy EN ISO 5555 a podléhá těmto analýzám v pořadí uvedeném ve vývojovém diagramu v příloze Ib nebo v jakémkoli jiném náhodném pořadí.
2.2 Pokud všechny výsledky analýzy vyhovují charakteristikám deklarované kategorie olivového oleje, celá šarže se prohlásí za vyhovující.
Pokud jediný z výsledků analýzy nevyhovuje charakteristikám deklarované kategorie olivového oleje, celá šarže se prohlásí za nevyhovující.
3. OVĚŘENÍ KATEGORIE ŠARŽE
3.1 Příslušný orgán může za účelem ověření kategorie šarže zvýšit počet dílčích vzorků odebraných v různých bodech šarže podle následující tabulky:
Tabulka 3
Počet dílčích vzorků podle velikosti šarže
|
Velikost šarže (v litrech) |
Počet dílčích vzorků |
|
Menší než 7 500 |
2 |
|
Od 7 500 do méně než 25 000 |
3 |
|
Od 25 000 do méně než 75 000 |
4 |
|
Od 75 000 do méně než 125 000 |
5 |
|
125 000 a více |
6 + 1 na každých dalších 50 000 litrů |
Každý díl představující dílčí vzorek musí být odebrán z průběžného místa v šarži; je třeba zaznamenat umístění každého dílčího vzorku a jednoznačně ho identifikovat.
Každý dílčí vzorek musí být vytvořen v souladu s postupy uvedenými v bodech 1.1 a 1.2.
Každý dílčí vzorek je pak podroben analýzám uvedeným v čl. 2 odst. 1.
3.2 Pokud některý z výsledků analýzy nejméně jednoho dílčího vzorku podle čl. 2 odst. 1 nevyhovuje charakteristikám deklarované kategorie olivového oleje, prohlásí se celá šarže, z níž byl vzorek odebrán, za nevyhovující.
PŘÍLOHA Ib
VÝVOJOVÝ DIAGRAM PRO OVĚŘENÍ SOULADU VZORKU OLIVOVÉHO OLEJE S DEKLAROVANOU KATEGORIÍ
Všeobecná tabulka
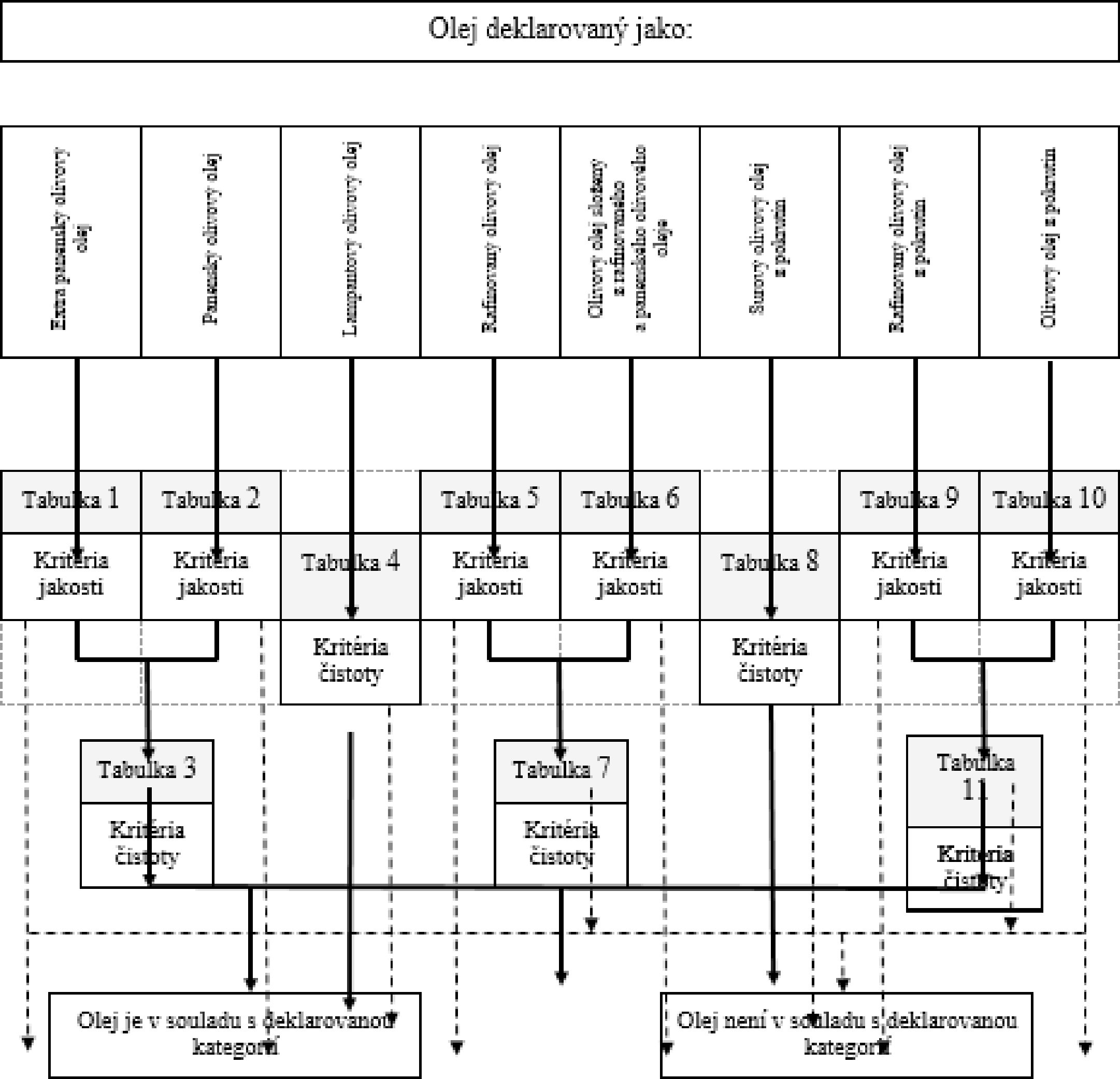
Tabulka 1 – Extra panenský olivový olej – kritéria jakosti
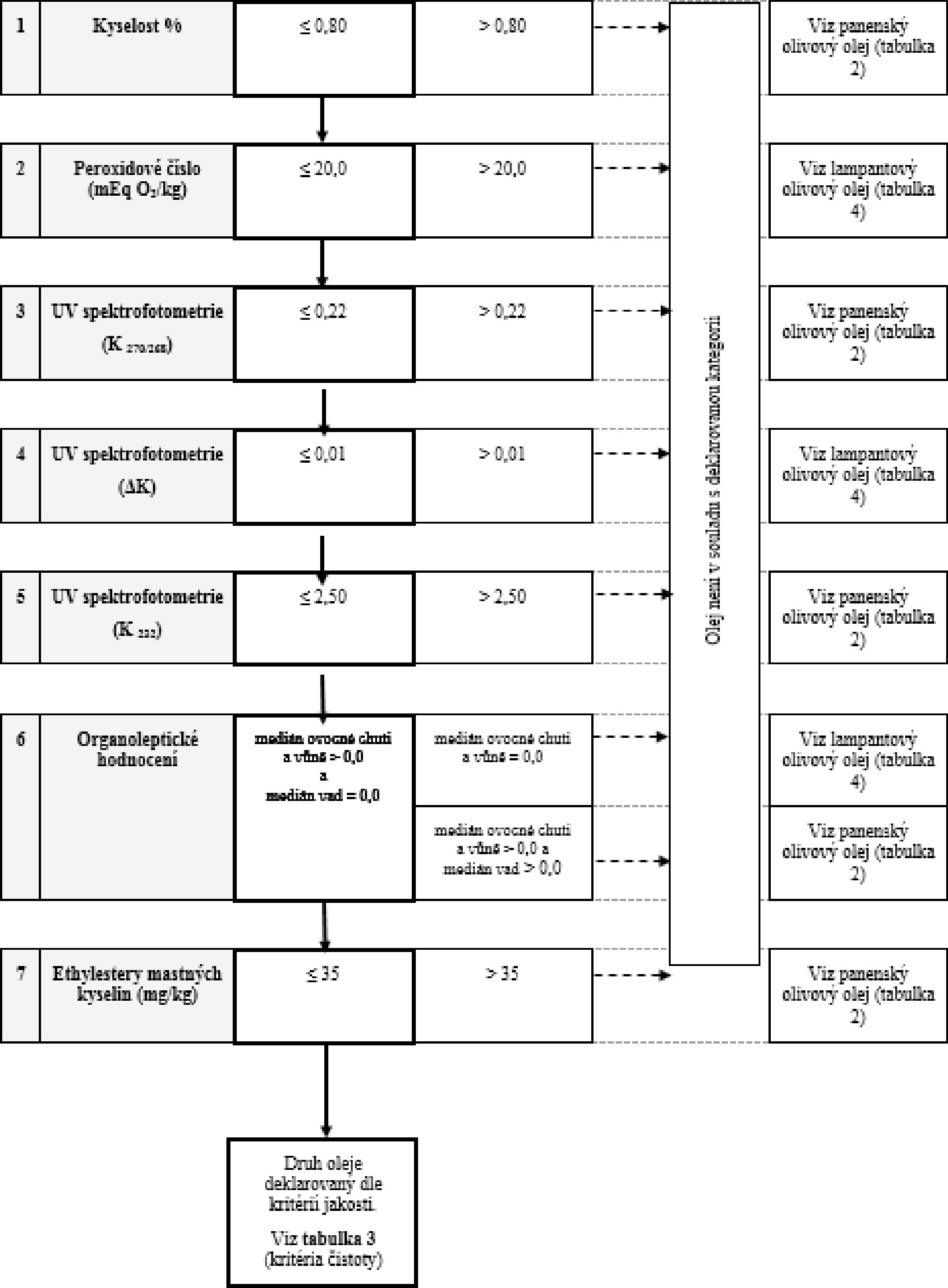
Tabulka 2 – Extra panenský olivový olej – kritéria jakosti
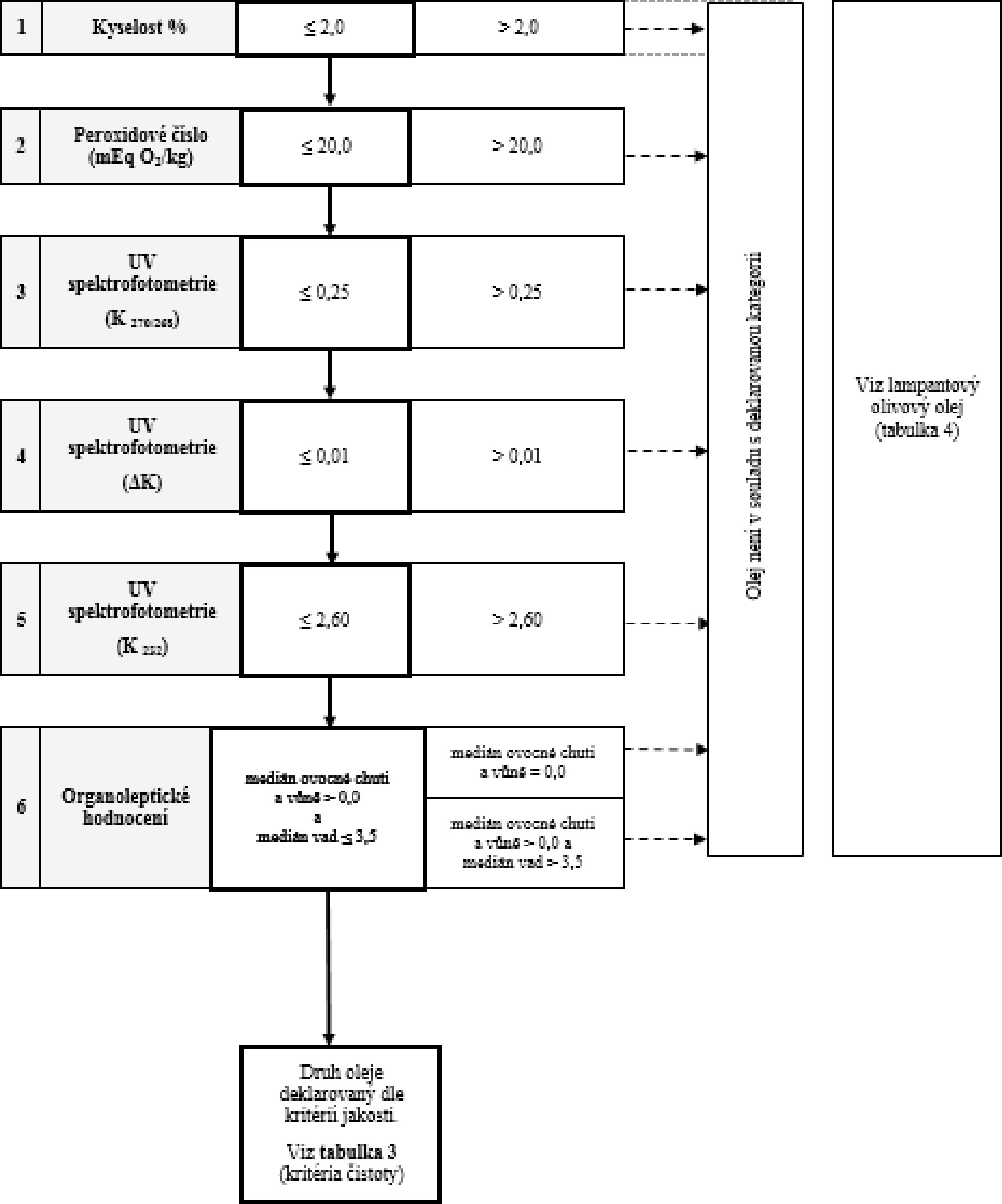
Tabulka 3 – Extra panenský olivový olej a panenský olivový olej – kritéria čistoty
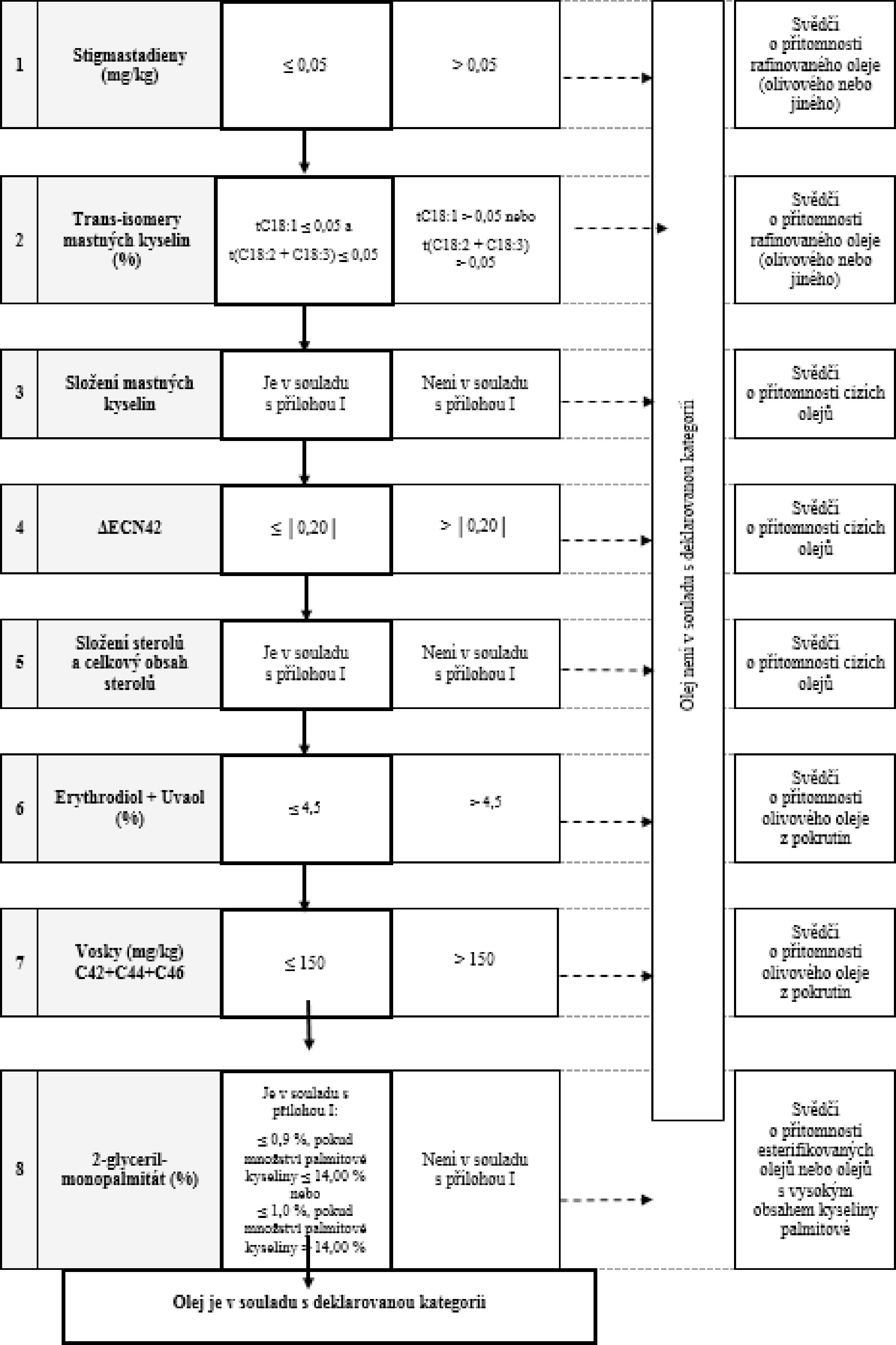
Tabulka 4 – Lampantový olivový olej – kritéria čistoty
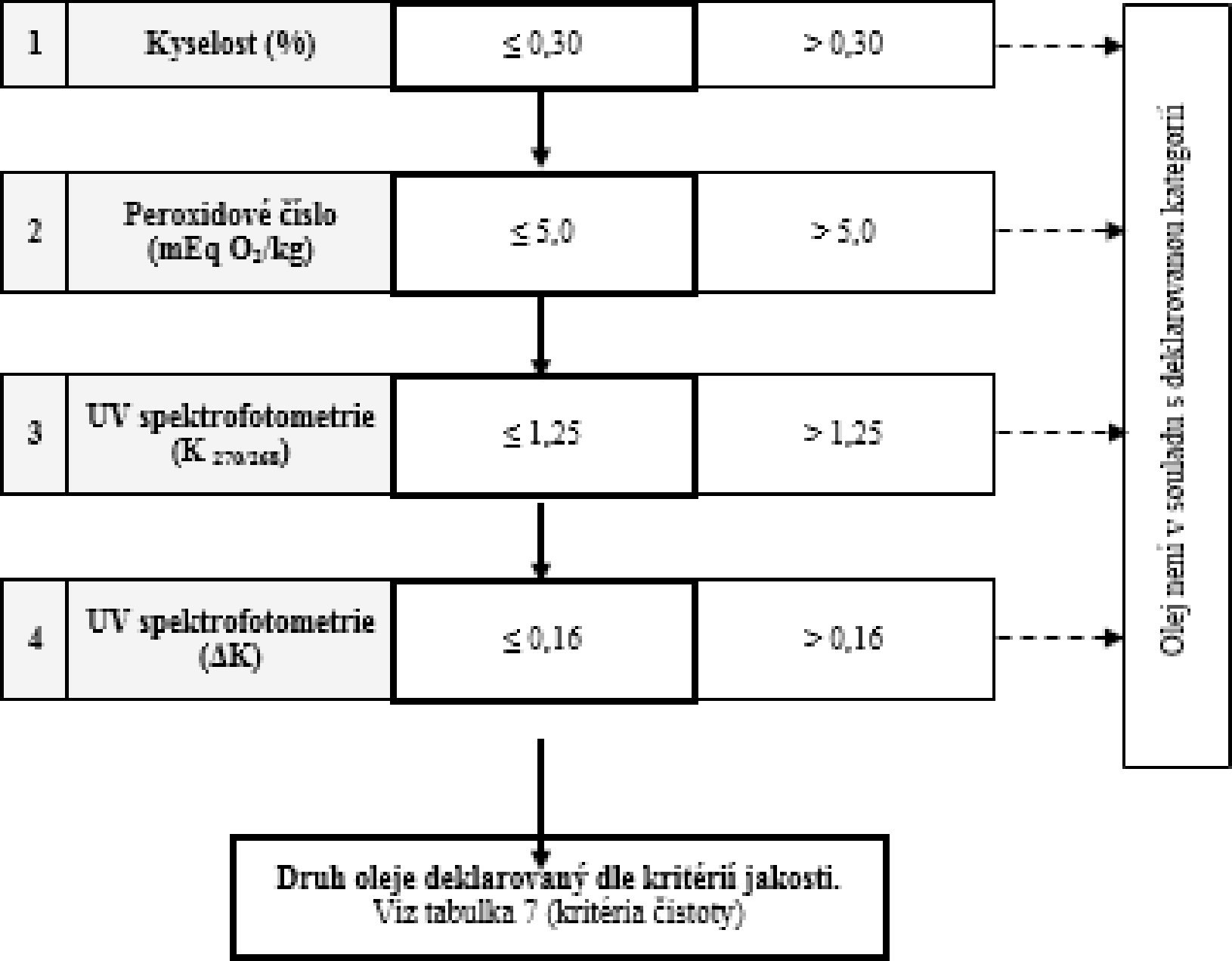
Tabulka 5 – Rafinovaný olivový olej – kritéria jakosti
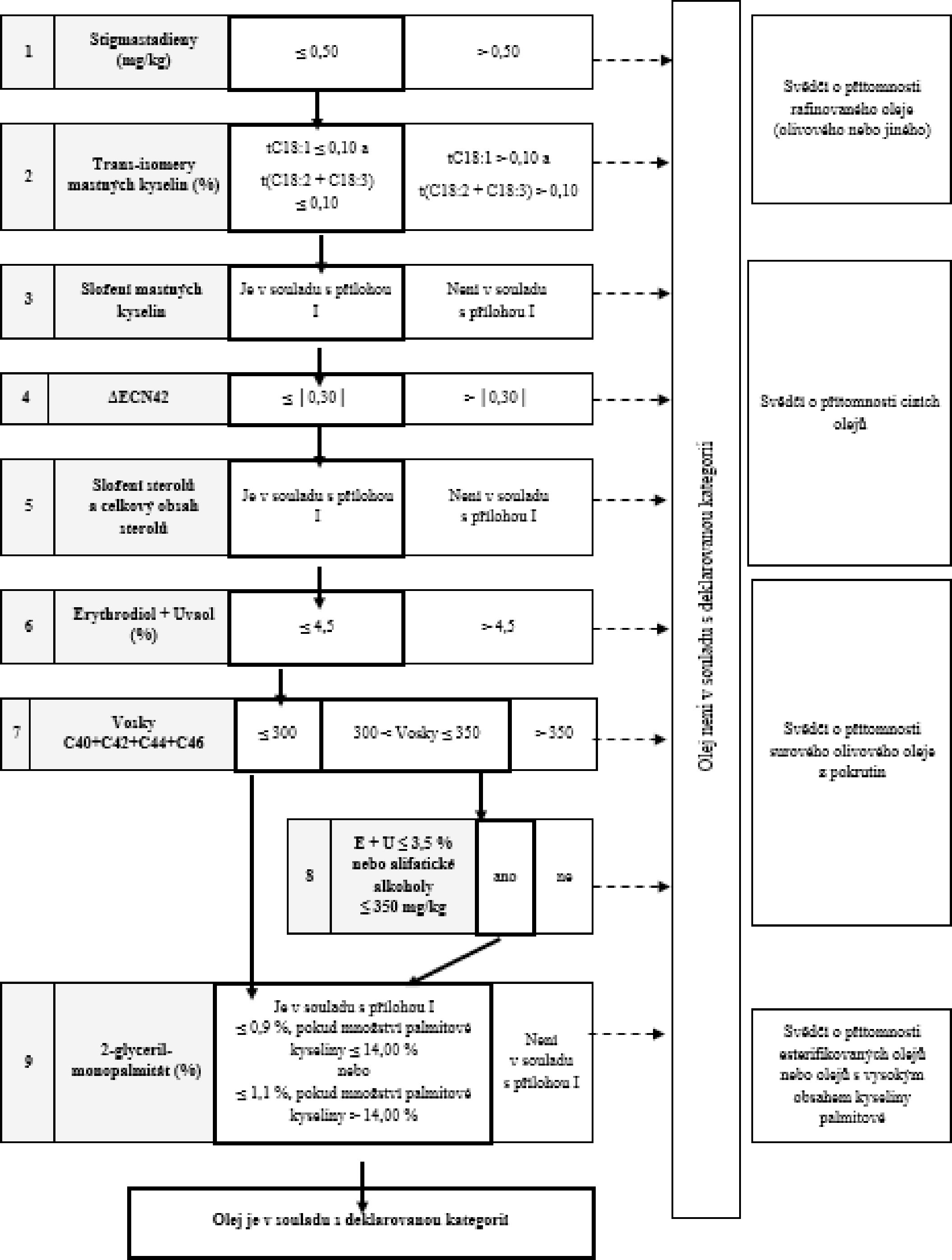
Tabulka 6 – Olivový olej (složený z rafinovaného a panenského olivového oleje) – kritéria jakosti
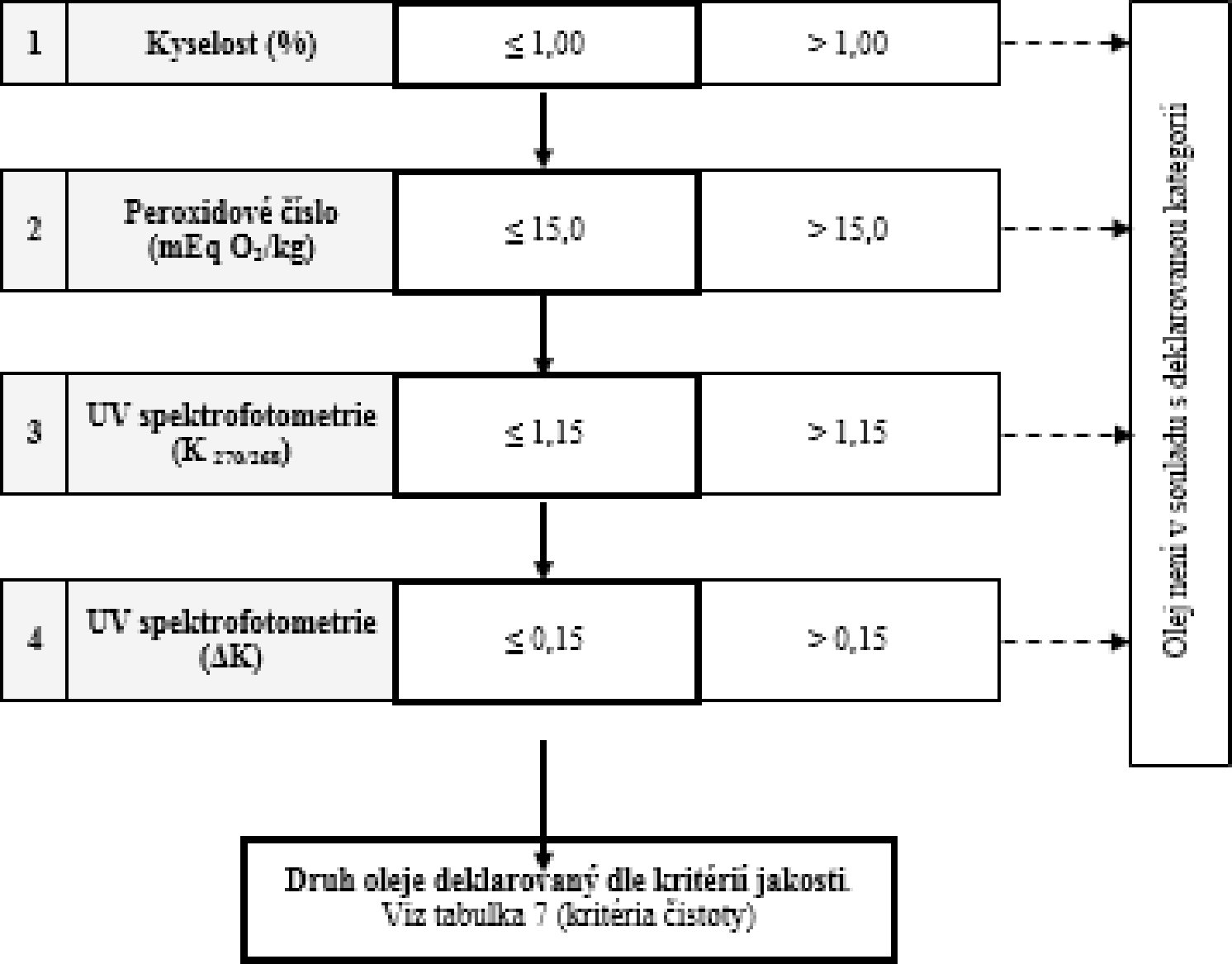
Tabulka 7 – Rafinovaný olivový olej a olivový olej složený z rafinovaného a panenského olivového oleje – kritéria čistoty
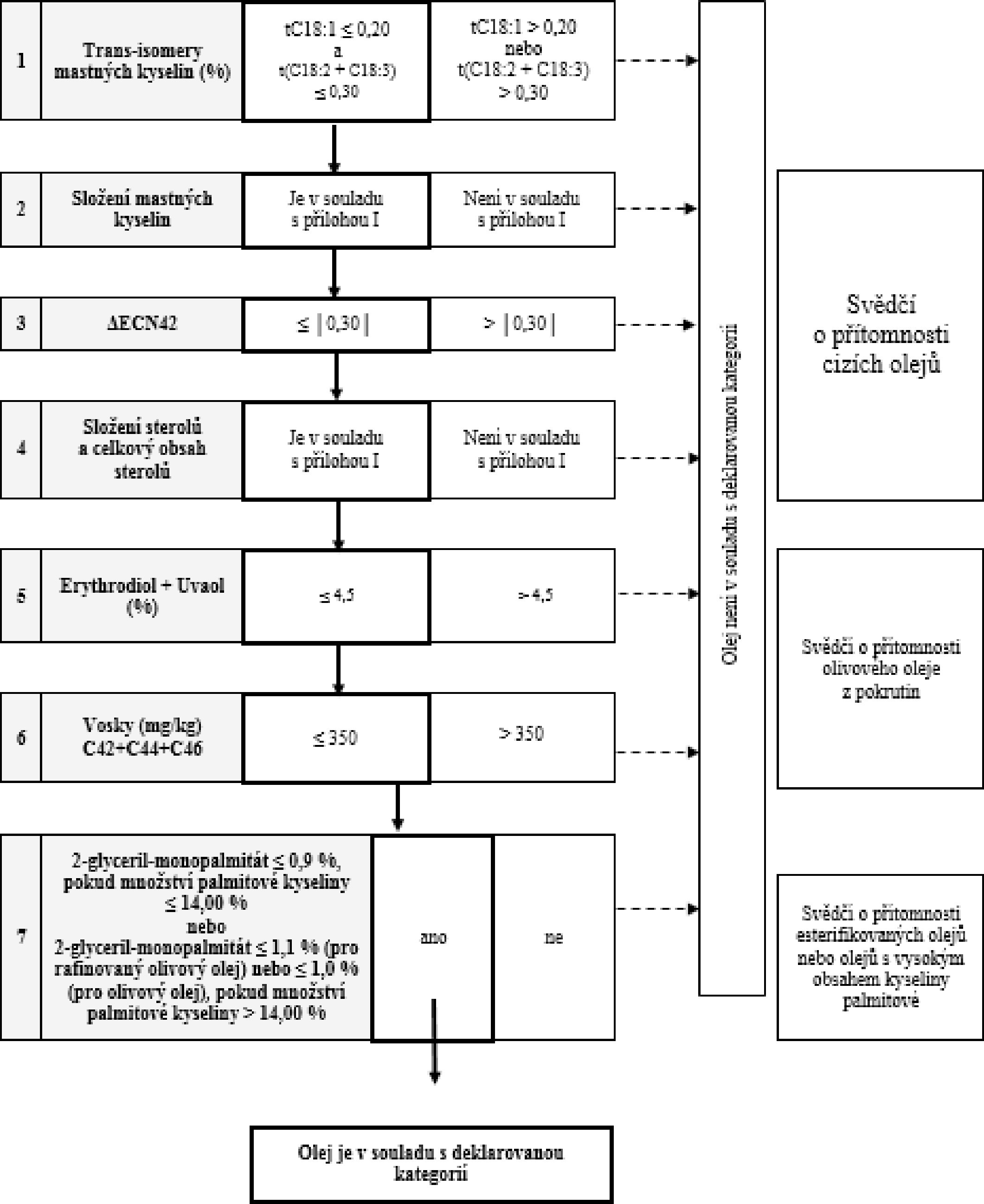
Tabulka 8 – Surový olivový olej z pokrutin – kritéria čistoty
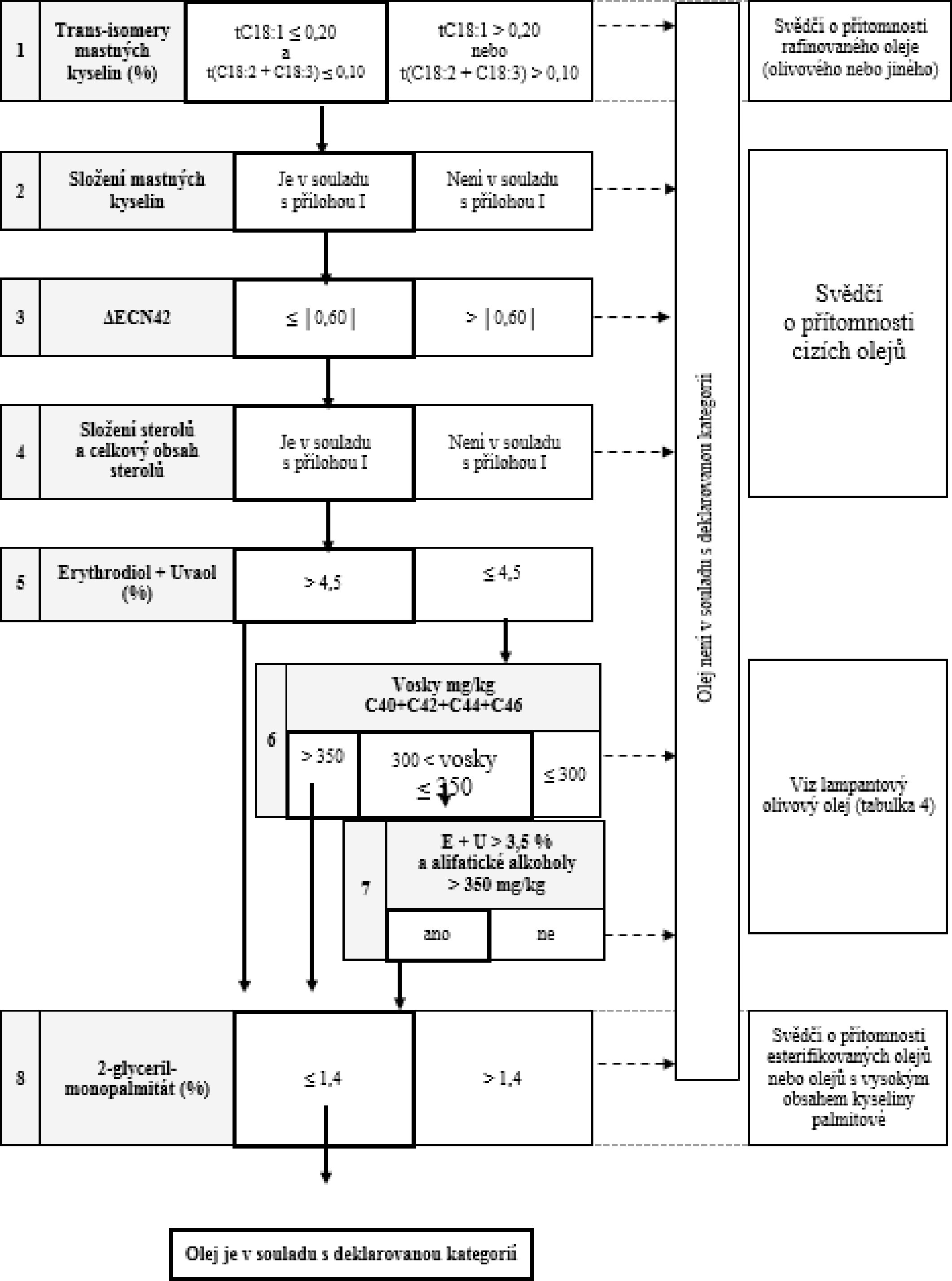
Tabulka 9 – Rafinovaný olivový olej z pokrutin – kritéria jakosti
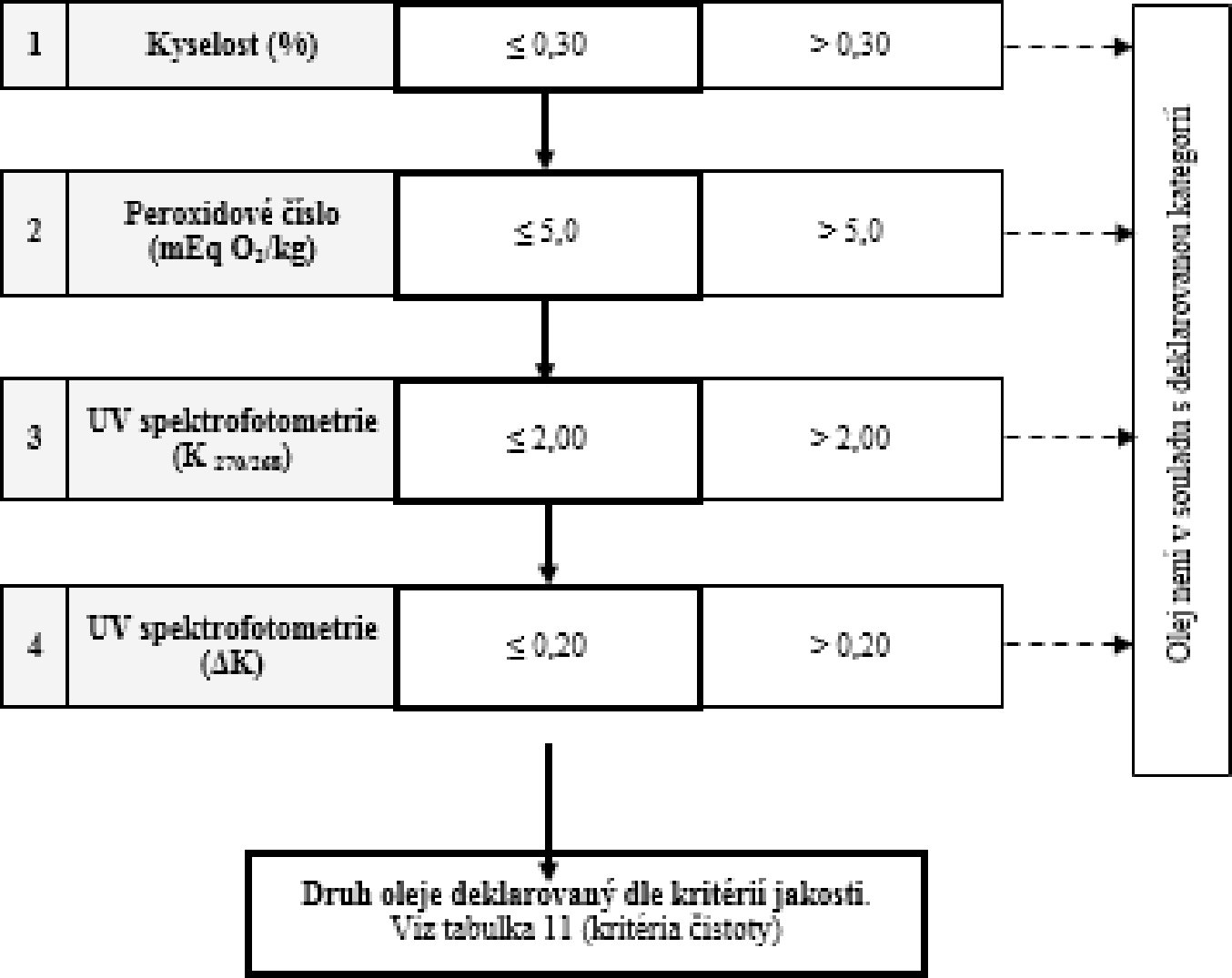
Tabulka 10 – Olivový olej z pokrutin – kritéria jakosti
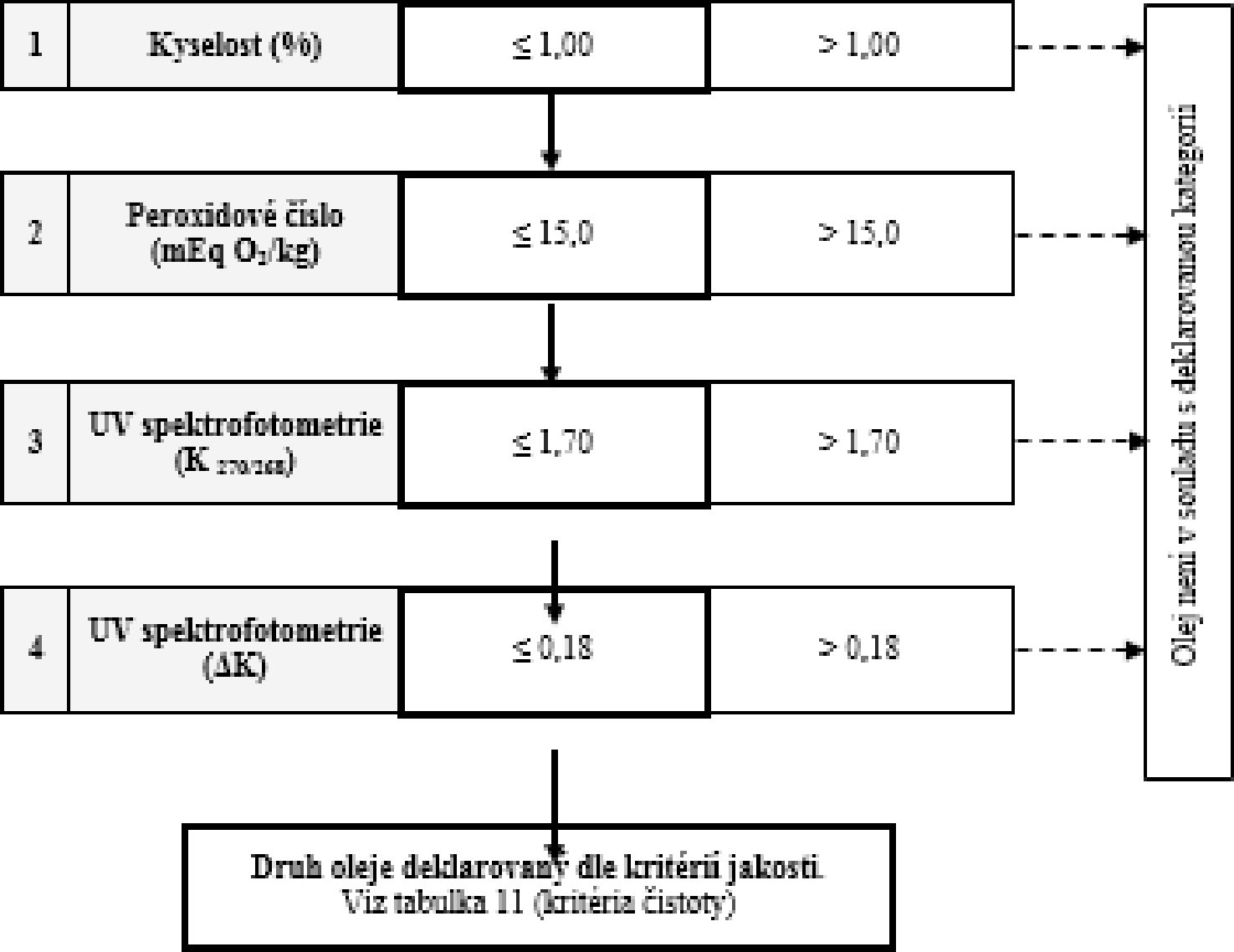
Tabulka 11 – Rafinovaný olivový olej z pokrutin a olivový olej z pokrutin – kritéria čistoty
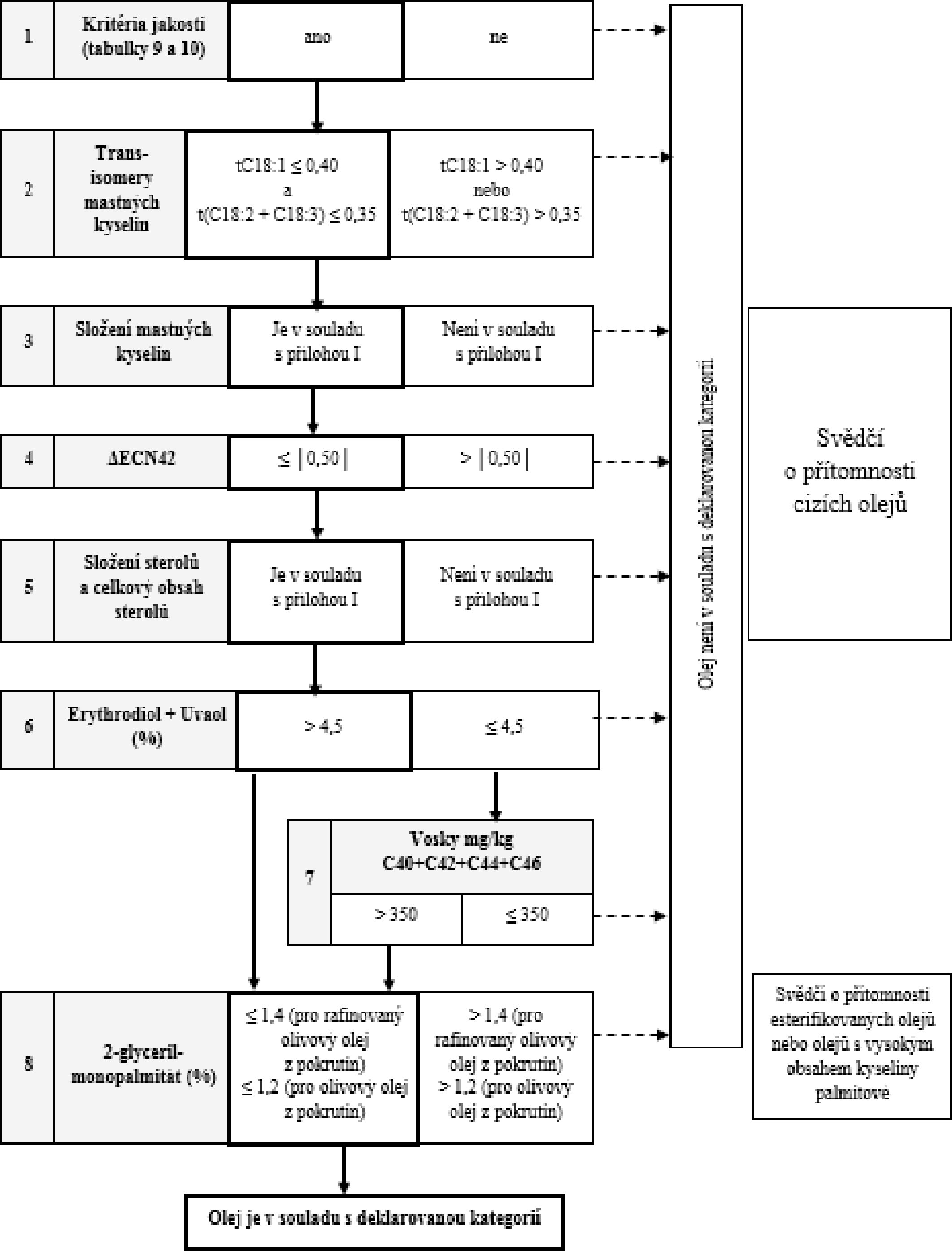
PŘÍLOHA II
STANOVENÍ VOLNÝCH MASTNÝCH KYSELIN, METODA ZA STUDENA
1. PŘEDMĚT A OBLAST POUŽITÍ
Tato metoda popisuje postup stanovování volných mastných kyselin v olivových olejích a olivových olejích z pokrutin. Obsah volných mastných kyselin je vyjádřen jako kyselost vypočtená v procentech kyseliny olejové.
2. PODSTATA METODY
Vzorek se rozpustí ve směsi rozpouštědel a přítomné volné mastné kyseliny se titrují roztokem hydroxidu draselného nebo hydroxidu sodného.
3. ČINIDLA
Všechna činidla by měla být čistoty p. a. a používaná voda by měla být destilovaná nebo odpovídající čistoty.
|
3.1 |
Směs diethyletheru a 95 % ethanolu 1:1 (V/V). Neutralizuje se těsně před použitím pomocí roztoku hydroxidu draselného (3.2) a přidá se 0,3 ml fenolftaleinového roztoku (3.3) na 100 ml směsi. Poznámka 1: Diethylether je vysoce hořlavý a může vytvářet výbušné peroxidy. Při jeho používání je třeba dbát zvýšené opatrnosti. Poznámka 2: Není-li možné použít diethylether, může se použít směsné rozpouštědlo obsahující ethanol a toluen. V případě potřeby může být ethanol nahrazen 2-propanolem. |
|
3.2 |
Hydroxid draselný nebo hydroxid sodný, titrační ethanolický nebo vodný roztok o koncentraci c(KOH) [nebo c(NaOH)] přibližně 0,1 mol/l, nebo v případě potřeby přibližně 0,5 mol/l. Dostupné jsou komerční roztoky. Přesná koncentrace roztoku hydroxidu draselného (nebo roztoku hydroxidu sodného) musí být známa a zkontrolována těsně před použitím. Použije se roztok připravený nejméně pět dnů před použitím a dekantovaný v láhvi z hnědého skla s gumovou zátkou. Roztok by měl být bezbarvý nebo pouze slabě zbarvený. Je-li při použití vodného roztoku hydroxidu draselného (nebo hydroxidu sodného) zpozorována separace fází, nahradí se vodný roztok roztokem ethanolickým. Poznámka 3: Stabilní bezbarvý roztok hydroxidu draselného (nebo hydroxidu sodného) je možné připravit takto: do varu se uvede 1 000 ml ethanolu nebo vody s 8 g hydroxidu draselného (nebo hydroxidu sodného) a 0,5 g hliníkových hoblin a nechá se jednu hodinu vařit pod zpětným chladičem. Okamžitě se destiluje. V destilátu se rozpustí požadované množství hydroxidu draselného (nebo hydroxidu sodného). Několik dnů se nechá stát a oddělí se čistá kapalina od usazeniny uhličitanu draselného (nebo uhličitanu sodného). Roztok je možné též připravit bez destilace takto: do 1 000 ml ethanolu (nebo vody) se přidají 4 ml aluminium-butylátu a směs se nechá několik dní stát. Odsazená kapalina se slije a v ní se rozpustí požadované množství hydroxidu draselného (nebo hydroxidu sodného). Roztok je připraven k použití. |
|
3.3 |
Fenolftalein, roztok o koncentraci 10 g/l v 95–96 % ethanolu (V/V), nebo alkalická modř 6B nebo thymolftalein, roztok o koncentraci 20 g/l v 95–96 % ethanolu (V/V). V případě silně zbarvených olejů se použije alkalická modř nebo thymolftalein. |
4. PŘÍSTROJE A POMŮCKY
Obvyklé laboratorní přístroje a pomůcky, mj.:
4.1 analytická váha;
4.2 Erlenmeyerova baňka na 250 ml;
4.3 byreta třídy A na 10 ml s hodnotou dílku 0,05 ml nebo odpovídající automatická byreta.
5. POSTUP
5.1 Příprava zkušebního vzorku.
Je-li vzorek zakalený, měl by být přefiltrován.
5.2 Zkušební vzorek
Hmotnost vzorku se řídí podle očekávané kyselosti (podle údajů v níže uvedené tabulce):
|
Očekávaná kyselost (kyselina olejová g/100 g) |
Hmotnost vzorku (g) |
Přesnost vážení (g) |
|
0 až 2 |
10 |
0,02 |
|
> 2 až 7,5 |
2,5 |
0,01 |
|
> 7,5 |
0,5 |
0,001 |
Vzorek se naváží v Erlenmeyerově baňce (4.2).
5.3 Stanovení
Vzorek (5.2) se rozpustí v 50 až 100 ml předem neutralizované směsi diethyletheru a ethanolu (3.1).
Titruje se za současného míchání roztokem hydroxidu draselného (nebo hydroxidu sodného) o koncentraci 0,1 mol/l (3.2) (viz poznámka 4), dokud nedojde k barevné změně indikátoru (zbarvení indikátoru musí trvat nejméně 10 sekund).
Poznámka 4: Pokud je k titraci potřeba více než 10 ml roztoku hydroxidu draselného (nebo hydroxidu sodného) o koncentraci 0,1 mol/l, použije se roztok o koncentraci 0,5 mol/l nebo se změní hmotnost vzorku podle očekávaného obsahu volných mastných kyselin a uvedené tabulky.
Poznámka 5: Pokud se během titrace roztok zakalí, přidá se dostačující množství rozpouštědel (3.1), aby byl roztok opět čirý.
Druhé stanovení se provádí pouze tehdy, je-li první výsledek vyšší než limit určený pro danou kategorii oleje.
6. VYJÁDŘENÍ VÝSLEDKŮ
Kyselost vyjádřená jako podíl kyseliny olejové v % hmotnostních se vypočítá podle vzorce:
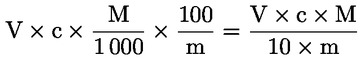
kde:
|
V |
= |
spotřeba titračního roztoku hydroxidu draselného (nebo hydroxidu sodného) v ml; |
|
c |
= |
přesná koncentrace titračního roztoku hydroxidu draselného (nebo hydroxidu sodného) v mol/l; |
|
M |
= |
282 g/mol, molární hmotnost kyseliny olejové v g na mol; |
|
m |
= |
hmotnost vzorku v g. |
Obsah kyseliny olejové se udává takto:
a) na dvě desetinná místa u hodnot 0 až 1 včetně;
b) na jedno desetinné místo u hodnot 1 až 100 včetně.
PŘÍLOHA III
STANOVENÍ PEROXIDOVÉHO ČÍSLA
1. Předmět
Tato příloha popisuje metodu pro stanovení peroxidového čísla živočišných a rostlinných olejů a tuků.
2. Definice
Peroxidové číslo je množství těchto látek ve vzorku, vyjádřené v miliekvivalentech aktivního kyslíku na kg, které oxidují jodid draselný za popsaných pracovních podmínek.
3. Podstata
Reakce vzorku s roztokem jodidu draselného v roztoku kyseliny octové a chloroformu. Titrace uvolněného jodu standardizovaným roztokem thiosíranu sodného.
4. Přístroje a pomůcky
Všechny použité předměty musí být zbaveny redukčních a oxidačních látek.
Poznámka 1: zábrusy nesmějí být mazány tukem.
4.1. Skleněná váženka o objemu 3 ml.
4.2. Baňky na 250 ml se zábrusy a zátkami, předem vysušené a naplněné čistým, suchým inertním plynem (dusíkem, nebo lépe oxidem uhličitým).
4.3. Byreta o objemu 5, 10 nebo 25 ml se stupnicí alespoň po 0,05 ml, pokud možno s automatickým seřízením na nulu, nebo rovnocenná automatická byreta.
4.4. Analytické váhy.
5. Reakční činidla
5.1. Chloroform čistoty p. a., zbavený kyslíku probubláváním proudem čistého suchého inertního plynu.
5.2. Ledová kyselina octová čistoty p. a., zbavená kyslíku probubláváním proudem čistého suchého inertního plynu.
5.3. Jodid draselný, nasycený vodný roztok, čerstvě připravený, zbavený jodu a jodičnanů. Přibližně 14 g jodidu draselného se nechá rozpustit v asi 10 ml vody při pokojové teplotě.
5.4. Thiosíran sodný, 0,01 mol/l (odpovídá 0,01 N), standardizovaný vodný roztok, připravený těsně před použitím.
Roztok thiosíranu sodného o koncentraci 0,01 mol/l se připraví denně před použitím ze standardního roztoku thiosíranu sodného o koncentraci 0,1 mol/l, nebo se určí přesná molarita. Ze zkušeností vyplývá, že stabilita je omezená a závisí na hodnotě pH a obsahu volného oxidu uhličitého. Pro roztok se použije pouze čerstvě převařená voda, případně se propláchne dusíkem.
Pro určení přesné molarity roztoku thiosíranu sodného se doporučuje následující postup:
S přesností na 0,001 g se do odměrné baňky (250 nebo 500 ml) odváží 0,27 až 0,33 g jodičnanu draselného (mKIO3) a rozpustí se v po rysku doplněné čerstvě převařené vodě (V2), která se předtím nechá ochladit na pokojovou teplotu. Pomocí pipety se 5 nebo 10 ml tohoto roztoku jodičnanu draselného (V1) přenese do Erlenmeyerovy baňky na 250 ml. Přidá se 60 ml čerstvě převařené vody, 5 ml kyseliny chlorovodíkové o koncentraci 4 mol/l a 25 až 50 mg jodidu draselného nebo 0,5 ml nasyceného roztoku jodidu draselného. Tento roztok se titruje roztokem thiosíranu sodného (V3) za účelem stanovení přesné molarity roztoku thiosíranu sodného.
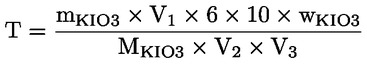
kde:
mKIO3 je hmotnost jodičnanu draselného v gramech
V1 je objem roztoku jodičnanu draselného v mililitrech (5 nebo 10 ml)
V2 je celkový objem roztoku jodičnanu draselného v mililitrech (250 nebo 500 ml)
V3 je objem roztoku thiosíranu sodného v mililitrech
wKIO3 je čistota jodičnanu draselného v g/100 g
MKIO3 je molekulová hmotnost jodičnanu draselného (214 g/mol)
T je přesná molarita roztoku thiosíranu sodného (mol/l).
5.5. Škrobový roztok, vodní disperze 10 g/l, čerstvě připravený z přírodního rozpustného škrobu. Lze použít i rovnocenná činidla.
6. Vzorek
Vzorek je nutno odebrat a skladovat v temnu a chladu ve zcela naplněných skleněných nádobách, hermeticky uzavřených zabroušenou skleněnou, nebo korkovou zátkou.
7. Postup
Stanovení musí být prováděno v rozptýleném denním světle, nebo při umělém osvětlení. Do skleněné váženky (4.1) nebo baňky (4.2) se s přesností na 0,001 g odváží vzorek o hmotnosti dle následující tabulky, podle předpokládaného peroxidového čísla:
|
Předpokládané peroxidové číslo (meq) |
Hmotnost zkušebního vzorku (g) |
|
0 až 12 |
5,0 až 2,0 |
|
12 až 20 |
2,0 až 1,2 |
|
20 až 30 |
1,2 až 0,8 |
|
30 až 50 |
0,8 až 0,5 |
|
50 až 90 |
0,5 až 0,3 |
Z baňky (4.2) se vyjme zátka a vloží se do ní váženka obsahující vzorek. Přidá se 10 ml chloroformu (5.1). Vzorek se rychle rozpustí zamícháním. Přidá se 15 ml kyseliny octové (5.2) a potom 1 ml roztoku jodidu draselného (5.3). Baňka se ihned uzavře, protřepá se po dobu jedné minuty a nechá se stát přesně 5 minut v temnu při teplotě 15 až 25 °C.
Přidá se přibližně 75 ml destilované vody. Uvolněný jod se za použití škrobového roztoku (5.5) jako indikátoru za silného protřepávání titruje roztokem thiosíranu sodného (5.4).
Z jednoho zkušebního vzorku se provádějí dvě stanovení.
Spolu s vlastním stanovením se provádí slepý pokus. Jestliže výsledky slepého pokusu přesáhnou 0,05 ml roztoku thiosíranu sodného o koncentraci 0,01 N (5.4), je nutno vyměnit znečištěná činidla.
8. Vyjádření výsledků
Peroxidové číslo (PV) vyjádřené v miliekvivalentech aktivního kyslíku na kilogram se vypočítá podle vzorce:
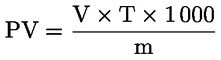
kde:
|
V |
= |
počet ml standardizovaného roztoku thiosíranu sodného (5.4) použitého pro stanovení, korigovaný s přihlédnutím k slepému pokusu. |
|
T |
= |
přesná molarita použitého roztoku thiosíranu sodného (5.4) v mol/l. |
|
m |
= |
hmotnost zkušebního vzorku v g. |
Za výsledek se považuje aritmetický průměr dvou stanovení.
Výsledek se zaokrouhlí na jedno desetinné místo.
PŘÍLOHA IV
STANOVENÍ OBSAHU VOSKU POMOCÍ KAPILÁRNÍ PLYNOVÉ CHROMATOGRAFIE
1. PŘEDMĚT
Tato metoda popisuje postup stanovení obsahu vosku v olivových olejích. Vosky se dělí podle počtu atomů uhlíku. Tuto metodu lze použít zejména k rozlišení mezi olivovým olejem získaným lisováním a extrakcí (olivový olej z pokrutin).
2. PODSTATA METODY
K tuku nebo oleji se přimísí vhodný vnitřní standard a poté se provádí chromatografická frakční destilace na hydratované silikagelové koloně. Získaná frakce eluovaná nejprve při testovacích podmínkách (jejíž polarita je menší než polarita triglyceridů) se přímo analyzuje pomocí kapilární plynové chromatografie.
3. PŘÍSTROJE A POMŮCKY
|
3.1 |
Kónická 25ml baňka. |
|
3.2 |
Skleněná chromatografická plynová kolona o vnitřním průměru 15 mm a délce 30 až 40 cm vybavená ventilem. |
|
3.3 |
Plynový chromatograf vhodný pro použití s kapilární kolonou a vybavený přímým vstřikováním, obsahující:
|
|
3.4 |
Mikrostříkačka na 10 μl pro přímý vstřik do kolony opatřená tvrzenou jehlou. |
|
3.5 |
Elektrický vibrátor. |
|
3.6 |
Rotační odpařovač. |
|
3.7 |
Muflová pec. |
|
3.8 |
Analytické váhy s přesností měření ± 0,1 mg. |
|
3.9 |
Běžné laboratorní skleněné nádoby. |
4. REAKČNÍ ČINIDLA
|
4.1 |
Silikagel s velikostí zrna od 60 do 200 μm. Silikagel se umístí alespoň na čtyři hodiny do pece o teplotě 500 °C. Nechá se ochladit a přidají se 2 % vody v poměru k odebranému množství silikagelu. Řádným protřepáním se směs homogenizuje. Před použitím se ponechá nejméně 12 hodin ve tmě. |
|
4.2 |
n-hexan pro chromatografii. |
|
4.3 |
Diethylether pro chromatografii. |
|
4.4 |
n-heptan pro chromatografii. |
|
4.5 |
Standardní roztok 0,1 % (m/V) laurylarachidátu v hexanu (vnitřní standard). (Lze též použít palmityl palmitát a myristyl stearát.)
|
|
4.6 |
Nosný plyn: vodík nebo čisté helium pro plynovou chromatografii. |
|
4.7 |
Pomocné plyny: — vodík, čistý pro plynovou chromatografii, — vzduch, čistý pro plynovou chromatografii. |
5. POSTUP
5.1 Příprava chromatografické kolony
Provede se suspenze 15 g silikagelu (4.1) v n-hexanu (4.2) a zavede se do kolony (3.2). Po spontánní sedimentaci se tato dokončí pomocí elektrického vibrátoru (3.5), aby byla chromatografická vrstva co nejhomogennější. Provede se perkolace 30 ml n-hexanu za účelem odstranění případných nečistot. Pomocí vah (3.8) se naváží přesně 500 mg vzorku do baňky (3.1) a přidá se vhodné množství vnitřního standardu (4.5) v závislosti na předpokládaném obsahu vosku. Např. 0,1 mg laurylu arachidátu v případě olivového oleje a 0,25 až 0,5 mg v případě olivového oleje z pokrutin. Získaný vzorek se za pomoci dvou dávek 2 ml n-hexanu (4.2) převede do chromatografické kolony.
Umožní se, aby hladina rozpouštědla poklesla tak, aby byla 1 mm nad horní úrovní absorbentu, a poté se provede perkolace 70 ml doplňkového n-hexanu za účelem odstranění n-alkanů, které jsou přirozeně přítomny. Poté se zahájí chromatografické eluování, odejme se 180 ml směsi n-hexanu/diethyletheru v poměru 99:1 při průtoku přibližně 15 kapek za 10 sekund. Eluování vzorku se musí provést při teplotě okolí 22 °C ± 4.
Poznámky:
— Směs n-hexanu s diethyletherem v poměru 99:1 se musí připravovat každý den.
— Aby bylo možno vizuálně kontrolovat správné eluování vosků, je možno do roztoku vzorku přidat 100 μl 1 % Sudanu v eluovací směsi. Jelikož má barvivo přechodnou vazbu mezi vosky a triglyceridy, je třeba eluci přerušit, jakmile zabarvení dosáhne spodní části chromatografické kolony, neboť všechny vosky byly eluovány.
Získaná frakce se usuší v rotačním odpařovači (3.6), až je téměř všechno rozpouštědlo odstraněno. Poslední 2 ml rozpouštědla se odstraní za pomoci slabého proudu dusíku; poté se přidá 2–4 ml n-heptanu.
5.2 Analýza plynovou chromatografií
5.2.1 Přípravné práce
Kolona se spojí s plynovým chromatografem (3.3), vstupní port se připojí ke kolonovému systému (on-column system) a výstupní port k detekčnímu činidlu. Poté se plynový chromatograf překontroluje (těsnost plynových vedení, funkce detekčního činidla a záznamníku atd.).
Kapilární kolony, které mají být použity poprvé, je nutno nejprve kondicionovat. Kapilární kolonou se nechá protékat malé množství nosného plynu, poté se zapne plynový chromatograf a nechá se postupně zahřívat. Postupně se zahřívá, dokud není zhruba po 4 hodinách dosaženo teploty 350 °C. Tato teplota se udržuje nejméně dvě hodiny a poté se provede regulace pracovních podmínek (regulace průtoku plynu, zapálení plamene, připojení elektronického zapisovače (3.3.4), nastavení teploty kolonové komory, detekčního činidla atd.) a signál se nastaví na citlivost, která činí nejméně dvojnásobek nejvyšší úrovně uvažované pro provedení analýzy. Základní linie záznamu musí být rovná, bez jakýchkoli píků nebo driftů.
Záporné drifty jsou známkou nedokonalé těsnosti spojů kolony, zatímco kladné svědčí o nedostatečné kondicionaci kolony.
5.2.2 Výběr pracovních podmínek
Pracovní podmínky, které je třeba všeobecně dodržovat, jsou tyto:
— teplota kolony:
—
|
|
20 °C/min |
|
5 °C/min |
|
20 °C/min |
|
|
nejdříve 80 °C (1′) |
→ |
240 °C |
→ |
325 °C (6′) |
→ |
340 °C (10′) |
— teplota detekčního činidla: 350 °C,
— množství nastříknuté látky: 1 μl roztoku n-heptanu (2–4 ml),
— nosný plyn: optimální lineární rychlost helia nebo vodíku u vybraného plynu (viz dodatek),
— citlivost přístroje: má odpovídat níže uvedeným podmínkám:
Tyto podmínky mohou být upraveny podle charakteristik kolony a plynového chromatografu s cílem oddělit všechny vosky, dosáhnout dostatečného rozpuštění píků (viz obrázek) a retenčního času vnitřního standardu C32, který musí být 18 ± 3 minuty. Nejreprezentativnější pík vosku musí dosáhnout nejméně 60 % z plného rozsahu.
Parametry pro integraci píků se nastaví tak, aby došlo ke správnému vyhodnocení ploch uvažovaných píků.
Poznámka:Vzhledem k tomu, že konečná teplota je vysoká, připouští se kladný drift, který nesmí přesáhnout 10 % z plného rozsahu.
5.3 Provedení analýzy
Do mikrostříkačky na 10 μl se natáhne 1 μl roztoku; píst mikrostříkačky se povytáhne tak, aby se vyprázdnila jehla. Jehla se zavede přes septum vstřikové komory a přibližně po jedné nebo dvou sekundách se rychle nastříkne roztok; přibližně po pěti sekundách se jehla pomalu vytáhne.
Zaznamenávání je prováděno, dokud vosky nejsou zcela eluovány.
Základní linie záznamu musí vždy odpovídat požadavkům.
5.4 Identifikace píků
Identifikace jednotlivých píků se provádí podle retenčních časů a porovnáním se směsmi vosků, jejichž retenční časy jsou známé a které byly analyzovány za stejných podmínek.
Chromatogram vosků panenského olivového oleje je znázorněn na obrázku.
5.5 Kvantitativní vyhodnocení
Pomocí integrátoru se vypočítají plochy píků odpovídajících vnitřnímu standardu a alifatickým esterům C40 až C46.
Obsah vosku jednotlivých esterů vyjádřený v mg/kg tukové látky se vypočítá podle vzorce:
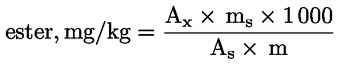
kde:
|
Ax |
= |
plocha píku každého esteru v milimetrech čtverečních; |
|
As |
= |
plocha píku vnitřního standardu v milimetrech čtverečních; |
|
ms |
= |
hmotnost přidaného vnitřního standardu v miligramech; |
|
m |
= |
hmotnost vzorku použitého pro stanovení v gramech. |
6. VYJÁDŘENÍ VÝSLEDKŮ
Uvádí se souhrn obsahů jednotlivých vosků C40 až C46v mg/kg tukové látky (ppm).
Poznámka:Sloučeniny, které je třeba kvantifikovat, se stanoví vzhledem k píkům esterů s počtem uhlíků mezi C40 a C46 podle příkladu chromatogramu vosků olivového oleje uvedeného na následujícím obrázku. Pokud se ester C46 objeví dvakrát, doporučuje se pro jeho identifikaci provést analýzu frakce vosků olivového oleje z pokrutin, v níž je pík C46 snadno rozpoznatelný, jelikož jasně převažuje.
Výsledky se vyjádří s přesností na jedno desetinné místo.
Obrázek
Chromatogram vosků olivového oleje ( 3 )
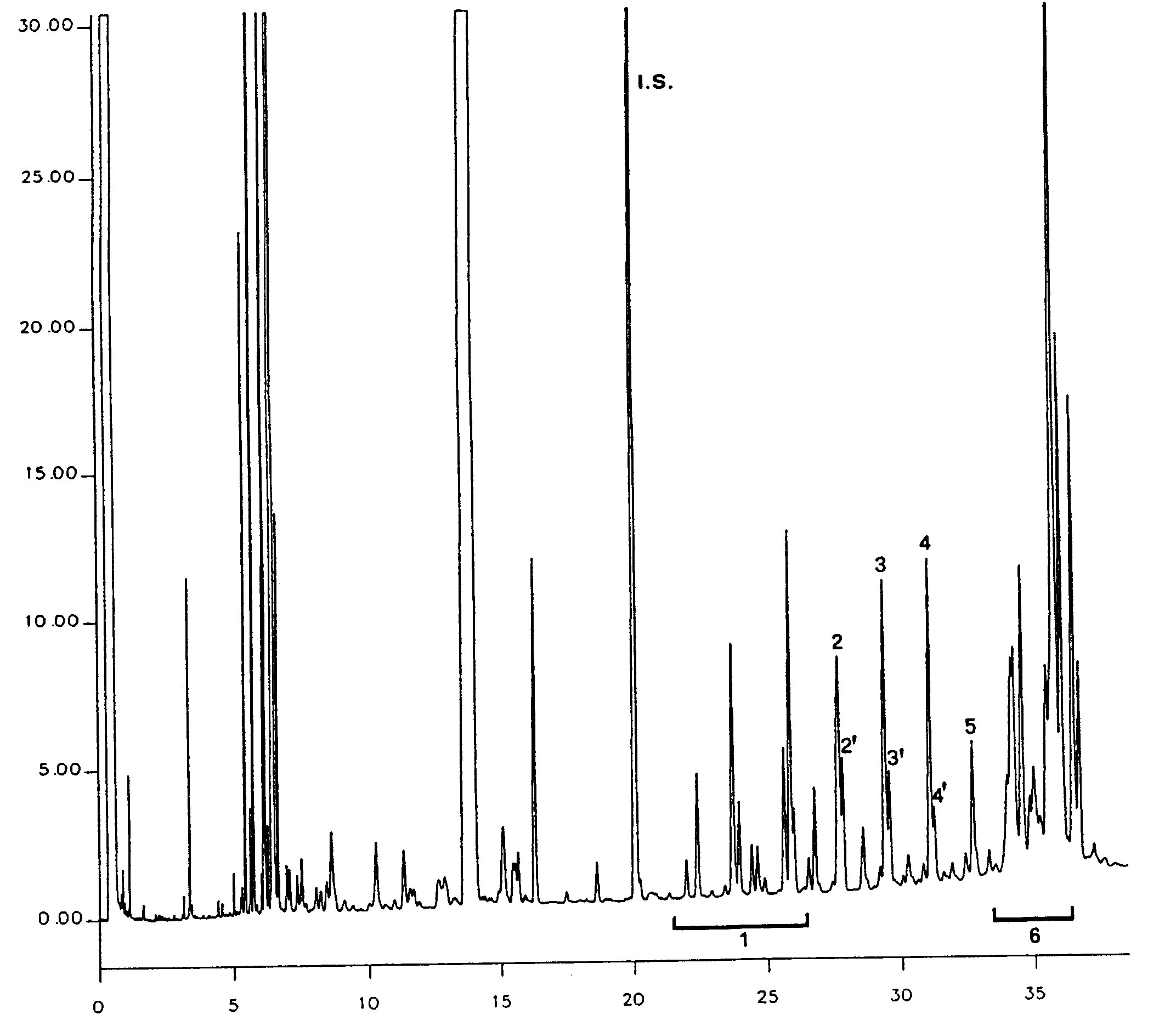
Vysvětlivky:
|
I.S. |
= |
Lauryl arachidátu; |
|
1 |
= |
Estery diterpenu; |
|
2 + 2′ |
= |
Estery C40; |
|
3 + 3′ |
= |
Estery C42; |
|
4 + 4′ |
= |
Estery C44; |
|
5 |
= |
Estery C46; |
|
6 |
= |
Estery sterolů a triterpenický alkohol. |
DODATEK
Stanovení lineární rychlosti plynu
Do plynového chromatografu nastaveného na normální pracovní podmínky se vstříkne 1 až 3 μl methanu nebo propanu. Změří se doba průchodu plynu kolonou od počátku vstřiku do okamžiku eluce píků (tM).
Lineární rychlost proudění v cm/s je dána poměrem L/tM, kde L je délka kolony v cm a (tM) je čas změřený v sekundách.
▼M32 —————
▼M26 —————
PŘÍLOHA VII
STANOVENÍ PROCENTUÁLNÍHO PODÍLU 2-GLYCERIL MONOPALMITÁTU
1. PŘEDMĚT A ROZSAH POUŽITÍ
Tato metoda popisuje analytický postup stanovení procentuálního podílu kyseliny palmitové ve 2. pozici triglyceridů hodnocením 2-glyceril monopalmitátu.
Tato metoda se použije na tekuté rostlinné oleje při teplotě okolí (20 °C).
2. PODSTATA METODY
Po přípravě se vzorek oleje podrobí účinku pankreatické lipázy. Parciální a specifická hydrolýza v pozici 1 a 3 molekuly triglyceridu způsobuje, že monoglyceridy se objeví na 2. pozici. Procentuální podíl 2-glyceril monopalmitátu v monoglycerické frakci se stanoví po silylaci pomocí kapilární plynové chromatografie.
3. PŘÍSTROJE A POMŮCKY
|
3.1 |
Kónická 25ml baňka. |
|
3.2 |
Kádinky na 100, 250 a 300 ml. |
|
3.3 |
Skleněná chromatografická kolona o vnitřním průměru 21–23 mm a délce 400 mm s vložkou ze sintrovaného skla a kohoutem. |
|
3.4 |
Nedělené pipety na 10, 50, 100 a 200 ml. |
|
3.5 |
Baňky na 100 a 250 ml. |
|
3.6 |
Rotační odpařovač. |
|
3.7 |
Centrifugační zkumavky s konickým dnem na 10 ml se zabroušenou skleněnou zátkou. |
|
3.8 |
Odstředivka pro zkumavky o obsahu 10 a 100 ml. |
|
3.9 |
Termostat umožňující udržet teplotu na 40 °C s přesností na 0,5 °C. |
|
3.10 |
Dělené pipety na 1 a 2 ml. |
|
3.11 |
Hypodermická stříkačka na 1 ml. |
|
3.12 |
Mikrostříkačka na 100 μl. |
|
3.13 |
Nálevka na 1 000 ml. |
|
3.14 |
Plynový chromatograf pro kapilární kolony se studeným nástřikovým zařízením on column pro přímé zavádění vzorku do kolony a s termostatem, který je schopen udržovat žádanou teplotu s přesností na 1 °C. |
|
3.15 |
Studené nástřikové zařízení on column pro přímé zavádění vzorku do kolony. |
|
3.16 |
Plameno-ionizační detektor a elektrometr. |
|
3.17 |
Integrátor se zapisovačem, vhodný pro elektrometr, s rychlostí odezvy nižší než 1 sekundu a s nastavitelnou rychlostí posunu papíru. |
|
3.18 |
Kapilární kolona, ze skla nebo taveného křemene, dlouhá 8 až 12 m s vnitřním průměrem 0,25 až 0,32 mm, obsahující methylpolysiloxan nebo 5 %-fenyl-methylpolysiloxan, o tloušťce 0,10 až 0,30 μm, kterou je možno použít při 370 °C. |
|
3.19 |
Mikrostříkačka na 10 μl pro přímý vstřik do kolony opatřená tvrzenou jehlou o délce min. 7,5 cm. |
4. CHEMIKÁLIE
|
4.1 |
Silikagel s velikostí zrna od 0,063 až po 0,200 mm (70/280 mesh), který se připraví takto: silikagel se položí do porcelánové misky, suší se v sušárně po dobu 4 hodin při teplotě 160 °C, pak se nechá ochladit v exsikátoru při pokojové teplotě. Přidá se voda o objemu shodném s 5 % váhy silikagelu takto: do 500 ml baňky se naváží 152 g silikagelu a přidá se 8 g destilované vody, baňka se zazátkuje a jemně protřepá, aby se voda rozložila rovnoměrně. Nechá se odstát na alespoň 12 hodin před použitím. |
|
4.2 |
n-hexan pro chromatografii. Hexan lze nahradit isooktanem (2,2,4-trimethylpentanem pro chromatografii) za předpokladu, že je dosaženo srovnatelných hodnot přesnosti. |
|
4.3 |
Isopropanol. |
|
4.4 |
Isopropanol, vodný roztok 1/1 (V/V). |
|
4.5 |
Pankreatická lipáza. Použitá lipáza musí mít aktivitu mezi 2,0 A 10 jednotkami lipázy na mg (v obchodě existují pankreatické lipázy s aktivitou mezi 2 a 10 jednotkami na mg enzymu). |
|
4.6 |
Tlumivý roztok trihydroxy-methyl-aminomethanu: 1 M vodný roztok upravený na pH 8 (kontrola potenciometrem) přidáním koncentrované kyseliny chlorovodíkové (1/1 V/V). |
|
4.7 |
Cholát sodný (enzymatické kvality), vodný roztok o koncentraci 0,1 % (tento roztok se musí použít do 15 dnů po přípravě). |
|
4.8 |
Chlorid vápenatý, vodný roztok o koncentraci 22 %. |
|
4.9 |
Diethylether pro chromatografii. |
|
4.10 |
Vyvíjecí rozpouštědlo: směs n-hexanu/diethyletheru (87/13) (V/V). |
|
4.11 |
Hydroxid sodný, roztok o koncentraci 12 % hmotnostních. |
|
4.12 |
Fenolftalein, 1 % roztok ethanolu. |
|
4.13 |
Nosný plyn: vodík nebo helium, pro plynovou chromatografii. |
|
4.14 |
Pomocné plyny: – vodík o minimální čistotě 99 %, bez vlhkosti a organických látek, a vzduch, pro plynovou chromatografii stejná čistota. |
|
4.15 |
Silanizační činidla: směs pyridinu/hexametyldisilazanu, trimetylchlorosilanu 9/3/1 (V/V/V) (Roztoky připravené k použití jsou na trhu. Mohou se použít jiná silylační činidla, zejména bis-trimethylsilyl trifluoracetamid + 1 % trimetylchlorosilan, zředěná stejným objemem bezvodého pyridinu.) |
|
4.16 |
Referenční vzorky: čisté monoglyceridy nebo směsi, které mají podobné procentuální složení jako vzorek. |
5. POSTUP
5.1 Příprava vzorku
|
5.1.1 |
Oleje, které mají volnou kyselost nižší než 3 %, nemusí být neutralizovány před chromatografií na silikagelové koloně. Oleje, které mají volnou kyselost vyšší než 3 %, musí být neutralizovány podle bodu 5.1.1.1.
|
|
5.1.2 |
Do kónické 25 ml baňky (3.1) se převede olej připravený podle výše uvedeného způsobu a rozpustí se ve vyvíjecím rozpouštědle (10 ml, 4.10). Roztok se nechá odstát na alespoň 15 minut před chromatografií na silikagelové koloně. Pokud je roztok kalný, odstředí se, aby byly zabezpečeny optimální podmínky pro chromatografii. (Mohou se použít kartony silikagelu SPE na 500 mg, které jsou připraveny k použití.) |
|
5.1.3 |
Příprava chromatografické kolony Do kolony se vlije (3.3) zhruba 30 ml vyvíjecího rozpouštědla (4.10) a pomocí skleněné tyčinky se do spodní části kolony zavede kousek vaty. Stlačí se, aby se odstranil vzduch. V kádince se připraví suspenze 25 g silikagelu (4.1) ve zhruba 80 ml vyvíjecího rozpouštědla a naleje se do kolony za pomoci nálevky. Ověří se, zda je celý silikagel zaveden do kolony; vymyje se vyvíjecím rozpouštědlem (4.10), otevře se kohoutek a hladina kapaliny se nechá dostoupit zhruba 2 mm nad horní úroveň silikagelu. |
|
5.1.4 |
Kolonová chromatografie Do 25 ml baňky (3.1) se naváží přesně 1,0 g připraveného vzorku podle bodu 5.1. Vzorek se rozpustí v 10 ml vyvíjecího rozpouštědla (4.10). Roztok se vlije do připravené chromatografické kolony podle bodu 5.1.3. Zabrání se tomu, aby se plocha kolony hýbala. Otevře se kohoutek a roztok vzorku se nechá odkapat, než dosáhne hladiny silikagelu. Rozpustí se pomocí 150 ml vyvíjecího rozpouštědla. Průtok se upraví na 2 ml/min (tak, aby 150 ml proteklo do kolony přibližně za 60–70 minut). Do baňky na 250 ml, která byla předtím zvážena, se odebere eluát. Rozpouštědlo se ve vakuu odpaří a jeho zbylé stopy se odstraní pod proudem dusíku. Baňka se zváží a vypočítá se množství získaného extraktu. (V případě použití už hotových silikagelových patron SPE se postupuje takto: zavede se 1 ml roztoku (5.1.2) do předem připravených patron s 3 ml n-hexanu. Po perkolování roztoku se vyvíjí 4 ml n-hexanu/dietyleteru v objemovém poměru 9/1 (V/V). Eluát se odebere do 10 ml zkumavky a odpařuje se pod proudem dusíku až do vysušení. Suché reziduum se podrobí pankreatické lipáze (5.2). Základem je ověřit složení mastných kyselin před a po přechodu patronou SPE.) |
5.2 Hydrolýza pankreatickou lipázou
|
5.2.1 |
Do centrifugační zkumavky se odváže 0,1 g oleje připraveného podle bodu 5.1. Přidá se 2 ml tlumivého roztoku (4.6), 0,5 ml roztoku cholátu sodného (4.7) a 0,2 ml roztoku chloridu vápenatého, přičemž po každém přidání se protřepe řádně směsí. Zkumavka se uzavře zábrusovou zátkou a umístí se do termostatu při teplotě 40 ± 0,5 °C. |
|
5.2.2 |
Přidá se 20 mg lipázy, opatrně se protřepe (tak, aby se nenamočila zátka), a zkumavka se dá do termostatu přesně na 2 minuty, potom se vybere, během 1 minuty důkladně protřepává a nechá se vychladit. |
|
5.2.3 |
Přidá se 1 ml dietyleteru, zazátkuje se a důkladně protřepe, potom se odstředí a pomocí mikrostříkačky se roztok etheru přenese do čisté a suché zkumavky. |
5.3 Příprava silanizovaných derivátů a plynová chromatografie
|
5.3.1 |
Pomocí mikrostříkačky se 100 μl roztoku (5.2.3) zavede do 10 ml zkumavky s kónickým dnem. |
|
5.3.2 |
Rozpouštědlo se odstraní pod slabým proudem dusíku, přidá se 200 μl silanizačního činidla (4.15), zkumavka se uzavře zátkou a nechá 20 minut odstát. |
|
5.3.3 |
Po 20 minutách se přidá 1 až 5 ml n-hexanu (v závislosti na chromatografických podmínkách): výsledný roztok je připravený pro plynovou chromatografii. |
5.4 Plynová chromatografie
Pracovní podmínky jsou tyto:
— teplota nástřikového zařízení (nástřikové zařízení on column) nižší než teplota varu rozpouštědla (68 °C),
— teplota detektoru: 350 °C,
— teplota kolony: nastavení teploty pece: 60 °C během 1 minuty, každou minutu se zvýší o 15 °C až do dosažení 180 °C, potom o 5 °C za minutu až do 340 °C, dále se udržuje 340 °C během 13 minut,
— nosný plyn: vodík nebo helium nastavené na lineární rychlost přiměřenou k dosažení rozlišení znázorněného na obrázku 1. Retenční čas triglyceridu C54 musí být 40 ± 5 minut (viz obrázek 2) (Výše uvedené podmínky postupu se uvádějí pouze orientačně. Každý subjekt je musí pro dosažení požadovaného rozlišení optimalizovat. Výška píku odpovídající 2-glyceril monopalmitátu musí dosáhnout alespoň 10 % rozsahu stupnice zapisovače.),
— množství nastříknuté látky: 0,5–1 μl roztoku (5 ml) n-hexanu (5.3.3).
5.4.1 Identifikace píků
Identifikace jednotlivých monoglyceridů se provádí podle retenčních časů a porovnáním se standardními směsmi monoglyceridů, které byly analyzovány za stejných podmínek.
5.4.2 Kvantitativní vyhodnocení
Plocha každého píku se vypočítá pomocí elektronického integrátoru.
6. VYJÁDŘENÍ VÝSLEDKŮ
Procentuální obsah glyceryl monopalmitátu se vypočítá na základě vztahu mezi plochou odpovídajícího píku a součtem ploch píků všech monoglyceridů (viz obrázek 2), podle vzorce:
Glycéril monopalmitate (%):
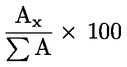
(Glycéril monopalmitate = glyceril monopalmitát)
kde:
|
Ax |
= |
plocha píku, který odpovídá glyceril monopalmitátu; |
|
ΣA |
= |
součet ploch všech píků, které odpovídají monoglyceridům; |
Výsledek se uvádí s přesností na jedno desetinné místo.
7. ZPRÁVA O ANALÝZE
Zpráva o analýze musí uvádět:
— odkaz na tuto metodu,
— všechny údaje potřebné k úplné identifikaci vzorku,
— výsledek analýzy,
— každý odklon od této metody, ať už pokud jde o rozhodnutí dotčených stran nebo z jiného důvodu,
— podrobné identifikační údaje o laboratoři, datum uskutečnění analýzy a podpis osob odpovědných za analýzu.
Obrázek 1
Chromatogram produktů z reakce silanizace, které byly získány lipázou na rafinovaném olivovém oleji s přidáním 20 % esterifikovaného oleje (100 %)
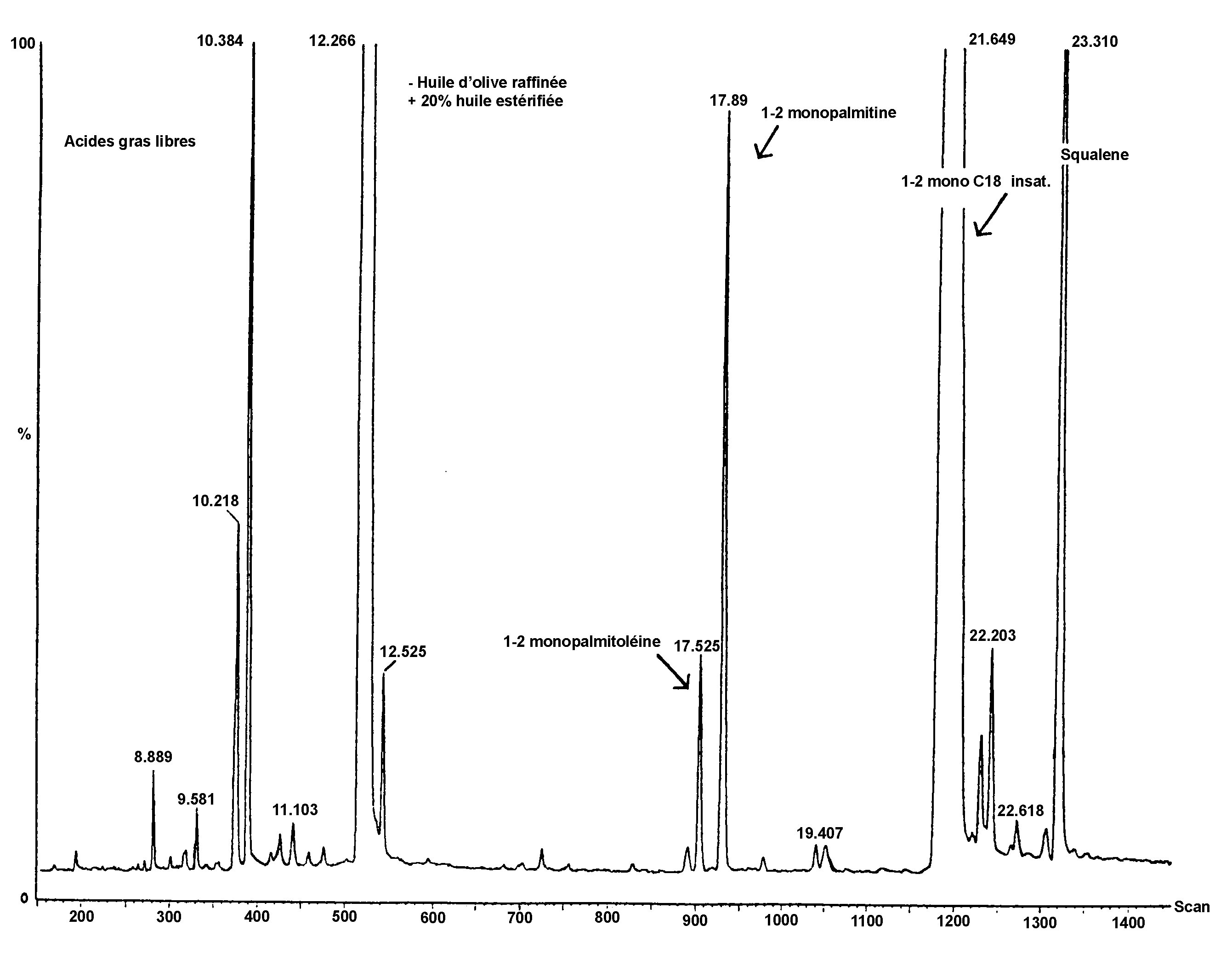
Vysvětlivky: „acides gras libres“ = volné mastné kyseliny; „Huile d’olive raffinée + 20 % huile estérifiée“ = rafinovaný olivový olej + 20 % esterifikovaný olej; „1-2 monopalmitoléine“ = 1-2 monopalmitolein; „1-2 mono C18 insat.“ = nenasycený 1-2 mono C18; „squalene“ = squalen.
Obrázek 2
Chromatogram:
|
A) |
neesterifikovaného olivového oleje po lipáze; po silanizaci; za těchto podmínek (kapilární kolona 8–12 m) je vosková frakce eluovaná zároveň s frakcí diglyceridu nebo krátce poté. Po lipáze by obsah triglyceridů neměl překročit 15 % 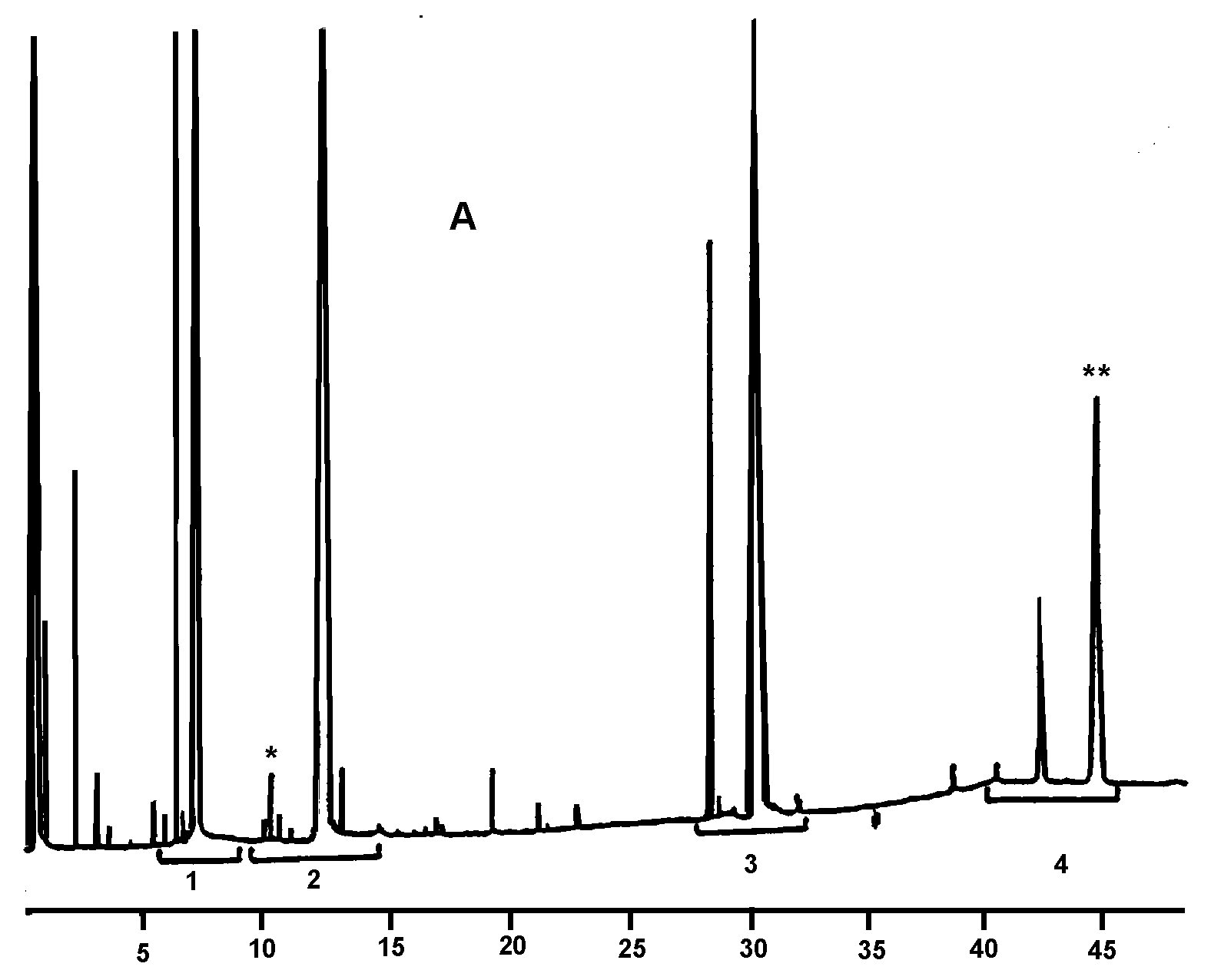
Vysvětlivky:
|
Chromatogram:
|
B) |
esterifikovaného olivového oleje po lipáze; po silanizaci; za těchto podmínek (kapilární kolona 8–12 m) je vosková frakce eluovaná zároveň s frakcí diglyceridu nebo krátce poté. Po lipáze by obsah triglyceridů neměl překročit 15 % 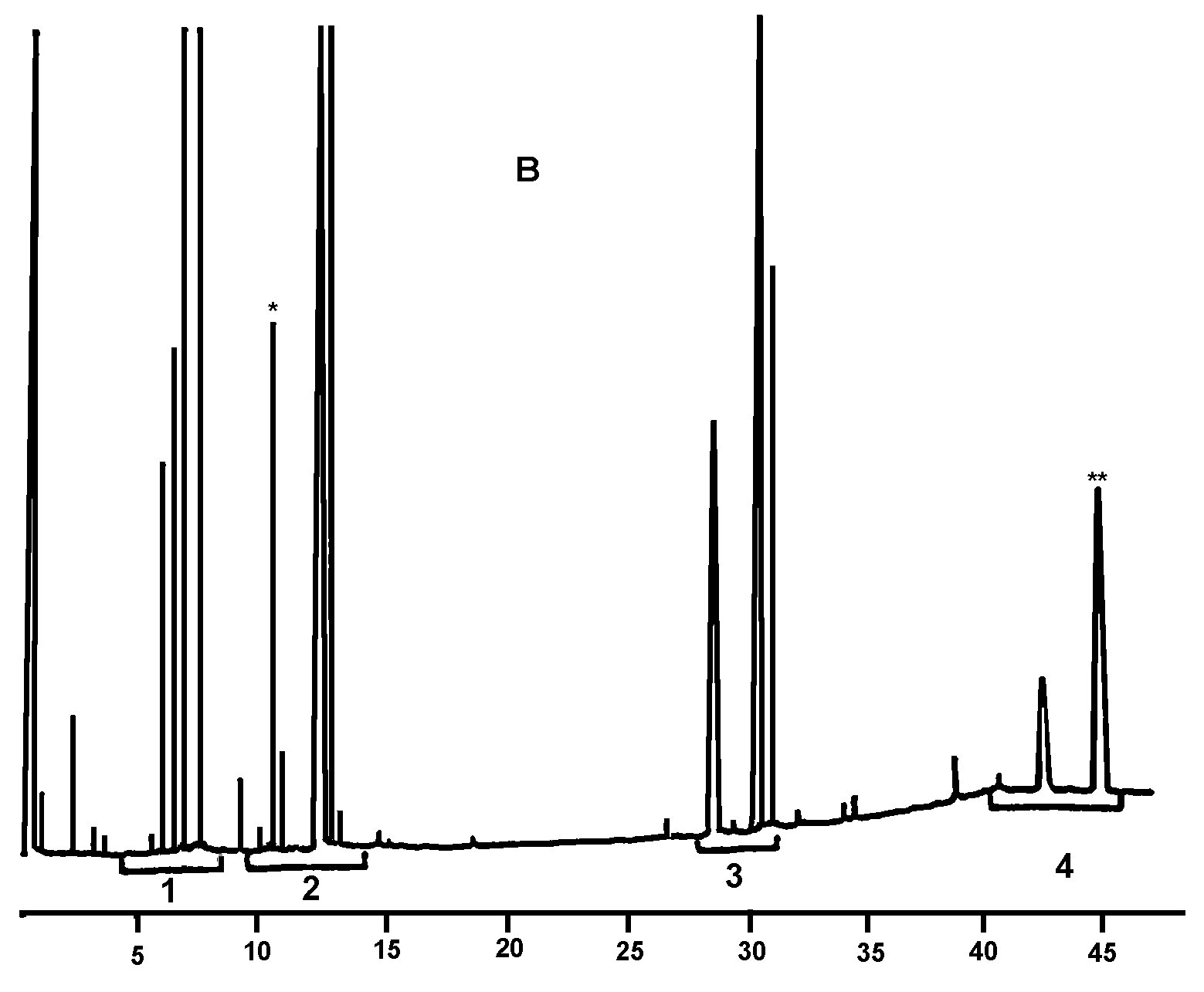
Vysvětlivky:
|
8. POZNÁMKY
PŘÍPRAVA LIPÁZY
Lipázy s dostatečnou aktivitou jsou obchodně dostupné. Je ale rovněž možné je připravit v laboratoři takto:
5 kg čerstvé vepřové slinivky břišní se vychladí na 0 °C; okolní sádlo a vazivová tkáň se oddělí a v míchadle se rozmělní, dokud nevznikne tekutá mazlavá pasta. Tato pasta se protřepe po dobu 4 až 6 hodin s 2,5 l bezvodého acetonu a poté odstředí. Zbytek se extrahuje třikrát pomocí stejného množství bezvodého acetonu, potom dvakrát pomocí směsi acetonu a diethyletheru 1:1 (V/V) a dvakrát pomocí diethyletheru.
Zbytek se po 48 hodin suší ve vakuu za účelem získání stabilního prášku, který je možné dlouho skladovat v ledničce chráněný před vlhkostí.
KONTROLA AKTIVITY LIPÁZY
Emulze olivového oleje se připraví takto:
V míchadle se po 10 minut protřepává směs složená ze 165 ml roztoku arabské gumy o koncentraci 100 g/l, 15 g drceného ledu a 20 ml předem neutralizovaného oleje.
Do 50 ml kádinky se zavede 10 ml této emulze, poté se postupně přidá 0,3 ml roztoku cholátu sodného o koncentraci 0,2 g/ml a 20 ml destilované vody.
Kádinka se vloží do termostatu udržovaného na teplotě 37 °C; vloží se elektrody pH metru a spirálovité míchadlo.
Pomocí byrety se po kapkách přidává roztok hydroxidu sodného 0,1 N, dokud hodnota pH nedosáhne 8,3.
Přidá se objem vodní suspenze lipázy (0,1 g/ml lipázy). Jakmile pH metr začne ukazovat pH 8,3, zapnou se stopky a po kapkách se přikapává roztok hydroxidu sodného takovou rychlostí, aby se udržovala hodnota pH 8,3. Zaznamená se objem spotřebovaného roztoku za minutu.
Údaje se zaznamenávají do grafového systému souřadnic tak, že osa nezávisle proměnných (x-ová osa) ponese časové údaje a na ose závisle proměnných se uvede počet mililitrů alkalického roztoku 0,1 N spotřebovaného na udržení konstantního pH. Výsledný graf musí být lineární.
Aktivita lipázy vyjádřená v lipázových jednotkách na 1 mg je pak dána tímto vzorcem:
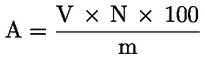
kde:
|
A |
je aktivita vyjádřená v jednotkách lipázy/mg; |
|
V |
je počet ml roztoku hydroxidu sodného 0,1 N za minutu (vypočítaný z grafu); |
|
N |
je molarita roztoku hydroxidu sodného; |
|
m |
je hmotnost zkušební lipázy v mg. |
Jednotka lipázy je definovaná jako množství enzymu, které uvolní 10 mikro-ekvivalentů kyseliny za minutu.
▼M20 —————
PŘÍLOHA IX
SPEKTROFOTOMETRICKÁ ANALÝZA V ULTRAFIALOVÉ OBLASTI SPEKTRA
ÚVOD
Spektrofotometrická analýza v ultrafialové oblasti spektra může poskytnout informace o jakosti tuku, stavu jeho zachovalosti a změnách způsobených technologickými procesy. Absorpce při vlnových délkách specifikovaných v metodě je dána přítomností systémů konjugovaných dienů a trienů, které jsou výsledkem procesů oxidace a/nebo rafinace. Tyto absorpce jsou vyjádřeny jako specifické extinkce
![]()
(extinkce vyvolaná 1 % roztokem tuku v předepsaném rozpouštědle, v 10mm kyvetě) obvykle označované písmenem K (též zmiňované jako „extinkční koeficient“).
1. OBLAST PŮSOBNOSTI
Tato příloha popisuje postup spektrofotometrické analýzy olivového oleje v ultrafialové oblasti spektra.
2. PODSTATA METODY
Vzorek se rozpustí v požadovaném rozpouštědle a změří se absorbance roztoku při určitých vlnových délkách proti čistému rozpouštědlu.
Vypočítá se specifická extinkce při 232 nm a 268 nm v isooktanu nebo při 232 nm a 270 nm v cyklohexanu pro 1 % hmotnostní koncentrace v 10mm kyvetě.
3. VYBAVENÍ
|
3.1. |
Spektrofotometr vhodný pro měření v ultrafialových vlnových délkách (mezi 220 nm a 360 nm) s možností odečítání jednotlivých nanometrických jednotek. Doporučuje se provádět pravidelné kontroly přesnosti a reprodukovatelnosti rozsahu absorbance a vlnových délek, jakož i rozptýleného světla.
|
|
3.2. |
Obdélníkové křemenné kyvety s víčky, vhodné pro měření v ultrafialových vlnových délkách (mezi 220 a 360 nm) o délce optické dráhy 10 mm. Naplněné vodou nebo jiným vhodným rozpouštědlem by se neměly vzájemně lišit o více než 0,01 jednotky extinkce. |
|
3.3. |
Odměrné baňky s jednou značkou o objemu 25 ml třídy A. |
|
3.4. |
Analytické váhy, které lze odečítat s přesností na 0,0001 g. |
4. ČINIDLA
Není-li uvedeno jinak, používají se během analýzy pouze činidla uznané analytické kvality a destilovaná nebo demineralizovaná voda nebo voda rovnocenné čistoty.
Rozpouštědlo: isooktan (2,2,4-trimethylpentan) pro měření při 232 nm a 268 nm a cyklohexan pro měření při 232 nm a 270 nm, s absorbancí nižší než 0,12 při 232 nm a nižší než 0,05 při 270 nm proti destilované vodě, měřeno v 10mm kyvetě.
5. POSTUP
|
5.1. |
Zkoumaný vzorek musí být dokonale homogenní a bez suspendovaných nečistot. Není-li tomu tak, musí být přefiltrován přes papír při teplotě přibližně 30 °C. |
|
5.2. |
Takto připraveného vzorku se odváží přibližně 0,25 g (s přesností na 1 mg) do odměrné baňky o objemu 25 ml, doplní se po rysku předepsaným rozpouštědlem a homogenizuje se. Výsledný roztok musí být dokonale čirý. Je-li roztok zakalený nebo obsahuje nečistoty, je nutno jej rychle přefiltrovat přes papír. POZNÁMKA: Pro měření absorbance panenských a extra panenských olivových olejů při při 232 nm a 268 nm zpravidla stačí hmotnost 0,25–0,30 g. Pro měření při 232 nm se obvykle požaduje vzorek o váze 0,05 g, takže se obvykle připraví dva různé roztoky. Pro měření absorbance olivového oleje z pokrutin, rafinovaného olivového oleje nebo kontaminovaného olivového oleje je vzhledem k jeho vyšší absorbanci zapotřebí menší část vzorku, např. 0,1 g. |
|
5.3. |
Je-li to třeba, proveďte korekci základní linie (220–290 nm) rozpouštědlem v obou křemenných kyvetách (ve vzorku i v referenční kyvetě), potom naplňte vzorkovou křemennou kyvetu zkušebním roztokem a změřte extinkce při 232, 268 nebo 270 nm proti rozpouštědlu, které bylo použito jako reference. Zaznamenané hodnoty extinkce musí být v rozmezí 0,1 až 0,8 nebo v rozsahu linearity spektrofotometru, kterou je nutno ověřit. Pokud tomu tak není, musí se měření zopakovat s koncentrovanějšími nebo případně zředěnějšími roztoky. |
|
5.4. |
Po měření absorbance při 268 nebo 270 nm, změřte absorbanci při λmax, λmax + 4 a λmax – 4. Tyto hodnoty absorbance se používají ke stanovení změny specifické extinkce (ΔΚ). POZNÁMKA: Má se za to, že λmax činí 268 nm pro isooktan použitý jako rozpouštědlo a 270 nm pro cyklohexan. |
6. VYJÁDŘENÍ VÝSLEDKŮ
|
6.1. |
Zaznamenávají se specifické extinkce (extinkční koeficienty) při různých vlnových délkách vypočtené ze vzorce:
kde:
vyjádřeno s přesností na dvě desetinná místa. |
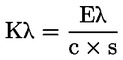
|
6.2. |
Změna specifické extinkce (ΔΚ) Změny absolutní hodnoty extinkce (ΔΚ) je dána vzorcem:
kde Km je specifická extinkce při vlnové délce pro maximální absorpce ve výši 270 nm a 268 nm v závislosti na použitém rozpouštědle, vyjádřeno s přesností na dvě desetinná místa. |
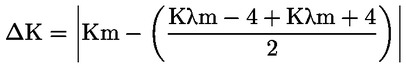
PŘÍLOHA X
STANOVENÍ METHYLESTERŮ MASTNÝCH KYSELIN PLYNOVOU CHROMATOGRAFIÍ
1. OBLAST PŮSOBNOSTI
Tato příloha obsahuje pokyny pro stanovení volných a vázaných mastných kyselin v rostlinných tucích a olejích plynovou chromatografií po jejich přeměně na methylestery mastných kyselin (FAME).
Vázané mastné kyseliny triacylglycerolů (TAG) a v závislosti na metodě esterifikace volné mastné kyseliny (FFA) se přeměňují na methylestery mastných kyselin (FAME), které se stanoví kapilární plynovou chromatografií.
Metoda popsaná v této příloze umožňuje stanovovat methylestery mastných kyselin od C12 po C24, včetně methylesterů nasycených, cis- a trans-mononenasycených a cis- a trans-polynenasycených mastných kyselin.
2. PODSTATA
Pro kvantitativní analýzu FAME se používá plynová chromatografie (GC). Methylestery mastných kyselin se připraví podle části A. Poté se vstříknou do injektoru a uvnitř něj se odpaří. Separace methylesterů mastných kyselin se provádí v analytických kolonách se specifickou polaritou a délkou. Pro detekci FAME se používá plameno-ionizační detektor (FID). Podmínky analýzy jsou uvedeny v části B.
V plynové chromatografii methylesterů mastných kyselin s FID lze jako nosný plyn (mobilní fáze) použít vodík nebo helium. Vodík urychluje separaci a zajišťuje výraznější píky. Stacionární fáze je mikroskopickou vrstvou tenkého tekutého filmu na inertním pevném povrchu z taveného křemene.
Když analyzované odtěkané sloučeniny procházejí kapilární kolonou, reagují se stacionární fází, jež je pokrytím na vnitřním povrchu kolony. Vzhledem k tomuto různému působení rozdílných sloučenin dochází k eluaci v různou dobu, která se nazývá retenčním časem sloučenin pro daný soubor parametrů analýzy. Srovnání retenčních časů se používá stanovení jednotlivých sloučenin.
ČÁST A
PŘÍPRAVA METHYLESTERŮ MASTNÝCH KYSELIN Z OLIVOVÉHO OLEJE A OLIVOVÉHO OLEJE Z POKRUTIN
1. OBLAST PŮSOBNOSTI
Tato část upřesňuje přípravu methylesterů mastných kyselin. Zahrnuje metody pro přípravu methylesterů mastných kyselin z olivového oleje a olivového oleje z pokrutin.
2. OBLAST POUŽITÍ
Příprava methylesterů mastných kyselin z olivového oleje a olivového oleje z pokrutin se provádí transesterifikací pomocí methanolového roztoku hydroxidu draselného při pokojové teplotě. Nezbytnost čištění vzorku před transesterifikací vzorku závisí na obsahu volných mastných kyselin ve vzorku a analytickém parametru, který má být stanoven, přičemž rozhodnout se lze podle následující tabulky:
|
Kategorie oleje |
Metoda |
|
Panenský olivový olej s kyselostí ≤ 2,0 % |
1. Mastné kyseliny 2. Trans-mastné kyseliny 3. ΔECN42 (po čištění pomocí silikagelu SPE) |
|
Rafinovaný olivový olej |
|
|
Olivový olej složený z rafinovaného a panenského olivového oleje |
|
|
Rafinovaný olivový olej z pokrutin |
|
|
Olivový olej z pokrutin |
|
|
Panenský olivový olej s kyselostí > 2,0 % Surový olivový olej z pokrutin |
1. Mastné kyseliny (po čištění pomocí silikagelu SPE) 2. Trans-mastné kyseliny (po čištění pomocí silikagelu SPE) 3. ΔECN42 (po čištění pomocí silikagelu SPE) |
3. METODIKA
3.1. Transesterifikace pomocí methanolového roztoku hydroxidu draselného při pokojové teplotě
3.1.1. Podstata
Methylestery se vytvářejí transesterifikací pomocí methanolového roztoku hydroxidu draselného jako mezistupeň před zmýdelněním.
3.1.2. Činidla
|
3.1.2.1. |
Methanol s obsahem maximálně 0,5 % (m/m) vody |
|
3.1.2.2. |
Hexan pro chromatografii |
|
3.1.2.3. |
Heptan pro chromatografii |
|
3.1.2.4. |
Diethylether, stabilizovaný pro účely analýzy |
|
3.1.2.5. |
Aceton pro chromatografii |
|
3.1.2.6. |
Eluční rozpouštědlo pro čištění oleje směsí hexanu/diethyletheru 87/13 (v/v) pro sloupcovou chromatografii/chromatografii SPE) |
|
3.1.2.7. |
Hydroxid draselný, přibližně 2M methanolový roztok: 11,2 g hydroxidu draselného se rozpustí ve 100 ml methanolu. |
|
3.1.2.8. |
Silikagelové patrony 1 g (6 ml) pro extrakci na pevnou fázi. |
3.1.3. Přístroje a pomůcky
|
3.1.3.1. |
Zkumavky se šroubovacím uzávěrem (o objemu 5 ml) opatřené spojem z PTFE |
|
3.1.3.2. |
Dělené nebo automatické pipety, 2 ml a 0,2 ml |
3.1.4. Čištění vzorků oleje
V případě potřeby budou vzorky oleje čištěny na silikagelových patronách pro extrakci na pevnou fázi. Silikagelová patrona (3.1.2.8) se vloží do vakuového elučního zařízení a promývá se s 6 ml hexanu (3.1.2.2), promývání se provádí mimo vakuum. Poté se do kolony vlije roztok oleje (přibližně 0,12 g) v 0,5 ml hexanu (3.1.2.2). Tento roztok se po průchodu kolonou eluuje s 10 ml hexanu/diethyletheru (87/13 v/v) (3.1.2.6). Kombinované eluáty se homogenizují a rozdělí na dva obdobné objemy. Alikvotní část se odpaří do sucha na rotační odparce za sníženého tlaku při pokojové teplotě. Residuum se rozpustí v 1 ml n-heptanu a roztok je připraven k analýze mastných kyselin plynovou chromatografií. Případně se odpaří druhá alikvotní část a residuum se rozpustí v 1 ml acetonu pro analýzu triglyceridů vysoce účinnou kapalinovou chromatografií (HPLC).
3.1.5. Postup
Do 5ml zkumavky se šroubovacím uzávěrem (3.1.3.1) se odváží přibližně 0,1 g vzorku oleje. Přidá se 2 ml heptanu (3.1.2.2) a vše se protřepe. Přidá se 0,2 ml roztoku hydroxidu draselného (3.1.2.7), přidá se uzávěr se spojem z PTFE, uzávěr se utěsní a důkladně protřepe po dobu 30 sekund. Nechá se odstát, dokud nebude svrchní vrstva roztoku čirá. Svrchní vrstva obsahující methylestery se slije. Roztok heptanu je připraven k nástřiku do plynového chromatografu. Do analýzy plynovou chromatografií je vhodné roztok uchovávat v ledničce. Nedoporučuje se skladovat roztok déle než 12 hodin.
ČÁST B
ANALÝZA METHYLESTERŮ MASTNÝCH KYSELIN PLYNOVOU CHROMATOGRAFIÍ
1. OBLAST PŮSOBNOSTI
Tato metoda popisuje použití plynové chromatografie v kapilární koloně pro stanovení kvalitativního a kvantitativního složení směsi methylesterů mastných kyselin získaných podle metody uvedené v části A.
Tato část není použitelná pro polymerní mastné kyseliny.
2. ČINIDLA
2.1. Nosný plyn
Inertní plyn (helium nebo vodík), dokonale vysušený a s obsahem kyslíku menším než 10 mg/kg.
Poznámka 1: Vodík umožňuje dvakrát zvýšit rychlost analýzy, ale je nebezpečný. Dostupná jsou bezpečnostní zařízení.
2.2. Pomocné plyny
|
2.2.1. |
Vodík (čistota ≥ 99,9 %), neobsahující organické nečistoty. |
|
2.2.2. |
Vzduch nebo kyslík, neobsahující organické nečistoty. |
|
2.2.3. |
Dusík (čistota > 99 %). |
2.3. Referenční standard
Směs methylesterů čistých mastných kyselin, nebo methylestery z tuku známého složení; výhodné je složení podobné analyzovanému tuku. Pro stanovení trans-isomerů nenasycených kyselin jsou užitečné cis- a trans-isomery methylesterů oktadecenové, oktadekadienové a oktadekatrienové kyseliny.
Pozornost je třeba věnovat ochraně polynenasycených mastných kyselin před oxidací.
3. PŘÍSTROJE A POMŮCKY
Uvedené pokyny se vztahují na obvyklá zařízení používaná pro plynovou chromatografii, která používají kapilární kolony a plameno-ionizační detektor.
3.1. Plynový chromatograf
Plynový chromatograf obsahuje tyto části:
3.1.1. Injektor
Injektor se používá pro kapilární kolonu, kde by injektor měl být konstruován přímo pro použití s touto kolonou. V tomto případě může být prováděn buď nástřik pomocí děliče (split type), nebo bez dělení přímo na kolonu (splitless type) nástřik „on column“.
3.1.2. Termostat
Termostat má mít schopnost vyhřát kolonu na teplotu nejméně 260 °C a udržovat žádanou teplotu s přesností na 0,1 °C. Poslední požadavek je zvlášť důležitý při použití trubice z taveného křemene.
Použití ohřevu s programovanou teplotou je doporučeno ve všech případech, zejména pro mastné kyseliny s méně než 16 atomy uhlíku.
3.1.3. Kapilární kolona
|
3.1.3.1. |
Trubice vyrobená z materiálu, který je inertní k analyzovaným látkám (obvykle sklo nebo tavený křemen). Vnitřní průměr se pohybuje mezi 0,20 mm až 0,32 mm. Vnitřní povrch musí být před pokrytím stacionární fází přiměřeným způsobem upraven (např. příprava povrchu, inaktivace). Pro cis- a trans-isomery mastných kyselin je dostatečná délka 60 m. |
|
3.1.3.2. |
U stacionární fáze jsou vhodné polární polysiloxany (kyanosilikony) vázané stacionární fáze (zesíťované). Poznámka 2: Při použití polárních polysiloxanů je nebezpečí obtížné identifikace a separace kyseliny linolenové a kyselin C20. Pokrytí má být tenké, tj. 0,1 μm až 0,2 μm. |
|
3.1.3.3. |
Montáž a kondicionace kolony Při montáži kapilární kolony, např. úpravě kolony v termostatu (nosič), výběru a provedení spojů (netěsné spoje), polohy konce kolony v injektoru a detektoru (zmenšení mrtvých prostorů) je nutno zachovat běžnou opatrnost. Kolonou se nechá protékat nosný plyn (např. 0,3 bar (30 kPa) pro kolonu délky 25 m a vnitřním průměru 0,3 mm). Kolona se ohřívá v termostatu za programované teploty rychlostí 3 °C/min od teploty okolí do teploty 10 °C pod limitní hodnotu, při které dochází k rozložení stacionární fáze. Termostat se udržuje při této teplotě po dobu jedné hodiny, dokud nedojde k ustálení základní linie. Potom se termostat ochladí na 180 °C a pracuje se za isotermních podmínek. Poznámka 3: Vhodné kondiciované kolony jsou obchodně dostupné. |
3.1.4. Plameno-ionizační detektor a konverzní zesilovač
3.2. Stříkačka
Stříkačka o maximálním objemu 10 μl, dělená po 0,1 μl.
3.3. Systém pro sběr dat
Systém pro sběr dat napojený online na detektory a používaný se softwarovým programem pro integraci píků a normalizaci.
4. POSTUP
Operace popsané v odstavcích 4.1 až 4.3 se týkají použití plameno-ionizačního detektoru.
4.1. Zkušební podmínky
4.1.1. Výběr optimálních pracovních podmínek u kapilárních kolon
Účinnost a permeabilita kapilárních kolon způsobují, že dělení složek a doba trvání analýzy jsou značně závislé na průtoku nosného plynu kolonou. Bude proto nezbytné optimalizovat pracovní podmínky působením na tyto parametry (nebo jednodušeji na tlakovou ztrátu na koloně) podle toho, zda je cílem zlepšit dělení, nebo zrychlit analýzu.
Tyto podmínky se ukázaly být vhodné pro separaci FAME (C4 až C26). Příklady chromatogramů jsou uvedeny v dodatku B:
|
Teplota injektoru: |
250 °C |
|
Teplota detektoru: |
250 °C |
|
Teplota termostatu: |
165 °C (8 min) až 210 °C při 2 °C/min |
|
Nosný plyn – vodík: |
tlak na koloně: 179 kPa, |
|
Celkový průtok: |
154,0 ml/min; |
|
Oddělovací poměr: |
1:100 |
|
Objem nástřiku: |
1 μl |
4.1.2. Stanovení rozlišení (viz dodatek A)
Rozlišení R dvou sousedních píků I a II se vypočte podle vzorce:
R = 2 × ((dr(II) – dr(I) )/(ω(I) + ω(II))) nebo R = 2 × ((tr(II) – tr(I) )/(ω(I) + ω(II))) (USP) (lékopis USA)
nebo
R = 1,18 × ((tr(II) – tr(I))/(ω0,5(I) + ω0,5(II))) (EP, BP, JP, DAB) (JP (lékopis Japonska), EP (Evropský lékopis), BP (lékopis UK))
kde:
d r(I) je retenční vzdálenost píku I;
d r(I) je retenční vzdálenost píku II;
t r(I) je retenční čas píku I;
t r(II) je retenční čas píku II;
ω(I) je šířka základu píku I;
ω(I) je šířka základu píku II;
ω0,5 je šířka píku uvedené sloučeniny, při střední výšce píku;
Pokud ω(I) ≈ ω(II), R se vypočte podle těchto vzorců:
R = (dr(II) – d r(I))/ω = (dr(II) – d r(I))/4σ
kde
σ je směrodatná odchylka (viz Dodatek A, obrázek 1).
Pokud se vzdálenost dr mezi dvěma píky d r(II) – d r(I) rovná 4σ, faktor rozlišení R = 1.
Nejsou-li dva píky úplně separovány, tečny vedené inflexními body dvou píků se protnou v bodě C. Pro úplnou separaci dvou píků musí být vzdálenost mezi dvěma píky:
d r(II) – d r(I) = 6 σ, odtud R = 1,5 (viz Dodatek A, obrázek 3).
5. VYJÁDŘENÍ VÝSLEDKŮ
5.1. Kvalitativní analýza
Provede se identifikace píků methylesterů pro vzorek z chromatogramu v dodatku B, obrázku 1, podle potřeby se interpoluje, nebo se porovná s píky referenčních směsí methylesterů (jak je uvedeno v bodě 2.3).
5.2. Kvantitativní analýza
5.2.1. Určení složení
Výpočet hmotnostního zlomku w i jednotlivých methylesterů mastných kyselin, vyjádřený v hmotnostních procentech methylesterů, se provede takto:
5.2.2. Metoda výpočtu
5.2.2.1. Obecný případ
Obsah složky i vyjádřený v hmotnostních procentech methylesterů se vypočítá určením plochy odpovídajícího píku vzhledem k součtu ploch všech píků podle vzorce:
wi = (Ai/ΣA) × 100
kde
Ai je plocha píku jednotlivého methylethylesteru mastných kyselin i;
ΣA je součet ploch všech píků všech jednotlivých methylesterů mastných kyselin.
Výsledky se vyjádří s přesností na dvě desetinná místa.
Poznámka 4: U tuků a olejů se hmotnostní zlomek methylesterů mastných kyselin rovná hmotnostnímu zlomku triacylglycerolů v gramech na 100 g. V případech, že tento předpoklad neplatí, viz 5.2.2.2.
5.2.2.2. Použití korekčních faktorů
V některých případech, zvláště za přítomnosti mastných kyselin s méně než osmi atomy uhlíku, nebo kyselin se sekundární skupinou se plochy opraví pomocí specifických korekčních faktorů (Fci). Tyto faktory se stanoví pro každý jednotlivý nástroj. Pro tento účel se použijí vhodné referenční materiály s certifikovaným složením mastných kyselin v odpovídajícím rozsahu.
Poznámka 5: Tyto korekční faktory nejsou shodné se teoretickými korekčními faktory FID, které jsou uvedeny v dodatku A, jelikož zahrnují rovněž výkonnost systému ve vztahu k injektoru atd. V případě větších rozdílů by měla být zkontrolována výkonnost celého systému.
Pro tyto referenční směsi se hmotnostní procenta FAME i vypočtou podle vzorce:
wi = (mi /Σm) × 100
kde
m i je hmotnost FAME i v referenční směsi,
Σm je celková hmotnost různých složek jako FAME v referenční směsi.
Z chromatogramu referenční směsi se vypočítá procentní podíl plochy pro FAME i takto:
wi = (Ai/ΣA) × 100
kde
Ai je plocha FAME i v referenční směsi,
ΣA součet všech ploch všech FAMW v referenční směsi.
Korekční faktor Fc je následně:
Fc = (mi × ΣA)/(Ai/Σm)
Pro vzorek činí hmotnostní procenta každého FAME i:
wi = (Fi × Ai)/Σ (Fi × Ai)
Výsledky se vyjádří s přesností na dvě desetinná místa.
Poznámka 6: Vypočtená hodnota odpovídá procentním procentům jednotlivých mastných kyselin vypočítaným jako triacylglyceroly na 100 g tuku.
5.2.2.3. Použití vnitřního standardu
V některých případech (např. když nejsou kvantifikovány všechny mastné kyseliny, k čemuž dochází, jsou-li přítomny kyseliny se čtyřmi a šesti atomy uhlíku současně s kyselinami s 16 a 18 atomy uhlíku, nebo když je třeba určit absolutní množství mastných kyselin ve vzorku) by měl být použit vnitřní standard. Jako vnitřní standard se používají nejčastěji methylestery mastných kyselin s 5, 15 nebo 17 atomy uhlíku. Pro vnitřní standard by měl být stanoven korekční faktor (pokud se použije).
Hmotnostní procenta složky i vyjádřená jako methylester lze vypočítat podle vzorce:
wi = (mIS × Fi × Ai)/(m × FIS × AIS)
kde:
A i je plocha FAME i,
A IS je plocha vnitřního standardu;
F i je korekční faktor mastné kyseliny i, vyjádřený jako FAME;
F IS je korekční faktor vnitřního standardu;
m je hmotnost zkušebního vzorku v miligramech,
m IS je hmotnost vnitřního standardu v miligramech.
Výsledky se vyjádří s přesností na dvě desetinná místa.
6. PROTOKOL O ZKOUŠCE
Protokol musí obsahovat odkaz na použitou metodu pro přípravu methylesterů a jejich stanovení plynovou chromatografií. Dále zahrnuje všechny pracovní podrobnosti nespecifikované v této standardní metodě nebo považované za volitelné, jakož i další okolnosti, které mohou ovlivnit výsledek.
Protokol musí obsahovat všechny informace nutné pro kompletní identifikaci vzorku.
7. PŘESNOST
7.1. Výsledky mezilaboratorní zkoušky
Podrobnosti k mezilaboratorní zkoušce týkající se přesnosti metody jsou uvedeny v příloze C normy IOC/T.20/Doc. No 33. Hodnoty odvozené z této mezilaboratorní zkoušky nelze použít na jiné než uvedené rozsahy koncentrace a matrice.
7.2. Opakovatelnost
Absolutní rozdíl mezi výsledky dvou samostatných jednotlivých zkoušek získanými stejnou metodou na stejném zkušebním materiálu v téže laboratoři týmž pracovníkem používajícím stejné zařízení krátce po sobě není u více než 5 % případů vyšší než R uvedené v tabulkách 1 až 14 přílohy C normy IOC/T.20/Doc. No 33.
7.3. Reprodukovatelnost
Absolutní rozdíl mezi výsledky dvou jednotlivých zkoušek získanými stejnou metodou na stejném zkušebním materiálu v různých laboratořích a různými pracovníky používajícími odlišná zařízení není u více než 5 % případů vyšší než R uvedené v tabulkách 1 až 14 přílohy C normy IOC/T.20/Doc. No 33.
Dodatek A
Obrázek 1
Šířka ω0,5 v poloviční výšce trojúhelníku (ABC) a šířka b v poloviční výšce trojúhelníku (NPM).
|
Obrázek 2 |
Obrázek 3 |
|
|
|
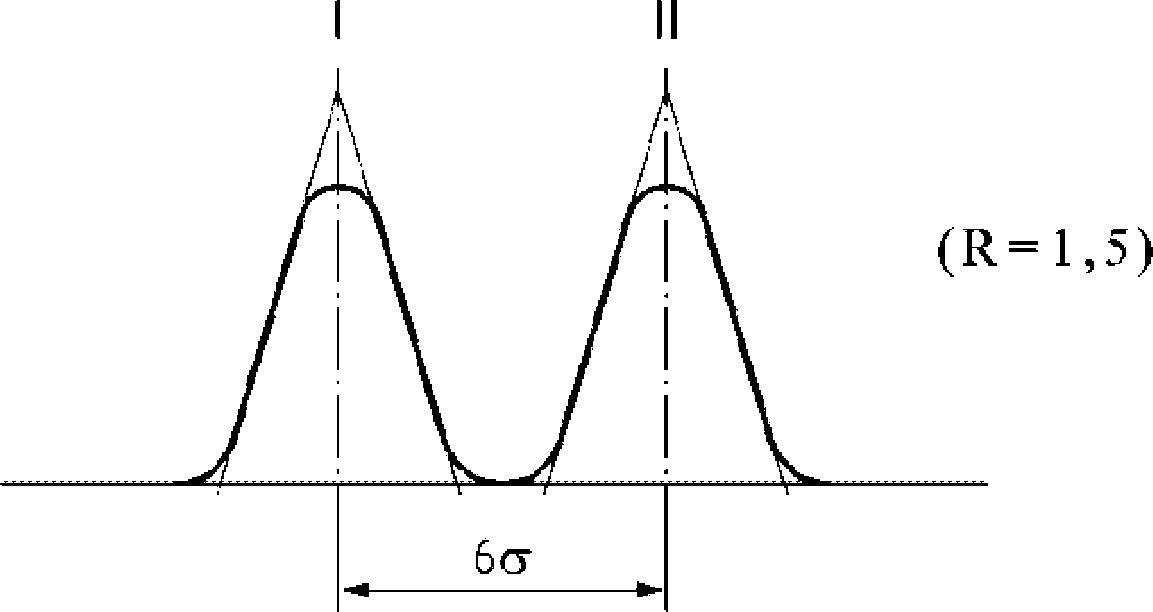
Dodatek B
Obrázek 1
Plynově-chromatografický profil získaný metodou studené methylace z olivového oleje z pokrutin
Chromatografické píky odpovídají methylesterům a ethylesterům, není-li uvedeno jinak.
PŘÍLOHA XI
STANOVENÍ OBSAHU TĚKAVÝCH HALOGENOVANÝCH ROZPOUŠTĚDEL V OLIVOVÉM OLEJI
1. PODSTATA
Tato metoda popisuje analýzu plynovou chromatografií s použitím techniky parního prostoru (head space).
2. PŘÍSTROJE A POMŮCKY
|
2.1 |
Plynový chromatograf vybavený detektorem elektronového záchytu (ECD). |
|
2.2 |
Zařízení pro přípravu vzorku v parním prostoru (head space). |
|
2.3 |
Skleněná kolona pro plynovou chromatografii o délce 2 m a průměru 2 mm se stacionární fází tvořenou 10 % OV101 nebo ekvivalentem, zachycenou v kyselinou prané a silanizované křemelině s velikostí částic 80-100 mesh. |
|
2.4 |
Nosný a pomocný plyn: dusík pro plynovou chromatografii, vhodný pro detektor elektronového záchytu. |
|
2.5 |
Skleněné baňky na 10 až 15 ml s teflonovým povlakem a hliníkovým uzávěrem s vybavením pro vpich stříkačky. |
|
2.6 |
Hermeticky těsnící svorky. |
|
2.7 |
Plynová stříkačka na 0,5 až 2 ml. |
3. CHEMIKÁLIE
Standard: halogenovaná rozpouštědla se stupněm čistoty vhodným pro plynovou chromatografii.
4. POSTUP
|
4.1 |
Do skleněné baňky se přesně naváží přibližně 3 g oleje (nesmí být použita opakovaně); baňka se hermeticky uzavře. Baňka se vloží na jednu hodinu do termostatu nastaveného na 70 °C. Pomocí stříkačky se opatrně odebere 0,2 až 0,5 ml z parního prostoru. Obsah stříkačky se nastříkne na kolonu chromatografu nastaveného takto: — teplota vstřiku: 150 °C, — teplota kolony: 70 až 80 °C, — teplota detektoru: 200 až 250 °C. Mohou být použity i jiné teploty, pokud se dosáhne ekvivalentních výsledků. |
|
4.2 |
Referenční roztoky: s použitím rafinovaného olivového oleje beze stop rozpouštědel se připraví standardní roztoky s různými koncentracemi těkavých halogenovaných rozpouštědel o hodnotách 0,05 mg/kg až 1 mg/kg. V případě potřeby se halogenovaná rozpouštědla rozředí pentanem. |
|
4.3 |
Kvantitativní vyhodnocení: porovnejte plochy nebo výšky píků vzorku a standardního roztoku, jehož koncentrace se nejvíce blíži předpokládané koncentraci. Je-li odchylka větší než 10 %, analýza musí být opakována, tj. musí být provedeno porovnání s dalším standardním roztokem, dokud nebude odchylka menší než 10 %. Obsah se stanoví na základě průměru jednotlivých vstřiků. |
|
4.4 |
Vyjádření výsledků: v mg/kg (ppm). Detekční limit činí u této metody 0,01 mg/kg. |
PŘÍLOHA XII
METODA MEZINÁRODNÍ RADY PRO OLIVOVÝ OLEJ PRO ORGANOLEPTICKÉ HODNOCENÍ PANENSKÉHO OLIVOVÉHO OLEJE
1. PŘEDMĚT A OBLAST POUŽITÍ
Cílem mezinárodní metody popsané v této příloze je stanovit postup hodnocení organoleptických vlastností panenského olivového oleje ve smyslu bodu 1 části VIII přílohy VII nařízení Evropského parlamentu a Rady (EU) č. 1308/2013 ( 4 ) a stanovit metodu jeho klasifikace na základě těchto vlastností. Metoda obsahuje rovněž pokyny týkající se nepovinného označování.
Popsaná metoda se vztahuje pouze na panenské olivové oleje a na klasifikaci nebo označování takovýchto olejů podle intenzity vnímaných vad a ovocné chuti a vůně, které stanoví skupina vybraných vyškolených a vyzkoušených posuzovatelů tvořících zkušební komisi.
Normy IOC uvedené v této příloze jsou použity v nejnovější dostupné verzi.
2. OBECNÝ ZÁKLADNÍ SLOVNÍK PRO SENZORICKOU ANALÝZU
Viz norma IOC/T.20/Doc. č. 4 „Senzorická analýza: obecný základní slovník“
3. SPECIFICKÝ SLOVNÍK
3.1 Negativní znaky
Zatuchlý / po kalném sedimentu Chuťově-čichový počitek typický pro olej získaný z oliv skladovaných na hromadách či v takových podmínkách, kde plody prošly pokročilým stupněm anaerobní fermentace, nebo pro olej získaný dekantací ze sedliny z kádí a podzemních nádrží, jež rovněž prošel procesem anaerobní fermentace.
Po plísních a vlhkosti, zemitý Chuťově-čichový počitek typický pro olivový olej získaný z plodů napadených plísněmi a kvasinkami v důsledku několikadenního uskladnění ve vlhku nebo pro olivový olej získaný z oliv sklizených s nánosem zeminy nebo bláta a neopraných.
Vinný/octový/kyselý/nakyslý Chuťově-čichový počitek typický pro některý olivový olej připomínající víno nebo ocet. Vzniká hlavně v důsledku procesu aerobního kvašení oliv nebo zbytků olivové pasty na lisovacích rohožích, které nebyly dobře umyté, což má za následek tvorbu kyseliny octové, ethylacetátu a ethanolu.
Žluklý Chuťově-čichový počitek typický pro všechny oleje, které prodělaly intenzivní proces autooxidace.
Po namrzlých olivách (po vlhkém dřevu) Chuťově-čichový počitek typický pro olivový olej získaný z oliv, které byly na stromě zasaženy mrazem.
3.1.1 Další negativní znaky
|
Přehřátý nebo připálený |
Chuťově-čichový počitek typický pro olej získaný nadměrným a/nebo dlouhodobým ohřevem během zpracování, zejména je-li pasta míchána za tepla a za nevhodných tepelných podmínek. |
|
Trávový/dřevový |
Chuťově-čichový počitek typický pro olivový olej získaný z vyschlých oliv. |
|
Hrubý |
Chuťově taktilní počitek vyvolaný některým starým olejem, který způsobuje v ústech hustý, kašovitý pocit. |
|
Olejový |
Chuťově-čichový počitek oleje připomínající naftu, mazací tuky nebo minerální olej. |
|
Po štávě z plodů |
Chuťově-čichový počitek, který olej získá při dlouhodobém kontaktu se šťávou z plodů, která prošla procesem fermentace. |
|
Slaný |
Chuťově-čichový počitek typický pro olivový olej získaný z oliv, které byly konzervovány v solném roztoku. |
|
Kovový |
Chuťově-čichový počitek připomínající kovy. Je typický pro olivový olej, který byl dlouhou dobu ve styku s kovovými povrchy během mletí, míchání, lisování nebo skladování. |
|
Espartový |
Chuťově-čichový počitek typický pro olivový olej získaný z oliv lisovaných v nových espartových rohožích. Může se lišit v závislosti na tom, zda byly rohože vyrobeny ze zeleného esparta nebo sušeného esparta. |
|
Po červech |
Chuťově-čichový počitek typický pro olivový olej získaný z oliv, které byly silně napadeny larvami mouchy olivové (Bactrocera oleae). |
|
Po okurkách |
Chuťově-čichový počitek vznikající v případě, že je olivový olej po delší dobu hermeticky uzavřen, zejména do plechovek, pocházející z 2,6-nonadienalu. |
3.2 Pozitivní znaky
|
Ovocná chuť a vůně |
Soubor čichových počitků typických pro olivový olej, které jsou závislé na odrůdě oliv a pocházejí ze zdravých a čerstvých oliv, zralých nebo nezralých. Jsou vnímány přímo a/nebo retronasální (pharyngonasální) metodou čichání. |
|
Hořký |
Základní charakteristická chuť oleje získaného ze zelených nebo nazelenalých oliv, kterou vnímáme hrazenými papilami, tvořícími na jazyku uskupení ve tvaru písmene V. |
|
Štiplavý |
Ostrý taktilní počitek typický pro olivové oleje vyrobené na začátku hospodářského roku převážně z ještě nezralých oliv. Možno jej vnímat v celé ústní dutině, zejména v hrdle. |
3.3 Nepovinná terminologie pro účely označování
Vedoucí zkušební komise může na požádání potvrdit, že hodnocené oleje splňují definice a rozpětí odpovídající výhradně následujícím termínům v závislosti na intenzitě a vnímání znaků.
Pozitivní znaky (ovocná chuť a vůně, hořký a štiplavý): podle intenzity vnímání:
— silný, pokud je medián tohoto znaku vyšší než 6,0,
— střední, pokud je medián tohoto znaku vyšší než 3,0 a nejvýše roven 6,0,
— mírný, pokud je medián tohoto znaku nejvýše roven 3,0.
|
Ovocná chuť a vůně |
Soubor čichových počitků typických pro olej, který závisí na odrůdě oliv a pochází ze zdravých a čerstvých oliv, u nichž nepřevažuje zralá ani nezralá ovocná chuť a vůně. Je vnímán přímo a/nebo retronasální (pharyngonasální) metodou čichání. |
|
Nezralá ovocná chuť a vůně |
Soubor čichových počitků typických pro olej, který připomíná nezralé plody, závisí na odrůdě oliv a pochází ze zelených, zdravých a čerstvých oliv. Je vnímán přímo a/nebo retronasální (pharyngonasální) metodou čichání. |
|
Zralá ovocná chuť a vůně |
Soubor čichových počitků typických pro olej, který připomíná zralé plody, závisí na odrůdě oliv a pochází ze zdravých, čerstvých oliv. Je vnímán přímo a/nebo retronasální (pharyngonasální) metodou čichání. |
|
Vyvážený |
Olej, jenž nevykazuje známky nevyváženosti, kterou se rozumí čichový, chuťový a hmatový vjem oleje, ve kterém medián znaku hořký a medián znaku štiplavý není o více než dva body vyšší než medián znaku ovocná chuť a vůně. |
|
Jemný olej |
Olej, ve kterém je medián znaku hořký a znaku štiplavý nejvýše roven 2,0. |
Seznam termínů podle intenzity vnímání:
|
Termíny podléhající předložení osvědčení o organoleptické zkoušce |
Medián znaku |
|
Ovocná chuť a vůně |
— |
|
Zralá ovocná chuť a vůně |
— |
|
Nezralá ovocná chuť a vůně |
— |
|
Mírná ovocná chuť a vůně |
≤ 3,0 |
|
Střední ovocná chuť a vůně |
3,0 < Me ≤ 6,0 |
|
Silná ovocná chuť a vůně |
> 6,0 |
|
Mírná zralá ovocná chuť a vůně |
≤ 3,0 |
|
Střední zralá ovocná chuť a vůně |
3,0 < Me ≤ 6,0 |
|
Silná zralá ovocná chuť a vůně |
> 6,0 |
|
Mírná nezralá ovocná chuť a vůně |
≤ 3,0 |
|
Střední nezralá ovocná chuť a vůně |
3,0 < Me ≤ 6,0 |
|
Silná nezralá ovocná chuť a vůně |
> 6,0 |
|
Mírná hořkost |
≤ 3,0 |
|
Střední hořkost |
3,0 < Me ≤ 6,0 |
|
Silná hořkost |
> 6,0 |
|
Mírná štiplavost |
≤ 3,0 |
|
Střední štiplavost |
3,0 < Me ≤ 6,0 |
|
Silná štiplavost |
> 6,0 |
|
Vyvážený olej |
Medián znaku hořký a medián znaku štiplavý není o více než 2,0 body vyšší než medián znaku ovocná chuť a vůně. |
|
Jemný olej |
Medián znaku hořký a medián znaku štiplavý není vyšší než 2,0. |
4. SKLENĚNÉ NÁDOBKY PRO HODNOCENÍ OLIVOVÉHO OLEJE
Viz norma IOC/T.20/Doc. č. 5 „Skleněné nádobky pro hodnocení olivového oleje“.
5. ZKUŠEBNÍ MÍSTNOST
Viz norma IOC/T.20/Doc. č. 6 „Návod na zřízení zkušební místnosti“
6. ZAŘÍZENÍ
Všechny kabiny se vybaví níže uvedeným zařízením, nezbytně nutným pro posuzovatele pro řádné splnění jejich úkolu a snadno dosažitelným:
— skleněné nádobky (normalizované) obsahující vzorky, označené číselným kódem, zakryté hodinovými skly a uchovávané při teplotě 28 °C ± 2 °C,
— profilový list (viz obrázek 1) v tištěné nebo elektronické podobě za předpokladu, že jsou splněny podmínky uvedené v profilovém listu, v případě potřeby i s pokyny k použití,
— pero nebo nesmazatelný inkoust,
— podnosy s nakrájenými jablky a/nebo vodou, sycenou vodou a/nebo suchary,
— skleněné nádobky s vodou o laboratorní teplotě,
— list papíru připomínající obecná pravidla uvedená v oddílech 8.4 a 9.1.1;
— plivátka.
7. VEDOUCÍ ZKUŠEBNÍ KOMISE A POSUZOVATELÉ
7.1 Vedoucí zkušební komise
Vedoucím zkušební komise musí být náležitě vyškolená osoba s odbornými znalostmi nezbytnými pro posuzování jednotlivých druhů olivových olejů, s nimiž v průběhu své práce přijde do styku. Je klíčovou postavou ve zkušební komisi a odpovídá za organizaci a řízení zkušebních činností.
Práce vedoucího zkušební komise vyžaduje základní odbornou přípravu v oblasti nástrojů senzorické analýzy, senzorického hodnocení, pečlivost při přípravě, organizaci a provádění zkoušek, jakož i zkušenost a trpělivost při vědeckém plánování a provádění zkoušek.
Je jedinou osobou, která odpovídá za výběr, odbornou přípravu a sledování posuzovatelů, aby se ubezpečil o jejich úrovni výkonnosti. Je proto odpovědný za hodnocení posuzovatelů, které musí být vždy objektivní a pro něž musí vypracovat konkrétní postupy založené na zkouškách a pevně stanovených kritériích výběru nebo vyřazení (viz norma IOC/T.20/Doc. č. 14 „Pokyny pro výběr, výcvik a sledování odborných posuzovatelů panenského olivového oleje“.
Vedoucí zkušební komise je odpovědný za výkon zkušební komise a tedy za její hodnocení, o kterém musí poskytnout spolehlivé a objektivní důkazy. V každém případě je povinen kdykoli prokázat, že metoda i posuzovatelé jsou pod kontrolou. Doporučuje se provádět pravidelnou kalibraci zkušební komise (IOC/T.20/Doc. č. 14, § 5).
Nese konečnou odpovědnost za vedení záznamů zkušební komise. Tyto záznamy musí být vždy dohledatelné, musí být v souladu s požadavky na zabezpečení a jakost stanovené v mezinárodních normách pro senzorickou analýzu a po celou dobu zajišťovat anonymitu vzorků.
Odpovídá za inventarizaci přístrojů a zařízení potřebných k dosažení souladu se specifikacemi této metody a za zajištění jejich řádného čištění a údržby, a o těchto skutečnostech, jakož i o dodržování zkušebních podmínek, uchovává písemné důkazy.
Odpovídá za příjem a skladování vzorků po jejich doručení do laboratoře, jakož i za jejich skladování po provedení zkoušky. Přitom musí za všech okolností zajistit anonymitu a řádné uskladnění vzorků, a za tímto účelem musí vypracovat písemné postupy s cílem zajistit, aby byl celý tento proces sledovatelný a poskytoval záruky.
Kromě toho odpovídá za přípravu, kódové označení vzorků a jejich předkládání posuzovatelům v daném pořadí hodnocení v souladu s předem připravenými protokoly, jakož i za sběr a statistické vyhodnocování dat získaných posuzovateli.
Odpovídá za vytvoření a vypracování jakýchkoli dalších postupů, které budou případně nezbytné k doplnění této normy a k řádnému fungování zkušební komise.
Musí hledat způsoby porovnávání výsledků zkušební komise panelu s výsledky získanými jinými zkušebními komisemi provádějícími analýzu panenského olivového oleje, aby se tak přesvědčil o řádném fungování zkušební komise.
Povinností vedoucího zkušební komise je motivovat členy zkušební komise a podněcovat jejich zájem, zvídavost a soutěživost. Za tímto účelem se důrazně doporučuje, aby zajistil obousměrný bezproblémový tok informací ke členům zkušební komise a průběžně je informoval o všech prováděných úkolech a dosahovaných výsledcích. Zdrží se rovněž vyjadřování svého mínění a musí zabezpečit, aby případné dominantní osobnosti neovlivňovaly ostatní posuzovatele.
Svolává posuzovatele v dostatečném předstihu a zodpovídá veškeré dotazy týkající se provádění zkoušek, avšak zdrží se jakéhokoli vyjadřování svého mínění o vzorku.
7.1.1. Zástupce vedoucího zkušební komise
Vedoucího zkušebního komise může na základě oprávněných důvodů nahradit zástupce, jenž ho může zastupovat v povinnostech souvisejícími s prováděním zkoušek. Tento zástupce musí mít veškeré potřebné dovednosti, které potřebuje vedoucí zkušební komise.
7.2 Posuzovatelé
Osoby působící jako posuzovatelé při organoleptických zkouškách olivového oleje tak musí činit dobrovolně. Je proto vhodné, aby uchazeči předkládali písemnou žádost. Uchazeče vybere, vyškolí a sleduje vedoucí zkušební komise podle jejich schopnosti rozlišovat mezi podobnými vzorky; je třeba vzít v úvahu, že jejich posuzovací schopnost se během odborné přípravy zlepší.
Posuzovatelé musí jednat jako skuteční senzoričtí pozorovatelé, nepřihlížet k vlastním osobním chutím a referovat pouze o počitcích, které vnímají. Proto musí vždy pracovat v tichu, uvolněně a beze spěchu a věnovat maximální smyslovou pozornost právě ochutnávanému vzorku.
Pro každou zkoušku je třeba 8 až 12 posuzovatelů, ačkoli je žádoucí mít několik rezervních posuzovatelů pro případ možných absencí.
8. PODMÍNKY ZKOUŠEK
8.1 Prezentace vzorku
Vzorek oleje pro analýzu se předkládá ve standardizovaných skleněných zkušebních nádobkách podle normy IOC/T.20/Doc. č. 5 „Skleněné nádobky pro hodnocení olivového oleje“.
Skleněná nádobka obsahuje 14–16 ml oleje nebo 12,8 až 14,6 g, pokud se vzorky váží, a je zakryta hodinovým sklem.
Každá nádobka musí být označena kódem složeným z náhodně vybraných číslic nebo kombinace písmen a číslic. Kód bude označen pomocí systému, který je bez zápachu.
8.2 Teplota při zkoušce a teplota vzorku
Vzorky oleje určené pro ochutnávání jsou uchovávány ve skleněných nádobkách při teplotě 28°C ± 2 °C po celou dobu zkoušky. Tato teplota byla zvolena proto, že usnadňuje zjišťování organoleptických rozdílů oproti teplotě okolí, a proto, že při nižších teplotách se zřídka uvolňují aromatické sloučeniny charakteristické pro tyto oleje, zatímco vyšší teploty vedou k výskytu těkavých sloučenin charakteristických pro zahřívané oleje. Metoda, která má být použita pro ohřev vzorků ve skleněné nádobce, je popsána v normě IOC/T.20/Doc. č. 5 „Skleněné nádobky pro hodnocení olivového oleje“.
Teplota ve zkušební místnosti se musí pohybovat mezi 20 °C a 25 °C (viz IOC/T.20/Doc. č. 6).
8.3 Doba zkoušek
Nejvhodnější dobou pro ochutnávání olejů je dopoledne. Je dokázáno, že čichově-chuťové smysly jsou v průběhu dne zvláště citlivé. Citlivost čichově-chuťových smyslů je před hlavním jídlem zvláště vysoká, zatímco po jídle slábne.
Toto kritérium však nesmí být přiváděno do extrému, aby posuzovatel nebyl rozptylován hladem a tím nebyla snížena jeho rozlišovací schopnost; proto se doporučuje, aby se ochutnávky konaly mezi 10. hodinou dopolední a polednem.
8.4 Posuzovatelé: obecná pravidla chování
Následující doporučení se týkají chování posuzovatelů během jejich práce.
Jsou-li posuzovatelé vyzváni vedoucím zkušební komise k účasti na organoleptické zkoušce, musí být připraveni dostavit se v stanoveném čase a přitom dodržet dále uvedená pravidla.
— 30 minut před stanoveným časem zkoušky je zakázáno kouření a pití kávy.
— Není povoleno používání vůní, kosmetiky ani mýdla, jejichž pach by přetrval do doby zkoušky. Ruce je nutno si umývat neparfémovaným mýdlem a je třeba si je oplachovat a sušit tak často, jak je nezbytné pro odstranění jakéhokoli pachu.
— Posuzovatelé nesmí jíst po dobu nejméně jedné hodiny před započetím zkoušky.
— Pokud by se posuzovatelé necítili fyzicky v pořádku, a zejména je-li ovlivněno jejich čichové nebo chuťové vnímání, nebo pokud jsou pod psychickým tlakem, jenž by omezoval jejich schopnost koncentrace, zdrží se ochutnávky a sdělí to vedoucímu zkušební komise.
— Pokud posuzovatelé splnili výše uvedené podmínky, zaujmou své místo v přidělené kabině co nejspořádaněji a co nejtišeji.
— Pozorně si přečtou pokyny v profilovém listu a nezačnou se zkoumáním vzorku, dokud nebudou plně připraveni pro úkol, který mají splnit (uvolněně a beze spěchu). V případě, že vyvstanou jakékoli pochybnosti, měli by se soukromě obrátit na vedoucího zkušební komise.
— Při plnění svých úkolů musí zachovávat ticho.
— Musí mít po celou dobu vypnutý mobilní telefon, aby nerušili soustředění a práci svých kolegů.
9. POSTUP ORGANOLEPTICKÉHO HODNOCENÍ A KLASIFIKACE PANENSKÉHO OLIVOVÉHO OLEJE
9.1 Technika ochutnávání
9.1.1 Posuzovatelé uchopí nádobku, drží ji přitom zakrytou hodinovým sklem a mírně nakloněnou a zakrouží nádobkou jednou úplně tak, aby se vnitřní stěny co nejvíce navlhčily. Jakmile dokončí tuto etapu, sejmou hodinové sklo a čichají ke vzorku pomalými a hlubokými nádechy, aby daný olej zhodnotili. Čichání by nemělo trvat déle než 30 sekund. Pokud během této doby nedojdou k závěru, před dalším pokusem si krátce odpočinou.
Po provedení čichové zkoušky posuzovatelé zhodnotí pocity v ústech (celkové retronasální čichově-chuťově-taktilní počitky). To provedou tak, že usrknou přibližně 3 ml oleje. Je velmi důležité rozprostřít olej po celé dutině ústní, od přední části úst a jazyka, podél lícních stěn až k zadní části a části podepírající patro a do krku, protože vnímání chutí a taktilních počitků se co do intenzity mění v závislosti na místu na jazyce, patře a v krku.
Je třeba zdůraznit, že pro posouzení je nebytné, aby se dostatečné množství oleje velmi pomalu rozprostřelo na zadní části jazyka směrem k části podepírající patro a ke krku, přičemž posuzovatel se soustřeďuje na pořadí, v němž se objeví hořký a štiplavý podnět. Pokud se tak nestane, oba tyto podněty mohou u některých olejů uniknout pozornosti nebo může být hořký podnět zastřen podnětem štiplavým.
Krátké opakované nádechy, při nichž je vzduch vtahován ústy, umožňují posuzovateli nejen plošně rozprostřít vzorek po celých ústech, ale též vnímat těkavé aromatické sloučeniny retronasální (pharyngonasální) metodou čichání vynuceným použitím tohoto kanálu.
Pozn.: Pokud posuzovatelé ve vzorku nevnímají ovocnou chuť a vůni a intenzita klasifikujícího negativního znaku dosahuje hodnoty 3,5 nebo nižší, může se vedoucí zkušební komise rozhodnout, že posuzovatelé vzorek zanalyzují ještě jednou za teploty okolí (IOC/T.20/Dok. č. 6/Rev. 1, září 2007, oddíl 3 – Obecné specifikace pro zřízení zkušební místnosti), přičemž upřesní okolnosti a pojem teploty okolí. Když vzorek dosáhne pokojové teploty, měli by jej posuzovatelé znovu zhodnotit, aby ověřili výhradně vnímání ovocné chuti a vůně. Pokud budou ovocnou chuť a vůni vnímat, zaznamenají její intenzitu na stupnici.
V úvahu se též bere taktilní počitek štiplavosti. Za tímto účelem je vhodné olej spolknout.
9.1.2 Při organoleptickém hodnocení panenského olivového oleje se doporučuje při každém sezení posuzovat maximálně ČTYŘI VZORKY a počet sezení by neměl překročit tři denně, aby se předešlo kontrastnímu účinku, jenž by mohl nastat při okamžitém posouzení dalších vzorků.
Jelikož postupné posuzování způsobuje únavu nebo ztrátu citlivosti způsobenou předchozími vzorky, je nutné použít něco, co může odstranit z úst zbytky olivového oleje z předchozího hodnocení.
Doporučuje se použít malý plátek jablka, který je možné po rozžvýkání odložit do plivátka. Poté se ústa vypláchnou malým množstvím vody o laboratorní teplotě. Mezi koncem jednoho sezení a začátkem dalšího musí uplynout nejméně 15 minut.
9.2 Používání profilového listu posuzovateli
Profilový list, který posuzovatelé použijí, je zobrazen na obrázku 1 této přílohy.
Každý posuzovatel ve zkušební komisi čichově a následně chuťově ( 5 ) zhodnotí předmětný olej. Následně uvede intenzitu, s níž vnímá každý z negativních a pozitivních znaků, do desetibodové hodnotící tabulky uvedené v obdrženém profilovém listu.
V případě, že posuzovatel vnímá jakékoli případné negativní znaky, které nejsou uvedeny v oddíle 4, musí je zaznamenat do rubriky „ostatní“ s použitím termínu nebo termínů, které dané znaky co nejpřesněji popisují.
9.3 Použití údajů vedoucími zkušební komise
Vedoucí zkušební komise shromáždí profilové listy vyplněné jednotlivými posuzovateli a zkontroluje intenzity přisouzené jednotlivým znakům. V případě, že zjistí jakoukoli nesrovnalost, vyzve posuzovatele, aby svůj profilový list revidoval a v případě nutnosti zkoušku zopakoval.
Vedoucí zkušební komise zadá údaje z hodnocení získané od jednotlivých členů zkušební komise do počítačového programu podobného tomu, který stanoví norma IOC/T.20/Doc. č.o15) za účelem statistického výpočtu výsledků analýzy na základě výpočtu jejich mediánu (viz oddíl 9.4 a dodatek k této příloze). Údaje týkající se daného vzorku se zadávají pomocí matice složené z devíti sloupců odpovídajících devíti organoleptickým vlastnostem a z n řádků odpovídajících n počtu posuzovatelů zkušební komise.
Pokud více než 50 % členů zkušební komise vnímalo určitou vadu a uvedlo ji v rubrice „jiné“, vypočítá vedoucí zkušební komise medián této vady a provede příslušnou klasifikaci.
Hodnota robustního variačního koeficientu, který vyjadřuje klasifikaci (vada s nejsilnější intenzitou a znakem ovocné chuti a vůně), musí být nižší nebo rovna 20 %.
Pokud tomu tak není, musí vedoucí komise opakovat hodnocení tohoto konkrétního vzorku při jiném sezení.
Pokud k takové situaci dochází často, doporučuje se, aby vedoucí zkušební komise poskytl posuzovatelům konkrétní doplňkovou odbornou přípravu (IOC/T.20/Doc. No 14, § 5) a pro kontrolu výkonnosti zkušební komise použil ukazatel opakovatelnosti a ukazatel odchylek (IOC/T.20/Doc. č. 14, § 6).
9.4 Klasifikace oleje
Olej se zařadí do následujících kategorií podle mediánu vad a mediánu znaku ovocné chuti a vůně. Medián vad se stanovuje jako medián vady vnímané s největší intenzitou. Medián vad a medián znaku ovocné chuti a vůně se vyjadřuje s přesností na jedno desetinné místo.
Klasifikace oleje se provádí porovnáváním hodnoty mediánu vad a mediánu znaku ovocné chuti a vůně s níže uvedenými referenčními intervaly. Při stanovování hranice těchto intervalů byla zohledněna chyba metody, proto se tyto hranice považují za absolutní. Počítačové programy umožňují zobrazení klasifikace v tabulce statistických údajů nebo na grafu.
a) Extra panenský olivový olej: medián vad se rovná 0,0 a medián znaku ovocné chuti a vůně je vyšší než 0,0.
b) Panenský olivový olej: medián vad je vyšší než 0,0, ale dosahuje nejvýše hodnoty 3,5, a medián znaku ovocné chuti a vůně je vyšší než 0,0.
c) Lampantový panenský olivový olej: medián vad je vyšší než 3,5 nebo je medián vad nejvýše roven 3,5 a medián ovocné chuti a vůně se rovná 0,0.
Poznámka 1: Pokud je medián znaku hořký a/nebo znaku štiplavý vyšší než 5,0, uvede vedoucí zkušební komise tuto skutečnost do protokolu o zkoušce.
V případě hodnocení, která se provádějí za účelem sledování shody, se provádí jedna zkouška. V případě oponentních hodnocení se musí analýza provést dvakrát při různých zkušebních sezeních. Výsledky duplicitní analýzy musí být statisticky homogenní. (Viz bod 9.5). Pokud tomu tak není, musí se vzorek znovu dvakrát analyzovat. Konečná hodnota mediánu klasifikačních znaků se vypočte na základě průměru obou mediánů.
9.5 Kritéria pro přijímání a zamítání duplikátů
K určení, zda jsou oba výsledky duplicitní analýzy homogenní nebo statisticky přijatelné, se použije níže definovaná normalizovaná chyba:
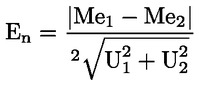
Kde Me1 a Me2 jsou mediány obou duplikátů (respektive první a druhé analýzy) a U1 a U2 jsou rozšířené nejistoty získané pro obě hodnoty, vypočtené následovně, jak je upřesněno v dodatku:
U
1 = c × s* and
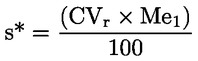
Pro rozšířenou nejistotu, c = 1,96; proto:
U1 = 0,0196 × CVr × Me1
kde CVr je robustní variační koeficient.
Aby bylo možno konstatovat, že obě získané hodnoty nejsou statisticky odlišné, se musí En rovnat 1,0 nebo být nižší než 1,0.
Obrázek 1
PROFILOVÝ LIST PANENSKÉHO OLIVOVÉHO OLEJE
|
Intenzita vnímání vad |
||||
|
Zatuchlý/po kalném sedimentu |
|
|
||
|
Plesnivý/vlhký/zemitý |
|
|
||
|
Vinný/octový Kyselý/nakyslý |
|
|
||
|
Po namrzlých olivách (po vlhkém dřevu) |
|
|
||
|
Žluklý |
|
|
||
|
Další negativní znaky: |
|
|
||
|
Deskriptor: |
Kovový □ Trávový □ Po červech □ Hrubý□ Slaný □ Přehřátý nebo připálený □ Po šťávě z plodů□ Espartový □ Po okurkách □ Olejový□ |
|||
|
Intenzita vnímání pozitivních znaků |
||||
|
Ovocná chuť a vůně |
|
|
||
|
|
Nezralý□ |
Zralý□ |
||
|
Hořký |
|
|
||
|
Štiplavý |
|
|
||
|
|
|
|
||
|
Jméno posuzovatele: |
|
Kód posuzovatele: |
||
|
Kód vzorku: |
Podpis: |
|||
|
Datum: |
||||
|
Poznámky: |
||||
Dodatek
Metoda výpočtu mediánu a intervaly spolehlivosti
Medián
![]()
Medián je definován jako reálné číslo Xm, pro které platí, že pravděpodobnost (p), že hodnoty rozdělení (X) jsou nižší než toto číslo (Xm), je nižší a rovna 0,5, a zároveň že pravděpodobnost (p), že hodnoty rozdělení (X) jsou nižší nebo rovné Xm, je vyšší a rovna 0,5. Praktičtější definice zní, že medián je padesátý percentil rozdělení čísel seřazených podle vzestupného pořadí. Zjednodušeně řečeno se jedná o prostřední hodnotu uspořádané množiny lichých čísel nebo průměr dvou prostředních hodnot uspořádané množiny sudých čísel.
Robustní směrodatná odchylka
Za účelem získání spolehlivého odhadu variability kolem průměru je třeba použít robustní směrodatné odchylky podle Stuarta a Kendalla (4). Tento vzorec uvádí robustní odchylku asymptotického typu, tj. robustní odhad variability zvažovaných údajů, kde N je počet pozorování a IQR je mezikvartilové rozpětí, které zahrnuje přesně 50 % případů daného rozdělení pravděpodobnosti.
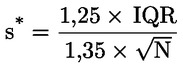
Výpočet mezikvartilového rozpětí se provádí výpočtem velikosti rozdílu odchylky mezi 75.. a 25.. percentilem.
![]()
Pokud je percentilem hodnota Xpc, pro kterou platí, že pravděpodobnost (p), že hodnoty rozdělení jsou nižší než Xpc, je nižší nebo rovna konkrétní setině, a zároveň že pravděpodobnost (p), že hodnoty rozdělení jsou nižší nebo rovny Xpc, je vyšší nebo rovna této konkrétní setině. Setina označuje zvolený fraktil rozdělení. V případě mediánu se rovná 50/100.
Pro praktické účely představuje percentil hodnotu rozdělení, která odpovídá určité ploše vyznačené od křivky rozdělení nebo hustoty. Například 25. percentil představuje hodnotu rozdělení odpovídající ploše, která se rovná 0,25 nebo 25/100.
Tato metoda slouží k výpočtu percentilů na základě reálných hodnot, které se vyskytují v datové matici (postup pro výpočet percentilů).
Robustní variační koeficient (%)
Robustní variační koeficient CVr % představuje bezrozměrné číslo, které udává procento variability analyzované množiny čísel. Z tohoto důvodu je tento koeficient velice užitečný pro ověření spolehlivosti posuzovatelů ve zkušební komisi.
95% intervaly spolehlivosti mediánu
95% intervaly spolehlivosti (hodnota chyby prvního druhu se rovná 0,05 nebo 5 %) představují interval, v němž by se hodnota mediánu mohla měnit za předpokladu, že by bylo možné pokus donekonečna opakovat. V praxi označuje interval proměnlivosti pokusu za přijatých provozních podmínek vycházející z předpokladu, že by se pokus mohl mnohokrát opakovat. Stejně jako v případě CVr% interval napomáhá při hodnocení spolehlivosti pokusu.
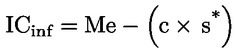
kde C = 1,96 v případě intervalu spolehlivosti na úrovni 95 %.
Příklad výpočetního listu je uveden v příloze I normy IOC/T 20/Doc. č. 15.
Odkazy
1) Wilkinson, L. 1990. Systat: The system for statistics. Evanston, IL. SYSTAT Inc.
2) Cicchitelli, G. 1984. Probabilità e Statistica. Maggioli Editore, Rimini.
3) Massart, D. L.; Vandeginste, B. G. M.; Deming, Y.; Michotte, L. 1988. Chemometrics. A textbook. Elsevier. Amsterdam.
4) Kendall, M. G.; Stuart, A. 1967. The advanced theory of statistics. Vol. 1. Hafner Publishing Co.
5) McGill, R.; Tukey, J. W.; Larsen, W. A. 1978. Variation of Box Plots. The American Statistician, 32, (2), 12-16.
6) IOC/T.28/Doc. č. 1, září 2007, Guidelines for the accreditation of sensory testing laboratories with particular reference to virgin olive oil according to standard ISO/IEC 17025:2005.
7) IOC/T.20/Doc. č. 14.
8) IOC/T.20/Doc. č. 15.
9) ISO/IEC 17025:05.
▼M20 —————
▼M19 —————
PŘÍLOHA XV
1. STANOVENÍ OBSAHU OLEJE V OLIVOVÝCH POKRUTINÁCH
1.1 Přístroje a pomcky
— vhodný extrakční přístroj, vybavený baňkami o objemu 200 ml až 250 ml,
— elektricky vyhřívaná lázeň (písková, vodní apod.) nebo topná deska,
— analytické váhy,
— laboratorní sušárna nastavitelná nejvýše na teplotu 80 °C,
— elektricky vyhřívaná sušárna s termostatickou kontrolou, schopná udržovat teplotu 103 ± 2 °C a zabezpečující provoz při atmosférickém nebo při sníženém tlaku,
— mechanický mlýnek, lehce čistitelný a umožňující rozmletí olivových pokrutin bez jejich zahřátí nebo významné změny vlhkosti a obsahu těkavých látek nebo obsahu oleje,
— extrakční patrony a bavlněná vata nebo filtrační papír, vše prosté látek rozpustných v hexanu,
— exsikátor,
— síto o velikosti otvorů 1 mm,
— pemza, ve formě drobných kousků, nebo jiný granulát zabraňující bouřlivému varu, předem vysušený.
1.2 Chemikálie
Technický hexan, jehož odparek nesmí být větší než 2 mg na 100 ml rozpouštědla.
2. POSTUP
2.1 Příprava zkušebního vzorku
V případě potřeby se k rozemletí laboratorního vzorku použije předem dobře vyčištěný mechanický mlýnek, aby částice zcela prošly sítem.
Asi jedna dvacetina vzorku se použije pro vyčištění mlýnku; tento rozemletý materiál se vyhodí. Zbytek se rozemele a shromáždí, důkladně promíchá a neprodleně analyzuje.
2.2 Velikost zkušebního vzorku
Po dokončení mletí se naváží asi 10 g vzorku s přesností na 0,01 g.
2.3 Příprava extrakční patrony
Zkušební vzorek se převede do extrakční patrony a utěsní se vatovým tamponem. Je-li použit filtrační papír, zkušební vzorek se do něj zabalí.
2.4 Vysušení předem
Jsou-li olivové pokrutiny velmi vlhké (tj. vlhkost a obsah těkavých látek je vyšší než 10 %), provede se vysušení předem umístěním naplněné extrakční patrony (nebo filtračního papíru) na vhodnou dobu do sušárny vyhřáté na teplotu nejvýše 80 °C, aby došlo ke snížení vlhkosti a obsahu těkavých látek na hodnotu menší než 10 %.
2.5 Příprava extrakční baňky
Extrakční baňka s několika kousky pemzy, která byla předem vysušena v sušárně při teplotě 103 ± 2 °C a zchlazena po dobu nejméně 1 hodiny v exsikátoru, se zváží s přesností na 1 mg.
2.6 První extrakce
Extrakční patrona (nebo filtrační papír) se zkušebním vzorkem se vloží do extrakčního přístroje. Do baňky se nalije potřebné množství hexanu. Extrakční baňka se nasadí na extrakční přístroj a to celé se umístí na elektricky vyhřívanou lázeň. Extrakční baňka se zahřívá tak, aby kondenzace extrakčního rozpouštědla byla nejméně 3 kapky za sekundu (tzn. mírný, ne prudký var). Extrahuje se 4 hodiny. Potom se extrakční přístroj nechá vychladnout. Extrakční patrona se vyjme z extrakčního přístroje a postaví se do proudu vzduchu, aby se odpařila většina rozpouštědla.
2.7 Druhá extrakce
Obsah extrakční patrony se vysype do mikromlýnku a co nejjemněji se rozemele. Rozemletá směs se převede zpět do extrakční patrony, která se vloží zpět do extrakčního přístroje.
Znovu se extrahuje další dvě hodiny do téže extrakční baňky, obsahující již prvou část extraktu.
Roztok obsažený v extrakční baňce musí být čirý. Není-li tomu tak, je nutno jej přefiltrovat přes filtrační papír, přičemž původní baňka a filtrační papír se několikrát propláchnou hexanem. Filtrát a proplachovací rozpouštědlo se převede do druhé předem vysušené baňky zvážené s přesností na 1 mg.
2.8 Odstranění rozpouštědla a vážení extraktu
Větší část rozpouštědla se z extrakční baňky odstraní oddestilováním na elektricky vyhřívané lázni. Poslední stopy rozpouštědla se odstraní zahříváním extrakční baňky po dobu 20 minut v sušárně při 103 ± 2 °C. Zbytku rozpouštědla se mohou odstranit proudem vzduchu, anebo výhodněji inertním plynem zaváděným do baňky po krátkou dobu, nebo pod vakuem.
Baňky s vyextrahovaným olejem se nechají vychladnout na okolní teplotu po dobu nejméně jedné hodiny a zváží se s přesností na 1 mg.
Zvážené baňky se za stejných podmínek znovu zahřívají po dobu 10 minut, opět se nechají vychladnout v exsikátoru a znovu se zváží.
Rozdíl mezi dvěma váženími nesmí být vyšší než 10 mg. Pokud je rozdíl vyšší, opakuje se zahřívání, vychlazení a vážení tak dlouho, dokud rozdíl mezi dvěma po sobě následujícími váženími je nejvýše 10 mg. Potom se zaznamená konečná hmotnost baňky.
Na stejném zkušebním vzorku se vždy provedou dvě zkoušky;.
3. VYJÁDŘENÍ VÝSLEDKŮ
3.1 Metoda výpočtu a vzorec
a) Extrakt z původního produktu vyjádřený jako procento hmotnostní se vypočítá takto:
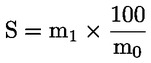
|
kde |
S = procento hmotnostní extraktu z původního produktu, m0 = hmotnost zkušebního vzorku v g, m1 = hmotnost extraktu po vysušení v g. |
Za výsledek se považuje aritmetický průměr dvou stanovení za předpokladu, že jsou splněny požadavky na opakovatelnost.
Výsledek se uvádí s přesností na jedno desetinné místo.
b) Extrakt jako procento hmotnostní v sušině se vypočítá takto:
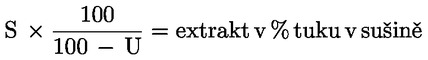
|
kde |
S = procento hmotnostní extraktu z půdního produktu, U = jeho vlhkost a obsah těkavých látek. |
3.2 Opakovatelnost
Absolutní rozdíl mezi dvěma jednotlivými na sobě nezávislými výsledky zkoušky, získanými stejným pracovníkem v rozmezí krátkého časového intervalu nebo souběžně může činit nejvýše 0,2 g hexanového extraktu na 100 g zkušebního vzorku.
Pokud je rozdíl větší, analýza se opakuje na dalších dvou zkušebních vzorcích. Pokud je rozdíl opět vyšší než 0,2 g, za výsledek se považuje aritmetický průměr všech čtyřech stanovení.
PŘÍLOHA XVI
STANOVENÍ JODOVÉHO ČÍSLA
1. OBLAST PŮSOBNOSTI
Tato mezinárodní norma specifikuje metodu stanovení jodového čísla v živočišných a rostlinných tucích a olejích, dále jen „tuky“.
2. DEFINICE
Pro účely této mezinárodní normy platí tato definice:
|
2.1 |
Jodové číslo: vyjadřuje hmotnost jodu vázaného na vzorek za pracovních podmínek specifikovaných touto mezinárodní normou. Jodové číslo je vyjádřeno v g jodu na 100 g vzorku. |
3. PODSTATA ZKOUŠKY
Zkušební vzorek se rozpustí v rozpouštědle a přidá se Wijsovo činidlo. Po stanoveném čase se přidá roztok jodidu draselného a voda a uvolněný jod se titruje roztokem thiosíranu sodného.
4. CHEMIKÁLIE
Všechna činidla musí mít čistotu p. a.
|
4.2 |
Jodid draselný, 100 g/l roztoku; nesmí obsahovat volný jod nebo jodičnany. |
|
4.3 |
Škrobový roztok. 5 g rozpustného škrobu se smísí se 30 ml vody a ke směsi se přidá 1 000 ml vroucí vody, vaří se tři minuty a roztok se nechá vychladnout. |
|
4.4 |
Thiosíran sodný, standardní odměrný roztok c(Na2S2O3.5H2O) = 0,1 mol/l, standardizovaný ne déle než 7 dnů před použitím. |
|
4.5 |
Rozpouštědlo připravené smícháním stejných objemů cyklohexanu a kyseliny octové. |
|
4.6 |
Wijsovo činidlo, obsahující jodmonochlorid v kyselině octové. Doporučuje se použít Wijsovo činidlo obchodně dostupné. Poznámka:Činidlo obsahuje 9 g ICl3 + 9 g I v kyselině octové. |
5. PŘÍSTROJE A POMŮCKY
Běžné laboratorní vybavení, a zejména:
|
5.1 |
Skleněná váženka, odpovídající hmotnosti zkušebního vzorku a hodící se do baňky (5.2). |
|
5.2 |
Erlenmeyerovy baňky na 500 ml se skleněnou zabroušenou zátkou, dokonale vysušené. |
6. PŘÍPRAVA ZKUŠEBNÍHO VZORKU
Homogenizovaný vzorek se vysuší síranem sodným a přefiltruje.
7. POSTUP ZKOUŠKY
|
7.1 |
Zkušební vzorek Hmotnost zkušebního vzorku se liší podle očekávaného jodového čísla, viz tabulka 1.
Tabulka 1
Zkušební vzorek se navažuje s přesností 0,1 mg do skleněné váženky (5.1). |
|
7.2 |
Stanovení Váženka se zkušebním vzorkem se vloží do 500ml baňky (6.2). Přidá se 20 ml rozpouštědla (4.5), a tuk se rozpustí. Přidá se přesně 25 ml Wijsova činidla (4.6), baňka se uzavře zátkou, obsah se promíchá a baňka se umístí na tmavé místo. Pipetování Wijsova činidla se neprovádí ústy. Stejným způsobem se připraví slepý pokus s přídavkem rozpouštědla a Wijsova činidla, ale vynechá se zkušební vzorek. Pro vzorky s jodovým číslem nižším než 150 se baňka ponechá 1 hodinu v temnotě, pro vzorky s jodovým číslem nad 150, pro polymerované výrobky a výrobky ve značné míře oxidované je nutno dobu prodloužit na 2 hodiny. Po této době se do každé baňky přidá 20 ml roztoku jodidu draselného (4.2) a 150 ml vody (4.1). Obsah baňky se titruje standardním odměrným roztokem thiosíranu sodného (4.4), dokud téměř nezmizí žluté zbarvení jodu. Potom se přidá několik kapek škrobového roztoku (4.3) a pokračuje se v titraci, dokud po intenzivním protřepání nezmizí modré zabarvení. Poznámka:Připouští se stanovení bodu ekvivalence potenciometrickou indikací. |
|
7.3 |
Počet stanovení Z jednoho zkušebního vzorku se provádějí dvě stanovení. |
8. VYJÁDŘENÍ VÝSLEDKŮ
Jodové číslo se vypočítá podle vzorce:
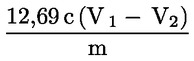
kde
|
c |
= |
přesná koncentrace standardního odměrného roztoku thiosíranu sodného (4.4) v mol/l, |
|
V1 |
= |
objem použitého standardního odměrného roztoku thiosíranu sodného (4.4) na slepý pokus v ml, |
|
V2 |
= |
objem použitého standardního odměrného roztoku thiosíranu sodného (4.4) pro vlastní stanovení v ml, |
|
m |
= |
hmotnost zkušebního vzorku v g (7.1). |
Jako výsledek se uvádí aritmetický průměr dvou stanovení za předpokladu, že je splněna podmínka opakovatelnosti.
PŘÍLOHA XVII
METODA PRO STANOVENÍ STIGMASTADIENOLŮ V ROSTLINNÝCH OLEJÍCH
1. ÚČEL
Stanovení stigmastadienolů v rostlinných olejích obsahujících nízké koncentrace těchto uhlovodíků, zejména v panenském olivovém oleji a surovém olivovém oleji z pokrutin.
2. OBLAST PŮSOBNOSTI
Tato norma může být použita pro všechny rostlinné oleje, ačkoliv měření jsou spolehlivá jedině tehdy, leží-li obsah těchto uhlovodíků mezi 0,01 a 4,0 mg/kg. Metoda je obzvlášť vhodná pro zjišťování přítomnosti rafinovaných rostlinných olejů (olivového oleje, olivového oleje z pokrutin, slunečnicového oleje, palmového oleje atd.) v panenském olivovém oleji, protože na rozdíl od panenských olejů rafinované oleje obsahují stigmastadienoly.
3. PODSTATA METODY
Izolace nezmýdelnitelného extraktu. Separace steroidové uhlovodíkové frakce kolonovou chromatografií na silikagelu a analýza pomocí kapilární plynové chromatografie.
4. PŘÍSTROJE A POMŮCKY
|
4.1 |
250 ml baňky pro použití se zpětným chladičem. |
|
4.2 |
Dělicí nálevky na 500 ml. |
|
4.3 |
100 ml baňky s kulatým dnem. |
|
4.4 |
Rotační odpařovač. |
|
4.5 |
Skleněná chromatografická kolona (vnitřní průměr 1,5 až 2,0 cm, délka 50 cm) s teflonovým kohoutem a se dnem se zátkou ze skelné vaty nebo s kotoučem ze sintrovaného skla. Za účelem připravení silikagelové kolony se do chromatografické kolony nalije hexan do výšky přibližně 5 cm a poté se kolonu naplní suspenzí tvořenou 15 g silikagelu a ve 40 ml hexanu pomocí dávek hexanu. Nechá se úplně usadit pomocí mírných vibrací. Přidá se bezvodý síran sodný do výšky přibližně 0,5 cm a nadbytečný hexan se nakonec eluuje. |
|
4.6 |
Plynový chromatograf s plameno-ionizačním detektorem, vstřikovacím systémem pro nástřik pomocí děliče nebo studeným vstřikovacím systémem typu on-column a termostatickou komorou, nastavitelnou na požadovanou teplotu s přesností ± 1 °C. |
|
4.7 |
Kapilární kolona z taveného křemene pro plynovou chromatografii (vnitřní průměr 0,25 nebo 0,32 mm, délka 25 m) potažená 5 % fenylmethylsilikonem o tloušťce filmu 0,25 mm. Poznámka 1: Mohou být použity jiné kolony s podobnou nebo nižší polaritou. |
|
4.8 |
Zapisovač s integrátorem s možností integrace mezi dvěma minimálními hodnotami. |
|
4.9 |
Mikrostříkačka na 5 až 10 ml pro plynovou chromatografii opatřená tvrzenou jehlou. |
|
4.10 |
Elektrický ohřívací plášť nebo topná deska. |
5. ČINIDLA
Všechna činidla musí být čistoty p. a., není-li uvedeno jinak. Používá se destilovaná voda nebo voda přinejmenším ekvivalentní čistoty.
|
5.1 |
Hexan nebo směs alkanů s bodem varu v rozmezí od 65 do 70 oC, destilované v rektifikační koloně. Hexan lze nahradit isooktanem (2,2,4-trimethylpentanem pro chromatografii) za předpokladu, že je dosaženo srovnatelných hodnot přesnosti. Lze zkontrolovat zbytek po odpaření 100 ml rozpouštědla. Rozpouštědla s vyšším bodem varu, než má n-hexan, se odpařují delší dobu. Z důvodu toxicity hexanu se však upřednostňují. |
|
5.2 |
96 % ethanol (V/V). |
|
5.3 |
Bezvodý síran sodný. |
|
5.4 |
10 % alkoholový roztok hydroxidu draselného. 10 ml vody se přidá k 50 g hydroxidu draselného, zamíchá se a doplní se ethanolem na objem 500 ml. Poznámka 3: Stáním alkoholový uhličitan draselný hnědne. Měl by být připraven každý den čerstvý a uchováván v dobře uzavřených lahvích z tmavého skla. |
|
5.5 |
Silikagel 60 pro kolonovou chromatografii, s velikostí částic 70-230 mesh (Merck, č. zboží 7734 apod.). Poznámka 4: Obvykle může být silikagel použit přímo z balení bez jakéhokoli úpravy. Některé šarže silikagelu však mohou vykazovat nízkou aktivitu, která vede k neuspokojivým chromatografickým separacím. V takových případech je nutno postupovat takto: silikagel se aktivuje nejméně 4hodinovým zahříváním na teplotu 550 °C. Po zahřátí se silikagel vloží do exsikátoru a po vychladnutí se převede do uzavíratelné baňky. Přidá se 2 % vody a protřepává se, dokud nezmizí hrudky a dokud se prášek volně nepřesýpá. Šarže silikagelů, jejichž výsledkem jsou chromatogramy se vzájemně se překrývajícími píky, je nutno upravit výše uvedeným způsobem. Jinou alternativou může být použití extra čistého silikagelu 60 (Merck, č. zboží 7754). |
|
5.6 |
Zásobní roztok (200 ppm) cholesta-3, 5-dienu (Sigma, 99 % čistoty) v hexanu (10 mg v 50 ml). |
|
5.7 |
Standardní roztok cholesta-3, 5-dienu v hexanu o koncentraci 20 ppm získaný naředěním výše uvedeného roztoku. Poznámka 5: Roztoky 5.6 a 5.7 uchovávané při teplotě do 4 °C jsou stabilní po dobu nejméně čtyř měsíců. |
|
5.8 |
Roztok n-nonakosanu v hexanu o koncentraci přibližně 100 ppm. |
|
5.9 |
Nosný plyn pro chromatografii: helium nebo vodík o 99,9990 % čistotě. |
|
5.10 |
Pomocné plyny pro plameno-ionizační detektor: vodík o 99,9990 % čistotě a čistý vzduch. |
6. POSTUP
6.1 Příprava nezmýdelnitelných látek
|
6.1.1 |
Do 250ml baňky (4.1) se naváží 20 ± 0, 1 g oleje, přidá se 1 ml standardního roztoku cholesta-3, 5-dienu (20 μg) a 75 ml 10 % alkoholového roztoku hydroxidu draselného, připojí se zpětný chladič a zahřívá se na mírný var po dobu 30 minut. Baňka obsahující vzorek se odstraní ze zdroje tepla a roztok se nechá mírně vychladnout (nenechá se vychladnout úplně, protože vzorek by se usadil). Přidá se 100 ml vody a roztok se převede do dělící nálevky (4.2) pomocí 100 ml hexanu. Směs se po 30 sekund energicky protřepe a nechá se stát až do separace fází. Poznámka 6: Pokud vznikne emulze, která hned znovu nezmizí, přidávají se malá množství ethanolu. |
|
6.1.2 |
Spodní vodná fáze se převede do druhé dělící nálevky a znovu se extrahuje 100 ml hexanu. Spodní fáze se nechá znovu vypustit a hexanové extrakty (smíchané v jiné dělící nálevce) se třikrát properou pokaždé 100 ml směsi ethanolu a vodu (1:1), dokud se nedocílí neutrální hodnoty pH. |
|
6.1.3 |
Hexanový roztok se nechá projít bezvodým síranem sodným (50 g), propere se 20 ml hexanu a odpaří se v rotačním odpařovači do sucha při 30 °C a za sníženého tlaku. |
6.2 Separace steroidové uhlovodíkové frakce
|
6.2.1 |
Zbytek se převede do rektifikační kolony pomocí dvou 1ml množství hexanu, vzorek se zavede do kolony tak, že se hladina roztoku nechá poklesnout na úroveň síranu sodného, a zahájí se chromatografická eluce s hexanem s rychlostí průtoku přibližně 1 ml/min. Prvních 25 až 30 ml eluátu se vyhodí a použije se dalších 40 ml frakce. Tato frakce se převede do 100ml baňky s kulatým dnem (4.3). Poznámka 7: První frakce obsahuje nasycené uhlovodíky (obrázek 1a) a druhá frakce steroidové uhlovodíky. Další eluce poskytuje squalen a příbuzné sloučeniny. Pro dosažení dobré separace nasycených a steroidových uhlovodíků je nutná optimalizace objemů frakcí. Za tímto účelem je nutno objem první frakce upravit tak, aby při analýze druhé frakce byly píky reprezentující nasycené uhlovodíky nízké (viz obrázek 1c). Pokud se žádné takové píky neobjeví, avšak standardní píky mají zároveň pouze omezenou velikost, je třeba objem první frakce snížit. Úplná separace složek první a druhé frakce navíc není nutná, poněvadž během analýzy plynovou chromatografií za podmínek popsaných v bodě 6.3.1 nedochází k žádnému překrývání píků. Optimalizace objemu druhé frakce není zpravidla nutná, protože další složky se dobře separují. Nicméně přítomnost většího píku s retenčním časem přibližně o 1,5 minuty nižším než u standardu je způsobena squalenem a svědčí o špatné separaci. |
|
6.2.2 |
Druhá frakce se zcela odpaří v rotačním odpařovači při 30 °C v mírném vakuu a zbytek se rozpustí neprodleně v 0,2 ml hexanu. Roztok se až do analýzy uchovává v ledničce. Poznámka 8: Zbytky 6.1.3 a 6.2.2 by neměly být přechovávány v suchém stavu a při pokojové teplotě. Jakmile jsou zbytky získány, je nutno k nim přidat neprodleně rozpouštědla a roztoky uchovávat v ledničce. |
6.3 Plynová chromatografie
|
6.3.1 |
Pracovní podmínky pro nástřik pomocí děliče: — teplota vstřikovací komůrky: 300 °C, — teplota detektoru: 320 °C, — integrátor se zapisovačem: parametry pro integraci by měly být zvoleny tak, aby se správně určily plochy píků. Doporučuje se integrace mezi dvěma minimy, — citlivost: zhruba 16násobek nejmenšího útlumu, — množství nastříknutého roztoku: 1 μl roztoku, — programu termostatu: 6minutová izoterma o hodnotě 235 °C, potom nárůst na teplotu 285 °C rychlostí 2 °C/min, — vstřikovací komůrka s rozdělovačem toku (dělicí poměr 1:15), — nosný plyn: helium nebo vodík o tlaku asi 120 kPa. Tyto podmínky mohou být upraveny podle specifikací chromatografu a kolony tak, aby poskytly chromatogramy splňující následující podmínky: pík vnitřního standardu se musí objevit během pěti minut před, nebo po čase uvedeném v bodu 6.3.2 a musí dosáhnout nejméně 80 % plného rozsahu stupnice. Systém plynové chromatografie je nutno přezkoušet nástřikem směsi zásobního roztoku cholestadienu (5.6) a roztoku n-nonakosanu (5.8). Pík cholesta-3, 5-dienu se musí objevit dříve, než pík n-nonakosanu (obrázek 1c). Pokud se neobjeví, je možno snížit teplotu termostatu a/nebo nahradit kolonu pro plynovou chromatografii kolonou s nižší polaritou. |
|
6.3.2 |
Identifikace píků Pík vnitřního standardu se objeví přibližně za 19 minut a píky stigmastadienolů s relativním retenčním časem přibližně 1,29 (viz obrázek 1b). Stigmasta-3, 5-dien se objevuje s malými množstvími izomeru a obvykle se obě tyto látky eluují současně jako jediný pík. Je-li však kolona příliš polární nebo vykazuje-li vysokou rozlišovací schopnost, může se izomer objevit jako malý pík těsně před píkem stigmasta-3, 5-dienu (obrázek 2). Aby byla zajištěna eluce stigmastadienolů jako jednoho píku, doporučuje se nahradit kolonu kolonou s nižší polaritou, nebo kolonou s větším vnitřním průměrem. Poznámka 9: Referenční chromatogram pro stigmastadienoly je možné získat analýzou rafinovaných rostlinných olejů s použitím menšího vzorku (1 až 2 g). Stigmastadienoly vytvářejí nápadný a snadno identifikovatelný pík. |
|
6.3.3 |
Kvantitativní analýza Obsah stigmastadienolů se stanoví podle vzorce: mg/kg stigmastadienolů =
Detekční limit: asi 0,01 mg/kg. Poznámka 10: Pokud se stigmastadienoly vyskytují v koncentraci vyšší než 4 mg/kg a je-li požadována kvantifikace, musí být použita metoda Mezinárodní rady pro olivy pro stanovení sterenů v rafinovaných olejích. |
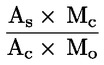
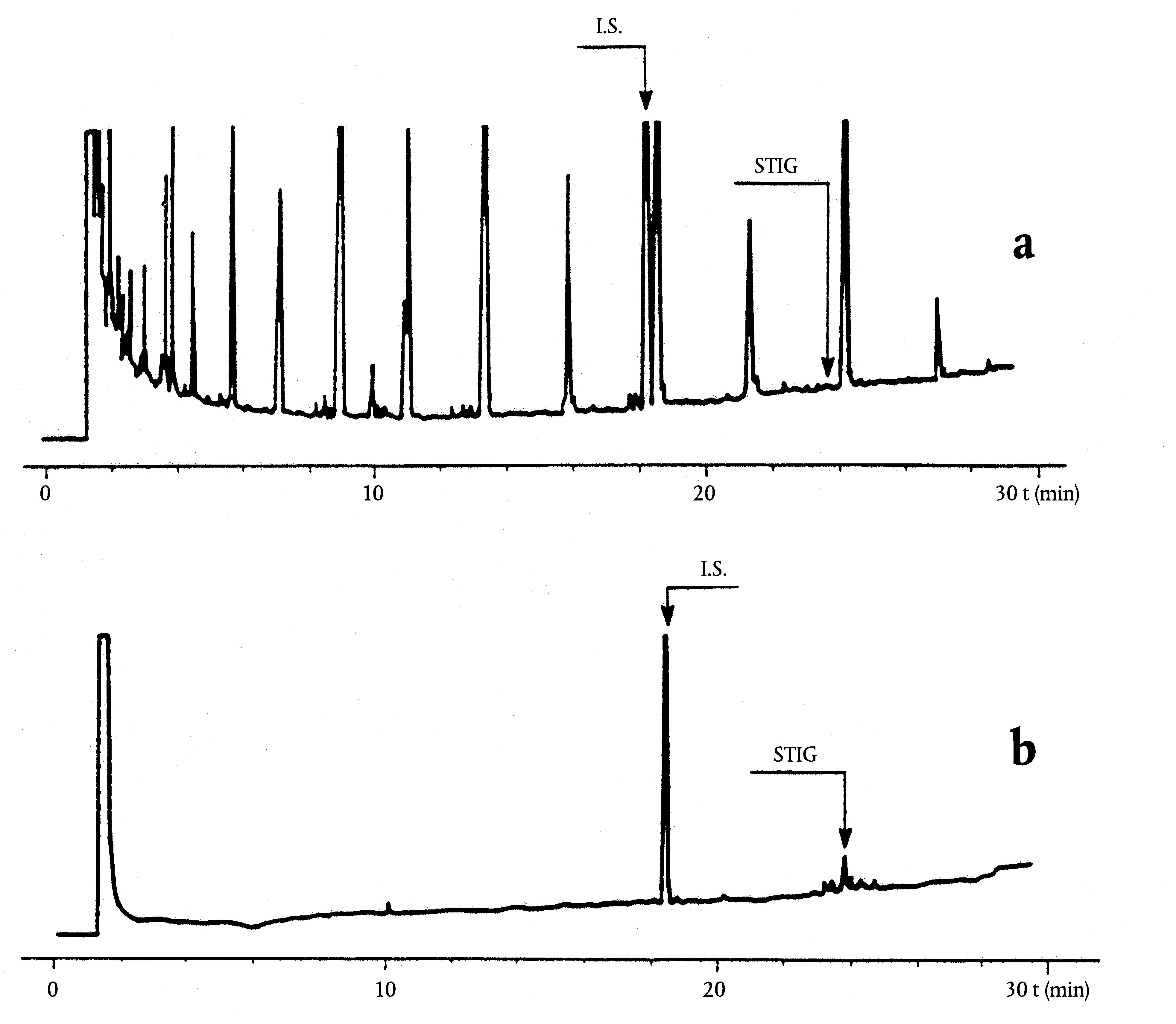
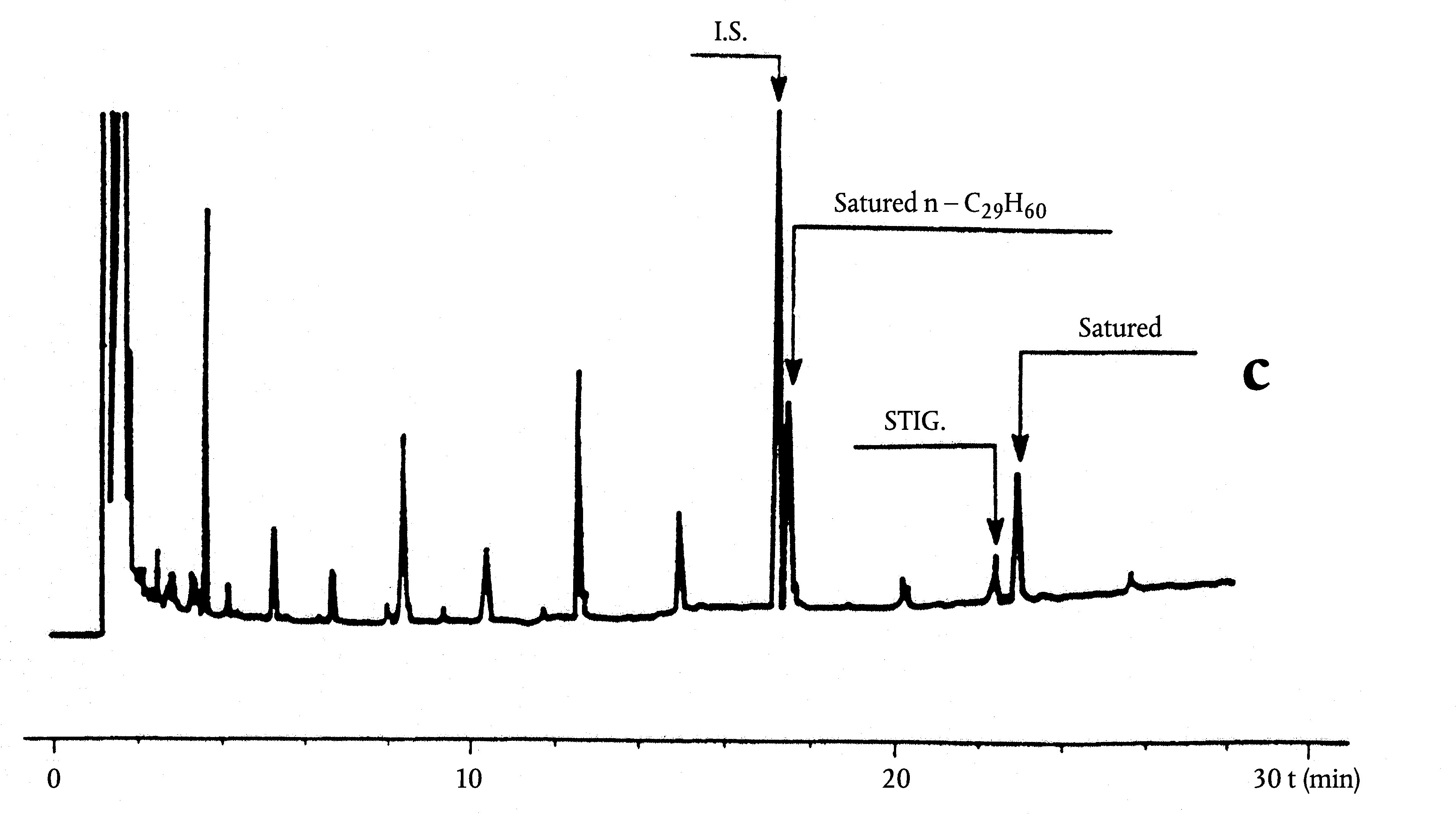 Obrázek č. 1
Obrázek č. 1
Plynové chromatogramy vzorků olivového oleje analyzovaných v kapilární koloně z taveného křemene (vnitřní průměr 0,25 mm, délka 25 m) potažené 5 % fenylmethylsilikonem o tloušťce filmu 0,25 μm.
a) první frakce (30 ml) z panenského olivového oleje se standardem,
b) druhá frakce (40 ml) z olivového oleje obsahujícího 0,10 mg/kg stigmastadienolů,
c) druhá frakce (40 ml) obsahující malou část první frakce.
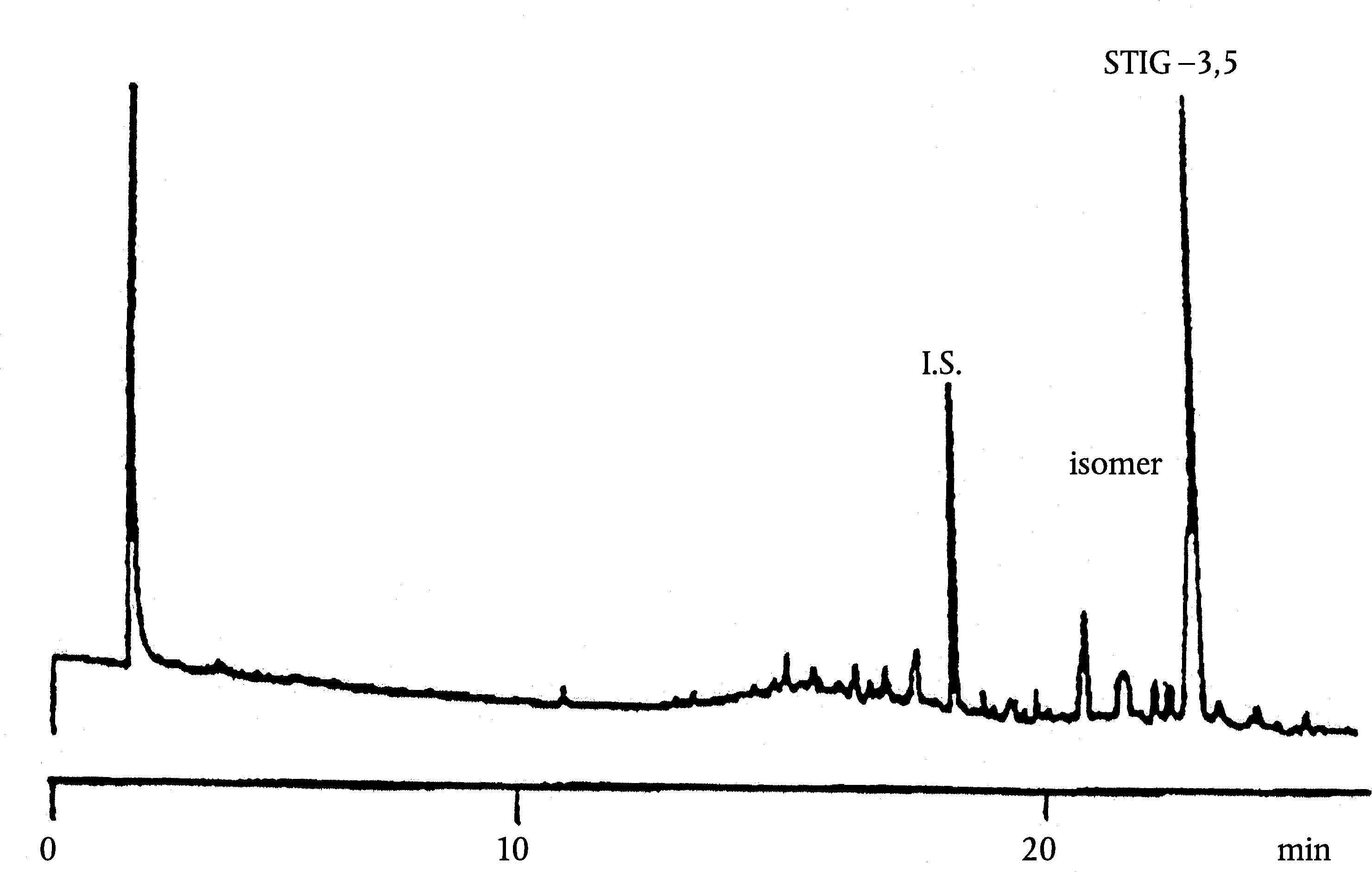 Obrázek č. 2
Obrázek č. 2
Plynový chromatogram získaný ze vzorku rafinovaného olivového oleje analyzovaného na koloně DB-5, vykazující izomer stigmasta-3, 5-dienu.
PŘÍLOHA XVIII
STANOVENÍ ROZDÍLU MEZI SKUTEČNÝM A TEORETICKÝM OBSAHEM TRIACYLGLYCEROLŮ S EKVIVALENTNÍM POČTEM UHLÍKOVÝCH ATOMŮ 42 (ECN 42)
1. OBLAST PŮSOBNOSTI
Stanovení absolutního rozdílu mezi experimentálními hodnotami triacylglycerolů (TAG) s ekvivalentním počtem atomů uhlíku 42, které byly v oleji stanoveny vysoce účinnou kapalinovou chromatografií (ECN42HPLC), a teoretickou hodnotou TAG s ekvivalentním počtem atomů uhlíku 42 vypočtenou ze složení mastných kyselin (ECN 42teoretická).
2. OBLAST POUŽITÍ
Tato metoda je použitelná pro olivové oleje. Metoda je použitelná pro určení přítomnosti malých množství semenného oleje (bohatého na linoleovou kyselinu) v olivových olejích všech tříd.
3. PRINCIP
Obsah triacylglycerolů ECN42 stanovený analyticky pomocí HPLC a teoretický obsah triacylglycerolů ECN42 (vypočítaný ze stanovení složení mastných kyselin pomocí GLC) odpovídá v jistém rozmezí pravému olivovému oleji. Pokud je rozdíl větší, než hodnoty přijaté pro každý druh oleje, znamená to, že olej obsahuje semenné oleje.
4. METODA
Metoda výpočtu teoretického obsahu triacylglycerolů s ECN 42 a rozdílu od údajů získaných HPLC je v podstatě provedená kombinací analytických údajů získaných jinými metodami. Dala by se rozdělit do tří fází: stanovení složení mastných kyselin kapilární plynovou chromatografií, výpočet teoretického složení triacylglycerolů s ECN 42, stanovení triacylglycerolů s ECN 42 pomocí HPLC.
4.1 Přístroje a pomůcky
|
4.1.1 |
Baňky s kulatým dnem (250 ml a 500 ml) |
|
4.1.2 |
Kádinky (100 ml) |
|
4.1.3 |
Skleněná chromatografická kolona o vnitřním průměru 21 mm, délce 450 mm, s kohoutem a standardně ukončená na horním konci spojkou s vnitřním zábrusem |
|
4.1.4 |
Dělicí nálevky (250 ml) s normalizovaným kohoutem na spodu kužele, vhodné ke spojení s horní částí kolony |
|
4.1.5 |
Skleněná tyčinka o délce 600 mm |
|
4.1.6 |
Skleněná nálevka o průměru 80 mm |
|
4.1.7 |
Odměrné baňky (50 ml) |
|
4.1.8 |
Odměrné baňky (20 ml) |
|
4.1.9 |
Rotační odparka |
|
4.1.10 |
Vysoce účinný kapalinový chromatograf s termostatickou regulací teploty kolony |
|
4.1.11 |
Dávkovače na nástřik 10 μl dávek |
|
4.1.12 |
Detektor: diferenciální refraktometr; schopnost měření v rozsahu nejméně 10–4 jednotky refrakčního indexu |
|
4.1.13 |
Kolona: korozivzdorná trubice o délce 250 mm a vnitřním průměru 4,5 mm plněná silikagelem o průměru částic 5 μm a obsahu uhlíku ve formě oktadecylsilanu 22 % až 23 % |
|
4.1.14 |
Programové vybavení pro zpracování dat |
|
4.1.15 |
Nádobky (přibližně 2 ml) se septy potaženými teflonem a šroubovými uzávěry |
4.2 Činidla
Reakční činidla by měla být analytické čistoty. Eluční rozpouštědla by měla být odplyněna a mohou být několikrát recyklována, aniž by ovlivnila separaci.
|
4.2.1 |
Petrolether pro chromatografii (40–60 oC) nebo hexan. Hexan lze nahradit isooktanem (2,2,4-trimethylpentanem pro chromatografii) za předpokladu, že je dosaženo srovnatelných hodnot přesnosti. Rozpouštědla s vyšším bodem varu, než má n-hexan, se odpařují delší dobu. Z důvodu toxicity hexanu se však upřednostňují. |
|
4.2.2 |
Čerstvě destilovaný diethylether bez peroxidu |
|
4.2.3 |
Eluční rozpouštědla pro čištění oleje směsí petroletheru/diethyletheru 87/13 (v/v) pro sloupcovou chromotografii |
|
4.2.4 |
Silikagel typu Merck 7734 o velikosti částic 70–230 s normalizovaným obsahem vody 5 % hmotnostních |
|
4.2.5 |
Skelná vata |
|
4.2.6 |
Aceton pro HPLC |
|
4.2.7 |
Acetonitril nebo propionitril pro HPLC |
|
4.2.8 |
Eluční rozpouštědlo pro HPLC: acetonitril + aceton (poměr se upraví podle požadované separace; začíná se se směsí o poměru 50:50) nebo propionitril |
|
4.2.9 |
Solubilizační rozpouštědlo: aceton |
|
4.2.10 |
Referenční triglyceridy: mohou se použít komerční triglyceridy (tripalmitin, triolein atd.); retenční časy se pak vynesou podle ekvivalentního počtu atomů uhlíku, nebo se mohou použít referenční chromatogramy pro sojový olej, směs sojového oleje a olivového oleje v poměru 30:70 a olivový olej (viz poznámky 1 a 2 a obrázky 1 až 4). |
|
4.2.11 |
Kolonka (6 ml) pro extrakci na pevnou fázi se silikagelem (1 g) |
|
4.2.12 |
Heptan pro chromatografii. Heptan lze nahradit isooktanem (2,2,4-trimethylpentanem pro chromatografii). |
4.3 Příprava vzorků
Vzhledem k celé řadě rušivých látek, které mohou vést k falešným pozitivním výsledkům, musí být vzorek vždy čištěn podle metody 2.507 IUPAC používané pro stanovení polárních sloučenin a tuků na smažení.
4.3.1 Příprava chromatografické kolony
Kolona (4.1.3) se naplní 30 ml elučního rozpouštědla (4.2.3). Pak se dovnitř kolony vsune trochu skelné vaty (4.2.5) a zatlačí se směrem dolů za pomocí skleněné tyčinky (4.1.5).
V 100 ml kádince se rozmíchá 25 g silikagelu (4.2.4) v 80 ml eluční směsi (4.2.3), poté se za pomocí skleněné nálevky (4.1.6) nalije do kolony.
Aby se veškerý silikagel vpravil do kolony, vypláchne se kádinka elučním roztokem a tento podíl se rovněž nalije do kolony.
Otevře se kohout a rozpouštědlo se nechá z kolony eluovat tak dlouho, dokud jeho hladina není přibližně 1 cm nad úrovní silikagelu.
4.3.2 Kolonová chromatografie
Olej se přefiltruje, homogenizuje a případně zbaví vody; poté se do 50 ml odměrné baňky (4.1.7) odváží 2,5 ± 0,1 g oleje s přesností na 0,001 g.
Olej se rozpustí přibližně ve 20 ml elučního rozpouštědla (4.2.3.). Pro snazší rozpouštění se případně lehce zahřeje. Poté se zchladí na pokojovou teplotu a objem se doplní elučním rozpouštědlem.
Za pomocí odměrné pipety se do kolony vpraví 20 ml roztoku připraveného podle bodu 4.3.1, otevře se kohout a rozpouštědlo se nechá eluovat na úroveň silikagelu.
Poté se eluuje 150 ml elučního rozpouštědla (4.2.3); průtok rozpouštědla se nastaví přibližně na 2 ml/min (150 ml projde kolonou přibližně za 60–70 minut).
Eluát se jímá do 250 ml baňky s kulatým dnem (4.1.1) předem vysušené v sušárně a přesně zvážené. Rozpouštědlo se odstraní za sníženého tlaku v rotační odparce (4.1.9) a residuum se zváží. Residuum se použije pro přípravu roztoku k analýze HPLC a pro přípravu methylesteru.
Výtěžek vzorku z kolony musí být nejméně 90 % pro kategorie extrapanenský, panenský, rafinovaný a olivový olej, a nejméně 80 % pro lampantový olej a olivový olej z pokrutin.
4.3.3 Přečištění SPE
Silikagelová kolona SPE se aktivuje průchodem 6 ml hexanu (4.2.3) ve vakuu, aby se zabránilo vysušení.
Množství 0,12 g se s přesností na 0,001 g odváží do 2 ml lahviček (4.1.15) a rozpustí se v 0,5 ml hexanu (4.2.3).
Roztok se vlije do kolony SPE a eluuje se ve vakuu s 10 ml roztoku hexan–diethylether (87:13 v/v) (4.2.3).
Shromážděné frakce se odpaří do sucha v rotačním odparce (4.1.9) za sníženého tlaku a při pokojové teplotě. K analýze triacylglycerolů (TAG) se reziduum rozpustí ve 2 ml acetonu (4.2.6).
4.4 Analýza HPLC
4.4.1 Příprava vzorků pro chromatografickou analýzu
5 % roztok analyzovaného vzorku se připraví odvážením 0,5 ± 0,001 g vzorku do a 10 ml odměrné baňky a doplněním solubilizačním rozpouštědlem (4.2.9) na objem 10 ml.
4.4.2 Postup
Nastavení chromatografického systému. Eluční rozpouštědlo (4.2.8) se pumpuje rychlostí 1,5 ml/min, aby se celý systém propláchl. Počká se, až se ustálí základní linie.
Velikost nástřiku vzorku připraveného podle bodu 4.3 je 10 μl.
4.4.3 Výpočet a vyjádření výsledků
Používá se metoda normalizace plochy, tj. předpokládá se, že součet ploch všech píků odpovídajících triacylglycerolům s ECN42 až ECN52 je roven 100 %.
Relativní procentní zastoupení jednotlivých triglyceridů se vypočítá podle vzorce:
![]()
.
Výsledky by měly být uvedeny alespoň na dvě desetinná místa.
Viz poznámky 1 až 4.
4.5 Výpočet triacylglycerolového složení (% molární) z údajů o složení mastných kyselin (% plochy)
4.5.1 Stanovení složení mastných kyselin
Složení mastných kyselin se určuje podle ISO 5508 pomocí kapilární kolony. Methylestery se připraví podle COI/T.20/Doc. No 24.
4.5.2 Mastné kyseliny pro výpočet
Glyceridy se seskupují podle ekvivalentního počtu uhlíkových atomů (ECN) a zohledňuje se následná ekvivalence mezi ECN a mastnými kyselinami. V úvahu se berou pouze mastné kyseliny s počtem uhlíkových atomů mezi 16 a 18, protože jedině takové mastné kyseliny jsou významné z hlediska olivového oleje. Mastné kyseliny se normalizují do 100 %.
|
Mastná kyselina |
Zkratka |
Molekulová hmotnost (MW) |
ECN |
|
palmitová kyselina |
P |
256,4 |
16 |
|
palmitoolejová kyselina |
Po |
254,4 |
14 |
|
stearová kyselina |
S |
284,5 |
18 |
|
olejová kyselina |
O |
282,5 |
16 |
|
linoleová kyselina |
L |
280,4 |
14 |
|
linolenová kyselina |
Ln |
278,4 |
12 |
4.5.3 Převod plochy (%) na moly pro všechny mastné kyseliny (1)
|
|
|
|
|
|
|
|
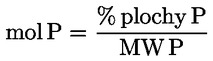
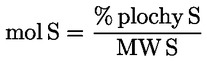
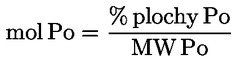
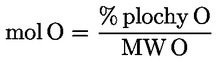
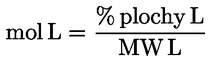
4.5.4 Normalizace molů mastných kyselin na 100 % (2)
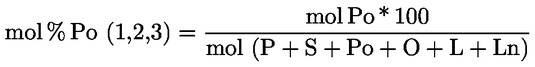
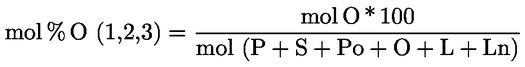
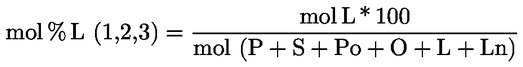
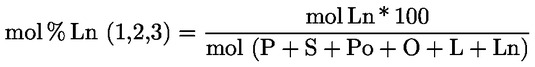
Výsledkem je procentní zastoupení každé mastné kyseliny v % molárních ve všech (1,2,3-) polohách triacylglycerolů.
Celkové množství nasycených mastných kyselin (SFA) P a S a nenasycených mastných kyselin (UFA) Po, O, L a Ln se vypočítá:
![]()
![]()
4.5.5 Výpočet složení mastných kyselin v pozicích 2- a 1,3- triacylglycerolů
Mastné kyseliny jsou rozděleny do tří skupin takto: jedna s polohou 2- a dvě stejné s polohou 1- a 3-, s různými koeficienty pro nasycené kyseliny (P a S) a nenasycené kyseliny (Po, O, L a Ln).
4.5.5.1 Nasycené mastné kyseliny s polohou 2- [P(2) a S(2)]
![]()
![]()
4.5.5.2 Nenasycené mastné kyseliny s polohou 2- [Po(2), O(2), L(2) a Ln(2)] (5):
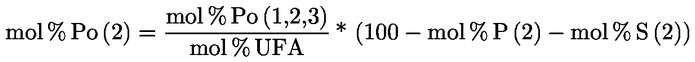
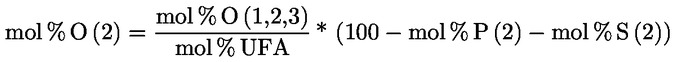
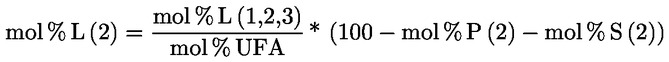
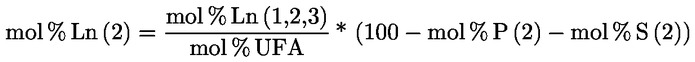
4.5.5.3 Mastné kyseliny s polohou 1,3- [P(1,3), S(1,3), Po(1,3), O(1,3), L(1,3) a Ln(1,3)] (6):
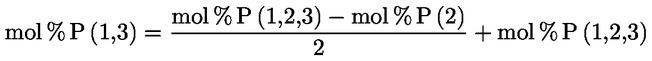
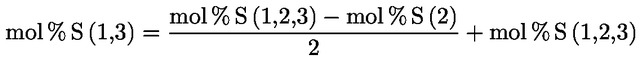
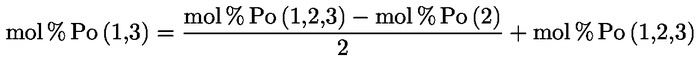
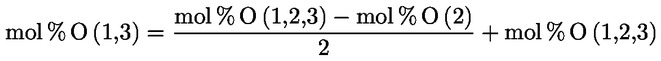
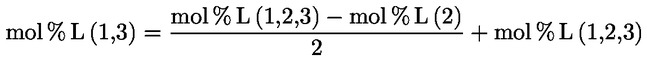
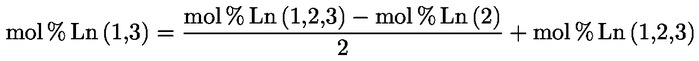
4.5.6 Výpočet triacylglycerolů
4.5.6.1 Triacylglyceroly s jednou mastnou kyselinou (AAA, zde LLL, PoPoPo) (7)
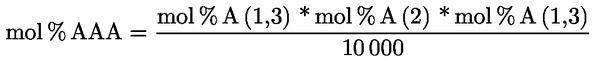
4.5.6.2 Triacylglyceroly se dvěma mastnými kyselinami (AAB, zde PoPoL, PoLL) (8)
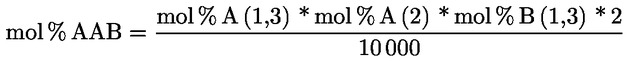
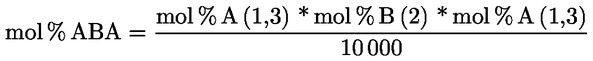
4.5.6.3 Triacylglyceroly se třemi různými mastnými kyselinami (ABC, zde OLLn, PLLn, PoOLn, PPoLn) (9)
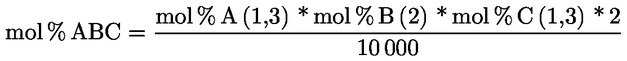
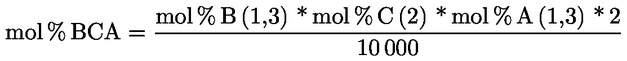
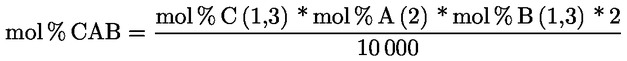
4.5.6.4 Triacylglyceroly s ekvivalentním počtem uhlíkových atomů 42
Triacylglyceroly s ECN42 se vypočítávají podle rovnic 7, 8 a 9 a jsou poté uvedeny v pořadí podle předpokládané eluce při HPLC (obvykle pouze tři píky).
LLL
PoLL a polohový izomer LPoL
OLLn a polohové izomery OLnL a LnOL
PoPoL a polohový izomer PoLPo
PoOLn a polohové izomery OPoLn a OLnPo
PLLn a polohové izomery LLnP a LnPL
PoPoPo
SLnLn a polohový izomer LnSLn
PPoLn a polohové izomery PLnPo a PoPLn
Triacylglyceroly s ECN42 se vyjádří jako součet devíti triacylglycerolů včetně jejich polohových izomerů. Výsledky by měly být uvedeny alespoň na dvě desetinná místa.
5. HODNOCENÍ VÝSLEDKŮ
Porovná se vypočtený teoretický obsah a obsah stanovený analýzou HPLC. Pokud je absolutní hodnota rozdílu výsledku z HPLC a výsledku určeného teoreticky vyšší než hodnota stanovená normou pro příslušnou kategorii oleje, vzorek obsahuje semenný olej.
Výsledky se uvádí na dvě desetinná místa.
6. PŘÍKLAD (ČÍSELNÉ ODKAZY SE VZTAHUJÍ NA ODDÍL TEXTU POPISUJÍCÍ METODU)
— 4.5.1 Výpočet % molárních mastných kyselin z údajů GLC (% normalizované oblasti)
Následující údaje o složení mastných kyselin byly získány pomocí GLC:
|
FA |
P |
S |
Po |
O |
L |
Ln |
|
MW |
256,4 |
284,5 |
254,4 |
282,5 |
280,4 |
278,4 |
|
% plochy |
10,0 |
3,0 |
1,0 |
75,0 |
10,0 |
1,0 |
— 4.5.3 Převedení plochy (%) na látkové množství (mol) pro všechny mastné kyseliny (viz vzorec (1))
|
mol P |
= |
|
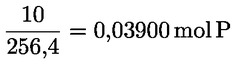
|
mol S |
= |
|
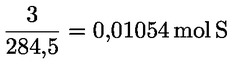
|
mol Po |
= |
|
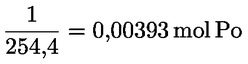
|
mol O |
= |
|
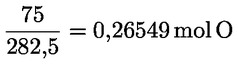
|
mol L |
= |
|
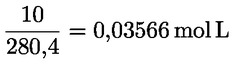
|
mol Ln |
= |
|
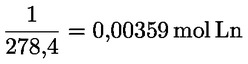
|
Celkem |
= |
0,35821 mol TAG |
— 4.5.4 Normalizace mastných kyselin (mol) na 100 % (viz vzorec (2))
|
mol % P(1,2,3) |
= |
|
|
mol % S(1,2,3) |
= |
|
|
mol % Po(1,2,3) |
= |
|
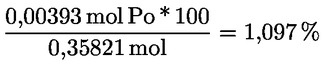
|
mol % O(1,2,3) |
= |
|
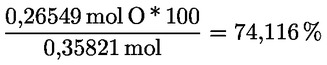
|
mol % L(1,2,3) |
= |
|
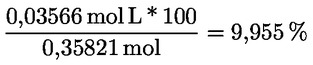
|
mol % Ln(1,2,3) |
= |
|
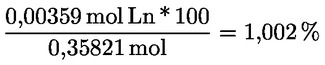
|
Celkem % molárních |
= |
100 % |
Součet nasycených a nenasycených mastných kyselin v poloze 1,2,3- TAG (viz vzorec (3):
![]()
![]()
— 4.5.5 Výpočet složení mastných kyselin v polohách 2- a 1,3- TAG
— 4.5.5.1 Nasycené mastné kyseliny v poloze 2- [P(2) a S(2)] (viz vzorec (4))
![]()
![]()
— 4.5.5.2 Nenasycené mastné kyseliny v poloze 2- [Po(1,3), O(1,3), L(1,3) a Ln(1,3)] (viz vzorec (5))
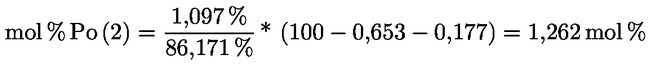
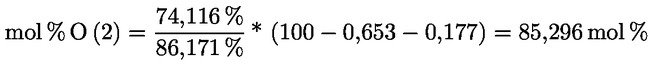
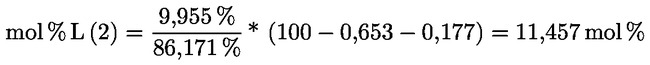
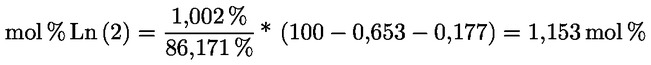
— 4.5.5.3 Mastné kyseliny v poloze 1,3- [P(1,3), S(1,3), Po(1,3), O(1,3), L(1,3) a Ln(1,3)] (viz vzorec (6))
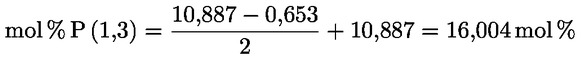
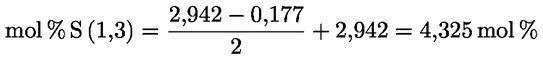
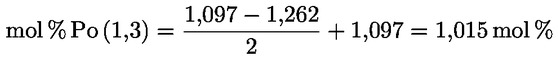
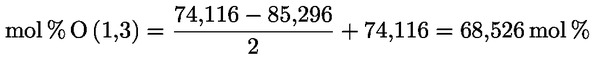
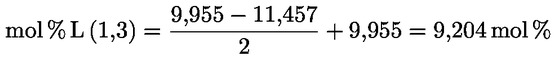
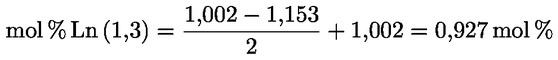
— 4.5.6 Výpočet triacylglycerolů
Z vypočítaného složení mastných kyselin v polohách 2- a 1,3-:
|
Mastné kyseliny v |
poloze 1,3- |
poloze 2- |
|
P |
16,004 % |
0,653 % |
|
S |
4,325 % |
0,177 % |
|
Po |
1,015 % |
1,262 % |
|
O |
68,526 % |
85,296 % |
|
L |
9,204 % |
11,457 % |
|
Ln |
0,927 % |
1,153 % |
|
Celkem |
100,0 % |
100,0 % |
se vypočítají následující triacylglyceroly:
LLL
PoPoPo
PoLL s 1 polohovým izomerem
SLnLn s 1 polohovým izomerem
PoPoL s 1 polohovým izomerem
PPoLn se 2 polohovými izomery
OLLn se 2 polohovými izomery
PLLn se 2 polohovými izomery
PoOLn se 2 polohovými izomery
— 4.5.6.1. Triacylglyceroly s jednou mastnou kyselinou (LLL, PoPoPo) (viz vzorec (7))
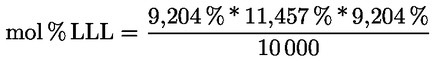 = 0,09706 mol LLL
= 0,09706 mol LLL
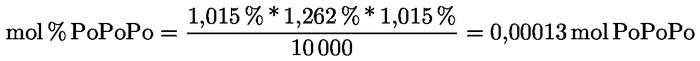
— 4.5.6.2 Triacylglyceroly se dvěma mastnými kyselinami (PoLL, SLnLn, PoPoL) (viz vzorec (8))
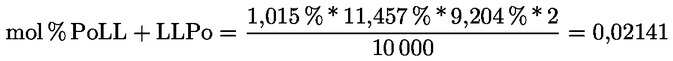
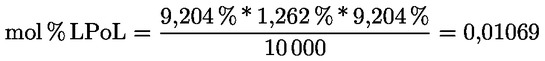
0,03210 mol PoLL
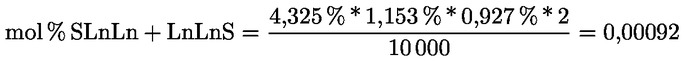
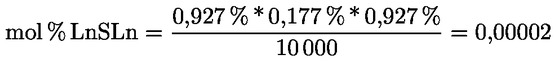
0,00094 mol SLnLn
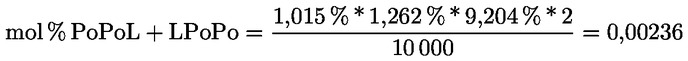
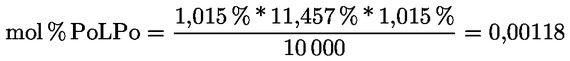
0,00354 mol PoPoL
— 4.5.6.3 Triacylglyceroly se třemi různými mastnými kyselinami (PoPLn, OLLn, PLLn, PoOLn) viz vzorec (9)
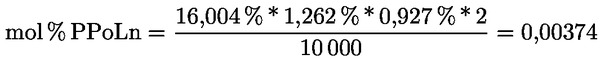
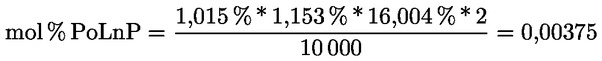
0,00761 mol PPoLn
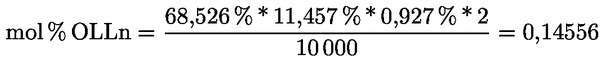
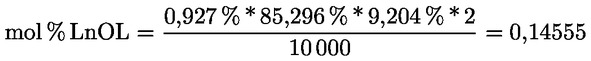
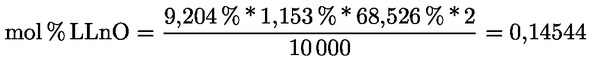
0,43655 mol OLLn
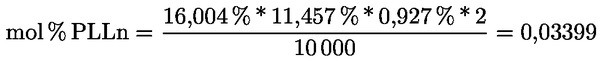
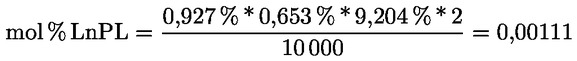
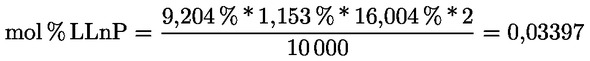
0,06907 mol PLLn
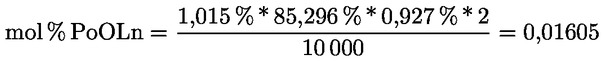
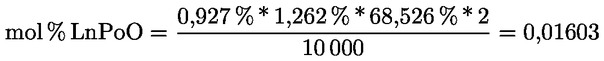
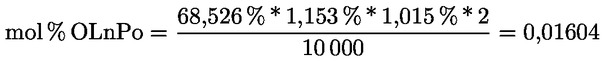
0,04812 mol PoOLn
ECN42 = 0,69512 mol TAG
Poznámka 1 Pořadí eluce je možné stanovit výpočtem ekvivalentního počtu atomů uhlíku, často definovaných jako
![]() , kde CN je počet atomů uhlíku a n je počet dvojných vazeb. Výpočet může být přesnější, pokud se vezme v úvahu původ dvojné vazby. Pokud jsou no, nl a nln počty dvojných vazeb náležejících olejové, linoleové resp. linolenové kyselině, ekvivalentní počet atomů uhlíku se může vypočítat použitím následujícího vzorce:
, kde CN je počet atomů uhlíku a n je počet dvojných vazeb. Výpočet může být přesnější, pokud se vezme v úvahu původ dvojné vazby. Pokud jsou no, nl a nln počty dvojných vazeb náležejících olejové, linoleové resp. linolenové kyselině, ekvivalentní počet atomů uhlíku se může vypočítat použitím následujícího vzorce:
![]()
kde se koeficienty do, dl a dln mohou vypočítat použitím referenčních triglyceridů. Za podmínek popsaných v této metodě platí přibližně tento vztah:
![]()
Poznámka 2 S několika referenčními triglyceridy je rovněž možné vypočítat rozlišení vzhledem k trioleinu:
![]()
trioleinza použití sníženého retenčního času
![]()
.
Grafické vyjádření závislosti log α na f (počtu dvojných vazeb) umožňuje určit retenční hodnoty všech triglyceridů mastných kyselin obsažených v referenčních triglyceridech – viz obrázek 1.
Poznámka 3 Účinnost kolony by měla umožnit jasné oddělení píku trilinoleinu od píků triglyceridů s blízkým retenčním časem RT. Eluce se zastaví po píku odpovídajícímu ECN 52.
Poznámka 4 Správnost měření ploch píků sledovaných v rámci tohoto stanovení je zajištěna, pokud druhý pík odpovídající ECN 50 je 50 % plného rozsahu záznamové stupnice.
Obrázek 1
Graf závislosti log α na f (počtu dvojných vazeb)
Počet dvojných vazeb
La: laurová kyselina; My: myristová kyselina; P: palmitová kyselina; S: stearová kyselina; O: olejová kyselina; L: linoleová kyselina; Ln: linolenová kyselina
Obrázek 2
Olivový olej s nízkým obsahem linoleové kyseliny
a
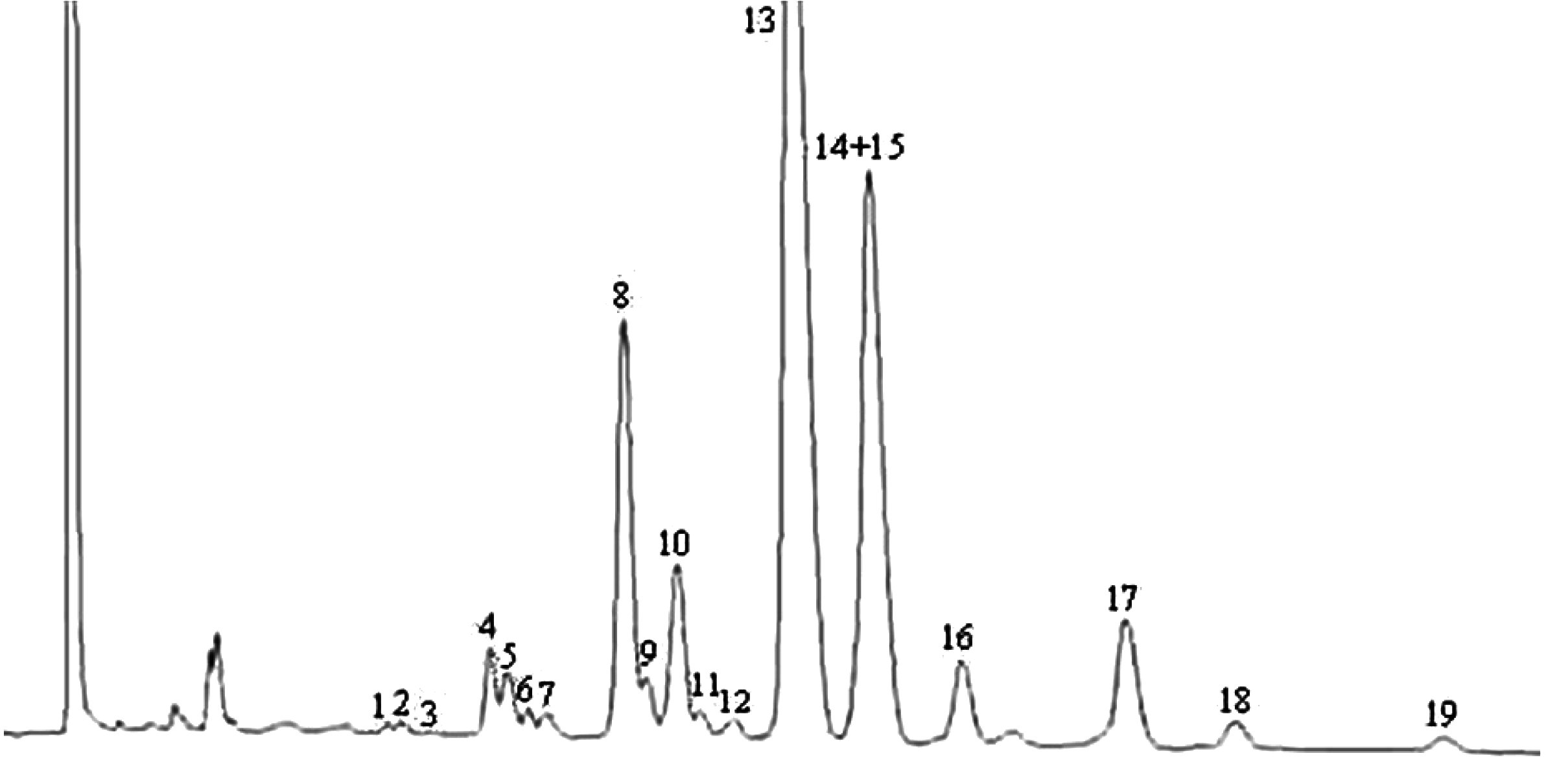
Rozpouštědlo: aceton/acetonitril
Profil a: hlavní složky chromatografických píků: ECN42: (1) LLL + PoLL; (2) OLLn + PoOLn; (3) PLLn; ECN44: (4) OLL + PoOL; (5) OOLn + PLL; (6) POLn + PPoPo; (7) OOL + PoOO; ECN46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; (12) PLP; ECN48: (13) OOO + PoPP; (14 + 15) SOL + POO; (16) POP; ECN50: (17) SOO; (18) POS + SLS.
b
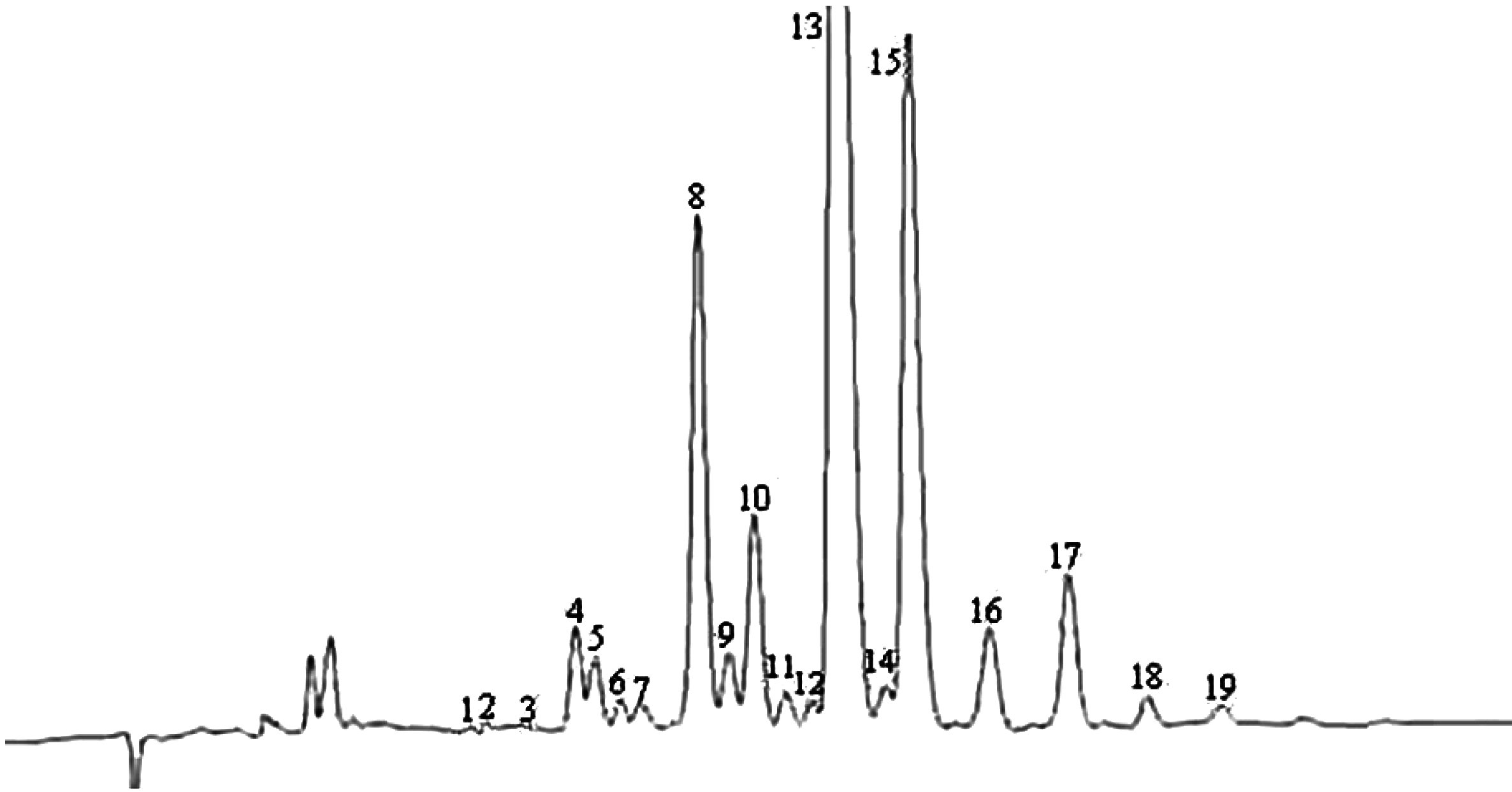
Rozpouštědlo: propionitril
Profil b: hlavní složky chromatografických píků: ECN42: (1) LLL; (2) OLLn + PoLL; (3) PLLn; ECN44: (4) OLL; (5) OOLn + PoOL; (6) PLL + PoPoO; (7) POLn + PPoPo + PPoL; ECN46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; (12) PLP; ECN48: (13) OOO + PoPP; (14) SOL; (15) POO; (16) POP; ECN50: (17) SOO; (18) POS + SLS
Obrázek 3
Olivový olej s vysokým obsahem linoleové kyseliny
a)
Rozpouštědlo: aceton/acetonitril (50:50)
Profil a: hlavní složky chromatografických píků: ECN42: (1‘) LLL + PoLL; (2‘) OLLn + PoOLn; (3‘) PLLn; ECN44: (4‘) OLL + PoOL; (5‘) OOLn + PLL; (6‘) POLn + PPoPo; ECN46: (7‘) OOL + PoOO; (8‘) PLO + SLL + PoOP; (9‘) PLP + PoPP; ECN48: (10‘) OOO; (11‘) POO + SLL + PPoO; (12‘) POP + PLS; ECN50: (13‘) SOO; (14‘) POS + SLS
b)
Rozpouštědlo: propionitril
Profil b: hlavní složky chromatografických píků: ECN42: (1) LLL; (2 + 2‘) OLLn + PoLL; (3) PLLn; ECN44: (4) OLL; (5) OOLn + PoOL; (6) PLL + PoPoO; (7) POLn + PPoPo + PPoL; ECN46: (8) OOL + LnPP; (9) PoOO; (10) SLL + PLO; (11) PoOP + SPoL + SOLn + SPoPo; ECN48: (12) PLP; (13) OOO + PoPP; (14) SOL; (15) POO; (16) POP; ECN50: (17) SOO; (18) POS + SLS; ECN52: (19) AOO.
PŘÍLOHA XIX
STANOVENÍ SLOŽENÍ A OBSAHU STEROLŮ A ALKOHOLOVÝCH SLOUČENIN KAPILÁRNÍ PLYNOVOU CHROMATOGRAFIÍ
1. OBLAST PŮSOBNOSTI
Tato metoda popisuje postup pro stanovení obsahu jednotlivých alkoholových sloučenin a celkový obsah alkoholových sloučenin v olivových olejích a olivových olejích z pokrutin a jejich směsích.
Alkoholové sloučeniny v olivových olejích a olivových olejích z pokrutin sestávají z alifatických alkoholů, sterolů a triterpenických dialkoholů.
2. PODSTATA METODY
Oleje, k nimž je přidán α-cholestanol a 1-ikosanol jako vnitřní standard, jsou zmýdelněny pomocí ethanolového roztoku hydroxidu draselného; nezmýdelnitelné látky se následně extrahují pomocí diethyletheru.
Jednotlivé sloučeniny alkoholu se od nezmýdelnitelných látek oddělí tenkovrstvou chromatografií na bazické silikagelové desce (referenční metoda) nebo vysoce účinnou kapalinovou chromatografií pomocí kolony naplněné silikagelem. Frakce oddělená ze silikagelu se převede na trimethylsilylethery a analyzuje se pomocí kapilární plynové chromatografie.
ČÁST 1
PŘÍPRAVA NEZMÝDELNITELNÝCH LÁTEK
1. OBLAST PŮSOBNOSTI
Tato část popisuje přípravu a extrakci nezmýdelnitelných látek. Zahrnuje přípravu a extrakci nezmýdelnitelných látek z olivového oleje a olivového oleje z pokrutin.
2. PODSTATA METODY
Vzorek se zmýdelní varem pod zpětným chladičem s ethanolovým roztokem hydroxidu draselného. Nezmýdelnitelné látky se extrahují pomocí diethyletheru.
3. PŘÍSTROJE A POMŮCKY
Obvyklé laboratorní vybavení a zejména:
3.1. Baňka s kulatým dnem na 250 ml vybavená zpětným chladičem se zábrusem.
3.2. Dělící nálevka na 500 ml.
3.3. Baňka na 250 ml.
3.4. Mikrostříkačky na 100 μl a 500 μl.
3.5. Válcová filtrační nálevka s pórovitou fritou G3 (pórovitost 15 až 40 μm) o průměru přibližně 2 cm a výšce 5 cm, vhodná pro vakuovou filtraci a s vnějším zábrusem.
3.6. Erlenmeyerova baňka na 50 ml s vnitřním zábrusem pro použití s filtrační nálevkou (3.5).
3.7. Zkumavka na 10 ml s kónickým dnem a skleněnou zátkou.
3.8. Exsikátor s chloridem vápenatým.
4. ČINIDLA
4.1. Hydroxid draselný, minimální titr 85 %.
4.2. Hydroxid draselný ve formě ethanolového roztoku o koncentraci přibližně 2 M.
130 g hydroxidu draselného (4.1) se za současného chlazení rozpustí ve 200 ml destilované vody a doplní se do jednoho litru ethanolem (4.7). Tento roztok je nutno uchovávat v dobře uzavřených skleněných láhvích z tmavého skla a skladovat po dobu maximálně dvou dnů.
4.3. Diethylether čistoty p.a.
4.4. Bezvodý síran sodný čistoty p.a.
4.5. Aceton čistoty pro chromatografii.
4.6. Diethylether čistoty pro chromatografii.
4.7. Ethanol čistoty p.a.
4.8. Ethylacetát čistoty p.a.
4.9. Vnitřní standard, α-cholestanol o čistotě vyšší než 99 % (čistota musí být kontrolována pomocí analýzy plynovou chromatografií).
4.10. Vnitřní standardní roztok α-cholestanolu, roztok (0,2 m/V) v ethylacetátu (4.8).
4.11. Roztok fenolftaleinu 10 g/l v ethanolu (bod 4.7).
4.12. 0,1 % roztok 1-ikosanolu v ethylacetátu (m/V) (vnitřní standard).
5. POSTUP
Pomocí mikrostříkačky na 500 μl (3.4) se do baňky na 250 ml (3.1) dá objem vnitřního standardního roztoku α-cholestanolu (4.10) a objem 1-ikosanolu (4.12) obsahující množství cholestanolu a ikosanolu rovnající se přibližně 10 % obsahu sterolu a alkoholu ve vzorku. Například při analýze olivového oleje se na 5 g vzorku olivového oleje přidá 500 μl roztoku α-cholestanolu (4.10) a 250 μl roztoku 1-ikosanolu (4.12). Při analýze olivového oleje z pokrutin se přidá 1 500 μl roztoku α-cholestanolu (4.10) a 1500 μl roztoku 1-ikosanolu (4.12). Vzorek se nechá odpařit do sucha v mírném proudu dusíku a v teplé vodní lázni. Po vychladnutí baňky se do stejné baňky naváží 5,00 ± 0,01 g vysušeného přefiltrovaného vzorku.
Poznámka 1: Živočišné nebo rostlinné oleje obsahující větší množství cholesterolu mohou vykazovat pík s retenčním časem identickým cholestanolu. Pokud k tomu dojde, musí být sterolová frakce analyzována jednou s vnitřním standardem a jednou bez něj.
Přidá se 50 ml ethanolového roztoku hydroxidu draselného o koncentraci 2 M (4.2) a malé množství pemzy, připojí se zpětný chladič a zahřeje se do mírného varu, dokud nedojde ke zmýdelnění (roztok se vyčeří). V zahřívání se pokračuje dalších 20 minut a poté se přes chladič přidá 50 ml destilované vody, chladič se pak odpojí a baňka se vychladí na teplotu přibližně 30 °C.
Obsah baňky se kvantitativně převede do dělicí nálevky na 500 ml (3.2) a propláchne se několika dávkami destilované vody (50 ml). Přidá se přibližně 80 ml diethyletheru (4.6), vše se důkladně protřepe po dobu přibližně 60 sekund a pravidelně se uvolňuje tlak obracením dělicí nálevky a otevíráním kohoutku. Nechá se odstát až do úplného oddělení obou fází (viz poznámka 2). Poté se co nejvíce mýdlového roztoku odčerpá do druhé dělicí nálevky. Stejným způsobem se provedou další dvě extrakce vodně-alkoholové fáze, vždy s použitím 60 až 70 ml diethyletheru (4.6).
Poznámka 2: Všechny emulze je možné odstranit přidáním malých množství ethanolu (4.7).
Všechny tři etherové extrakty se smíchají v jedné dělicí nálevce obsahující 50 ml vody. Dále se promývá vodou (50 ml), dokud se promývací voda při přidání kapky roztoku fenolftaleinu nepřestane zbarvovat růžově (4.11). Odstraní se vodná fáze, etherová fáze se přefiltruje na bezvodém síranu sodném (4.4) do předem zvážené baňky na 250 ml, přičemž nálevka a filtr se promyjí malými množstvími diethyletheru (4.6).
Rozpouštědlo se nechá odpařit destilací ve vakuu v rotačním odpařovači při 30 °C. Přidá se 5 ml acetonu (4.5) a těkavé rozpouštědlo se zcela odstraní mírným proudem dusíku. Zbytek se suší v sušárně při 103 ± 2 °C po dobu 15 min. Po vychlazení v exsikátorech se zváží s přesností na 0,1 mg.
ČÁST 2
SEPARACE FRAKCÍ ALKOHOLOVÝCH SLOUČENIN
1. OBLAST PŮSOBNOSTI
Nezmýdelnitelné látky, jejichž příprava je popsána v části 1, se nechají frakcionovat v různých alkoholových sloučeninách, alifatických alkoholech, sterolech a triterpenických dialkoholech (erythrodiolu a uvaolu).
2. PODSTATA METODY
Nezmýdelnitelné látky lze frakcionovat pomocí základní tenkovrstvé chromatografie (referenční metoda), zviditelnit a odpovídající pásma seškrábat a extrahovat. Alternativní metodou separace je vysoce účinná kapalinová chromatografie za použití kolony se silikagelem a UV detektoru, jejíž pomocí lze získat jednotlivé frakce. Alifatické a triterpenické alkoholy, sterol a triterpenické dialkoholy se společně izolují.
3. PŘÍSTROJE A POMŮCKY
Obvyklé laboratorní vybavení a zejména:
3.1. Kompletní zařízení pro tenkovrstvou chromatografii se skleněnými deskami o rozměrech 20 × 20 cm.
3.2. Ultrafialová lampa s vlnovou délkou 366 nebo 254 nm.
3.3. Mikrostříkačky na 100 μl a 500 μl.
3.4. Válcová filtrační nálevka s pórovitou fritou G3 (pórovitost 15 až 40 μm) o průměru přibližně 2 cm a výšce 5 cm, vhodná pro vakuovou filtraci a s vnějším zábrusem.
3.5. Erlenmeyerova baňka na 50 ml s vnitřním zábrusem pro použití s filtrační nálevkou (3.4).
3.6. Zkumavka na 10 ml s kónickým dnem a skleněnou zátkou.
3.7. Exsikátor s chloridem vápenatým.
3.8. Systém vysoce účinné kapalinové chromatografie složený z:
3.8.1. Binárního čerpadla.
3.8.2. Ručního nebo automatického vstřikovače s 200μl vstřikovací smyčkou.
3.8.3. Integrovaného odplynovací zařízení.
3.8.4. Detektoru UV-VIS nebo IR.
3.9. Kolony pro vysoce účinnou kapalinovou chromatografii (25 cm x 4 mm vnitřní průměr) se silikagelem 60 (velikost částic 5 μm).
3.10. Injekčního filtru, 0,45 μm.
3.11. Erlenmeyerovy baňky na 25 ml.
4. ČINIDLA
4.1. Hydroxid draselný, minimální titr 85 %.
4.2. Hydroxid draselný ve formě ethanolového roztoku o koncentraci přibližně 2 M.
130 g hydroxidu draselného (4.1) se za současného chlazení rozpustí ve 200 ml destilované vody a doplní se do jednoho litru ethanolem (4.9). Tento roztok je nutno uchovávat v dobře uzavřených skleněných láhvích z tmavého skla a skladovat po dobu maximálně dvou dnů.
4.3. Diethylether čistoty p.a.
4.4. Hydroxid draselný ve formě ethanolového roztoku o koncentraci přibližně 0,2 M.
13 g hydroxidu draselného (4.1) se rozpustí ve 20 ml destilované vody a doplní se do jednoho litru ethanolem (4.9).
4.5. Silikagelem potažené skleněné desky (20 x 20) bez fluorescenčního indikátoru o tloušťce 0,25 mm (obchodně dostupné, připraveny k použití).
4.6. Aceton čistoty pro chromatografii.
4.7. n-hexan čistoty pro chromatografii.
4.8. Diethylether čistoty pro chromatografii.
4.9. Ethanol čistoty p.a.
4.10. Ethylacetát čistoty p.a.
4.11. Referenční roztok pro tenkovrstvou chromatografii: cholesterol, fytosteroly, alkoholy a erythrodiol – 5 % roztok v ethylacetátu (4.10).
4.12. Roztok 2,7-dichlor-fluoresceinu, 0,2 % v ethanolovém roztoku. Mírně se alkalizuje přidáním několika kapek 2M alkoholového roztoku hydroxidu draselného (4.2).
4.13. n-hexan (4.7)/diethylether (4.8) směs 65:35 (V/V).
4.14. Mobilní fáze vysoce účinné kapalinové chromatografie: n-hexan (4.7)/diethylether (4.8) směs 1:1 (V/V).
5. REFERENČNÍ METODA SEPARACE ALKOHOLOVÝCH SLOUČENIN POMOCÍ ZÁKLADNÍ TENKOVRSTVÉ CHROMATOGRAFICKÉ DESKY
Příprava bazických desek pro tenkovrstvou chromatografii. Silikagelové desky (4.5) se na 10 sekund ponoří nebo namočí zhruba 4 cm hluboko do ethanolového roztoku hydroxidu draselného o koncentraci 0,2 M (4.4), poté se nechají dvě hodiny sušit v digestoři a nakonec na jednu hodinu umístí do sušárny vyhřáté na 100 °C.
Desky se vyjmou ze sušárny a až do okamžiku použití uloží do exsikátoru s chloridem vápenatým (3.7) (takto upravené desky musí být použity během patnácti dnů).
Vyvíjecí komora se naplní směsí hexanu a diethyletheru (4.13) (viz poznámka 3) do výšky přibližně 1 cm. Vyvíjecí komora se uzavře vhodným krytem a nejméně půl hodiny ponechá v klidu a chladnu, aby se mohla vytvořit rovnováha mezi parami a kapalinou. Na vnitřní povrch vyvíjecí komory je možné připojit proužky filtračního papíru namočené v eluční směsi za účelem zkrácení doby vyvolávání přibližně o jednu třetinu a docílení stejnoměrnější, pravidelné eluce složek.
Poznámka 3: Vyvíjecí roztok musí být pro každou analýzu vyměněn, aby bylo dosaženo dokonale reprodukovatelných podmínek vyvíjení. Lze použít alternativní rozpouštědlo 50:50 (V/V) n-hexanu a diethyletheru.
Připraví se přibližně 5 % roztok nezmýdelnitelné látky, jejíž příprava je popsána v části 1, v ethylacetátu (4.10) a 0,3 ml tohoto roztoku se pomocí mikrostříkačky na 100 μl (3.3) nastříkne jako úzký a rovnoměrný pruh k dolnímu okraji (2 cm) chromatografické desky (4.5). Ve stejné úrovni se nanese 2 až 3 μl referenčního roztoku materiálu (4.11) za účelem stanovení sterolového pásma a pásma triterpenických dialkoholů a alkoholů po dokončení vyvíjení.
Deska se umístí do vyvíjecí komory (3.1). Teplota okolí se udržuje mezi 15 a 20 °C (viz poznámka 4). Vyvíjecí komora se okamžitě uzavře a vzorek se nechá eluovat, dokud čelo rozpouštědla nedosáhne přibližně 1 cm od horního okraje desky. Deska se poté vyjme z vyvíjecí komory a rozpouštědlo se nechá odpařit pod proudem horkého vzduchu nebo se deska na chvíli nechá v digestoři.
Poznámka 4: Vyšší teplota může zhoršit oddělování.
Deska se lehce a rovnoměrně postříká roztokem 2,7-dichlor-fluoresceinu (4.12) a nechá se uschnout. Při pozorování pod UV lampou (3.2) je možné stanovit sterolové pásmo a pásmo triterpenických dialkoholů a alkoholů porovnáním skvrn vytvořených referenčním roztokem (bod 4.11). Obrysy pásem podél okrajů fluorescence se vyznačí černým značkovačem (viz obrázek tenkovrstvé desky č. 1).
Silikagel se z označené oblasti seškrábe pomocí kovové stěrky. Odebraná hmota se rozmělní na malé části a převede do filtrační nálevky (3.4). Přidá se 10 ml horkého ethylacetátu (4.10). Obsah se důkladně promíchá kovovou stěrkou a přefiltruje (v případě potřeby ve vakuu). Filtrát se shromáždí v Erlenmeyerově baňce (3.5) připojené na filtrační nálevku.
Zbytek v baňce se třikrát propláchne diethyletherem (4.3) (přibližně 10 ml při každém propláchnutí). Filtrát se přitom sbírá do stejné baňky připojené k nálevce. Filtrát se odpaří na objem přibližně 4 až 5 ml a zbytkový roztok se převede do předem zvážené zkumavky na 10 ml (3.6). Zkumavka se vysuší pod mírným proudem dusíku. Zbytek se znovu rozpustí několika kapkami acetonu (4.6) a opět vysuší. Zbytek ve zkumavce je tvořen sterolem a triterpenickýcmi dialkoholy nebo frakcemi alkoholů a triterpenických dialkoholů.
6. SEPARACE ALKOHOLOVÉ FRAKCE POMOCÍ VYSOCE ÚČINNÉ KAPALINOVÉ CHROMATOGRAFIE
Nezmýdelnitelné látky získané podle postupu v části 1 se rozpustí ve 3 ml mobilní fáze (4.14), roztok se přefiltruje přes injekční filtr (3.10) a uchová se.
200 μl přefiltrovaného roztoku nezmýdelnitelných látek se vstříkne do zařízení vysoce účinné kapalinové chromatogrofie (3.8).
Nechá se proběhnout separace vysoce účinnou kapalinovou chromatografií při toku 0,8 ml/min. Prvních pět minut se nezohlední; do 25 ml Erlenmeyerových baněk (3.11) se zachytí alifatické a triterpenické alkoholy odseparované mezi 5. a 10. minutou a steroly a erytrodiol a uvaol odseparované mezi 11. a 25. minutou (viz poznámka 5).
Separaci lze sledovat UV detektorem při vlnové délce 210 nm nebo detektorem indexu lomu (viz obrázek 6).
Frakce se odpaří do sucha a připraví se na chromatografickou analýzu.
Poznámka 5: Je potřeba pečlivě sledovat tlak čerpadla vysoce účinné kapalinové chromatografie, protože diethylether může způsobit zvýšení tlaku – pro udržení tlaku pod kontrolou je zapotřebí upravit průtok.
ČÁST 3
ANALÝZA FRAKCÍ ALKOHOLOVÝCH SLOUČENIN PLYNOVOU CHROMATOGRAFIÍ
1. OBLAST PŮSOBNOSTI
Tato část popisuje použití plynové chromatografie v kapilární koloně pro stanovení kvalitativního a kvantitativního složení alkoholových sloučenin izolovaných podle metody uvedené v části 2.
2. PODSTATA METODY
Frakce získané z nezmýdelnitelných látek pomocí tenkovrstvé chromatografie nebo vysokoúčinné kapalinové chromatografie se derivatizují na trimethylsilylethery a analyzují pomocí kapilární plynové chromatografie opatřené nástřikovým zařízením s děličem a plameno-ionizačním detektorem.
3. PŘÍSTROJE A POMŮCKY
Obvyklé laboratorní vybavení a zejména:
3.1. Zkumavka na 10 ml s kónickým dnem a skleněnou zátkou.
3.2. Plynový chromatograf vhodný pro použití s kapilární kolonou, opatřený nástřikovým zařízením s děličem složený z:
3.2.1. termostatické kolonové komory schopné udržovat požadovanou teplotu s přesností ± 1 °C;
3.2.2. vstřikovací jednotky s termostatem (vstřikové komory) s povlakem ze silanizovaného skla a s dělicím systémem;
3.2.3. plameno-ionizačního detektoru (FID);
3.2.4. systému pro sběr dat vhodného pro provoz s detektorem FID (3.10.3) s možností manuální integrace.
3.3. Kapilární kolona z taveného křemene dlouhá 20 až 30 m, s vnitřním průměrem 0,25 až 0,32 mm, potažená 5 % difenyl – 95 % dimethylpolysiloxanem (stacionární fáze SE-52 nebo SE-54 nebo ekvivalentní), o jednotné tloušťce 0,10 až 0,30 μm.
3.4. Mikrostříkačka na 10 μl pro plynovou chromatografii opatřená tvrzenou jehlou pro nástřik pomocí děliče.
4. ČINIDLA
4.1. Bezvodý pyridin pro chromatografii.
4.2. Hexamethyl disilazan čistoty p.a.
4.3. Trimethyl-chlorsilan čistoty p.a.
4.4. Zkušební roztoky sterol(trimethylsilyl)etherů. Připraví se ze sterolů a z erythrodiolu získaných z olejů, které je obsahují, těsně před použitím.
4.5. Standardní roztoky trimethylsilyletherů alifatických alkoholů C20 až C28. Mohou se připravit ze směsí čistých alkoholů v době požadovaného použití.
4.6. Nosný plyn: vodík nebo helium, čisté pro plynovou chromatografii.
4.7. Pomocné plyny: vodík, helium, dusík a vzduch, čisté pro plynovou chromatografii.
4.8. Činidlo pro silylaci tvořené směsí pyridinu, hexamethyl-disilazanu a trimethyl-chlorsilanu v poměru 9:3:1 (V/V/V).
4.9. n-hexan čistoty pro chromatografii.
5. PŘÍPRAVA TRIMETHYLSILYLETHERŮ
Do zkumavky (3.1) obsahující frakci alkoholových sloučenin se přidá činidlo pro silylaci (4.8) (viz poznámka 6), v množství 50 μl na každý mg alkoholové sloučeniny. Přitom je nutné zabránit jakékoli absorpci vlhkosti (viz poznámka 7).
Poznámka 6: Roztoky připravené k použití jsou obchodně dostupné. Dostupná jsou rovněž další silylační činidla, jako například N,O-bis-(trimethylsilyl)trifluoracetamid + 1 % trimethyl-chlor-silan pro smíchání se stejným objemem bezvodého pyridinu. Pyridin lze nahradit stejným množstvím acetonitrilu.
Poznámka 7: Vznik slabé opalescence je normální a nezpůsobuje žádnou anomálii. Tvorba bílých vloček nebo existence růžového zabarvení jsou známkami přítomné vlhkosti nebo degradace činidla. V tomto případě se zkouška musí opakovat (pouze v případě, že byl použit hexamethyl-disilazan/trimethyl-chlorsilan).
Zkumavka (3.1) se uzavře a pečlivě protřepe bez převracení, dokud se sloučeniny nerozpustí. Poté se nechá v klidu nejméně 15 minut při laboratorní teplotě a následně několik minut odstřeďuje. Čirý roztok je připraven pro plynovou chromatografickou analýzu.
6. ANALÝZA PLYNOVOU CHROMATOGRAFIÍ
6.1. Přípravné operace a kondicionace kapilární kolony
Kapilární kolona se připojí (3.3) k plynovému chromatografu tak, že vstup kolony se připojí k nástřikovému zařízení s děličem a výstup kolony se připojí k detektoru.
Provede se kompletní kontrola plynového chromatografu (těsnost plynových okruhů, účinnost detektoru, účinnost oddělovacího a zapisovacího systému atd.).
Kolonu, která má být použita poprvé, je nutno nejprve kondicionovat. Kolonou se nechá protékat malé množství plynu, poté se zapne plynový chromatograf a nechá se postupně zahřívat, dokud není dosaženo teploty nejméně o 20 °C vyšší, než je provozní teplota (viz poznámka 8). Tato teplota se udržuje nejméně dvě hodiny a poté se systém převede do pracovního režimu (regulace průtoku plynu, oddělování, zapálení plamene, připojení výpočetního systému, nastavení teploty kolony, detektoru, injektoru atd.) a zaznamená se signál s citlivostí, která činí nejméně dvojnásobek citlivosti uvažované pro provedení analýzy. Základní linie záznamu musí být rovná, bez jakýchkoli píků nebo driftů. Záporné drifty jsou známkou nedokonalé těsnosti spojů kolony, zatímco kladné svědčí o nedostatečné kondicionaci kolony.
Poznámka 8: Teplota kondicionace musí být nejméně o 20 °C nižší než maximální teplota použitelná pro uvažovanou stacionární fázi.
6.2. Provozní podmínky
Je zapotřebí optimalizovat teplotní program a průtok nosného plynu tak, aby se získaly chromatogramy podobné chromatogramům na obrázcích 3 až 6.
Byly testovány a shledány jako užitečné následující parametry:
6.2.1. Alifatické alkoholy
|
Program pece |
180 °C (8 min.) → 260 °C (při 5 °C/min.) → 260 °C (15 min.) |
|
Teplota injektoru |
280 °C |
|
Teplota detektoru: |
290 °C |
|
Lineární rychlost nosného plynu: |
helium (20 až 30 cm/s), vodík (30 až 50 cm/s) |
|
Oddělovací poměr: |
1:50 až 1:100 |
|
Vstříknutý objem: |
0,5 až 1 μl trimethylsilyletherového roztoku |
6.2.2. Sterol a triterpenické dialkoholy
|
Program pece |
260 ± 5 °C isotermický |
|
Teplota injektoru |
280 – 300 °C |
|
Teplota detektoru |
280 – 300 °C |
|
Lineární rychlost nosného plynu |
helium (20 až 30 cm/s), vodík (30 až 50 cm/s) |
|
Oddělovací poměr: |
1:50 až 1:100 |
|
Vstříknutý objem: |
0,5 až 1 μl trimethylsilyletherového roztoku |
Tyto podmínky se mohou měnit podle charakteristik kolony a plynového chromatografu s cílem zajistit, aby chromatogramy splňovaly tyto požadavky:
— retenční čas alkoholu C26 musí být 18 ± 5 minut,
— pík alkoholu C22 musí být u olivového oleje 80 ± 20 % plného rozsahu a u oleje z pokrutin 40 ± 20 % plného rozsahu,
— retenční čas β-sitosterolu by měl být 20 ± 5 minut,
— pík kampesterolu musí být u olivového oleje (střední obsah 3 %) 20 ± 5 % plného rozsahu,
— všechny přítomné steroly musí být odděleny. Kromě separace musí být píky též zcela rozlišeny, tj. dráha záznamu píku se musí vrátit na základní linii dříve, než se zvedne do dalšího píku. Neúplné rozlišení je však tolerováno za předpokladu, že pík s relativním retenčním časem 1,02 (sitostanol) je možné kvantifikovat pomocí kolmé úsečky.
6.3. Analytický postup
Do mikrostříkačky na 10 μl (3.4) se natáhne 1 μl hexanu, poté 0,5 μl vzduchu a nakonec 0,5 až 1 μl zkušebního roztoku. Píst mikrostříkačky se povytáhne tak, aby se vyprázdnila jehla. Jehla se zavede přes membránu injektoru a přibližně po jedné nebo dvou sekundách se rychle nastříkne roztok. Přibližně po pěti sekundách se jehla pomalu vytáhne. Lze užít rovněž automatický injektor.
Zaznamenávání je prováděno, dokud přítomná trimethylsilyletherová směs příslušných alkoholových sloučenin není zcela eluována. Základní linie musí i nadále splňovat požadavky odpovídajících provozních podmínek (6.2.1 nebo 6.2.2).
6.4. Identifikace píků
Identifikace jednotlivých píků se provádí podle retenčních časů a porovnáním se směsí alifatických a triterpenických alkoholů nebo sterolu a triterpenických dialkoholů TMSE, analyzovanou za stejných podmínek. Chromatogram frakce alifatických a triterpenických dialkoholů je znázorněn na obrázku 3 a odpovídající chromatogramy sterolů a triterpenických dialkoholů na obrázku 2.
Alifatické alkoholy se eluují v tomto pořadí: C20-ol (V.S.), C22-ol, C23-ol, C24-ol, C25-ol, C26-ol, C27-ol a C28-ol.
Steroly a triterpenické dialkoholy se eluují v tomto pořadí: cholesterol, brassikasterol, ergosterol, 24-methylen-cholesterol, kampesterol, kampestanol, stigmasterol, Δ7-kampesterol, Δ5,23-stigmastadienol, chlerosterol, β-sistosterol, sitostanol, Δ5-avenasterol, Δ5,24-stigmastadienol, Δ7-stigmastenol, Δ7-avenasterol, erythrodiol a uvaol.
6.5. Kvantitativní vyhodnocení
Pomocí systému sběru dat se vypočítají plochy píků 1-ikosanolu a alifatických alkoholů C22, C24, C26 a C28. Faktor odezvy 1-eikosanolu by se měl považovat za rovný 1.
Pomocí výpočetního systému se vypočítá plocha píků α-cholestanolu, sterolů a triterpenických dialkoholů. Píky jakýchkoli sloučenin, které nejsou mezi sloučeninami uvedenými v tabulce 1 (výpočet pro ergosterol se provádět nesmí) se ignorují. Faktor odezvy α-cholestanolu by se měl považovat za rovný 1.
Obsah jednotlivých alkoholových sloučenin vyjádřený v mg/kg tuku se vypočítá podle vzorce:
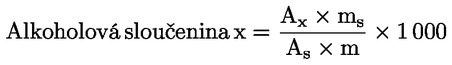
kde:
|
Ax |
= |
plocha píku alkoholové sloučeniny x vypočtená dle výpočetního systému. |
|
As |
= |
plocha píku 1-ikosanolu/α-cholestanolu vypočtená dle výpočetního systému. |
|
ms |
= |
množství přidaného 1-ikosanolu/α-cholestanolu v mg. |
|
m |
= |
hmotnost vzorku použitého pro stanovení v g. |
7. VYJÁDŘENÍ VÝSLEDKŮ
Uvádí se obsah jednotlivých alifatických a triterpenických alkoholů v mg/kg tuku a jejich souhrn jako „celkový obsah alifatických alkoholů“. Celkový obsah je součet C22, C24, C26 a C28.
Složení jednotlivých alkoholových sloučenin by se mělo vyjadřovat s přesností na jedno desetinné místo.
Celková koncentrace sterolů by se měla vyjádřit v celých číslech.
Procentní podíl jednotlivých sterolů z poměru plochy příslušného píku k celkové ploše píků sterolů se vypočítá podle vzorce:
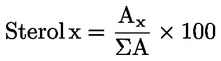
kde:
|
Ax |
= |
plocha píku sterolu x, |
|
ΣA |
= |
celková plocha píků sterolů. |
Zjevný β -sitosterol: Δ5,23-stigmastadienol + chlerosterol + β-sitosterol + sitostanol + Δ5-avenasterol + Δ5,24-stigmastadienol.
Vypočítá se procentní podíl erythrodiolu a uvaolu:
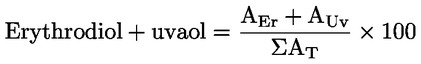
kde:
|
AEr |
= |
plocha erythrodiolu vypočtená dle výpočetního systému, |
|
AUv |
= |
plocha uvaolu vypočtená dle výpočetního systému. |
|
Σ AT |
= |
součet ploch píků sterolu + erythrodiolu + uvaolu vypočtených dle výpočetního systému. |
Kromě výpočtu relativního podílu jednotlivých sterolů a triterpenických dialkoholů a celkové koncentrace sterolů se musí vypočítat také koncentrace erythrodiolu a uvaolu a jejich součet v mg/kg tuku, a to podle následujících vzorců:
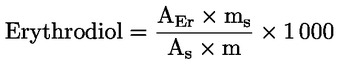
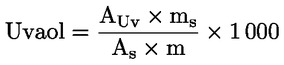
kde:
|
Aer |
= |
plocha píku erythrodiolu vypočtená dle výpočetního systému, |
|
Auv |
= |
plocha uvaolu vypočtená dle výpočetního systému, |
|
As |
= |
plocha píku α-cholestanolu vypočtená dle výpočetního systému, |
|
ms |
= |
množství přidaného α-cholestanolu v mg. |
|
m |
= |
hmotnost vzorku použitého pro stanovení v g. |
Dodatek
|
|
1 Uhlovodíky 2 α-tokoferol 3 Prenoly 4 Triterpenické alkoholy 5 Alifatické alkoholy 6 Metylsteroly 7 Steroly 8 Triterpenické dialkoholy |

Obrázek 1 – tenkovrstvá chromatografie nezmýdelnitelné frakce z olivového oleje z pokrutin dvakrát eluované s hexanem:diethyletherem (65:35), vyvinuté s SO4H2 (50 %) a zahřáté. Pásma, která je třeba seškrábat, se nachází v obdélníku; 1 jsou pásma alifatických alkoholů a 2 sterolů a triterpenických dialkoholů.
Tabulka I – Relativní retenční časy sterolů
|
Pík |
Identifikace |
Relativní retenční čas |
||
|
Kolona SE 54 |
Kolona SE 52 |
|||
|
1 |
Cholesterol |
Δ-5-cholesten-3β-ol |
0,67 |
0,63 |
|
2 |
Cholestanol |
5α-cholestan-3β-ol |
0,68 |
0,64 |
|
3 |
Brassikasterol |
[24S]-24-methyl-Δ-5,22-cholestadien-3β-ol |
0,73 |
0,71 |
|
* |
Ergosterol |
[24S]-24-methyl-Δ-5,7,22 cholestatrien-3β-ol |
0,78 |
0,76 |
|
4 |
24-methylen-cholesterol |
24-methylen-Δ-5,24-cholestadien-3β-o1 |
0,82 |
0,80 |
|
5 |
Kampesterol |
(24R)-24-methyl-Δ-5-cholesten-3β-ol |
0,83 |
0,81 |
|
6 |
Kampestanol |
(24R)-24-methyl-cholestan-3β-ol |
0,85 |
0,82 |
|
7 |
Stigmasterol |
(24S)-24-ethyl-Δ-5,22-cholestadien-3β-ol |
0,88 |
0,87 |
|
8 |
Δ-7-kampesterol |
(24R)-24-methyl-Δ-7-cholesten-3β-ol |
0,93 |
0,92 |
|
9 |
Δ-5,23-stigmastadienol |
(24R,S)-24-ethyl-Δ-5,23-cholestadien-3β-ol |
0,95 |
0,95 |
|
10 |
Chlerosterol |
(24S)-24-ethyl-Δ-5,25-cholestadien-3β-ol |
0,96 |
0,96 |
|
11 |
β-sitosterol |
(24R)-24-ethyl-Δ-5-cholesten-3β-ol |
1,00 |
1,00 |
|
12 |
Sitostanol |
24-ethyl-cholestan-3β-ol |
1,02 |
1,02 |
|
13 |
Δ-5-avenasterol |
(24Z)-24-ethyliden-Δ-cholesten-3β-ol |
1,03 |
1,03 |
|
14 |
Δ-5,24-stigmastadienol |
(24R,S)-24-ethyl-Δ-5,24-cholestadien-3β-ol |
1,08 |
1,08 |
|
15 |
Δ-7-stigmastenol |
(24R,S)-24-ethyl-Δ-7-cholesten-3β-ol |
1,12 |
1,12 |
|
16 |
Δ-7-avenasterol |
(24Z)-24-ethyliden-Δ-7-cholesten-3β-ol |
1,16 |
1,16 |
|
17 |
Erythrodiol |
5α-olean-12-en-3β,28-diol |
1,41 |
1,41 |
|
18 |
Uvaol |
Δ12-ursen-3β,28-diol |
1,52 |
1,52 |
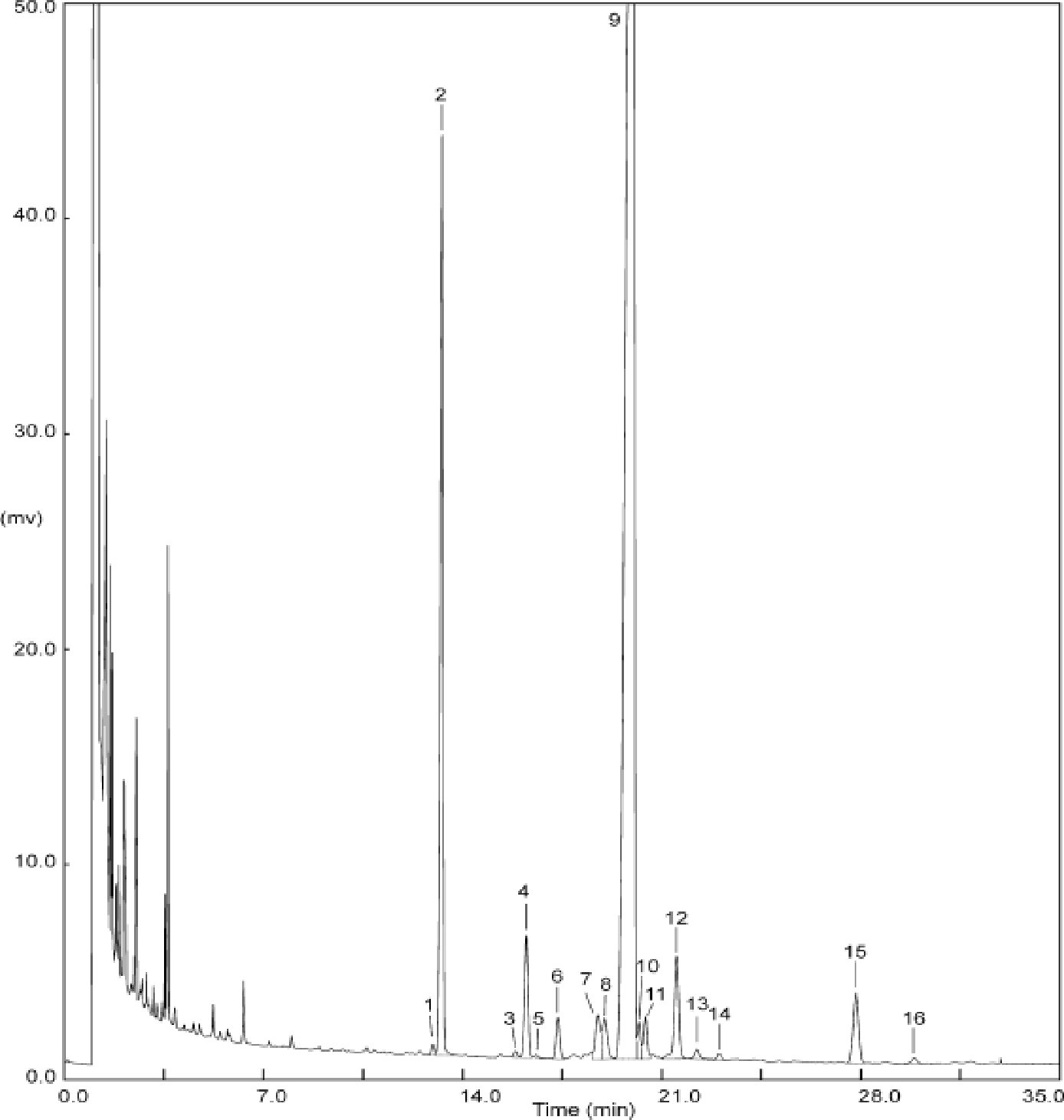
Obrázek 2 – GC-FID chromatografický profil sterolu a triterpenických dialkoholů z rafinovaného olivového oleje. (1) Cholesterol, (2) α-cholestanol (V.S.), (3) 24-methylencholesterol, (4) kampesterol, (5) kampestanol, (6) stigmasterol, (7) Δ5,23-stigmastadienol, (8) chlerosterol, (9) β-sitosterol, (10) sitostanol, (11) Δ5-avenasterol, (12) Δ5,24-stigmastadienol, (13) Δ7-stigmastenol, (14) Δ7-avenasterol, (15) erythrodiol, (16) uvaol.
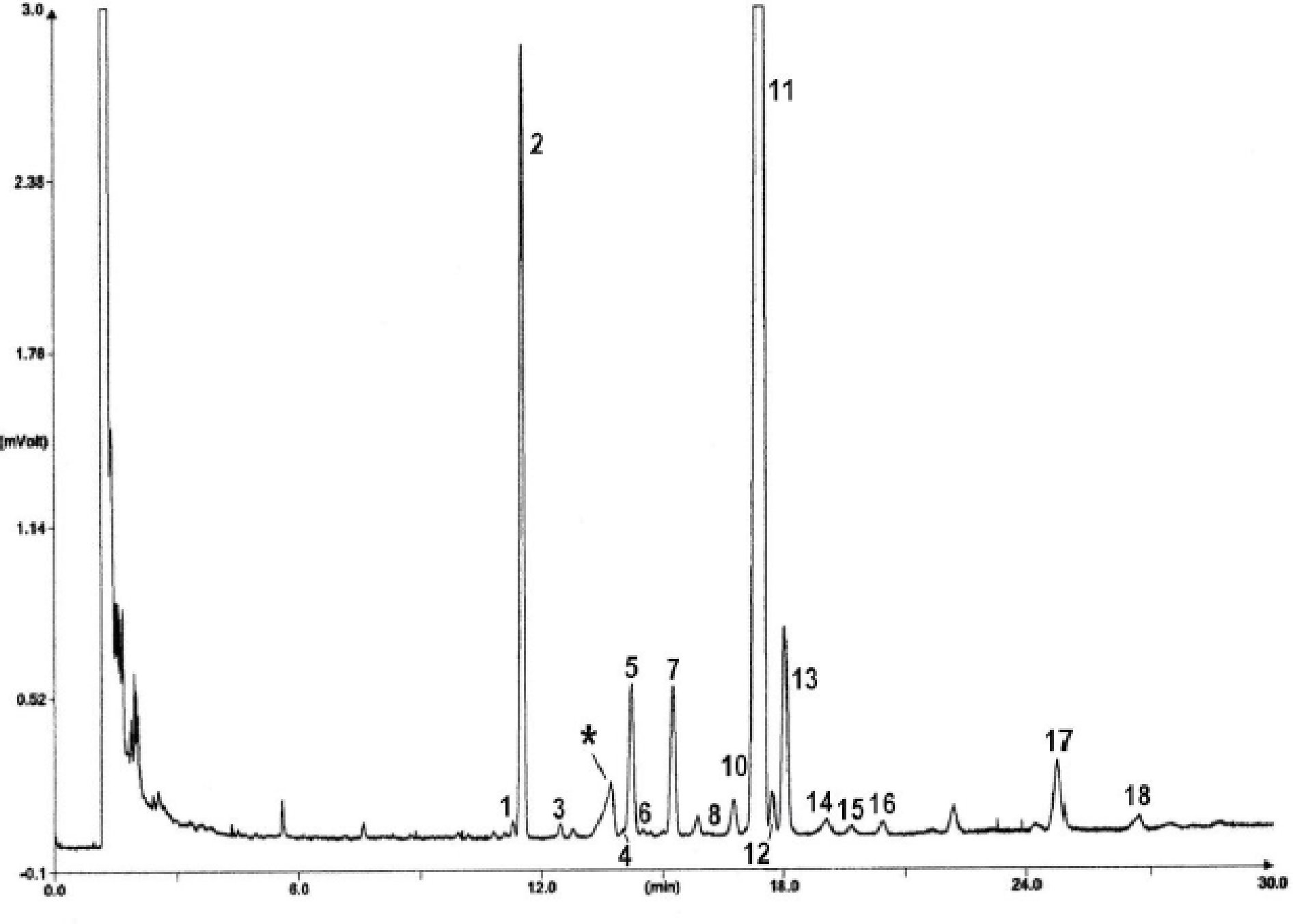
Obrázek 3 – GC-FID chromatografický profil sterolu a triterpenických dialkoholů z lampantového olivového oleje. (1) Cholesterol, (2) α-cholestanol, (3) brassikasterol, (4) 24-methylencholesterol, (5) kampesterol, (6) kampestanol, (7) stigmasterol, (8) Δ7-kampesterol, (9) Δ5,23-stigmastadienol, (10) chlerosterol, (11) β-sitosterol, (12) sitostanol, (13) Δ5-avenasterol, (14) Δ5,24-stigmastadienol, (15) Δ7-stigmastenol, (16) Δ7-avenasterol, (17) erythrodiol, (18) uvaol.
Obrázek 4 – GC-FID chromatografický profil alifatických alkoholů a triterpenických alkoholů z olivového oleje. (V.S.) C20-ol, (1) C22-ol, (2) C24-ol, (3) C26-ol, (4) C28-ol, (5) triterpenické alkoholy.
Obrázek 5 – GC-FID chromatografický profil alifatických alkoholů a triterpenických alkoholů z rafinovaného olivového oleje a olivového oleje po druhém odstřeďování. (V.S.) C20-ol, (1) C22-ol, (2) C24-ol, (3) C26-ol, (4) C28-ol, (5) triterpenické alkoholy.
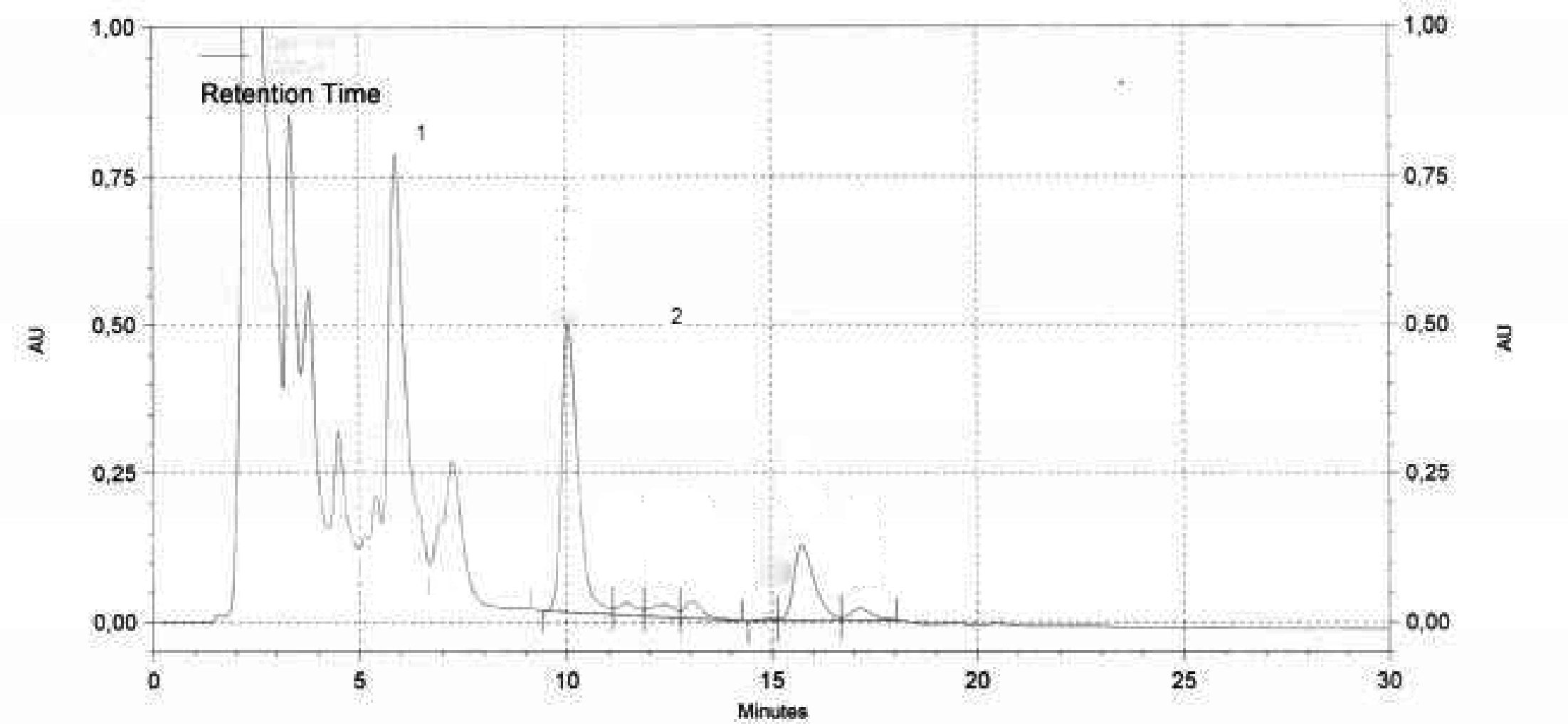
Obrázek 6 – Chromatogram nezmýdelnitelných látek z olivového oleje separovaných vysoce účinnou kapalinovou chromatografií za použití UV detektoru. (1) Alifatické a triterpenické alkoholy, (2) steroly a triterpenické dialkoholy.
PŘÍLOHA XX
Metoda stanovení obsahu vosků, methylesterů mastných kyselin a ethylesterů mastných kyselin kapilární plynovou chromatografií
1. ÚČEL
Tato metoda slouží ke stanovení obsahu vosků, methylesterů mastných kyselin a ethylesterů mastných kyselin v olivových olejích. Jednotlivé vosky a alkylestery jsou separovány podle počtu atomů uhlíku. Metoda je doporučeným nástrojem pro rozlišení mezi olivovým olejem a olivovým olejem z pokrutin a stanoví jakostní parametr pro extra panenský olivový olej, což umožňuje rozeznat podvodně smíchané extra panenské olivové oleje s oleji nižší kvality, ať už se jedná o panenské, lampantové či jiné dezodorizované oleje.
2. PODSTATA METODY
K oleji se přimísí vhodný vnitřní standard a poté se provádí chromatografická frakční destilace na hydratované silikagelové koloně. Získaná frakce eluovaná při testovacích podmínkách (jejíž polarita je menší než polarita triglyceridů) se přímo analyzuje pomocí kapilární plynové chromatografie.
3. ZAŘÍZENÍ
|
3.1. |
Kónická 25ml baňka. |
|
3.2. |
Skleněná kolona pro kapalnou chromatografii o vnitřním průměru 15 mm a délce 30 až 40 cm vybavená vhodným kohoutem. |
|
3.3. |
Plynový chromatograf, vhodný pro použití s kapilární kolonou a vybavený systémem pro přímý nástřik na kolonu, obsahující následující části:
|
|
3.4. |
mikrostříkačka na 10 μl pro přímý vstřik do kolony opatřená tvrzenou jehlou; |
|
3.5. |
elektrický vibrátor; |
|
3.6. |
rotační odparka; |
|
3.7. |
muflová pec; |
|
3.8. |
analytická váha pro vážení s přesností ± 0,1 mg; |
|
3.9. |
běžné laboratorní sklo. |
4. ČINIDLA
|
4.1. |
Silikagel, 60–200 μm mesh. Silikagel se umístí alespoň na čtyři hodiny do muflové pece o teplotě 500 °C. Nechá se ochladit a přidají se 2 % vody v závislosti na množství použitého silikagelu. Řádným protřepáním se směs homogenizuje a před použitím se ponechá nejméně 12 hodin v exsikátoru. |
|
4.2. |
n-hexan pro chromatografii nebo pro residuální analýzu. Hexan lze nahradit isooktanem (2,2,4-trimethylpentanem pro chromatografii) za předpokladu, že je dosaženo srovnatelných hodnot přesnosti. Rozpouštědla s vyšším bodem varu, než má n-hexan, se odpařují delší dobu. Z důvodu toxicity hexanu se však upřednostňují. Musí být kontrolována čistota – lze například zkontrolovat zbytek po odpaření 100 ml rozpouštědla. POZOR! – Výpary se mohou vznítit. Chraňte před zdroji tepla, jiskrami či přímým plamenem. Ujistěte se, že jsou láhve vždy řádně uzavřeny. Při použití zajistěte řádné větrání. Zabraňte hromadění výparů a eliminujte veškeré příčiny možného vzniku požáru, jako jsou topná tělesa a elektrická zařízení, jež nejsou vyrobena z nehořlavého materiálu. Vdechnutí je škodlivé, neboť může dojít k poškození nervových buněk. Zabraňte vdechnutí výparů. V případě nutnosti použijte vhodný respirační přístroj. Zabraňte styku s očima a s pokožkou. Isooktan je hořlavá kapalina představující nebezpečí požáru. Meze výbušnosti v ovzduší odpovídají 1,1 % až 6,0 % (obsahu v % objemových). Při požití a vdechnutí je toxický. Při práci s tímto rozpouštědlem použijte digestoř v dobrém provozním stavu. |
|
4.3. |
Ethylether pro chromatografii
POZOR! - Vysoce hořlavý a mírně jedovatý. Dráždí pokožku. Vdechnutí je škodlivé. Může způsobit poškození zraku. Účinky mohou být opožděné. Může vytvářet výbušné peroxidy. Výpary se mohou vznítit. Chraňte před zdroji tepla, jiskrami či přímým plamenem. Ujistěte se, že jsou láhve vždy řádně uzavřeny. Při použití zajistěte řádné větrání. Zabraňte hromadění výparů a eliminujte veškeré příčiny možného vzniku požáru, jako jsou topná tělesa a elektrická zařízení, jež nejsou vyrobena z nehořlavého materiálu. Neodpařujte do sucha nebo téměř do sucha. Přidání vody nebo vhodného redukčního prvku může omezit tvorbu peroxidů. Nepijte. Zabraňte vdechnutí výparů. Zabraňte dlouhotrvajícímu či opakovanému styku s pokožkou. |
|
4.4. |
N-heptan čistoty pro chromatografii nebo isooktan. POZOR! - Hořlavý. Vdechnutí je škodlivé. Chraňte před zdroji tepla, jiskrami či přímým plamenem. Ujistěte se, že jsou láhve vždy řádně uzavřeny. Při použití zajistěte řádné větrání. Zabraňte vdechnutí výparů. Zabraňte dlouhotrvajícímu či opakovanému styku s pokožkou. |
|
4.5. |
Standardní roztok 0,05 % (m/V) laurylcharidátu (poznámka 3) v heptanu (vnitřní standard pro vosky). Poznámka 3: Lze také použít palmityl-palmitát, myristyl-stearát či arachidyl-laureát. |
|
4.6. |
Standardní roztok 0,02 % (m/V) methylheptakaprinátu v heptanu (vnitřní standard pro methylestery a ethylestery). |
|
4.7. |
Sudan 1 (1-fenylazo-2-naftol). |
|
4.8. |
Nosný plyn: vodík či helium, čistý pro plynovou chromatografii. POZOR! Vodík. Pod tlakem vysoce hořlavý. Chraňte před zdroji tepla, jiskrami, přímým plamenem a uchovávejte mimo dosah elektrických přístrojů z hořlavého materiálu. Ujistěte se, zda je ventil láhve po použití vodíku vždy uzavřen. Používejte vždy s redukčním ventilem. Před otevřením ventilu láhve uvolněte pnutí pružiny redukčního ventilu. Při otvírání ventilu nestůjte před výpustným otvorem láhve. Při použití zajistěte řádné větrání. Nepřepouštějte vodík z jedné láhve do druhé. Nemíchejte plyn v láhvi. Ujistěte se, že láhve nelze převrhnout. Chraňte je před slunečním světlem a před zdroji tepla. Skladujte v nekorozivním prostředí. Nepoužívejte poškozené nebo neoznačené láhve. Helium. Vysoce stlačený plyn. Snižuje množství kyslíku pro dýchání. Uchovávejte v zavřené láhvi. Při použití zajistěte řádné větrání. Nevstupujte do skladovacích prostor, pokud nejsou řádně větrány. Používejte vždy s redukčním ventilem. Před otevřením ventilu láhve uvolněte pnutí pružiny redukčního ventilu. Nepřepouštějte plyn z jedné láhve do druhé. Ujistěte se, že láhve nelze převrhnout. Při otvírání ventilu nestůjte před výpustným otvorem láhve. Chraňte je před slunečním světlem a před zdroji tepla. Skladujte v nekorozivním prostředí. Nepoužívejte poškozené nebo neoznačené láhve. Nevdechujte. Používejte výhradně pro technické účely. |
|
4.9. |
Pomocné plyny: — Vodík, čistý pro plynovou chromatografii. — Vzduch, čistý pro plynovou chromatografii. POZOR! Vzduch. Vysoce stlačený plyn. Dbejte opatrnosti při použití v přítomnosti hořlavých látek, protože teplota pro samovznícení většiny organických složek vzduchu je při vysokém tlaku znatelně nižší. Ujistěte se, že je ventil láhve po použití vždy uzavřen. Vždy používejte redukční ventil. Před otevřením ventilu láhve uvolněte pnutí pružiny redukčního ventilu. Při otvírání ventilu nestůjte před výpustným otvorem láhve. Nepřepouštějte plyn z jedné láhve do druhé. Nemíchejte plyn v láhvi. Ujistěte se, že láhve nelze převrhnout. Chraňte je před slunečním světlem a před zdroji tepla. Skladujte v nekorozivním prostředí. Nepoužívejte poškozené nebo neoznačené láhve. Vzduch pro technické účely nesmí být vdechován nebo používán v respiračních přístrojích. |
5. POSTUP
5.1. Příprava chromatografické kolony
15 g silikagelu (bod 4.1) se suspenduje v n-hexanu (bod 4.2) a zavede do kolony (bod 3.2). Po spontánní sedimentaci se tato dokončí pomocí elektrického vibrátoru, aby byla chromatografická vrstva co nejhomogennější. Provede se perkolace 30 ml n-hexanu za účelem odstranění případných nečistot. Do 25ml baňky (bod 3.1) se na analytické váze (bod 3.8) naváží přesně 500 mg vzorku a přidá se vhodné množství vnitřního standardu (bod 4.5) v závislosti na předpokládaném obsahu vosku, např. 0,1 mg laurylcharidátu v případě olivového oleje, 0,25 až 0,5 mg v případě olivového oleje z pokrutin a 0,05 mg methylheptakaprinátu v případě olivových olejů (bod 4.6).
Získaný vzorek se za pomoci dvou dávek 2 ml n-hexanu (bod 4.2) převede do chromatografické kolony.
Umožní se, aby hladina rozpouštědla poklesla tak, aby byla 1 mm nad horní úrovní absorbentu. Perkoluje se další n-hexan/diethyleter (99:1) a odebere se 220 ml při průtoku přibližně 15 kapek za 10 sekund. (Tato frakce obsahuje methylestery, ethylestery a vosky.) (poznámka 4) (poznámka 5)
Poznámka 4: Každý den by se měla připravit čerstvá směs n-hexanu/diethyleteru (99:1).
Poznámka 5: Do vzorku roztoku lze přidat 100 μl eluční směsi s 1 % barviva Sudan I pro účely vizuální kontroly řádné eluce vosků.
Retenční doba barviva je mezi retenční dobou vosků a triglycerolů. Když tedy barvivo dosáhne dna chromatografické kolony, musí být eluce suspendována, protože veškeré vosky byly eluovány.
Získaná frakce se usuší v rotačním odpařovači, až je téměř všechno rozpouštědlo odstraněno. Poslední 2 ml rozpouštědla se odstraní za pomoci slabého proudu dusíku. Odebere se frakce obsahující methylestery a ethylestery tak, že se naředí 2 až 4 ml n-heptanu nebo isooktanu.
5.2. Plynová chromatografická analýza
5.2.1. Přípravné práce
Kolona se spojí s plynovým chromatografem (bod 3.3), vstupní port se připojí ke kolonovému systému (on-column system) a výstupní port k detektoru. Poté se plynový chromatograf překontroluje (těsnost plynových vedení, funkce detektoru a záznamníku atd.).
Kolonu, která má být použita poprvé, je nutno nejprve kondicionovat. Kolonou se nechá protékat malé množství nosného plynu, poté se zapne plynový chromatograf. Nechá se postupně zahřívat, dokud není po přibližně čtyřech hodinách dosaženo teploty 350 °C.
Tato teplota se udržuje nejméně dvě hodiny a poté se systém převede do předepsaných podmínek (regulace průtoku plynu, zapálení plamene, připojení elektronického zapisovače (bod 3.3.4), nastavení teploty kolonové komory, detektoru, atd.). Signál se nastaví na citlivost, která činí nejméně dvojnásobek nejvyšší úrovně uvažované pro provedení analýzy. Základní linie záznamu musí být rovná, bez jakýchkoli píků nebo driftů.
Záporné drifty jsou známkou nedokonalé těsnosti spojů kolony, zatímco kladné svědčí o nedostatečné kondicionaci kolony.
5.2.2. Výběr pracovních podmínek pro vosky, methylestery a ethylestery (poznámka 6).
Všeobecné pracovní podmínky jsou tyto:
|
— |
Teplota kolony : 20 °C/min 5 °C/min počáteční teplota 80 °C (1′) |
|
— |
Teplota detektoru : 350 °C. |
|
— |
Množství nastříknuté látky : 1 μl roztoku n-heptanu (2 až 4 ml). |
|
— |
Nosný plyn : helium nebo vodík při optimální lineární rychlosti pro zvolený plyn (viz dodatek A). |
|
— |
Citlivost přístroje : vhodná pro dosažení výše uvedených podmínek. |
Poznámka 6: Z důvodu vysoké konečné teploty je povolen kladný drift, nesmí však přesáhnout 10 % plného rozsahu.
Tyto podmínky mohou být upraveny podle charakteristik kolony a plynového chromatografu s cílem oddělit veškeré vosky, methylestery mastných kyselin a ethylestery mastných kyselin a dosáhnout přijatelné separace píků (viz obrázky 2, 3 a 4) a retenčního času vnitřního standardu laurylcharidátu 18 ± 3 minuty. Nejreprezentativnější pík vosků musí dosáhnout 60 % z plného rozsahu, vnitřní standard methylheptakaprinát pro methylestery a ethylestery musí dosáhnout plného rozsahu.
Parametry pro integraci píků se nastaví tak, aby došlo ke správnému vyhodnocení ploch uvažovaných píků.
5.3. Provedení analýzy
Do 10μl mikrostříkačky se natáhne 10 μl roztoku; píst mikrostříkačky se povytáhne tak, aby se vyprázdnila jehla. Jehla se zavede do vstřikové komory a po jedné nebo dvou sekundách se rychle nastříkne roztok. Přibližně po pěti sekundách se jehla pomalu vytáhne.
Zaznamenávání je prováděno, dokud vosky nebo stigmastadieny nejsou zcela eluovány v závislosti na analyzované frakci.
Základní linie záznamu musí vždy odpovídat požadovaným podmínkám.
5.4. Identifikace píků
Identifikace jednotlivých píků se provádí podle retenčních časů a porovnáním se směsmi vosků, jejichž retenční časy jsou známy a které byly analyzovány za stejných podmínek. Alkylestery se identifikují ze směsí methylesterů a ethylesterů hlavních mastných kyselin v olivových olejích (palmitová a olejová).
Chromatogram vosků panenského olivového oleje je znázorněn na obrázku 1. Obrázky 2 a 3 zobrazují chromatogramy dvou maloobchodně prodávaných extra panenských olivových olejů, jeden obsahuje methylestery a ethylestery, v druhém obsaženy nejsou. Na obrázku 4 jsou chromatogramy extra panenského olivového oleje nejvyšší třídy a stejného oleje uměle obohaceného 20 % dezodorizovaného oleje.
5.5. Kvantitativní vyhodnocení vosků
Pomocí integrátoru se vypočítají plochy píků odpovídajících vnitřnímu standardu laurylcharidátu a alifatickým esterům C40 až C46.
Celkový obsah vosků se vypočítá sečtením jednotlivých vosků v mg/kg tukové látky takto:
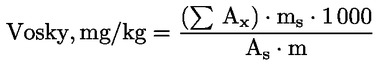
kde:
|
Ax |
= |
plocha píku příslušného esteru vypočtená počítačem |
|
As |
= |
plocha píku vnitřního standardu laurylcharidátu vypočtená počítačem |
|
ms |
= |
hmotnost přidaného vnitřního standardu laurylcharidátu v mg; |
|
m |
= |
hmotnost vzorku použitého pro stanovení v gramech. |
5.5.1. Kvantitativní vyhodnocení methylesterů a ethylesterů
Pomocí integrátoru se vypočítají plochy píků odpovídajících vnitřnímu standardu methylheptakaprinátu, methylesterů mastných kyselin C16 a C18 a ethylesterů mastných kyselin C16 a C18.
Obsah každého alkylesteru v mg/kg tukové látky se vypočítá takto:
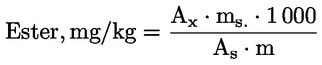
kde:
|
Ax |
= |
plocha píku příslušného esteru C16 a C18 vypočtená počítačem |
|
As |
= |
plocha píku vnitřního standardu methylheptakaprinátu vypočtená počítačem |
|
ms |
= |
hmotnost přidaného vnitřního standardu methylheptakaprinátu v mg; |
|
m |
= |
hmotnost vzorku použitého pro stanovení v gramech. |
6. VYJÁDŘENÍ VÝSLEDKŮ
Uvádí se souhrn obsahů jednotlivých vosků C40 až C46 (poznámka 7) v mg/kg tukové látky.
Uvádí se souhrn obsahů jednotlivých methylesterů a ethylesterů C16 až C18 a celkový součet methylesterů a ethylesterů.
Výsledky se zaokrouhlují na celé mg/kg.
Poznámka 7: Prvky kvantitativního vyhodnocení se vztahují k píkům esterů se sudým karbonovým číslem C40 až C46 podle vzorového chromatogramu vosků v olivovém oleji na přiloženém obrázku. Pokud se ester C46 rozštěpí, doporučuje se pro účely identifikace analyzovat frakci vosku olivového oleje z pokrutin, kde lze pík C46 rozlišit, neboť zřetelně převládá.
Uvádí se poměr mezi ethylestery a methylestery.
Obrázek 1
Příklad plynového chromatogramu frakce vosků olivového oleje ( 6 )
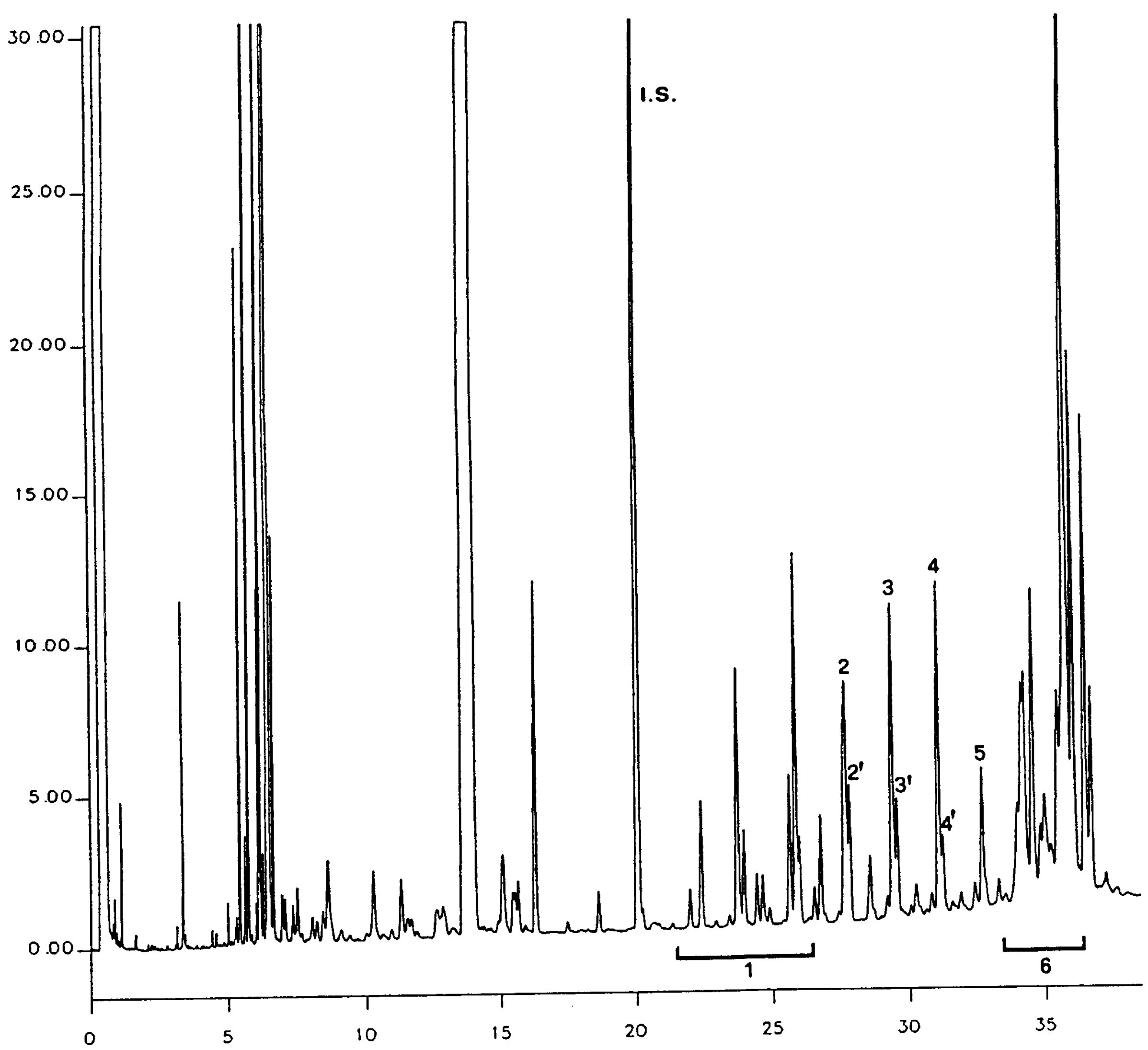
Píky methylesterů a ethylesterů mastných kyselin s retenční dobou 5 až 8 minut.
Vysvětlivky:
|
I.S. |
= |
(vnitřní standard) Laurylcharidát |
|
1 |
= |
Estery diterpenu |
|
2+2′ |
= |
Estery C40 |
|
3+3′ |
= |
Estery C42 |
|
4+4′ |
= |
Estery C44 |
|
5 |
= |
Estery C46 |
|
6 |
= |
Estery sterolů a triterpenické alkoholy |
Obrázek 2
Methylestery, ethylestery a vosky v panenském olivovém oleji
Vysvětlivky:
|
1 |
– |
Methyl C16 |
|
2 |
– |
Ethyl C16 |
|
3 |
– |
Vnitřní standard methylheptakaprinát |
|
4 |
– |
Methyl C18 |
|
5 |
– |
Ethyl C18 |
|
6 |
– |
Squalene |
|
7 |
– |
Vnitřní standard laurylcharidát |
|
A |
– |
Estery diterpenů |
|
B |
– |
Vosky |
|
C |
– |
Estery sterolů a triterpenů |
Obrázek 3
Methylestery, ethylestery a vosky v extra panenském olivovém oleji
Vysvětlivky:
|
1 |
– |
Vnitřní standard methylheptakaprinát |
|
2 |
– |
Methyl C18 |
|
3 |
– |
Ethyl C18 |
|
4 |
– |
Squalen |
|
5 |
– |
Vnitřní standard laurylcharidát |
|
A |
– |
Estery diterpenů |
|
B |
– |
Vosky |
|
C |
– |
Estery sterolů a triterpenů |
Obrázek 4
Část chromatogramu extra panenského olivového oleje a stejného oleje uměle obohaceného dezodorizovaným olejem
Vysvětlivky:
|
1 |
– |
Vnitřní standard methyl-myristát |
|
2 |
– |
Methyl-palmitát |
|
3 |
– |
Ethyl-palmitát |
|
4 |
– |
Vnitřní standard methylheptakaprinát |
|
5 |
– |
Methyl-linoleát |
|
6 |
– |
Methyl-oleát |
|
7 |
– |
Methyl-stearát |
|
8 |
– |
Ethyl-linoleát |
|
9 |
– |
Ethyl-oleát |
|
10 |
– |
Ethyl-stearát |
Dodatek A
Stanovení lineární rychlosti plynu
Do plynového chromatografu nastaveného na normální pracovní podmínky se nastříkne 1:3 μl methanu (nebo propanu). Pomocí stopek se změří doba průchodu plynu kolonou od počátku vstřiku do okamžiku vzniku píků (tM).
Lineární rychlost proudění v cm/s je dána poměrem L/tM, kde L je délka kolony v cm a tM je čas v sekundách, změřený stopkami.
▼M28 —————
PŘÍLOHA XXI
Výsledky kontroly shody prováděné v odvětví olivového oleje uvedené v čl. 8 odst. 2
|
|
Označování |
Chemické ukazatele |
Organoleptické vlastnosti (4) |
Závěr |
|||||||||||||
|
Vzorek |
Kategorie |
Země původu |
Místo, kde byla inspekce provedena (1) |
Oficiální název |
Označení původu |
Podmínky skladování |
Chybné informace |
Čitelnost |
C/NC (3) |
Parametry mimo limity ANO/NE |
Pokud ano, označte který (které) (2) |
C/NC (3) |
Medián závad |
Medián znaku ovocná chuť a vůně |
C/NC (3) |
Požadované opatření |
Sankce |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) Vnitřní trh (lisovny, stáčírny, maloobchodní prodej), vývoz, dovoz. (2) Všechny charakteristiky olivového oleje uvedené v příloze I musí mít kód. (3) Odpovídá/neodpovídá. (4) Není požadováno u olivového oleje a oleje z pokrutin. |
|||||||||||||||||
( 1 ) Úř. věst. L 228, 1.9.2009, s. 3.
( 2 ) Směrnice Evropského parlamentu a Rady 2011/91/EU ze dne 13. prosince 2011 o údajích nebo značkách určujících šarži, ke které potravina patří (Úř. věst. L 334, 16.12.2011, s. 1).
( 3 ) Po eluování esterů sterolů musí být chromatografická linie bez významných píků (triglyceridů).
( 4 ) Nařízení Evropského parlamentu a Rady (EU) č. 1308/2013 ze dne 17. prosince 2013, kterým se stanoví společná organizace trhů se zemědělskými produkty a zrušují nařízení Rady (EHS) č. 922/72, (EHS) č. 234/79, (ES) č. 1037/2001 a (ES) č. 1234/2007 (Úř. věst. L 347, 20.12.2013, s. 671).
( 5 ) Může ochutnávání oleje odmítnout, pokud při přímém čichovém vnímání zaznamená, že některý z negativních znaků je extrémně intenzivní. Tuto výjimečnou okolnost zaznamená do profilového listu.
( 6 ) Po eluaci esterů sterolů nesmí chromatogram vykazovat žádné výrazné píky (triacylglyceroly).