82/434/EHSDruhá směrnice Komise 82/434/EHS ze dne 14. května 1982 o sbližování právních předpisů členských států týkajících se metod analýzy nezbytných pro kontrolu složení kosmetických prostředků
| Publikováno: | Úř. věst. L 185, 30.6.1982, s. 1-28 | Druh předpisu: | Směrnice |
| Přijato: | 14. května 1982 | Autor předpisu: | Evropská komise |
| Platnost od: | 30. června 1982 | Nabývá účinnosti: | 7. června 1982 |
| Platnost předpisu: | Ano | Pozbývá platnosti: | |
Text aktualizovaného znění s celou hlavičkou je dostupný pouze pro registrované uživatele.
Tento dokument je třeba brát jako dokumentační nástroj a instituce nenesou jakoukoli odpovědnost za jeho obsah
|
DRUHÁ SMĚRNICE KOMISE ze dne 14. května 1982 o sbližování právních předpisů členských států týkajících se metod analýzy nezbytných pro kontrolu složení kosmetických prostředků (Úř. věst. L 185, 30.6.1982, p.1) |
Ve znění:
|
|
|
Úřední věstník |
||
|
No |
page |
date |
||
|
L 108 |
92 |
28.4.1990 |
||
DRUHÁ SMĚRNICE KOMISE
ze dne 14. května 1982
o sbližování právních předpisů členských států týkajících se metod analýzy nezbytných pro kontrolu složení kosmetických prostředků
(82/434/EHS)
KOMISE EVROPSKÝCH SPOLEČENSTVÍ,
s ohledem na Smlouvu o založení Evropského hospodářského společenství,
s ohledem na směrnici Rady 76/768/EHS ze dne 27. července 1976 o sbližování právních předpisů členských států týkajících se kosmetických prostředků ( 1 ) ve znění směrnice 79/661/EHS ( 2 ), a zejména na čl. 8 odst. 1 uvedené směrnice,
vzhledem k tomu, že se ve směrnici 76/768/EHS stanoví úřední zkoušení kosmetických prostředků s cílem zajistit dodržení podmínek předepsaných předpisy Společenství, které se týkají složení kosmetických prostředků;
vzhledem k tomu, že by všechny nezbytné metody analýzy měly být stanoveny co nejdříve; že první krok k dosažení tohoto cíle již byl učiněn definováním některých metod ve směrnici Komise 80/1335/EHS ( 3 ) a druhý krok spočívá v definování metod důkazu některých oxidačních činidel a stanovení peroxidu vodíku v prostředcích pro péči o vlasy, důkazu a semikvantitativního stanovení určitých oxidačních barviv v barvách na vlasy, důkazu a stanovení dusitanů, důkazu a stanovení volného formaldehydu, stanovení resorcinolu v šamponech a vlasových lotionech a stanovení methanolu vzhledem k ethanolu nebo propan-2-olu;
vzhledem k tomu, že opatření této směrnice jsou v souladu se stanoviskem Výboru pro přizpůsobení směrnice 76/768/EHS technickému pokroku,
PŘIJALA TUTO SMĚRNICI:
Článek 1
Členské státy učiní všechny nezbytné kroky k zajištění toho, aby se při úředním zkoušení kosmetických prostředků:
— důkaz oxidačních činidel a stanovení peroxidu vodíku v prostředcích pro péči o vlasy,
— důkaz a semikvantitativní stanovení určitých oxidačních barviv v barvách na vlasy,
— důkaz a stanovení dusitanů,
— důkaz a stanovení volného formaldehydu,
— stanovení resorcinolu v šamponech a vlasových lotionech,
— stanovení methanolu vzhledem k ethanolu nebo propan-2-olu
prováděly metodami popsanými v příloze.
Článek 2
Členské státy uvedou v účinnost právní a správní předpisy nezbytné pro dosažení souladu s touto směrnicí nejpozději do 31. prosince 1983.
Neprodleně o nich uvědomí Komisi.
Článek 3
Tato směrnice je určena členským státům.
PŘÍLOHA
I. DŮKAZ OXIDAČNÍCH ČINIDEL A STANOVENÍ PEROXIDU VODÍKU V PROSTŘEDCÍCH PRO PÉČI O VLASY
ROZSAH A OBLAST POUŽITÍ
Jodometrické stanovení peroxidu vodíku v kosmetice je možné pouze za nepřítomnosti jiných oxidačních činidel vytvářejících jod z jodidů. Z tohoto důvodu je před jodometrickým stanovením peroxidu vodíku nutné zjistit a identifikovat jakákoli jiná přítomná oxidační činidla. Tento důkaz se dělí do dvou fází; první zahrnuje peroxodisírany, bromičnany a peroxid vodíku, druhá peroxid barnatý.
A. DŮKAZ PEROXODISÍRANŮ, BROMIČNANŮ A PEROXIDU VODÍKU
1. PODSTATA METODY
Peroxodisíran sodný, peroxodisíran draselný a peroxodisíran amonný, bromičnan draselný, bromičnan sodný a peroxid vodíku – bez ohledu na to, zda pochází či nepochází z peroxidu barnatého – se dokazují pomocí sestupné papírové chromatografie, při které se používá dvou vyvíjecích rozpouštědel.
2. REAKČNÍ ČINIDLA
Všechna použitá reakční činidla musí být čistoty p.a.
|
2.1 |
0,5 % (m/v) vodné referenční roztoky následujících sloučenin:
|
|
2.2 |
Vyvíjecí rozpouštědlo A, 80 % (v/v) ethanol |
|
2.3 |
Vyvíjecí rozpouštědlo B, benzen – methanol – 3-methyl-butan-1-ol – voda (34: 38: 18: 10 objemově) |
|
2.4 |
Detekční činidlo A, 10 % (m/v) vodný roztok jodidu draselného |
|
2.5 |
Detekční činidlo B, 1 % (m/v) vodný roztok škrobu |
|
2.6 |
Detekční činidlo C, 10 % (m/m) kyselina chlorovodíková |
|
2.7 |
4 N kyselina chlorovodíková |
3. PŘÍSTROJE A POMŮCKY
|
3.1 |
Chromatografický papír (papír Whatman č. 3 nebo č. 4 nebo jejich ekvivalenty) |
|
3.2 |
Mikropipeta na 1 μl |
|
3.3 |
Odměrné baňky na 100 ml |
|
3.4 |
Skládané filtry |
|
3.5 |
Zařízení pro sestupnou papírovou chromatografii |
4. PŘÍPRAVA VZORKU
4.1 Výrobky rozpustné ve vodě
Z každého vzorku se připraví dva roztoky rozpuštěním 1 g a 5 g výrobku ve 100 ml vody. 1 μl každého z těchto roztoků se použije pro provedení papírové chromatografie popsané v oddílu 5.
4.2 Výrobky mírně rozpustné ve vodě
|
4.2.1. |
Naváží se 1 g a 5 g vzorku a rozptýlí se v 50 ml vody, v obou případech se doplní vodou na 100 ml a promíchají se. Obě suspenze se přefiltrují přes skládaný filtr (3.4) a z každého z filtrátů se použije 1 μl pro provedení papírové chromatografie popsané v oddílu 5. |
|
4.2.2 |
Ještě jednou se připraví dvě suspenze vzorků rozptýlením 1 g a 5 g v 50 ml vody, okyselí se zředěnou kyselinou chlorovodíkovou (2.7), doplní se vodou na 100 ml a promíchají se. Suspenze se přefiltrují přes skládaný filtr (3.4) a 1 μl každého z těchto roztoků se použije pro provedení papírové chromatografie popsané v oddílu 5. |
4.3 Krémy
5 g a 20 g každého výrobku se rozptýlí ve 100 ml vody a suspenze se použijí pro provedení papírové chromatografie popsané v oddílu 5.
5. METODA
|
5.1 |
Přiměřené množství rozpouštědel A (2.2) a B (2.3) se umístí do dvou oddělených chromatografických komor, aby se provedla sestupná papírová chromatografie. Chromatografické komory se sytí párami rozpouštědla alespoň 24 hodin. |
|
5.2 |
1 μl roztoku vzorku a všech referenčních roztoků připravených podle oddílu 4 a bodu 2.1 se nanese na startovní body proužku chromatografického papíru (Whatman č. 3 nebo ekvivalentní) 40 cm dlouhého a 20 cm širokého (3.1) nebo jiných vhodných rozměrů a rozpouštědlo se nechá odpařit na vzduchu. |
|
5.3 |
Chromatografický proužek (5.2) se umístí do chromatografické kolony naplněné vyvíjecím rozpouštědlem A (5.1) a vyvíjí se, dokud čelo rozpouštědla nepostoupí o 35 cm (asi 15 hodin). |
|
5.4 |
Postup popsaný v bodech 5.2 a 5.3 se opakuje za použití chromatografického papíru (Whatman č. 4 nebo ekvivalentní) (3.1) a vyvíjecího roztoku B. Chromatografie probíhá, dokud čelo rozpouštědla nepostoupí o 35 cm (asi pět hodin). |
|
5.5 |
Po ukončení vyvíjení se chromatogramy vyjmou a usuší se na vzduchu. |
|
5.6 |
Skvrny se objeví po následném postříkání:
|
|
5.7 |
Za výše zmíněných podmínek vztahujících se k vyvíjecím rozpouštědlům A (2.2) a B (2.3) jsou hodnoty Rf referenčních látek (2.1) přibližně následující:
|
B. STANOVENÍ PEROXIDU BARNATÉHO
1. PODSTATA METODY
Peroxid barnatý se dokazuje tvorbou peroxidu vodíku po okyselení vzorku (A.4.2) a přítomností barnatých iontů:
— v nepřítomnosti peroxodisíranů (A) přídavkem zředěné kyseliny sírové do části kyselého roztoku vzorku (B.4.1), čímž vznikne bílá sraženina síranu barnatého. Přítomnost barnatých iontů ve vzorku (B.4.1) se opět potvrdí papírovou chromatografií způsobem popsaným níže (B.5),
— ve vzorcích, ve kterých jsou peroxid barnatý a peroxodisírany přítomny současně (B.4.2), vyloužením zbytku z roztoku (B.4.2) v alkalickém prostředí; po rozpuštění taveniny (B.4.2.3) v kyselině chlorovodíkové se přítomnost barnatých iontů potvrdí papírovou chromatografií a/nebo vysrážením síranu barnatého.
2. REAKČNÍ ČINIDLA
|
2.1 |
Methanol |
|
2.2 |
36 % (m/m) koncentrovaná kyselina chlorovodíková |
|
2.3 |
6 N kyselina chlorovodíková |
|
2.4 |
4 N kyselina sírová |
|
2.5 |
Disodná sůl kyseliny rhodizonové (5,6-dihydroxycyklohex-5-en1,2,3,4-tetron, sodná sůl) |
|
2.6 |
Chlorid barnatý (BaCl2·2H2O) |
|
2.7 |
Bezvodý uhličitan sodný |
|
2.8 |
1 % (m/v) vodný roztok chloridu barnatého |
|
2.9 |
Vyvíjecí rozpouštědlo skládající se z methanolu, koncentrované kyseliny chlorovodíkové (koncentrace 36 %) a vody (80: 10: 10 objemově) |
|
2.10 |
Detekční činidlo, 0,1 % (m/v) vodný roztok disodné soli kyseliny rhodizonové, připravuje se bezprostředně před použitím. |
3. PŘÍSTROJE A POMŮCKY
|
3.1 |
Mikropipeta na 5 μl |
|
3.2 |
Platinové kelímky |
|
3.3 |
Odměrné baňky na 100 ml |
|
3.4 |
Chromatografický papír Schleicher and Schüll 2043 b nebo ekvivalentní. Papír se vyčistí tak, že se přes noc nechá vyvíjet v komoře pro sestupnou chromatografii (A.3.5) obsahující vyvíjecí rozpouštědlo (B.2.9) a poté se usuší. |
|
3.5 |
Skládaný filtrační papír |
|
3.6 |
Běžné zařízení pro provádění vzestupné papírové chromatografie |
4. PŘÍPRAVA VZORKU
4.1 Výrobky, ve kterých nejsou přítomny peroxodisírany
|
4.1.1 |
2 g výrobku se rozptýlí v 50 ml vody a pH suspenze se upraví přibližně na hodnotu 1 kyselinou chlorovodíkovou (B.2.3). |
|
4.1.2 |
Suspenze se pomocí vody převede do odměrné baňky na 100 ml, doplní se po rysku vodou a promíchá se. Tato suspenze se použije pro analýzu papírovou chromatografií popsanou v oddílu 5 a pro důkaz barya vysrážením síranu. |
4.2 Výrobky, ve kterých jsou přítomny peroxodisírany
|
4.2.1 |
2 g výrobku se rozptýlí ve 100 ml vody a přefiltruje se. |
|
4.2.2 |
K vysušenému zbytku se přidá množství uhličitanu sodného odpovídající sedmi až desetinásobku hmotnosti zbytku (B.2.7), promíchá se a směs se půl hodiny taví v platinovém kelímku (B.3.2). |
|
4.2.3 |
Ochladí se na pokojovou teplotu, tavenina se rozpustí v 50 ml vody a přefiltruje se (B.3.5). |
|
4.2.4 |
Zbytek z taveniny se rozpustí v kyselině chlorovodíkové (B.2.3) a doplní se vodou na 100 ml. Tento roztok se použije pro analýzu papírovou chromatografií popsanou v oddílu 5 a pro důkaz barya vysrážením síranu. |
5. METODA
|
5.1 |
Přiměřené množství vyvíjecího rozpouštědla (B.2.9) se umístí do komory pro vzestupnou chromatografii a komora se sytí alespoň 15 hodin. |
|
5.2 |
Na kus chromatografického papíru – předem upraveného tak, jak je popsáno v bodu B.3.4 – se nanese do třech startovních bodů 5 μl každého z roztoků připravených podle bodů B.4.1.2 a B.4.2.4 a referenční roztok B.2.8. |
|
5.3 |
Vzorek a referenční skvrny se vysuší na vzduchu. Chromatogram se vyvíjí, dokud čelo rozpouštědla nepostoupí o 30 cm. |
|
5.4 |
Chromatogram se vyjme z komory a usuší na vzduchu. |
|
5.5 |
Skvrny na chromatogramu se objeví po postříkání papíru detekčním činidlem B.2.10. V přítomnosti barya se na chromatogramu objeví červené skvrny s hodnotou Rf asi 0,10. |
C. STANOVENÍ PEROXIDU VODÍKU
1. PODSTATA METODY
Jodometrické stanovení peroxidu vodíku je založeno na následující reakci:
![]()
Tato konverze probíhá pomalu, může však být urychlena přidáním molybdenanu amonného. Vzniklý jod se stanoví titračně thiosíranem sodným a je mírou obsahu peroxidu vodíku.
2. DEFINICE
Obsah peroxidu vodíku změřený níže popsaným způsobem se vyjádří v hmotnostních procentech výrobku (% m/m).
3. REAKČNÍ ČINIDLA
Všechna reakční činidla musí být čistoty p.a.
|
3.1 |
2 N kyselina sírová |
|
3.2 |
Jodid draselný |
|
3.3 |
Molybdenan amonný |
|
3.4 |
0,1 N thiosíran sodný |
|
3.5 |
10 % (m/v) roztok jodidu draselného, připravuje se bezprostředně před použitím |
|
3.6 |
20 % (m/v) roztok molybdenanu amonného |
|
3.7 |
1 % (m/v) roztok škrobu |
4. PŘÍSTROJE A POMŮCKY
|
4.1 |
Kádinky na 100 ml |
|
4.2 |
Byreta na 50 ml |
|
4.3 |
Odměrné baňky na 250 ml |
|
4.4 |
Odměrné válce na 25 a 100 ml |
|
4.5 |
Nedělené pipety na 10 ml |
|
4.6 |
Erlenmeyerovy baňky na 250 ml |
5. METODA
|
5.1 |
Do kádinky na 100 ml se naváží 10 g (m gramů) výrobku obsahujícího asi 0,6 g peroxidu vodíku. Obsah se pomocí vody převede do odměrné baňky na 250 ml, doplní se po rysku vodou a promíchá se. |
|
5.2 |
10 ml roztoku vzorku (5.1) se odpipetuje do Erlenmeyerovy baňky na 250 ml (4.6) a postupně se přidá 100 ml 2 N kyseliny sírové (3.1), 20 ml roztoku jodidu draselného (3.5) a tři kapky roztoku molybdenanu amonného (3.6). |
|
5.3 |
Vzniklý jod se okamžitě titruje 0,1 N roztokem thiosíranu sodného (3.4) a těsně před dosažením bodu ekvivalence se přidá několik mililitrů roztoku škrobu (3.7) jako indikátoru. Zaznamená se spotřeba 0,1 N roztoku thiosíranu sodného (3.4) v mililitrech (V). |
|
5.4 |
Způsobem popsaným v bodech 5.2 a 5.3 se provede slepý pokus, přičemž se nahradí 10 ml roztoku vzorku 10 ml vody. Zaznamená se spotřeba 0,1 N roztoku thiosíranu sodného na slepý pokus (V0 v ml). |
6. VÝPOČET
Obsah peroxidu vodíku ve výrobku vyjádřený v hmotnostních procentech (% m/m) se vypočte pomocí následujícího vzorce:
|
% peroxidu vodiku |
= V − Vo × 1,7008 × 250 × 100m × 10 × 1 000 |
|
= V − Vo × 4,252m |
kde
|
m |
= |
množství analyzovaného výrobku (5.1) v gramech, |
|
V0 |
= |
spotřeba 0,1 N roztoku thiosíranu sodného na slepý pokus (5.4) v mililitrech, |
|
V |
= |
spotřeba 0,1 N roztoku thiosíranu sodného při titraci roztoku vzorku (5.3) v mililitrech. |
7. OPAKOVATELNOST ( 4 )
Pro výrobky obsahující asi 6 % (m/m) peroxidu vodíku by neměla absolutní hodnota rozdílu dvou stanovení provedených současně se stejným vzorkem překročit 0,2 %.
II. DŮKAZ A SEMIKVANTITATIVNÍ STANOVENÍ URČITÝCH OXIDAČNÍCH BARVIV V BARVÁCH NA VLASY
1. ROZSAH A OBLAST POUŽITÍ
Tato metoda je vhodná pro důkaz a semikvantitativní stanovení následujících látek v barvách na vlasy ve formě krému nebo kapaliny:
|
Látky |
Symbol |
|
Fenylendiaminy |
|
|
o-Fenylendiamin |
(OPD) |
|
m-Fenylendiamin |
(MPD) |
|
p-Fenylendiamin (příloha V) |
(PPD) |
|
Methylfenylendiaminy |
|
|
4-Methyl-1,2-fenylendiamin (toluen-3,4-diamin) |
(OTD) |
|
4-Methyl-1,3-fenylendiamin (toluen-2,4-diamin) |
(MTD) |
|
2-Methyl-1,4-fenylendiamin (toluen-2,5-diamin) |
(PTD) |
|
Diaminofenoly |
|
|
2,4-Diaminofenol |
(DAP) |
|
Hydrochinon |
|
|
Benzen-1,4-diol |
(H) |
|
a-Naftol |
(αN) |
|
Pyrogallol |
|
|
1,2,3-trihydroxybenzen |
(P) |
|
Resorcinol |
|
|
1,3-dihydroxybenzen |
(R) |
2. PODSTATA METODY
Oxidační barviva se extrahují z barev ve formě krému nebo kapaliny při pH 10 96 % ethanolem a dokazují se chromatografií na tenké vrstvě, a to jedno nebo dvourozměrnou.
K semikvantitativnímu stanovení těchto látek se chromatogram vzorků porovná pomocí čtyř vyvíjecích systémů s chromatogramy referenčních látek vyvíjenými současně a za co nejpodobnějších podmínek.
3. REAKČNÍ ČINIDLA
Všechna reakční činidla musí být čistoty p.a.
|
3.1 |
Ethanol, bezvodý |
|
3.2 |
Aceton |
|
3.3 |
Ethanol, 96 % (v/v) |
|
3.4 |
Roztok amoniaku, 25 %
|
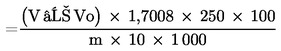
|
3.5 |
Kyselina L(+)askorbová |
|
3.6 |
Chloroform |
|
3.7 |
Cyklohexan |
|
3.8 |
Dusík, technický |
|
3.9 |
Toluen |
|
3.10 |
Benzen |
|
3.11 |
n-Butanol |
|
3.12 |
Butan-2-ol |
|
3.13 |
Kyselina fosforná, 50 % roztok (v/v) |
|
3.14 |
Diazotační činidlo. Buď: — 3-nitro-1-benzendiazoniumchlorbenzensulfonát (ve formě stabilní soli), jako např. v červeni 2 JN – Francolor, — 2-chlor-4-nitro-1-benzendiazoniumnaftalenbenzoát (ve formě stabilní soli), jako např. v činidle NNCD – referenční č. 74 150 FLUKA, nebo ekvivalentní. |
|
3.15 |
Dusičnan stříbrný |
|
3.16 |
p-Dimethylaminobenzaldehyd |
|
3.17 |
2,5-Dimethylfenol |
|
3.18 |
Chlorid železitý hexahydrát |
|
3.19 |
Kyselina chlorovodíková, 10 % roztok (m/v) |
|
3.20 |
Referenční látky Referenční látky jsou sloučeniny uvedené v oddílu 1 „Rozsah a oblast použití“. V případě aminosloučenin musí být referenční sloučenina buď ve formě hydrochloridu (mono nebo dihydrochloridu), nebo jako volná báze. |
|
3.21 |
Referenční roztoky 0,5 % (m/v) Připraví se 0,5 % roztoky (m/v) referenčních látek uvedených v bodu 3.20. Naváží se 50 ± 1 mg referenční látky do odměrné baňky na 10 ml. Přidá se 5 ml 96 % ethanolu (3.3) a 250 ml kyseliny askorbové (3.5). Roztok se alkalizuje přidáním roztoku amoniaku (3.4) na hodnotu pH 10 (zjišťuje se indikátorovým papírkem). Doplní se na 10 ml 96 % ethanolem (3.3) a promíchá se. Roztoky je možno uchovávat po dobu jednoho týdne v chladu a temnu. V některých případech může po přidání kyseliny askorbové a roztoku amoniaku vzniknout sraženina. Před dalším postupem je třeba ji nechat usadit. |
|
3.22 |
Vyvíjecí rozpouštědla
|
||||||||||||||||
|
3.23 |
Detekční činidla 3.23.1 Diazotační činidlo Připraví se 5 % (m/v) vodný roztok zvoleného činidla (3.14). Tento roztok se musí připravit bezprostředně před použitím. 3.23.2 Ehrlichovo činidlo 2 g p-dimethylaminobenzaldehydu (3.16) se rozpustí ve 100 ml 10 % vodného roztoku kyseliny chlorovodíkové (m/v) (3.19). 3.23.3 2,5-dimethylfenol – chlorid železitý hexahydrát Roztok 1: Rozpustí se 1 g dimethylfenolu (3.17) ve 100 ml 96 % ethanolu (3.3). Roztok 2: Rozpustí se 4 g hexahydrátu chloridu železitého (3.18) ve 100 ml 96 % ethanolu (3.3). Při vyvolání chromatogramu se těmito roztoky provádí postřik samostatně, nejprve roztokem 1, poté roztokem 2. 3.23.4 Amoniakální dusičnan stříbrný 25 % amoniak (3.4) se přidá k 5 % (m/v) vodnému roztoku dusičnanu stříbrného (3.15), až se sraženina rozpustí. Toto reakční činidlo se připravuje bezprostředně před použitím. Nelze jej uchovávat. |
4. PŘÍSTROJE A POMŮCKY
|
4.1 |
Běžné laboratorní vybavení pro chromatografii na tenké vrstvě.
|
|
4.2 |
Odstředivka, 4000 ot./min. |
|
4.3 |
Centrifugační zkumavky na 10 ml se šroubovými uzávěry potaženými PTFE nebo ekvivalentní. |
5. POSTUP
5.1 Příprava vzorků k analýze
První 2 až 3 cm krému vytlačeného z tuby se odstraní.
Do centrifugační zkumavky (4.3) předem propláchnuté dusíkem se převede: 300 mg kyseliny askorbové se 3 g krému nebo 3 g homogenizované kapaliny.
Přidává se po kapkách 25 % amoniak (3.4) až pH dosáhne hodnoty 10. Doplní se na 10 ml 96 % ethanolem (3.3).
Homogenizuje se pod atmosférou dusíku (3.8), zazátkuje se a centrifuguje se po 10 minut při 4000 ot./min.
Použije se supernatant.
5.2 Chromatografie
5.2.1 Nanášení na desku
Pod atmosférou dusíku (3.8) se na chromatografickou desku (4.1.3) nanese po 1 μl každé shora uvedené referenční látky v devíti bodech vzdálených od sebe 1,5 cm podél linie vzdálené asi 1,5 cm od okraje desky.
Referenční látky se nanášejí v tomto uspořádání:
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
|
R |
P |
H |
PPD |
DAP |
PTD |
OPD |
OTD |
MPD |
|
MTD |
a-N |
Dále se v bodech 10 a 11 nanesou vždy 2 μl zkoumaných roztoků získaných podle bodu 5.1.
Deska se uchovává pod atmosférou dusíku (3.8) až do okamžiku, kdy je zahájeno vyvíjení chromatogramu.
5.2.2 Vyvíjení
Deska se vloží do komory předem propláchnuté dusíkem (3.8) a nasycené jedním ze čtyř rozpouštědel (3.22) a nechá se vyvíjet při pokojové teplotě (20 až 25 °C) ve tmě, dokud čelo rozpouštědla nedosáhne vzdálenosti asi 15 cm od startovací linie.
Deska se vyjme a usuší pod atmosférou dusíku (3.8) při pokojové teplotě.
5.2.3 Vyvolání
Deska se postříká jedním ze čtyř roztoků uvedených v bodu 3.23.
5.2.4 Důkaz
Porovnají se hodnoty Rf a barvy skvrn vzorku a chromatografovaných referenčních látek.
V tabulce 1 jsou uvedeny příklady hodnot Rf a barev skvrn pro jednotlivé látky v závislosti na použitém rozpouštědle a indikátoru.
Nejistý důkaz je někdy možné potvrdit tak, že se přidá roztok odpovídající referenční látky k extraktu vzorku.
5.2.5 Semikvantitativní odhad
Intenzita skvrn jednotlivých látek dokázaných v bodu 5.2.4 se porovnává vizuálně s odpovídajícím rozsahem koncentrací referenčních látek.
Při nadměrně vysoké koncentraci jedné nebo více látek nalezených ve vzorku se extrakt vzorku zředí a měření se opakuje.
TABULKA 1
Hodnoty Rf a barvy získané bezprostředně po vyvolání
|
Referenční látka (3.20) |
Vyvíjecí rozpouštědla |
Detekční činidla |
||||||
|
Rf Hodnoty |
Výsledné barvy |
|||||||
|
(3.22.1) |
(3.22.2) |
(3.22.3) |
(3.22.4) |
Diazotační činidlo (3.23.1) |
Ehrlichovo činidlo (3.23.2) |
Dimethylfenol (3.23.3) |
AgNO3 (3.23.4) |
|
|
OPD |
0,62 |
0,60 |
0,30 |
0,57 |
světle hnědá |
— |
— |
světle hnědá |
|
MPD |
0,40 |
0,60 |
0,47 |
0,48 |
fialově hnědá (*) |
žlutá |
světle hnědá |
světle hnědá |
|
PPD |
0,20 |
0,50 |
0,30 |
0,48 |
hnědá |
jasně červená (*) |
fialová |
šedá |
|
OTD |
0,60 |
0,60 |
0,53 |
0,60 |
hnědá (*) |
světle oranžová |
světle hnědá |
šedavě hnědá |
|
MTD |
0,40 |
0,67 |
0,45 |
0,60 |
červenavě hnědá (*) |
žlutá |
hnědá |
černá |
|
PTD |
0,33 |
0,65 |
0,37 |
0,70 |
hnědá |
oranžová |
fialová (*) |
šedá |
|
DAP |
0,07 |
— |
0 |
0,05 |
hnědá (*) |
oranžová |
fialová |
hnědá |
|
H |
0,50 |
0,35 |
0,80 |
0,20 |
— |
oranžová |
fialová |
černá (*) |
|
αN |
0,90 |
0,80 |
0,90 |
0,75 |
oranžově hnědá |
— |
fialová (*) |
černá |
|
P |
0,37 |
— |
0,67 |
0,05 |
hnědá |
velmi světle fialová |
velmi světle hnědá |
hnědá (*) |
|
R |
0,50 |
0,37 |
0,80 |
0,17 |
oranžová (*) |
světle fialová |
velmi světle hnědá |
světle hnědá |
| Poznámka 1. OPD se ukazuje pouze slabě; musí se použít rozpouštědlo (3.22.3), aby se jasně odlišil od OTD. 2. (*) Označuje nejlépe vyvolané barvy. |
||||||||
6. ZKOUŠKA DVOUROZMĚRNOU CHROMATOGRAFIÍ NA TENKÉ VRSTVĚ
Tato metoda dvourozměrné chromatografie vyžaduje použití dalších standardů a reakčních činidel.
6.1 Další referenční roztoky a látky
|
6.1.1 |
β-naftol (β N) |
|
6.1.2 |
2-aminofenol (OAP) |
|
6.1.3 |
3-aminofenol (MAP) |
|
6.1.4 |
4-aminofenol (PAP) |
|
6.1.5 |
2-nitro-1,4-fenylendiamin (2-NPPD) |
|
6.1.6 |
4-nitro-1,2-fenylendiamin (4-NOPD) Připraví se 0,5 % (m/v) roztoky všech dalších referenčních látek způsobem popsaným v bodu 3.21. |
6.2 Další vyvíjecí rozpouštědlo
|
6.2.1 |
Ethylacetát – cyklohexan – 25 % roztok amoniaku (65: 30: 0,5 objemově) |
6.3 Další detekční systém
Do vyvíjecí komory pro chromatografii na tenké vrstvě se vloží skleněná nádobka, přidají se asi 2 g krystalického jodu a komora se uzavře odpovídajícím víkem.
6.4 Chromatografie
|
6.4.1 |
Podle obrázku 1 se na desce pro chromatografii na tenké vrstvě (4.1.3) vyznačí na straně s absorbentem dvě linky. |
|
6.4.2 |
V dusíkové atmosféře (4.1.1) se nanese 1 až 4 μl extraktu (5.1) do základního bodu 1 (obr. 1), který leží asi 2 cm od obou okrajů. Množství extraktu závisí na intenzitě skvrn na chromatogramech (5.2). |
|
6.4.3 |
Mezi body 2 a 3 (obr. 1) se nanesou oxidační barviva dokázaná nebo pokládaná za dokázaná podle bodu 5.2 (vzdálenost mezi body je 1,5 cm). Nanáší se 2 μl každého referenčního roztoku – kromě DAP, který je nutno nanést v objemu 6 μl. Nanášení se provádí v atmosféře dusíku (6.4.2). |
|
6.4.4 |
Postup podle bodu 6.4.3 se opakuje v základních bodech 4 a 5 (obr. 1) a deska se uchovává pod dusíkem až do zahájení vyvolávání chromatogramu (vzdálenost mezi body je 1,5 cm). |
|
6.4.5 |
Chromatografická komora se propláchne dusíkem (3.8) a umístí se do ní vhodné množství vyvíjecího rozpouštědla 3.22.2. Deska (6.4.4) se vloží do komory a vyvíjí se ve tmě v prvním vyvíjecím směru (obr. 1). Vyvíjí se, dokud čelo rozpouštědla nedosáhne linie vyznačené na desce (přibližně 13 cm). |
|
6.4.6 |
Deska se z komory vyjme a umístí do jiné chromatografické komory, předem propláchnuté dusíkem, aby se vyvíjecí rozpouštědlo odpařovalo alespoň 60 minut. |
|
6.4.7 |
Pomocí dělené zkumavky se do komory předem propláchnuté dusíkem (3.8) převede vhodné množství vyvíjecího rozpouštědla (6.2), do komory se vloží deska otočená o 90° (6.4.6) a chromatogram se vyvíjí ve druhém směru (opět ve tmě), dokud čelo rozpouštědla nedosáhne linie vyznačené na straně s adsorbentem. Deska se vyjme z komory a rozpouštědlo se nechá odpařit na vzduchu. |
|
6.4.8 |
Deska se na 10 minut umístí do chromatografické komory obsahující páry jodu (6.3) a dvourozměrný chromatogram se interpretuje s využitím hodnot Rf a barev současně chromatografovaných referenčních látek (hodnoty Rf a barvy skvrn jsou uvedeny v tabulce II). Poznámka Nejlepšího vybarvení skvrn se dosáhne tehdy, ponecháli se po vyvíjení chromatogram v atmosféře po dobu půl hodiny. |
|
6.4.9 |
Přítomnost oxidačních barviv nalezených podle bodu 6.4.8 je možno definitivně potvrdit opakováním postupu popsaného v bodech 6.4.1 až 6.4.8, kdy se v základním bodu 1 přes množství extraktu podle bodu 6.4.2 nanese 1 μl referenčních látek identifikovaných v bodu 6.4.8. Neobjeví-li se oproti chromatogramu získanému podle bodu 6.4.8 žádná jiná skvrna, je interpretace chromatogramu podle bodu 6.4.8 správná. |
TABULKA II
Barvy referenčních látek po chromatografii a vyvíjení parami jodu
|
Referenční látky |
Barvy po vyvíjení parami jodu |
|
R |
béžová |
|
P |
hnědá |
|
α-N |
fialová |
|
β-N |
světle hnědá |
|
H |
fialově hnědá |
|
MPD |
žlutavě hnědá |
|
PPD |
fialově hnědá |
|
MTD |
tmavě hnědá |
|
PTD |
žlutavě hnědá |
|
DAP |
tmavě hnědá |
|
OAP |
oranžová |
|
MAP |
žlutavě hnědá |
|
PAP |
fialově hnědá |
|
2NPPD |
hnědá |
|
4NOPD |
oranžová |
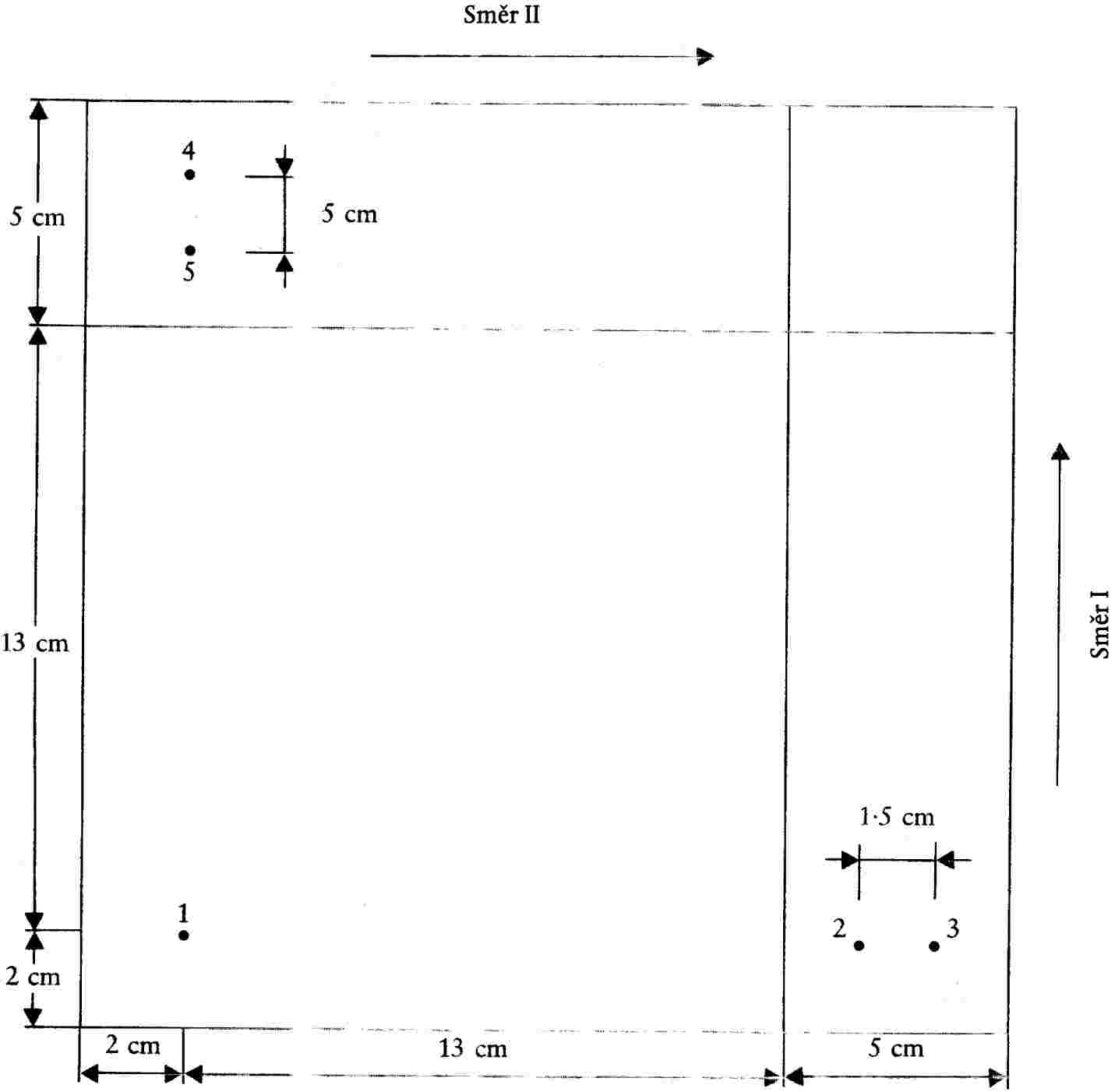
Obrázek 1
III. DŮKAZ A STANOVENÍ DUSITANŮ
A. DŮKAZ
1. ROZSAH A OBLAST POUŽITÍ
Tato metoda je vhodná k důkazu dusitanů v kosmetických prostředcích, zejména v krémech a pastách.
2. PODSTATA METODY
Přítomnost dusitanu indikuje vznik zbarvených derivátů s 2-aminobenzaldehydfenylhydrazonem (Nitrin®).
3. REAKČNÍ ČINIDLA
Všechna reakční činidla musí být čistoty p.a.
|
3.1 |
Zředěná kyselina sírová: 2 ml koncentrované kyseliny sírové (
|
|
3.2 |
Zředěná kyselina chlorovodíková: 1 ml koncentrované kyseliny chlorovodíkové (
|
|
3.3 |
Methanol |
|
3.4 |
Roztok 2-aminobenzaldehydfenylhydrazonu (reakční činidlo Nitrin®) v methanolu. Naváží se 2,0 g Nitrinu® a převede se kvantitativně do odměrné baňky na 100 ml. Po kapkách se přidají 4 ml zředěné kyseliny chlorovodíkové (3.2) a protřepe se. Doplní se po rysku methanolem a míchá, dokud se roztok úplně nevyčeří. Roztok se uchovává v hnědé skleněné lahvi (4.3). |
4. PŘÍSTROJE A POMŮCKY
|
4.1 |
Kádinky na 50 ml |
|
4.2 |
Odměrná baňka na 100 ml |
|
4.3 |
Hnědá skleněná láhev na 125 ml |
|
4.4 |
Skleněná deska 10 × 10 cm |
|
4.5 |
Špachtle z plastu |
|
4.6 |
Filtrační papír 10 × 10 cm |
5. POSTUP
|
5.1 |
Část analyzovaného vzorku se rovnoměrně rozetře na skleněnou desku (4.4) tak, aby byl povrch pokryt vrstvou nejvýše 1 cm silnou. |
|
5.2 |
List filtračního papíru (4.6) se nasákne destilovanou vodou. Položí se na vzorek a přitiskne špachtlí z plastu (4.5). |
|
5.3 |
Počká se asi jednu minutu a do středu filtračního papíru se nanesou: — 2 kapky zředěné kyseliny sírové (3.1), — poté dvě kapky roztoku Nitrinu® (3.4). |
|
5.4 |
Po 5 až 10 sekundách se filtrační papír odstraní a prohlédne se proti dennímu světlu. Červenofialové zbarvení indikuje přítomnost dusitanů. Je-li obsah dusitanů nízký, změní se červenofialové zbarvení během 5 až 15 sekund na žluté. Je-li přítomno velké množství dusitanů, dojde k této barevné změně až po jedné nebo dvou minutách. |
6. POZNÁMKA
Intenzita tmavočerveného zbarvení a doba, která uplyne před změnou na žlutou, naznačují obsah dusitanů ve vzorku.
B. STANOVENÍ
1. ROZSAH
Metoda popisuje stanovení dusitanů v kosmetických prostředcích.
2. DEFINICE
Obsah dusitanů ve vzorku stanovený touto metodou se vyjádří v hmotnostních procentech dusitanu sodného.
3. PODSTATA METODY
Po zředění vzorku vodou a vyčeření se nechá proběhnout reakce přítomných dusitanů se sulfanilamidem a N-1-naftylethylendiaminem a absorbance vzniklého zbarvení se měří při 538 nm.
4. REAKČNÍ ČINIDLA
Všechna reakční činidla musí být čistoty p.a.
|
4.1 |
Čeřidla: tato reakční činidla nelze používat déle než jeden týden po přípravě.
|
|
4.2 |
Roztok dusitanu sodného: 0,500 g dusitanu sodného se rozpustí v destilované vodě v odměrné baňce na 1000 ml a doplní se vodou po rysku. 10,0 ml tohoto zásobního standardního roztoku se zředí na 500 ml; 1,0 ml výsledného roztoku = 10 mikrogramů NaNO2. |
|
4.3 |
1 N roztok hydroxidu sodného |
|
4.4 |
0,2 % roztok sulfanilamidhydrochloridu: 2,0 g sulfanilamidu se za zahřívání rozpustí v 800 ml vody. Po ochlazení se za míchání přidá 100 ml koncentrované kyseliny chlorovodíkové. Zředí se vodou na 1000 ml. |
|
4.5 |
5 N kyselina chlorovodíková |
|
4.6 |
N-1-naftylové činidlo: Tento roztok se musí připravit v den použití. Rozpustí se 0,1 g dihydrochloridu N-1-naftylethylendiaminu ve vodě a zředí se vodou na 100 ml. |
5. PŘÍSTROJE A POMŮCKY
|
5.1 |
Analytické váhy |
|
5.2 |
Odměrné baňky na 100, 250, 500 a 1000 ml |
|
5.3 |
Nedělené nebo dělené pipety |
|
5.4 |
Odměrné válce na 100 ml |
|
5.5 |
Skládaný filtrační papír, bez dusitanů, průměr 15 cm |
|
5.6 |
Vodní lázeň |
|
5.7 |
Spektrofotometr a kyvety s optickou drahou 1 cm |
|
5.8 |
pHmetr |
|
5.9 |
Mikrobyreta na 10 ml |
|
5.10 |
Kádinky na 250 ml |
6. POSTUP
|
6.1 |
S přesností na 0,1 mg se naváží asi 0,5 g (m gramů) homogenizovaného vzorku, kvantitativně se převede horkou destilovanou vodou do kádinky na 250 ml (5.10) a doplní se horkou destilovanou vodou asi na 150 ml. Kádinka (5.10) se na půl hodiny vloží do vodní lázně (5.6) vyhřáté na 80 °C. Během této doby se obsah občas protřepe. |
|
6.2 |
Ochladí se na pokojovou teplotu a postupně se za míchání přidají 2 ml Carrezova činidla I (4.1.1) a 2 ml Carrezova činidla II (4.1.2). |
|
6.3 |
Přidává se 1 N roztok hydroxidu sodného (4.3), dokud se pH neupraví na 8,3. (Použije se pHmetr (5.8)). Obsah se kvantitativně převede do odměrné baňky na 250 ml (5.2) a doplní destilovanou vodou po rysku. |
|
6.4 |
Obsah se promíchá a zfiltruje přes skládaný papírový filtr (5.5). |
|
6.5 |
Do odměrné baňky na 100 ml (5.2) se odpipetuje (5.3) odpovídající alikvotní část (V mililitrů) čirého filtrátu, avšak ne více než 25 ml, a doplní se destilovanou vodou na objem 60 ml. |
|
6.6 |
Po promíchání se přidá 10,0 ml roztoku sulfanilamid–hydrochloridu (4.4) a poté 6,0 ml 5 N kyseliny chlorovodíkové (4.5). Promíchá se a ponechá stát pět minut. Přidají se 2,0 ml N-1-naftylového činidla (4.6), promíchá se a ponechá stát tři minuty. Zředí se vodou po rysku a promíchá se. |
|
6.7 |
Připraví se slepý pokus opakováním operací popsaných v bodech 6.5 a 6.6 bez přídavku N1-naftylového činidla (4.6). |
|
6.8 |
Změří se (5.7) absorbance roztoku připraveného podle bodu 6.6 při 538 nm proti roztoku slepého pokusu (6.7). |
|
6.9 |
Z kalibračního grafu (6.10) se odečte obsah dusitanu sodného v mikrogramech na 100 ml roztoku (m1 mikrogramů), odpovídající absorbance změřené podle bodu 6.8. |
|
6.10 |
S využitím roztoku dusitanu sodného o koncentraci 10 μg na ml (4.2) se sestrojí kalibrační graf pro koncentrace 0, 20, 40, 60, 80 a 100 μg dusitanu sodného ve 100 ml. |
7. VÝPOČET
Obsah dusitanu sodného ve vzorku vyjádřený v hmotnostních procentech se vypočte pomocí následujícího vzorce:
![]()
kde:
|
m |
= |
hmotnost vzorku odebraného pro analýzu v gramech (6.1), |
|
m1 |
= |
obsah dusitanu sodného v mikrogramech zjištěný podle bodu 6.9, |
|
V |
= |
počet mililitrů filtrátu použitého pro měření (6.5). |
8. OPAKOVATELNOST ( 5 )
Pro obsah asi 0,2 % (m/m) dusitanu sodného by neměla absolutní hodnota rozdílu dvou stanovení provedených současně se stejným vzorkem překročit 0,005 %.
IV. DŮKAZ A STANOVENÍ VOLNÉHO FORMALDEHYDU
1. ROZSAH A OBLAST POUŽITÍ
Tato metoda popisuje důkaz a dva způsoby stanovení v závislosti na tom, zda jsou či nejsou přítomny donory formaldehydu. Je použitelná pro všechny kosmetické prostředky.
1.1 Důkaz
1.2 Obecné kolorimetrické stanovení pomocí pentan-2,4-dionu
Tato metoda se používá v případě, je-li použit formaldehyd samotný nebo s konzervačními přísadami, které nejsou donory formaldehydu.
V opačném případě, a pokud výsledek přesáhne nejvyšší povolenou koncentraci, musí se použít následující metoda pro potvrzení.
1.3 Stanovení za přítomnosti donorů formaldehydu
Při použití výše uvedené metody (1.2) se při derivatizaci donory formaldehydu štěpí, což vede k příliš vysokým výsledkům (vázaný formaldehyd a formaldehyd v polymerní formě).
Je nezbytné oddělit volný formaldehyd kapalinovou chromatografií.
2. DEFINICE
Obsah volného formaldehydu ve vzorku stanovený touto metodou se vyjadřuje v hmotnostních procentech.
3. DŮKAZ
3.1 Podstata metody
Volný a vázaný formaldehyd poskytuje v prostředí kyseliny sírové za přítomnosti Schiffova činidla růžové nebo lila zbarvení.
3.2 Reakční činidla
Všechna reakční činidla musí být čistoty p.a. a použije se demineralizovaná voda.
|
3.2.1 |
Fuchsin; |
|
3.2.2 |
Siřičitan sodný heptahydrát; |
|
3.2.3 |
Koncentrovaná kyselina chlorovodíková (d = 1,19); |
|
3.2.4 |
Kyselina sírová, asi 1M; |
|
3.2.5 |
Schiffovo činidlo: Do kádinky se naváží 100 mg fuchsinu (3.2.1) a rozpustí se v 75 ml vody o teplotě 80 °C. Po ochlazení se přidá 2,5 g siřičitanu sodného heptahydrátu (3.2.2) a 1,5 ml kyseliny chlorovodíkové (3.2.3). Objem se doplní do 100 ml. Je použitelné do dvou týdnů. |
3.3 Postup
|
3.3.1 |
Do kádinky na 10 ml se naváží 2 g vzorku. |
|
3.3.2 |
Přidají se dvě kapky kyseliny sírové (3.2.4) a 2 ml Schiffova činidla (3.2.5). Toto činidlo musí být při použití naprosto bezbarvé. Obsah se protřepe a nechá stát 5 minut. |
|
3.3.3 |
Objeví-li se do pěti minut růžové nebo lila zbarvení, je přítomen formaldehyd v množství vyšším než 0,01 % a musí být stanoven volný a vázaný formaldehyd metodou podle bodu 4 a v případě potřeby metodou podle bodu 5. |
4. OBECNÉ KOLORIMETRICKÉ STANOVENÍ POMOCÍ PENTAN2,4DIONU
4.1 Podstata metody
Formaldehyd reaguje s pentan2,4-dionem v přítomnosti octanu amonného za tvorby 3,5-diacetyl-1,4-dihydrolutidinu. Ten se extrahuje butan1olem a absorbance extraktu se měří při 410 nm.
4.2 Reakční činidla
Všechna reakční činidla musí být čistoty p.a. a použije se demineralizovaná voda.
|
4.2.1 |
Bezvodý octan amonný; |
|
4.2.2 |
Koncentrovaná kyselina octová, d4 20= 1,05; |
|
4.2.3 |
Pentan2,4dion, čerstvě předestilovaný za sníženého tlaku 25 mm Hg při 25 °C – nesmí vykazovat žádnou absorpci při 410 nm; |
|
4.2.4 |
Butan1ol; |
|
4.2.5 |
Kyselina chlorovodíková, 1M; |
|
4.2.6 |
Kyselina chlorovodíková, asi 0,1M; |
|
4.2.7 |
Hydroxid sodný, 1M; |
|
4.2.8 |
Roztok škrobu, čerstvě připravený (1 g/50 ml vody) podle Evropského lékopisu, 2. vydání 1980, část I-VII-1-1; |
|
4.2.9 |
Formaldehyd, 37 % až 40 % (m/V); |
|
4.2.10 |
Odměrný roztok jodu, 0,05 M; |
|
4.2.11 |
Odměrný roztok thiosíranu sodného, 0,1 M; |
|
4.2.12 |
Činidlo obsahující pentan-2,4-dion: V odměrné baňce na 1000 ml se rozpustí: — 150 g octanu amonného (4.2.1), — 2 ml pentan-2,4-dionu (4.2.3), — 3 ml kyseliny octové (4.2.2). Doplní se na 1000 ml vodou (pH roztoku asi 6,4). Toto činidlo musí být připraveno čerstvě. |
|
4.2.13 |
Činidlo (4.2.12) bez přídavku pentan-2,4-dionu; |
|
4.2.14 |
Standardní roztok formaldehydu: zásobní roztok 5 g roztoku formaldehydu (4.2.9) se převede do odměrné baňky na 1000 ml a doplní se vodou na 1000 ml. Koncentrace tohoto roztoku se stanoví takto: Odebere se 10,00 ml; přidá se 25,00 ml odměrného roztoku jodu (4.2.10) a 10,00 ml roztoku hydroxidu sodného (4.2.7). Nechá se stát pět minut. Okyselí se 11,00 ml HCl (4.2.5) a přebytek jodu se titruje odměrným roztokem thiosíranu sodného (4.2.11), jako indikátor se použije roztok škrobu (4.2.8). Spotřeba 1 ml 0,05M odměrného roztoku jodu (4.2.10) odpovídá 1,5 mg formaldehydu; |
|
4.2.15 |
Standardní roztok formaldehydu: zředěný roztok Zásobní roztok formaldehydu se zředí vodou nejprve v poměru 1/20 a poté v poměru 1/100. 1 ml výsledného roztoku obsahuje asi 1 μg formaldehydu. Přesný obsah se vypočte. |
4.3 Přístroje a pomůcky
|
4.3.1 |
Běžné laboratorní vybavení; |
|
4.3.2 |
Filtr pro separaci fází, Whatman 1 PS (nebo ekvivalentní); |
|
4.3.3 |
Odstředivka; |
|
4.3.4 |
Vodní lázeň nastavená na teplotu 60 °C; |
|
4.3.5 |
Spektrofotometr; |
|
4.3.6 |
Skleněné kyvety s délkou optické dráhy 1 cm. |
4.4 Postup
4.4.1 Roztok vzorku:
Do odměrné baňky na 100 ml se s přesností na 0,001 g naváží množství zkušebního vzorku (v gramech) odpovídající očekávanému obsahu formaldehydu asi 150 μg.
Doplní se po rysku vodou a zamíchá se (roztok S).
(Je nutno ověřit, zda je hodnota pH asi 6; v opačném případě se přidá roztok kyseliny chlorovodíkové (4.2.6).)
Do Erlenmeyerovy baňky na 50 ml se odměří
— 10,00 ml roztoku S,
— 5,00 ml činidla obsahujícího pentan-2,4-dion (4.2.12),
— demineralizovaná voda na konečný objem 30 ml.
4.4.2 Referenční roztok
Možný rušivý vliv základního zbarvení zkušebního vzorku se odstraní použitím tohoto referenčního roztoku:
Do Erlenmeyerovy baňky na 50 ml se odměří
— 10 ml roztoku S,
— 5 ml činidla (4.2.13),
— demineralizovaná voda na konečný objem 30 ml.
4.4.3 Roztok pro slepý pokus
Do Erlenmeyerovy baňky na 50 ml se přidá:
— 5 ml činidla obsahujícího pentan-2,4-dion (4.2.13),
— demineralizovaná voda na konečný objem 30 ml.
4.4.4 Stanovení
|
4.4.4.1 |
Směsi připravené podle bodů 4.4.1, 4.4.2 a 4.4.3 se protřepou. Erlenmeyerovy baňky se přesně na 10 minut vloží do vodní lázně vyhřáté na 60 °C. Nechají se vychladnout dvě minuty v lázni s ledovou vodou. |
|
4.4.4.2 |
Obsah se převede do dělicí nálevky na 50 ml obsahující 10 ml butan-1-olu (4.2.4). Každá baňka se vypláchne 3 až 5 ml vody. Směs se přesně 30 sekund důkladně třepe. Fáze se nechají oddělit. |
|
4.4.4.3 |
Obsah se přefiltruje do měřicích kyvet (4.3.2) přes filtr pro separaci fází. Fáze lze rovněž odstředit (při 3000 ot./min po dobu 5 minut). |
|
4.4.4.4 |
Absorbance (A1) extraktu roztoku vzorku podle bodu 4.4.1 proti extraktu referenčního roztoku 4.4.2 se změří při 410 nm V. |
|
4.4.4.5 |
Obdobně se změří absorbance (A2) extraktu roztoku slepého pokusu podle bodu 4.4.3 proti butan-1-olu. Poznámka: Všechny tyto operace musí být provedeny do 25 minut od okamžiku, kdy byly Erlenmeyerovy baňky vloženy do lázně vyhřáté na 60°C. |
4.4.5 Kalibrační křivka
|
4.4.5.1 |
Do Erlenmeyerovy baňky na 50 ml se přidá: — 5 ml zředěného standardního roztoku podle bodu 4.2.15, — 5 ml činidla obsahujícího pentan-2,4-dion (4.2.12), — demineralizovaná voda na celkový objem 30 ml. |
|
4.4.5.2 |
Postupuje se tak, jak je popsáno v bodu 4.4.4, a měří se absorbance proti butan-1-olu (4.2.4). |
|
4.4.5.3 |
Postup se opakuje s 10, 15, 20 a 25 ml zředěného standardního roztoku (4.2.15). |
|
4.4.5.4 |
Nulová hodnota (odpovídající zabarvení reakčních činidel) se získá postupem podle bodu 4.4.4.5. |
|
4.4.5.5 |
Po odečtení nulové hodnoty od každé hodnoty absorbance získané podle bodu 4.4.5.1 a 4.4.5.3 se sestrojí kalibrační křivka. Beerův zákon platí až do 30 μg formaldehydu. |
4.5 Výpočty
|
4.5.1 |
Od hodnoty A1 se odečte hodnota A2 a z kalibrační křivky (4.4.5.5) se odečte množství C formaldehydu v roztoku vzorku (4.4.1) v μg. |
|
4.5.2 |
Obsah formaldehydu ve vzorku (% m/m) se vypočte pomocí vzorce
kde
|
4.6 Opakovatelnost ( 6 )
Při obsahu formaldehydu 0,2 % by neměl rozdíl dvou stanovení provedených současně se stejným vzorkem překročit 0,005 % pro kolorimetrickou metodu s pentan-2,4-dionem.
Vede-li kolorimetrické stanovení volného formaldehydu k výsledkům vyšším, než je nejvyšší koncentrace podle směrnice 76/768/EHS, tj.
a) 0,05 % až 0,2 % ve výrobku, u něhož není obsah formaldehydu uveden na etiketě;
b) více než 0,2 % ve výrobku s uvedením obsahu formaldehydu, nebo bez něho,
musí být použito postupu podle bodu 5.
5. STANOVENÍ ZA PŘÍTOMNOSTI DONORŮ FORMALDEHYDU
5.1 Podstata metody
Oddělený formaldehyd se převede na žlutý derivát lutidinu reakcí s pentan2,4dionem v reaktoru za kolonou a absorbance vzniklého derivátu se měří při 420 nm.
5.2 Reakční činidla
Všechna použitá reakční činidla musí být čistoty p.a. a použije se demineralizovaná voda.
|
5.2.1 |
Voda čistoty pro HPLC nebo voda ekvivalentní kvality; |
|
5.2.2 |
Bezvodý octan amonný; |
|
5.2.3 |
Koncentrovaná kyselina octová; |
|
5.2.4 |
Pentan-2,4-dion (uchovává se při 4 °C); |
|
5.2.5 |
Bezvodý hydrogenfosforečnan disodný; |
|
5.2.6 |
Kyselina trihydrogenfosforečná, 85 % (d = 1,7); |
|
5.2.7 |
Methanol čistoty pro HPLC; |
|
5.2.8 |
Dichlormethan; |
|
5.2.9 |
Formaldehyd, 37 % až 40 % (m/V); |
|
5.2.10 |
Hydroxid sodný, 1M; |
|
5.2.11 |
Kyselina chlorovodíková, 1M; |
|
5.2.12 |
Kyselina chlorovodíková, 0,002M; |
|
5.2.13 |
Roztok škrobu čerstvě připravený podle Evropského lékopisu (viz 4.2.8); |
|
5.2.14 |
Odměrný roztok jodu, 0,05M; |
|
5.2.15 |
Odměrný roztok thiosíranu sodného, 0,1M; |
|
5.2.16 |
Mobilní fáze: Vodný roztok hydrogenfosforečnanu disodného (5.2.5), 0,006 M, upravený na pH 2,1 kyselinou trihydrogenfosforečnou (5.2.6); |
|
5.2.17 |
Činidlo do reaktoru za kolonou: V odměrné baňce na 1000 ml se rozpustí — 62,5 g octanu amonného (5.2.2), — 7,5 ml kyseliny octové (5.2.3), — 5 ml pentan2,4dionu (5.2.4). Objem se doplní po rysku vodou (5.2.1). Roztok se uchovává ve tmě. Skladovatelnost: nejvýše tři dny při 25 °C. Barva roztoku by se neměla měnit. |
|
5.2.18 |
Standardní roztok formaldehydu: zásobní roztok 10 g roztoku formaldehydu (5.2.9) se převede do odměrné baňky na 1000 ml a doplní se vodou na 1000 ml. Koncentrace tohoto roztoku se stanoví takto: Odebere se 5 ml; přidá se 25 ml odměrného roztoku jodu (5.2.14) a 10 ml roztoku hydroxidu sodného (5.2.10). Nechá se stát pět minut. Okyselí se 11 ml HCl (5.2.11) a přebytek jodu se titruje odměrným roztokem thiosíranu sodného (5.2.15), jako indikátor se použije roztok škrobu (5.2.13). Spotřeba 1 ml odměrného roztoku jodu (5.2.14) odpovídá 1,5 mg formaldehydu. |
|
5.2.19 |
Standardní roztok formaldehydu: zředěný roztok Zásobní roztok formaldehydu se zředí v poměru 1/100 mobilní fází (5.2.16). 1 ml výsledného roztoku obsahuje asi 37 μg formaldehydu. Přesný obsah se vypočte. |
5.3 Přístroje a pomůcky
|
5.3.1 |
Běžné laboratorní vybavení; |
|
5.3.2 |
Bezpulsní čerpadlo pro HPLC; |
|
5.3.3 |
Nízkotlaké bezpulsní čerpadlo (případně druhé čerpadlo pro HPLC); |
|
5.3.4 |
Nastřikovací ventil se smyčkou o objemu 10 μl; |
|
5.3.5 |
Reaktor za kolonou obsahující následující prvky: + jednolitrová tříhrdlá baňka, + jednolitrové topné hnízdo, + dvě kolony Vigreux o minimálně 10 patrech, vzduchem chlazené, + trubice z korozivzdorné oceli (sloužící jako výměník tepla) 1,6 mm, vnitřní průměr 0,23 mm, délka 400 mm, + teflonová trubice 1,6 mm, vnitřní průměr 0,30 mm, délka 5 m (francouzské pletivo – viz doplněk 1), + jeden díl ve tvaru T bez mrtvého objemu (Valco nebo ekvivalentní), + tři spojky bez mrtvého objemu, nebo: jeden modul za kolonou Applied Biosystems PCRS 520 nebo ekvivalentní s reaktorem o objemu 1 ml. |
|
5.3.6 |
Membránový filtr, velikost pórů 0,45 μm; |
|
5.3.7 |
Patrona SEPPAKR C18 nebo ekvivalentní; (C18 se píše s 18 jako spodním indexem) |
|
5.3.8 |
Hotové kolony: — Bischoff hypersil RP 18 (typ NC číslo C 25.46 1805), — (5 μm, délka 250 mm, vnitřní průměr 4,6 mm), — nebo DuPont, Zorbax ODS — (5 μm, délka 250 mm, vnitřní průměr 4,6 mm), — nebo Phase SEP, spherisorb ODS 2 — (5 μm, délka 250 mm, vnitřní průměr 4,6 mm). |
|
5.3.9 |
Předkolona Bischoff K1 hypersil RP 18 (typ K1 G 6301 1805) (5 μm, délka 10 mm), nebo ekvivalentní. |
|
5.3.10 |
Kolona a předkolona se spojí systémem Ecotube (typ A 15020508 Bischoff) nebo ekvivalentním. |
|
5.3.11 |
Aparatura (5.3.5) se sestaví podle blokového schématu v doplňku 2. Spoje za nastřikovacím ventilem musí být co nejkratší. V tomto případě má trubka z korozivzdorné oceli mezi výstupem z reaktoru a vstupem do detektoru ochladit směs před detekcí; teplota detektoru není známa, ale je konstantní. |
|
5.3.12 |
Detektor pro viditelnou a UV oblast; |
|
5.3.13 |
Zapisovač; |
|
5.3.14 |
Odstředivka; |
|
5.3.15 |
Ultrazvuková lázeň; |
|
5.3.16 |
Vibrační míchadlo (Vortex nebo ekvivalentní). |
5.4 Postup
5.4.1 Kalibrační křivka
Sestrojí se vynesením výšek píků jako funkce koncentrace zředěných formaldehydových kalibračních roztoků.
Kalibrační roztoky se připraví zředěním standardního roztoku formaldehydu (5.2.19) mobilní fází (5.2.16):
— 1 ml roztoku (5.2.19) se zředí na 20 ml (asi 185 μg/100 ml)
— 2 ml roztoku (5.2.19) se zředí na 20 ml (asi 370 μg/100 ml)
— 5 ml roztoku (5.2.19) se zředí na 25 ml (asi 740 μg/100 ml)
— 5 ml roztoku (5.2.19) se zředí na 20 ml (asi 925 μg/100 ml).
Kalibrační roztoky se ponechají asi jednu hodinu stát při pokojové teplotě a musí být čerstvě připraveny.
Kalibrační křivka je lineární pro koncentrace od 1,00 do 15,00 μg/ml.
5.4.2 Příprava vzorků
5.4.2.1 Emulze (krémy, make-up, oční stíny)
Do odměrné baňky na 100 ml s uzávěrem se s přesností na 0,001 g naváží zkušební vzorek (m gramů) odpovídající očekávanému obsahu 100 μg formaldehydu. Přidá se přesně 20 ml dichlormethanu (5.2.8) a 20 ml kyseliny chlorovodíkové (5.2.12). Promíchá se pomocí vibračního míchadla (5.3.16) a v ultrazvukové lázni (5.3.15). Fáze se oddělí odstředěním (dvě minuty při 3000 g). Mezitím se promyje patrona (5.3.7) 2 ml methanolu (5.2.7) a kondicionuje se 5 ml vody (5.2.1).
Kondicionovanou patronou se nechají protéci 4 ml vodné fáze extraktu, první dva mililitry se odstraní a následující podíl se zachytí.
5.4.2.2 Lotiony, šampony
Do odměrné baňky na 100 ml s uzávěrem se s přesností na 0,001 g naváží zkušební vzorek (m gramů) odpovídající očekávanému obsahu 500 μg formaldehydu.
Doplní se po rysku mobilní fází (5.2.16).
Roztok se zfiltruje přes filtr (5.3.6) a nastříkne se nebo nechá protéci patronou (5.3.7) předem kondicionovanou podle bodu 5.4.2.1. Všechny roztoky musí být nastříknuty hned po přípravě.
5.4.3 Chromatografické podmínky
— Průtok mobilní fáze: 1 ml/min,
— Průtok činidla: 0,5 ml/min,
— Celkový průtok na výstupu z detektoru: 1,5 ml/min,
— Objem nástřiku: 10 μl,
— Eluční teplota: u obtížných separací se kolona ponoří do ledové lázně a vyčká se do ustálení teploty (15 až 20 minut),
— Teplota reakce za kolonou: 100 °C,
— Detekce: při 420 nm.
Poznámka: Celý chromatografický systém a systém za kolonou je nutno po použití propláchnout vodou (5.2.1). Nepoužije-li se systém do dvou dnů, musí po tomto proplachu následovat ještě propláchnutí methanolem (5.2.7). Před novým kondicionováním se systémem nechá protéci voda, aby se zabránilo rekrystalizaci.
5.5 Výpočet
Emulze: (5.4.2.1):
Obsah formaldehydu v % (m/m):
![]()
Lotiony a šampony:
V tomto případě platí vzorec:
![]()
kde
|
m |
= |
hmotnost analyzovaného vzorku v gramech (5.4.2.1), |
|
C |
= |
koncentrace formaldehydu v μg/100 ml odečtená z kalibrační křivky (5.4.1). |
5.6 Opakovatelnost ( 7 )
Při obsahu formaldehydu 0,05 % by neměl rozdíl výsledků dvou stanovení provedených současně se stejným vzorkem překročit 0,001 %.
Při obsahu formaldehydu 0,2 % by neměl rozdíl výsledků dvou stanovení provedených současně se stejným vzorkem překročit 0,005 %.
V. STANOVENÍ RESORCINOLU V ŠAMPONECH A VLASOVÝCH LOTIONECH
1. ROZSAH A OBLAST POUŽITÍ
V této metodě je specifikováno stanovení resorcinolu v šamponech a vlasových lotionech plynovou chromatografií. Metoda je vhodná pro koncentrace od 0,1 do 2,0 hmotnostních procent vzorku.
2. DEFINICE
Obsah resorcinolu ve vzorku stanovený touto metodou se vyjádří v hmotnostních procentech.
3. PODSTATA METODY
Resorcinol a 3,5dihydroxytoluen (5methylresorcinol) přidávaný jako vnitřní standard se ze vzorku oddělí chromatografií na tenké vrstvě. Obě sloučeniny se izolují seškrabáním jejich skvrn z horní vrstvy desky a extrakcí methanolem. Nakonec se vyextrahované sloučeniny vysuší, podrobí silylaci a stanoví se plynovou chromatografií.
4. REAKČNÍ ČINIDLA
Všechna reakční činidla musí být čistoty p.a.
|
4.1 |
25 % kyselina chlorovodíková (m/m) |
|
4.2 |
Methanol |
|
4.3 |
Ethanol 96 % (v/v) |
|
4.4 |
Silikagelové desky pro chromatografii na tenké vrstvě připravené k použití (plastové nebo hliníkové) s fluorescenčním indikátorem. Desky se deaktivují následujícím postupem: běžná silikagelová deska se nastříká vodou, dokud se povrch neleskne. Nastříkané desky se nechají schnout při pokojové teplotě jednu až tři hodiny. Poznámka Bez deaktivace může docházet ke ztrátám resorcinolu v důsledku ireverzibilní adsorpce na silikagelu. |
|
4.5 |
Vyvíjecí rozpouštědlo: aceton – chloroform – kyselina octová (20: 75: 5 objemově). |
|
4.6 |
Standardní roztok resorcinolu: rozpustí se 400 mg resorcinolu ve 100 ml 96 % ethanolu (4.3) (1 ml odpovídá 4000 μg resorcinolu). |
|
4.7 |
Roztok vnitřního standardu: rozpustí se 400 mg 3,5dihydroxytoluenu (DHT) ve 100 ml 96 % ethanolu (4.3) (1 ml odpovídá 4000 μg DHT). |
|
4.8 |
Směs standardů: v odměrné baňce na 100 ml se smíchá 10 ml roztoku 4.6 a 10 ml roztoku 4.7, doplní se po rysku 96 % ethanolem (4.3) a promíchá (1 ml roztoku odpovídá 400 μg resorcinolu a 400 μg DHT). |
|
4.9 |
Silylační činidla:
|
5. PŘÍSTROJE A POMŮCKY
|
5.1 |
Běžné vybavení pro chromatografii na tenké vrstvě a plynovou chromatografii |
|
5.2 |
Skleněné laboratorní nádobí |
6. POSTUP
6.1 Příprava vzorku
|
6.1.1 |
Do kádinky na 150 ml se přesně naváží zkušební vzorek (m gramů) výrobku, který obsahuje asi 20 až 50 mg resorcinolu. |
|
6.1.2 |
Okyselí se kyselinou chlorovodíkovou (4.1) do kyselé reakce (je potřeba přibližně 2 až 4 ml), přidá se 10 ml (40 mg DHT) roztoku vnitřního standardu (4.7) a promíchá se. Ethanolem (4.3) se převede do odměrné baňky na 100 ml, doplní se ethanolem po rysku a promíchá. |
|
6.1.3 |
250 μl roztoku (6.1.2) se nanese na deaktivovanou desku se silikagelem (4.4) jako nepřerušovaná linie o délce přibližně 8 cm. Je třeba dbát na to, aby linie byla co nejužší. |
|
6.1.4 |
Na stejnou desku se stejným způsobem (6.1.3) nanese 250 μl směsi standardů (4.8). |
|
6.1.5 |
Do dvou bodů startovací linie se nanese po 5 μl roztoků 4.6 a 4.7 k usnadnění lokalizace skvrn po vyvolání. |
|
6.1.6 |
Deska se vyvíjí v nenasycené komoře naplněné vyvíjecím rozpouštědlem 4.5, dokud čelo rozpouštědla nedosáhne vzdálenosti 12 cm od startovací linie; obvykle to trvá asi 45 minut. Deska se usuší na vzduchu a zóna resorcinolu/DHT se lokalizuje pod krátkovlnným UV světlem (254 nm). Obě tyto sloučeniny mají přibližně stejnou hodnotu Rf. Poloha pásů se vyznačí tužkou ve vzdálenosti 2 mm od vnějšího tmavého rozhraní pásů. Tyto zóny se z desky odstraní a adsorbenty každého pásu se shromáždí v lahvi na 10 ml. |
|
6.1.7 |
Adsorbent obsahující vzorek a absorbent obsahující směs standardů se extrahují následujícím způsobem: přidají se 2 ml methanolu (4.2) a extrahuje se jednu hodinu za neustálého míchání. Směs se zfiltruje a extrakce se opakuje po dobu dalších 15 minut se 2 ml methanolu. |
|
6.1.8 |
Extrakty se spojí a rozpouštědlo se nechá přes noc odpařit ve vakuovém exsikátoru naplněném vhodným sušicím prostředkem. Nezahřívá se. |
|
6.1.9 |
Odparky (6.1.8) se silylují způsobem popsaným v bodu 6.1.9.1 nebo 6.1.9.2.
|
6.2 Plynová chromatografie
6.2.1 Chromatografické podmínky
Kolona musí poskytovat rozlišení R rovné nebo lepší než 1,5:
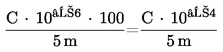
kde:
|
r1 a r2 |
= |
retenční časy dvou píků v minutách, |
|
w1 a w2 |
= |
šířky píků v polovině výšky píků v mm, |
|
d’ |
= |
rychlost posuvu papíru v mm za minutu. |
Následující kolona a podmínky pro plynovou chromatografii se ukázaly jako vhodné:
|
Materiál |
kolony: |
korozivzdorná ocel |
|
Délka |
200 cm |
|
|
Vnitřní průměr: |
˜ 3 mm |
|
|
Náplň: |
10 % OV17 na Chromosorbu WAW, 100 až 120 mesh |
|
|
Plamenový ionizační detektor |
||
|
Teplotní režim |
||
|
Kolona: |
185 °C (izotermální) |
|
|
Detektor: |
250 °C |
|
|
Nástřik: |
250 °C |
|
|
Nosný plyn: |
dusík |
|
|
Průtok: |
45 ml/min |
|
Průtoky vodíku a vzduchu se nastaví podle pokynů výrobce.
|
6.2.2 |
Do plynového chromatografu se nastříkne 1 až 3 μl roztoků připravených podle bodu 6.1.9. Pro každý roztok (6.1.9) se provede pět nástřiků, změří se plochy píků, vypočte se průměr a poměr ploch píků: S = plocha píku resorcinolu/plocha píku DHT. |
7. VÝPOČET
Koncentrace resorcinolu ve vzorku vyjádřená v hmotnostních procentech (% m/m) je dána vztahem:
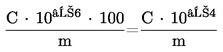
kde:
|
M |
= |
hmotnost zkušebního vzorku v gramech (6.1.1), |
|
Svzorku |
= |
průměrná plocha píku podle bodu 6.2.2 pro roztok vzorku, |
|
Sstandardní směsi |
= |
průměrná plocha píku podle bodu 6.2.2 pro směs standardů. |
8. OPAKOVATELNOST ( 8 )
Pro obsah resorcinolu asi 0,5 % by neměla absolutní hodnota rozdílu dvou stanovení provedených současně se stejným vzorkem překročit 0,025 %.
VI. STANOVENÍ METHANOLU VZHLEDEM K ETHANOLU NEBO PROPAN-2-OLU
1. ROZSAH A OBLAST POUŽITÍ
V metodě je popsána analýza methanolu ve všech typech kosmetických prostředků (včetně aerosolů) plynovou chromatografií.
Metodou je možno stanovit relativní hladiny od 0 do 10 %.
2. DEFINICE
Obsah methanolu stanovený podle této metody se vyjadřuje v hmotnostních procentech methanolu vzhledem k ethanolu nebo propan-2-olu.
3. PODSTATA METODY
Stanovení se provádí plynovou chromatografií.
4. REAKČNÍ ČINIDLA
Používají se reakční činidla čistoty p.a.
|
4.1 |
Methanol |
|
4.2 |
Absolutní ethanol |
|
4.3 |
Propan-2-ol |
|
4.4 |
Chloroform zbavený alkoholů promytím vodou |
5. PŘÍSTROJE A POMŮCKY
|
5.1 |
Plynový chromatograf: s tepelně vodivostním detektorem pro aerosolové vzorky, s plamenovým ionizačním detektorem pro jiné než aerosolové vzorky. |
|
5.2 |
Odměrné baňky na 100 ml |
|
5.3 |
Pipety na 2 ml, 20 ml a 0 až 1 ml |
|
5.4 |
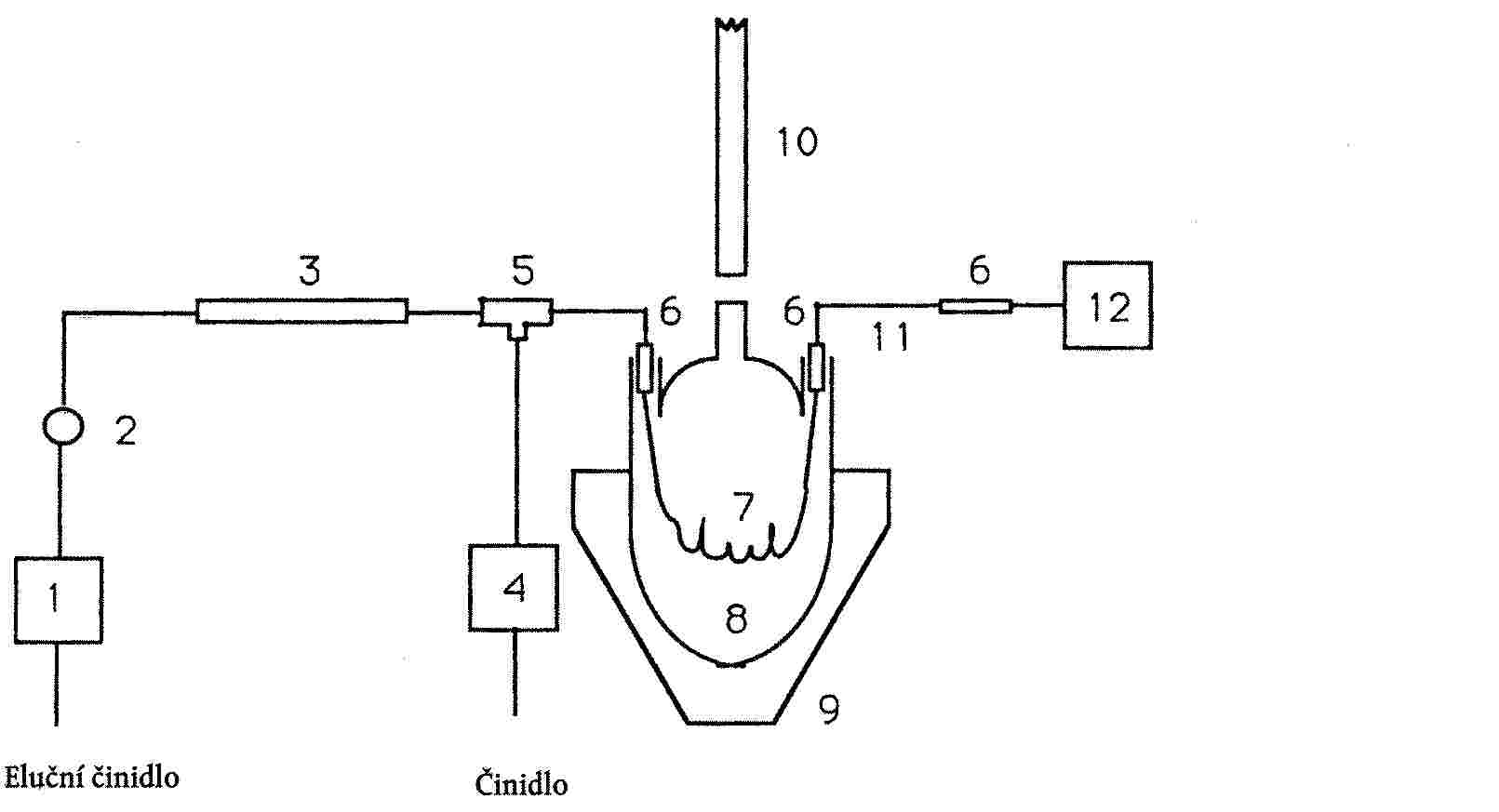
Mikrostříkačky na 0 až 100 μl a 0 až 5 μl a (pouze pro aerosolové vzorky) speciální plynotěsná injekční stříkačka s posuvným ventilem (viz obrázek 5 ( 9 ), postup odběru vzorků). |
6. POSTUP
6.1 Příprava vzorku
|
6.1.1 |
Vzorky z výrobků v obalech na aerosoly se odebírají postupem podle kapitoly II přílohy ke směrnici Komise 80/1335/EHS ze dne 22. prosince 1980 ( 10 ) a poté se analyzují plynovou chromatografií za podmínek popsaných v bodu 6.2.1. |
|
6.1.2 |
Vzorky výrobků, které nejsou v obalech na aerosoly, odebírané postupem podle výše uvedené kapitoly II se zředí vodou na koncentraci 1 až 2 % ethanolu či propan-2-olu a poté se analyzují plynovou chromatografií za podmínek popsaných v bodu 6.2.2. |
6.2 Plynová chromatografie
|
6.2.1 |
Pro aerosolové vzorky se použije tepelně vodivostní detektor.
|
||||||||||||||||||||||||||||||||||||||||||||||
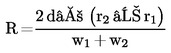
|
6.2.2 |
Jiné než aerosolové vzorky:
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
7. KALIBRAČNÍ GRAF
|
7.1 |
Pro plynovou chromatografii podle bodu 6.2.1 (náplň kolony Hallcomid M18) se použijí následující standardní směsi. Tyto směsi se připraví odměřením pomocí pipet, ale přesné množství se stanoví zvážením pipety nebo baňky po každém přidání.
Do chromatografu se nastřikují 2 až 3 μl za podmínek uvedených v bodu 6.2.1. Pro každou směs se vypočtou poměry ploch píků (methanol/ethanol) nebo (methanol/propan2ol). Sestrojí se kalibrační graf vynesením:
|
|
7.2 |
Pro plynovou chromatografii podle bodu 6.2.2 (náplň kolony Porapak QS nebo Chromosorb 105) se použijí následující standardní směsi. Tyto směsi se připraví odměřením pomocí pipet, ale přesné množství se stanoví zvážením pipety nebo baňky po každém přidání.
Do chromatografu se nastřikují 2 až 3 μl za podmínek uvedených v bodu 6.2.2. Pro každou kalibrační směs se vypočtou poměry ploch píků (methanol/ethanol) nebo (methanol/propan2ol) a sestrojí se kalibrační graf vynesením:
|
|
7.3 |
Kalibrační graf musí být přímkový. |
8. OPAKOVATELNOST ( 11 )
Při relativním obsahu 5 % methanolu vzhledem k ethanolu nebo propan2olu by neměl rozdíl dvou stanovení provedených současně se stejným vzorkem překročit 0,25 %.
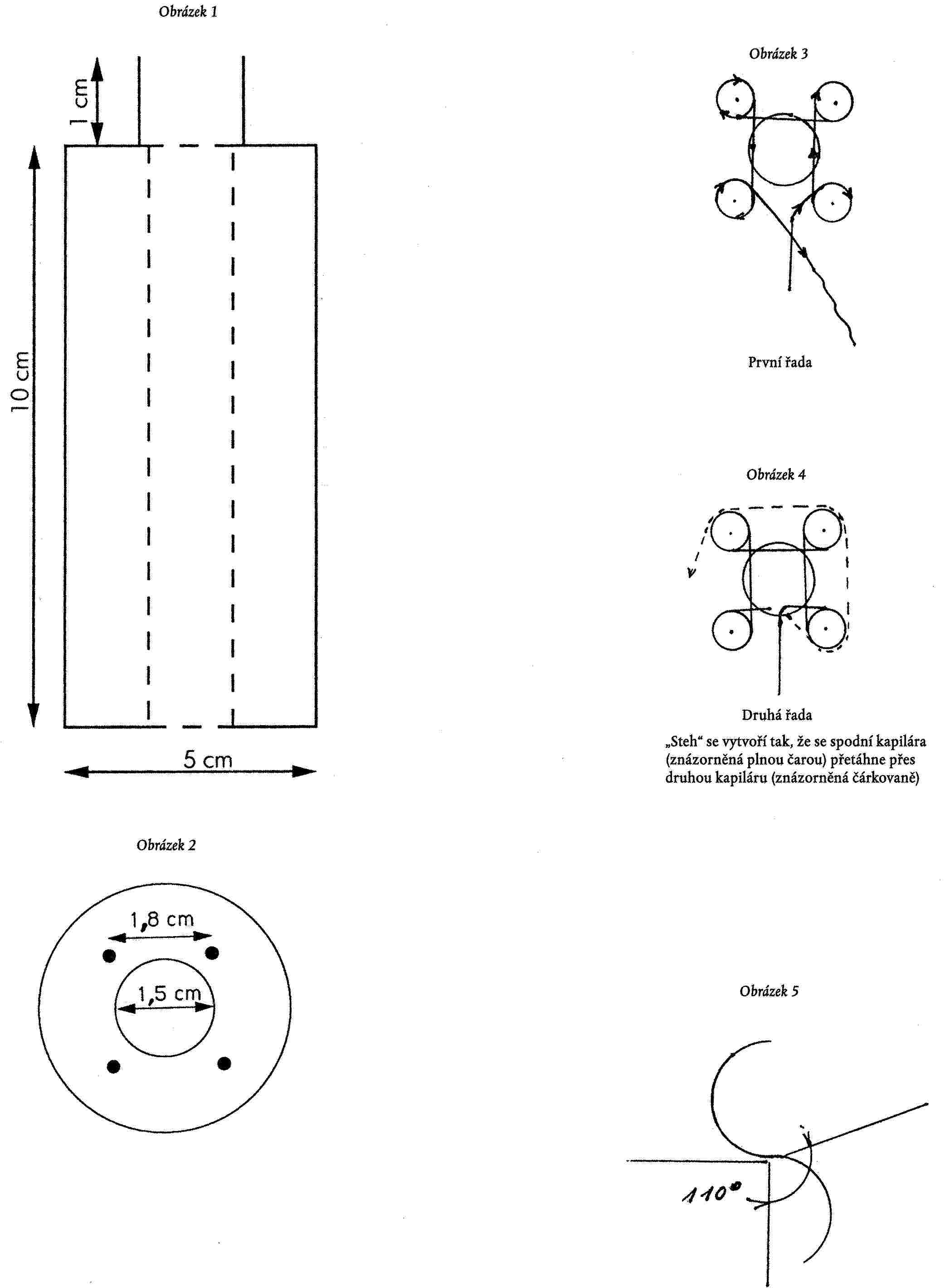
Dodatek 1
POKYNY PRO ZHOTOVENÍ „FRANCOUZSKÉHO PLETIVA“
NEZBYTNÉ PŘÍSLUŠENSTVÍ
— Dřevěná navíjecí cívka:
— vnější průměr 5 cm s otvorem o průměru 1,5 cm uprostřed. Cívka se opatří čtyřmi ocelovými hřebíky (podle obrázků 1 a 2). Vzdálenost mezi dvěma hřebíky musí být 1,8 cm a musí být ve vzdálenosti 0,5 cm od středového otvoru;
— jedna pevná jehla (háček) k protahování teflonové kapiláry;
— 5 m teflonové kapiláry o průměru 1,6 mm, vnitřního průměru 0,3 mm.
POSTUP:
Před zhotovením „francouzského pletiva“ musí být teflonová kapilára protažena středovým otvorem od horního konce cívky k dolnímu konci (asi 10 cm kapiláry se nechá vyčnívat z dolního konce cívky, což umožní protáhnout řetízek během pletení); poté se kapilára ovine kolem čtyř hřebíků, jak je znázorněno na obrázku 3.
Horní a dolní část pletiva bude chráněna kovovými kroužky a stahovacími šrouby; při utahování je třeba dbát na to, aby se teflon nepoškodil. Kapilára se podruhé ovine kolem každého hřebíku a následujícím způsobem se vytvoří „stehy“:
— spodní část kapiláry se háčkem přetáhne přes horní část kapiláry (obrázek 4). Toto se postupně opakuje u každého hřebíku (1, 2, 3 a 4 proti směru hodinových ručiček), dokud se nezhotoví 5 m nebo požadovaná délka pletiva.
Je nutno ponechat asi 10 cm kapiláry k uzavření řetízku. Kapilára se provlékne každou ze čtyř smyček a lehce se utáhne tak, aby se řetízek uzavřel.
Poznámka: Francouzské pletivo pro reaktory za kolonou je komerčně dostupné (Supelco).
Schématické vyobrazení cívky
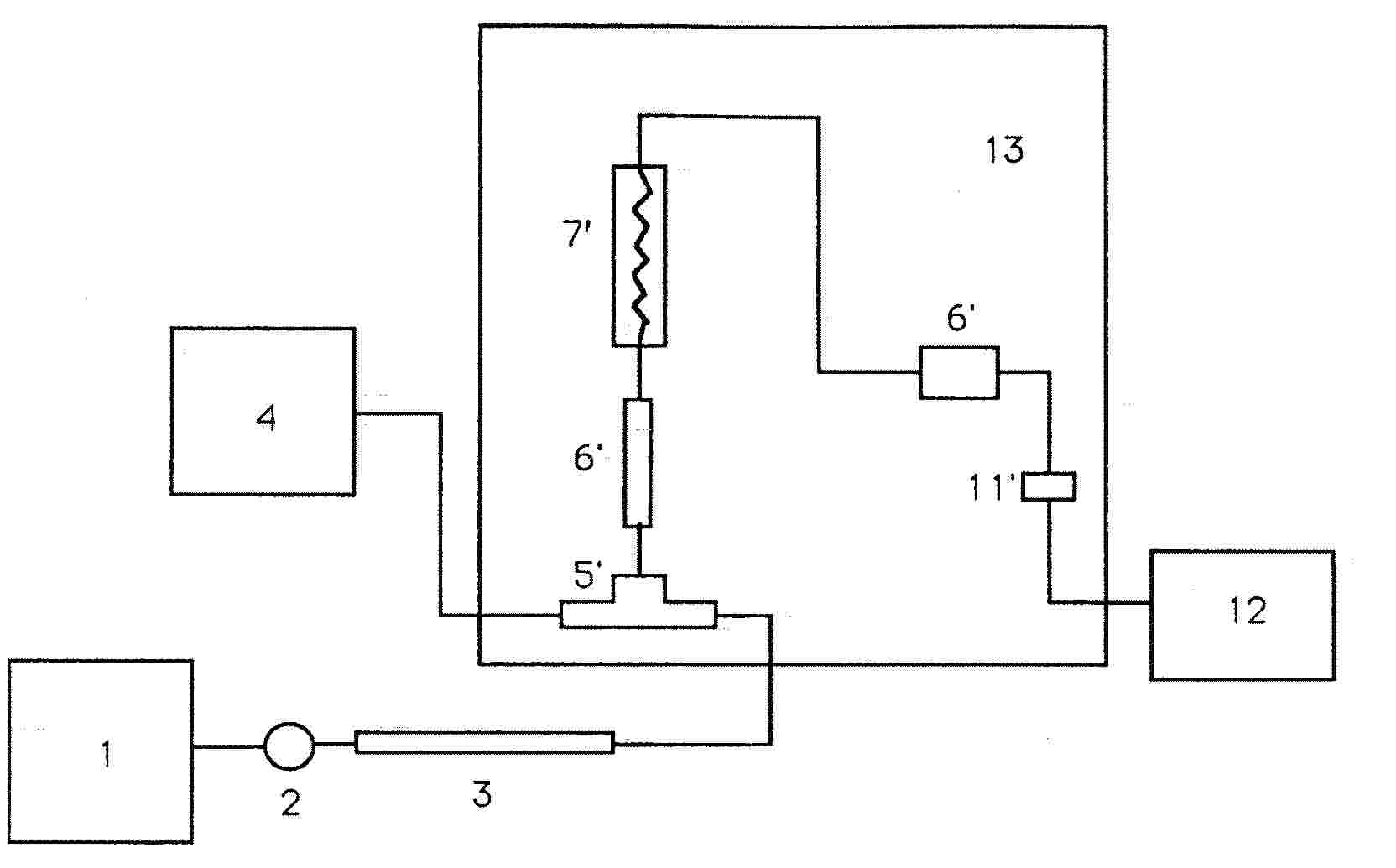
Dodatek 2
|
1 |
= |
Čerpadlo pro HPLC |
|
2 |
= |
nastřikovací ventil |
|
3 |
= |
kolona s předkolonou |
|
4 |
= |
čerpadlo pro činidla |
|
5 |
= |
díl ve tvaru T bez mrtvého objemu |
|
5‘ |
= |
díl ve tvaru T (Vortex) |
|
6 6‘ |
= |
spojka bez mrtvého objemu |
|
7 |
= |
„francouzské pletivo“ |
|
7‘ |
= |
reaktor |
|
8 |
= |
tříhrdlá baňka s vroucí vodou |
|
9 |
= |
topné hnízdo |
|
10 |
= |
chladič |
|
11 |
= |
výměník tepla z trubky z korozivzdorné oceli |
|
11‘ |
= |
výměník tepla |
|
12 |
= |
detektor pro viditelnou a UV oblast |
|
13 |
= |
modul za kolonou PCRS 520 |
( 1 ) Úř. věst. L 262, 27.9.1976, s. 169.
( 2 ) Úř. věst. L 192, 31.7.1979, s. 35.
( 3 ) Úř. věst. L 383, 31.12.1980, s. 27.
( 4 ) Viz norma ISO 5725.
( 5 ) Viz norma ISO 5725.
( 6 ) ISO 5725.
( 7 ) ISO 5725.
( 8 ) Viz norma ISO 5725.
( 9 ) Úř. věst. L 383, 31.12.1980, s. 27.
( 11 ) Viz norma ISO 5725.